
Revealing the promoting effect of Zn on Ni-based CO2 hydrogenation catalysts†
Liang
Shen
,
Wenhao
Zhang
,
Yifei
Feng
,
Jing
Xu
and
Minghui
Zhu
 *
*
State Key Laboratory of Chemical Engineering, School of Chemical Engineering, East China University of Science and Technology, 130 Meilong Road, Shanghai 200237, China. E-mail: minghuizhu@ecust.edu.cn
First published on 15th March 2023
Abstract
Hydrogenation of CO2 to produce high-value chemicals is a response to increasing environmental and energy concerns. The target products of CO2 hydrogenation, methane and CO, are both important energy sources and raw materials for the production of higher hydrocarbons. Therefore, it is very important to understand the mechanism of selectivity control of catalysts. In this work, we aim to elucidate the selectivity regulation of Zn on Ni catalysts, which has been extensively studied in the literature without reaching a consensus. We have prepared a series of catalysts with different Zn content and systematically investigated the relationship between their structural evolution and selectivity. It is found that the introduction of Zn preferentially forms an alloy with Ni and at higher loadings is present as ZnOx, which participates in the strong metal–support interaction (SMSI). The conversion of the active sites into a Ni–Zn alloy hinders further hydrogenation of the adsorbed CO intermediates and makes the catalyst CO-selective. The presence of the Ni–ZnOx interface changes the CO2 activation mechanism from an association pathway to a redox pathway.
1. Introduction
The annual increase in greenhouse gas emissions has led to a number of serious environmental problems, such as ocean acidification and global warming.1–3 Carbon dioxide (CO2), produced by burning fossil fuels, is the main source of greenhouse gasses. Hydrogenation of CO2 into high-value chemicals such as carbon monoxide (CO), methane (CH4), methanol (CH3OH), and higher hydrocarbons or alcohols is a potential pathway for reducing CO2 emissions.4–6 At atmospheric pressure, CO2 can be hydrogenated to CO via the reverse water gas shift reaction (RWGS, CO2 + H2 → CO + H2O) or CH4via the Sabatier reaction (CO2 + 4H2 → CH4 + 2H2O). CO is a versatile feedstock and can be used to produce higher hydrocarbons by Fischer–Tropsch synthesis.7–9 CH4 is an important energy source that can be conveniently stored and transported through well-developed pipelines.10,11 An important goal of CO2 hydrogenation research is the rational design of catalysts to improve the yield of target products.A variety of noble metals (Ru, Rh, Pd) and base metals (Ni, Cu, Fe, Co) have been investigated for CO2 hydrogenation.12–18 Among them, Ni has attracted much attention due to its relatively low cost and promoting activity in catalyzing CO2 hydrogenation under mild conditions.19 The selectivity of Ni-based catalysts shows a dependence on nanoparticle size, with larger nanoparticles producing mainly CH4 and smaller nanoparticles producing mainly CO.16,20,21 Promoters can regulate the selectivity of catalysts.22,23 Zn doping has been shown to alter the CO2 hydrogenation selectivity of Ni-based catalysts, making them CO-selective.24–27 Wang et al. found a strong metal–support interaction (SMSI) induced by hydrogen over Ni/ZnO catalysts. Electron transfer between Ni and Zn leads to an electron-rich state of Ni. The amount and strength of CO adsorption on electron-rich Ni are greatly weakened, which hinders the hydrogenation of *CO to CH4.25 Lin et al. believed that the introduction of Zn leads to the formation of a Ni–Zn alloy. Through a combination of density functional theory calculation (DFT) and characterization, they proposed that the Niδ−–Znδ+ pair decreases both the CO adsorption capacity and H2 dissociation ability.26 However, the in situ X-ray absorption structure spectra (XANES) performed by Wang et al. show a Ni–O–Zn structure over NiZn/ZrO2, which makes the positive valence state of Ni (Niδ+) stable during the reaction, while *CO is difficult to hydrogenate to CH4 on Niδ+.27 Regardless of the unclear mechanism regulating the selectivity, there is still controversy about the nature of the active sites.
In this study, we systematically investigate the influence of different loadings of Zn on Ni-based catalysts. The CO2 and CO hydrogenation activities of catalysts with different Zn contents are evaluated. High resolution transmission electron microscopy (HRTEM), X-ray diffraction (XRD) and quasi in situ X-ray photoelectron spectroscopy (XPS) are used to obtain the structural properties of the catalyst. In situ diffuse reflectance infrared spectroscopy (DRIFTS) and kinetic studies are performed to determine the relationship between structure and performance. Our results show that Ni–Zn alloy and SMSI can occur simultaneously. The reason for the high CO selectivity of the Zn-containing catalyst is the difficulty of further hydrogenation of *CO at the Ni–Zn alloy sites.
2. Experimental details
2.1 Catalyst preparation
Supported NiZn/Al2O3 catalysts with NiO loading of 20 wt% were prepared by a deposition–precipitation method. The loading of ZnO varied from 0 to 20 wt%. In general, Al2O3 support (AEROXIDE Alu130, Evonik, 99.6%) was suspended in 150 ml deionized water, after which a desired amount of nickel nitrate hexahydrate (Ni(NO3)2·6H2O, General Reagent, AR) and zinc nitrate hexahydrate (Zn(NO3)2·6H2O, Sinopharm Inc., AR) were added. Ammonium carbonate solution ((NH4)2CO3, Aladdin, AR, 0.5 M), as a precipitant, was added dropwise with stirring. The solution was stirred continuously for 1 h, then separated by centrifugation and washed thoroughly with deionized water. The solid precursor obtained was oven dried at 60 °C for 12 h and then calcined at 450 °C for 3 h with a heating ramp of 2 °C min−1. The catalysts thus prepared were designated as 20Ni, 20Ni0.5Zn, 20Ni1Zn, 20Ni2Zn, and 20Ni20Zn. For comparison, the Zn/Al2O3 catalyst containing 20 wt% ZnO but no Ni was prepared by the same method.2.2 Catalyst characterizations
XRD patterns were acquired using a Bruker D8-Advance X-ray powder diffractometer with Cu Kα radiation (λ = 0.154 nm) at 40 kV and 40 mA in the 2θ range of 10–80° with a scan speed of 10° min−1. To characterize the activated catalyst, we first reduced the catalyst at 500 °C for 2 h under an atmosphere of 60% H2/N2 (Air Liquide).Quasi in situ XPS spectra were recorded using a Thermo Fisher ESCALAB250Xi spectrometer with a monochromatic Al Kα X-ray source (1486.68 eV). A pretreatment chamber was used to activate the catalyst. The catalyst was first reduced at 500 °C for 2 h under an atmosphere of 60% H2/N2 (Air Liquide). The sample was then vacuumed and transferred directly to the analytical chamber. Charge calibration was performed by setting the C 1s peak to 284.8 eV.
HRTEM studies were performed in a Talos F200X at an accelerating voltage of 200 kV. First, the catalyst was suspended in ethanol. Then 2–3 drops of this slurry were applied to the grid of the copper microscope and dried with an infrared lamp.
In situ DRIFTS was performed using an FT-IR spectrometer (PerkinElmer Frontier) equipped with an in situ diffuse reflection cell with a small cavity volume (TC-DRS-K01, Jiaxing Puxiang Tech. Ltd.). Prior to testing, the sample was first heated to 500 °C in Ar atmosphere (Air Liquide) and then reduced at 500 °C for 2 h under an atmosphere of 60% H2/N2. The sample was purged with Ar (Air Liquide) for 15 min and then cooled to room temperature. Ar background spectra were collected every 50 °C from 450 to 100 °C. The reaction gas (3% CO2/12% H2/85% inert gas for CO2 hydrogenation and 4% CO/12% H2/84% inert gas for CO hydrogenation, Air Liquide) was then purged and spectra were collected 5 min after reaching each set temperature to ensure steady state. For the CO-DRIFTS experiment, the sample was first heated to 500 °C in Ar (Air Liquide) and then reduced at 500 °C for 2 h under an atmosphere of 60% H2/N2. The sample was purged with Ar for 15 min and then cooled to 200 °C. Then 1% CO/Ar (Air Liquide) was injected, and spectra were recorded.
The H2 pulse experiment was performed in a fixed bed plug flow reactor equipped with an online thermal conductivity detector (TCD). Ar (Air Liquide) was used as the carrier gas in this experiment. Prior to the test, the sample was reduced with 60% H2/N2 at 500 °C for 2 h. The samples were then purged with Ar for 15 min and cooled to 25 °C, then 10% H2/Ar (Air Liquide) pulses were performed at 25 °C until adsorption saturation. Dispersion is defined as the ratio between the number of surface metallic nickel sites and the theoretical total nickel content.
2.3 Activity test
Steady-state CO2 hydrogenation activities were tested in a fixed-bed plug flow reactor, and the product was analyzed by online gas chromatography (GC2060, Ruimin) equipped with a TDX-01 column, an FID, and a TCD. Typically, 50 mg of fresh catalyst was added to a U-shaped quartz tube and fixed with quartz wool. Before testing, the catalyst was first reduced at 500 °C for 2 h under 25 sccm 60% H2/N2. Catalytic performance was evaluated at 250 °C and 275 °C, either under 25 sccm 15% CO2/60% H2/25% N2 for CO2 hydrogenation or under 20% CO/60% H2/20% N2 for CO hydrogenation.For the kinetic measurements, the catalyst was diluted with silicon carbide (SiC, Aladdin, 99.9%) to achieve a CO2 conversion of less than 10% and to ensure that the test was performed within the kinetic range. Internal and external diffusion was eliminated by adjusting the particle size of the catalysts and the space velocity of the reaction gas. The test was performed at 275 °C. To determine the H2 order, the H2 partial pressure was varied from 0.3 to 0.6 bar with a fixed CO2 partial pressure of 0.15 bar. To determine CO2 order, CO2 partial pressure was varied from 0.075 to 0.15 bar, with a fixed H2 partial pressure of 0.6 bar. The activation energy was measured in the temperature range from 250 to 280 °C.
3. Results and discussion
3.1 The catalytic performance of the NiZn/Al2O3 catalysts
The catalytic performance of CO2 hydrogenation over NiZn/Al2O3 catalysts was evaluated at a weight hourly space velocity (WHSV) of 30 L h−1 g−1 at 250 and 275 °C (Fig. 1a and S1†). Ni/Al2O3 shows high activity in CO2 methanation. The CH4 formation rate reaches 17.10 μmol g−1 s−1 at 275 °C, with an extremely high CH4 selectivity of 97.72%. Zn inhibits the formation of CH4 and to some extent increases the production rate of CO. 1% ZnO reduces the CH4 formation rate from 17.10 to 9.02 μmol g−1 s−1 at 275 °C, accompanied by an increase in the CO formation rate from 0.40 to 0.49 μmol g−1 s−1. Almost no CH4 is observed on 20Ni20Zn, but the CO formation rate reaches 1.81 μmol g−1 s−1 at 275 °C with a CO selectivity of 99.1%. It is worthwhile to mention that the comparison of 20Ni20Zn with other RWGS catalysts (Table S1†) shows the possibility of designing efficient Ni-based RWGS catalysts through the introduction of Zn.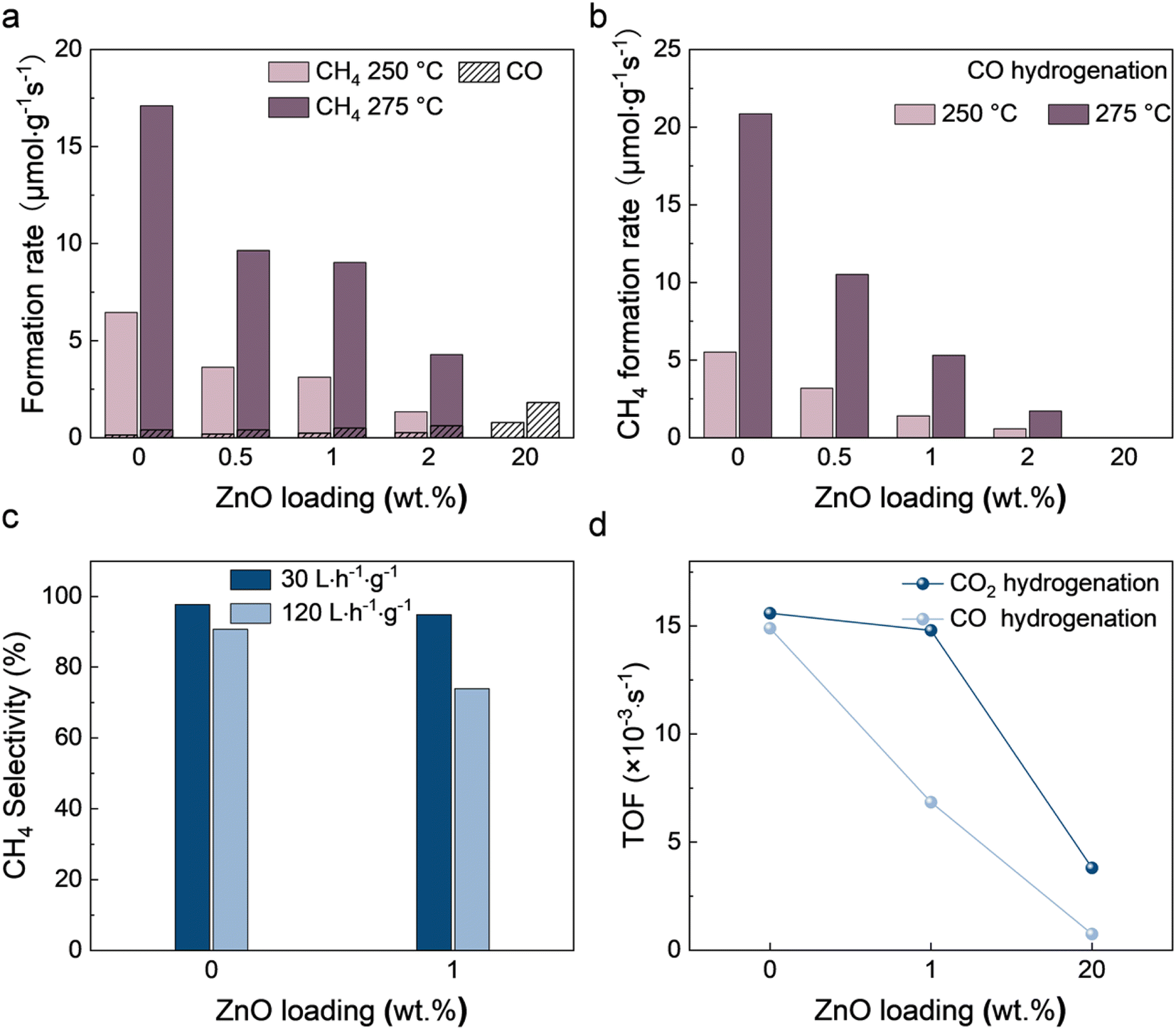 | ||
| Fig. 1 Catalytic performance of (a) CO2 and (b) CO hydrogenation, (c) space velocity dependency evaluation results, and (d) TOF of NiZn/Al2O3 catalysts. | ||
We also studied the CO hydrogenation activity over NiZn/Al2O3 catalysts (Fig. 1b and S1†). The addition of Zn has a similar inhibitory effect on CO hydrogenation. The CH4 formation rate over Ni/Al2O3 is 20.86 μmol g−1 s−1 at 275 °C, and the selectivity of CH4 is 96.62%. 1% ZnO greatly reduced the CH4 formation rate to 5.31 μmol g−1 s−1. Almost no methane is observed on the 20Ni20Zn catalyst at 250 and 275 °C. It is worth noting that the reaction pathway of CO2 methanation on many Ni/Al2O3 catalysts is the activation of CO2 to *CO and further methanation of *CO.28–30 The space velocity dependency results in our study show the same pathway on our catalysts (Fig. 1c). Therefore, it is suggested that the suppression of CH4 production on catalysts by Zn occurs via inhibition of further hydrogenation of CO.
We selected three typical catalysts, 20Ni, 20Ni1Zn, and 20Ni20Zn, and measured the dispersion based on the H2 pulse experiment (Table S2†). The dispersion percentages of 20Ni, 20Ni1Zn, and 20Ni20Zn were found to be 14.3%, 8.2%, and 5.9%, respectively. The turnover frequency (TOF) of the catalysts was determined based on the dispersion measured by the H2 pulse experiment (see Fig. 1d and Table S2†). The introduction of Zn to the Ni/Al2O3 catalysts decreased the TOF for both CO hydrogenation and CO2 hydrogenation, with the reduction being more significant for the former. For instance, compared to 20Ni, the TOF for CO2 hydrogenation of 20Ni20Zn decreased by 75.6% from 15.6 × 10−3 to 3.8 × 10−3 s−1, while the TOF for CO hydrogenation decreased by 94.9% from 14.9 × 10−3 to 0.8 × 10−3 s−1. These results suggest that the addition of Zn alters the properties of the active sites and that the effect cannot be explained purely by geometry.
3.2 Structure of the NiZn/Al2O3 catalysts
The XRD patterns of the as-prepared catalysts are shown in Fig. 2a. In all patterns, diffraction peaks at 2θ = 37.6, 45.9, and 67.0° can be seen, which are assigned to the Al2O3 support. For 20Ni and 20Ni1Zn, no diffraction peak associated with the Ni species can be observed. For 20Ni20Zn, a diffraction peak is seen at 2θ = 43.2° corresponding to the (2 0 0) plane of NiO. The largest NiO particles formed on 20Ni20Zn could be due to the low dispersion of NiO caused by the lowest Al2O3 support content (60 wt%) and are consistent with the results of the H2 pulse experiment (Table S2†). The catalysts were reduced under the same preactivation conditions as in the activity test, and the reduced catalysts were designated as 20Ni-H, 20Ni1Zn-H, and 20Ni20Zn-H. The absence of Ni/NiO diffraction peaks on 20Ni-H and 20Ni1Zn-H shows the remarkable dispersion of Ni species over these two catalysts (Fig. 2b). However, on 20Ni20Zn-H, a distinct diffraction peak appears at 2θ = 43.7°, which is lower than 44.5° of the Ni (1 1 1) facet. This probably means that Zn penetrates into the lattice of metallic Ni under the induction of a reducing atmosphere and forms a Ni–Zn alloy.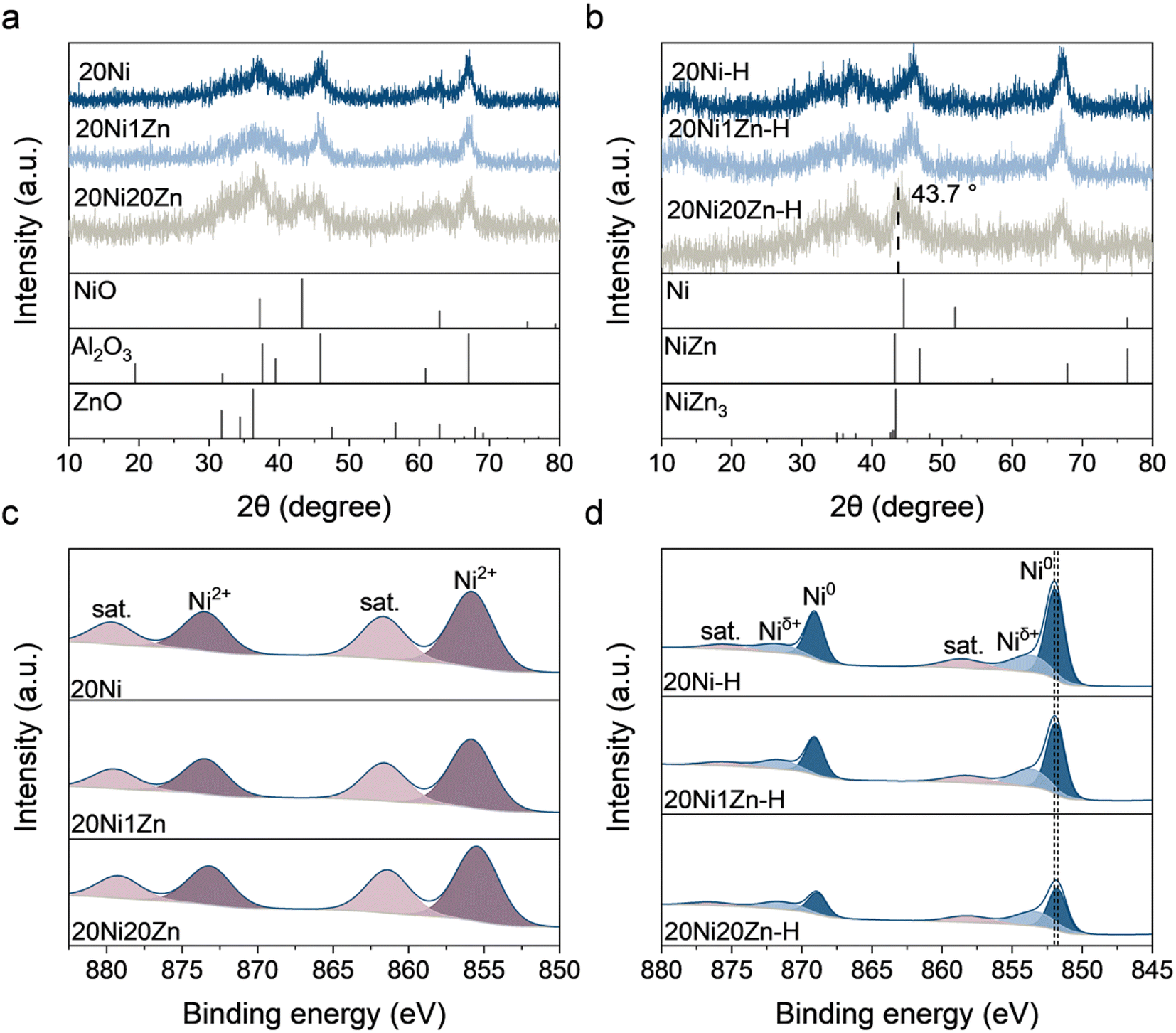 | ||
| Fig. 2 XRD patterns of (a) as-prepared and (b) reduced catalysts and XPS spectra of Ni 2p for (c) as-prepared and (d) reduced catalysts. | ||
Quasi in situ XPS further characterized the chemical state of the surface of the catalysts. Ni2+ species associated with binding energies at 855.8 and 873.5 eV, with satellite peaks at 861.7 and 879.6 eV can be observed on all three fresh catalysts (Fig. 2c).24,31–33 While the Ni 2p spectra of the reduced catalysts show the peak of metallic Ni0 at about 852.0 eV and another shoulder peak of Niδ+ at about 853.8 eV.34–36 The presence of the Niδ+ peak indicates a strong Ni–Al interaction, which is consistent with the literature.37,38 With the increase of ZnO content, the proportion of Ni0 in the total Ni species in the reduced catalysts gradually decreases (Table S3†), which shows the lower reduction degree of Ni species. It is worth mentioning that Zn also changes the binding energy of Ni species. The binding energy of the peak attributed to Ni0 decreases from 852.0 eV at 20Ni-H to 851.8 eV at 20Ni20Zn-H, indicating that Zn modification leads to an electron-rich state of Ni0 by electron interaction.
For all Zn LMM Auger electron spectra, the Zn2+ peak can be observed with a kinetic energy of 986–987 eV and the Znδ+ peak at 990–991 eV (Fig. S2†).39–41 The fitting results (Table S3†) show that the reduction process leads to an increase in the Znδ+ content. For 20Ni1Zn, the catalyst reduction process slightly increases the Znδ+ content from 35.13 to 35.81%, but for 20Ni20Zn, the Znδ+ content increases significantly from 23.78 to 31.54% during the reduction process. The reduction of Zn2+ increases the Znδ+ content, but the anomaly is that the increase of Znδ+ is more significant at 20Ni20Zn than at 20Ni1Zn. Two factors may contribute to the increase in Znδ+ content: (1) the formation of Ni–Zn alloy and (2) the formation of ZnOx. The Ni–Zn alloy with Niδ−–Znδ+ structure can be formed due to the higher electronegativity of Ni (1.9) than of Zn (1.6), which corresponds to the shift of the nickel species in the Ni 2p spectra to lower binding energies (Fig. 2d) and has been demonstrated by previous DFT calculations.26 The formation of ZnOx is closely related to the strong metal–support interaction (SMSI) that leads to the encapsulation of active metals by the Zn species in the form of anoxic oxides and has been widely reported in the CuZnAl catalytic system.42,43
To better understand the role of Zn in the catalysts, the structures of 20Ni1Zn-H and 20Ni20Zn-H were studied by HRTEM (Fig. 3, S3 and S4†). In addition to lattice fringes of 0.203 nm corresponding to the Ni (1 1 1) facet, lattice fringes of 0.212 nm corresponding to the NiZn3 (3 1 5) facet are also seen on 20Ni1Zn-H (Fig. 3a and S3†). No ZnO overlayer is observed, suggesting that the increase in Znδ+ on 20Ni1Zn-H is due to the formation of a small amount of Ni–Zn alloy rather than SMSI. On 20Ni20Zn-H, on the other hand, not only many lattice fringes belonging to NiZn3 (2 3 3) crystal facet are found, but also the phenomenon of encapsulation is observed (framed area), indicating that Ni–Zn alloy and SMSI are formed simultaneously on the 20Ni20Zn catalyst (Fig. 3b and S4†). The encapsulation of the metallic Ni sites by SMSI could also be responsible for the significant decrease in dispersion measured in the H2 pulse experiment (Table S2†). The absence of an overlayer on 20Ni1Zn suggests that Zn is preferentially alloyed with Ni and SMSI can only occur at high Zn content.
 | ||
| Fig. 3 HRTEM images of (a) 20Ni1Zn-H and (b) 20Ni20Zn-H. | ||
3.3 Structure–performance relationship of the NiZn/Al2O3 catalysts
As mentioned above, the reaction pathway of CO2 methanation on our catalysts has been shown to proceed via *CO intermediates. Thus, CO2 activation and further hydrogenation of *CO are two key factors that determine the hydrogenation activity of CO2. There are two CO2 activation pathways, the redox mechanism and the association mechanism.44 In the redox mechanism, CO2 is dissociated directly into *CO, and the rate-determining step (RDS) is the reduction of oxygen sites on the surface by *H.45,46 In the association mechanism, CO2 is activated by formate intermediates supported by *H, and its rate can be determined by H2 dissociation, the interaction of *H with *CO2, or the reduction of *OH by *H.47 Therefore, the RDS of the association mechanism is controversial. For 20Ni, we observed reaction orders of 0.79 and −0.27 for the formation of CO2 and H2 to CO, respectively (Table 1). For 20Ni20Zn, the CO2 order decreases to 0.42, while the H2 order assumes a positive value of 0.21. A significant increase in sensitivity to *H concentration for 20Ni20Zn indicates the conversion of the CO2 activation pathway to a redox mechanism after Zn addition.| Catalyst | Reaction order of CH4 formation | Reaction order of CO formation | E a (kJ mol−1) | |||
|---|---|---|---|---|---|---|
| CO2 | H2 | CO2 | H2 | CH4 | CO | |
| 20Ni | 0.25 | 0.53 | 0.79 | −0.27 | 83.69 | 91.36 |
| 20Ni1Zn | 0.09 | 0.42 | 0.71 | −0.18 | 92.46 | 75.92 |
| 20Ni20Zn | — | — | 0.42 | 0.21 | — | 79.91 |
To investigate the adsorbed species (especially *CO) and their tendency to change during the reaction process, in situ DRIFTS spectra were collected during the CO2 hydrogenation reaction (Fig. 4). At 150 °C or below, CO2 adsorbed on the surface of the catalyst produces bicarbonate species with the characteristic bands at 1637–1657 cm−1.48–50 When the temperature reaches 200 °C, formate species appear at 1575–1590 cm−1 and various types of adsorbed CO bands in the range 1800–2100 cm−1.51–54 Among them, the bands at 1846 and 1913 cm−1 are assigned to bridged *CO and the band at 2022 cm−1 to linear *CO.50,55,56 At higher temperatures, the methane peak at 3014 cm−1 can be observed,57,58 along with the decrease of *CO, which also provides more evidence for the formation of methane by *CO intermediates. At 200 °C, a shoulder peak at 2070 cm−1, which belongs to subcarbonyl species, can be observed above 20Ni and 20Ni1Zn. This peak is attributed to CO adsorption on low-coordinated Ni.59 The absence of this peak on 20Ni20Zn may be due to the formation of a large number of NiZn alloy. Using in situ DRIFTS, reaction mechanisms of CO2 hydrogenation on our catalysts can be proposed, including activation of CO2 to *CO via formate intermediates or direct cleavage and further hydrogenation of *CO to CH4. It is worth noting that the strength of formate species for 20Ni and 20Ni1Zn drops sharply at 250–300 °C, which corresponds to the appearance of the CH4 peak (Fig. 4d and e). However, in the case of 20Ni20Zn, there is no clear correlation between the formation of methane and the presence of formate species (Fig. 4f). This observation suggests a shift in the CO2 activation pathway toward a redox mechanism.
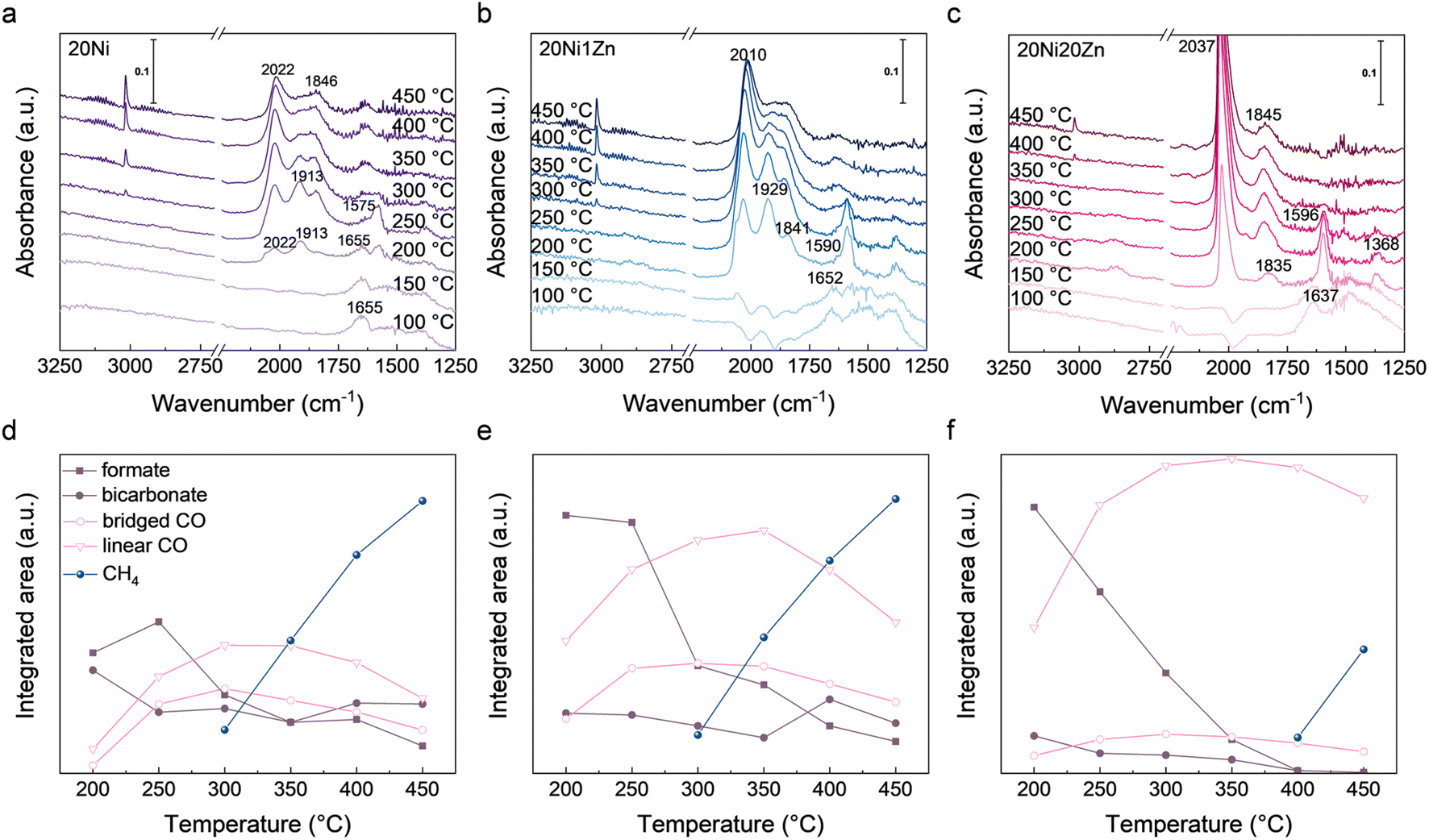 | ||
| Fig. 4 In situ DRIFTS spectra of (a) 20Ni, (b) 20Ni1Zn, and (c) 20Ni20Zn during CO2 hydrogenation. Evolution of species in in situ DRIFTS of (d) 20Ni, (e) 20Ni1Zn, and (f) 20Ni20Zn. | ||
The introduction of Zn has been reported to cause a selectivity change in the CO2 hydrogenation reaction by weakening the adsorption strength of CO, resulting in easy desorption of CO and difficulty in further hydrogenation.24,26,27 Although the amount of CO adsorbed on the Zn-containing catalyst is indeed less, as shown in Fig. S5,† a strong linear *CO peak exists for 20Ni20Zn during the CO2 hydrogenation process, as shown in Fig. 4c. The adsorption strength of *CO as an intermediate appears to be negatively correlated with the selectivity of CH4. However, the high intensity of the linear *CO peaks suggests that the difficulty of CO adsorption is not the main reason for the selectivity change in CO2 hydrogenation. During the reaction, the accumulation of intermediate species reflects the relative rate of generation and consumption. A strong *CO peak indicates that the CO generation rate over 20Ni20Zn is much larger than the consumption rate. Although it is difficult to determine the intrinsic CO adsorption capacity of the active sites during the reaction, the evidence suggests that the selectivity change is not solely due to the weakening of CO adsorption on the Zn-containing catalyst. We also performed the in situ DRIFTS investigation of CO hydrogenation directly. The intensity of CO adsorption is still reasonable on both 20Ni1Zn and 20Ni20Zn at different temperatures (Fig. S6†), which excludes the possibility that differences in CO generation rate are responsible for regulating the product selectivity. Therefore, it can be concluded that the decrease in CH4 selectivity is not due to the difficulty of CO adsorption, but rather to the inability of the adsorbed CO to react further.
By combining the mechanism study and structure characterization, the structure–performance relationship can be determined. The introduction of Zn preferentially forms an alloy with Ni and occurs at higher loading than ZnOx, leading to SMSI. The Ni–ZnOx interface induced by SMSI can promote the redox activation of CO2 due to the abundant oxygen vacancies.60 Although the Ni–Zn alloy phase has the ability to adsorb CO, it is difficult to further convert it to CH4, which ultimately affects the product selectivity. To further verify this, we measured the CO2 hydrogenation activity and selectivity over a wider temperature range from 250 to 450 °C (Fig. 5). 20Ni20Zn is still a CO-selective catalyst at 400 °C, but becomes CH4-selective at 450 °C. From a thermodynamic point of view, the CO2 methanation reaction is strongly exothermic (ΔH298 K = −164 kJ mol−1), while the RWGS reaction is endothermic (ΔH298 K = 41.2 kJ mol−1), and therefore increasing the reaction temperature is generally unfavorable for CH4 selectivity. Therefore, the change in selectivity from CO to CH4 at 400 °C to 450 °C may be due to overcoming the activation energy for further hydrogenation of *CO. The further hydrogenation of CO is just considered as a rate-determining step (RDS) for CO2 methanation over Ni/Al2O3 catalysts.30 The phenomenon that the TOF values of CO2 and CO hydrogenation decrease with the introduction of Zn also supports the change in the properties of the active phase, i.e., from metallic Ni to Ni–Zn alloy. Thus, for the Ni–Zn alloy, the kinetic limitation of this RDS hinders the further conversion of *CO and reverses the selectivity of CO2 hydrogenation.
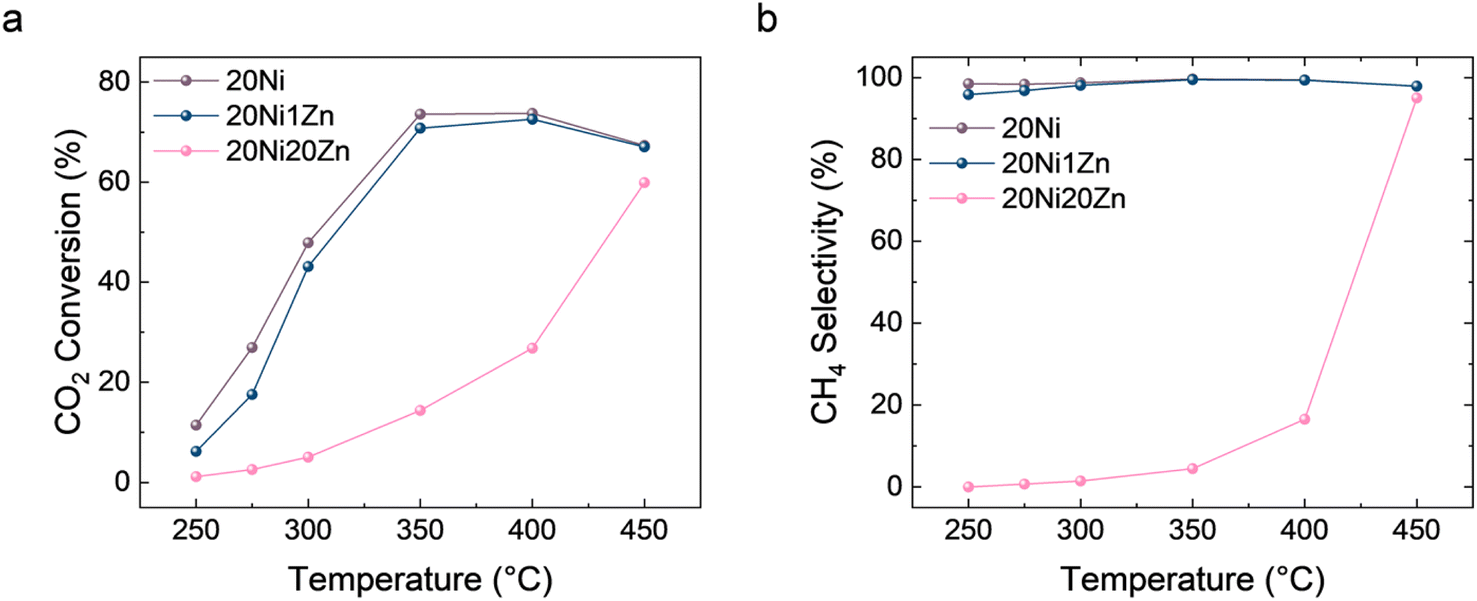 | ||
| Fig. 5 CO2 hydrogenation activity of NiZn/Al2O3 catalysts over a wider range of temperatures. | ||
4. Conclusions
Overall, we systematically investigated the effect of Zn on a Ni-based CO2 hydrogenation catalyst and its structure–activity relationship. The NiZn/Al2O3 supported catalysts with different ZnO loading were prepared by a deposition–precipitation method. The introduction of Zn decreases both the CO2 and CO hydrogenation activities of the catalyst and leads to a reversal of CO2 hydrogenation selectivity from CH4 to CO, which opens up new possibilities for rational design of efficient RWGS catalysts. Structural characterization based on XRD, XPS and TEM shows that Zn is preferentially alloyed with Ni. SMSI occurs when the Zn content is further increased. The Ni–ZnOx interface contributes to the CO2 activation pathway changing from an associative mechanism to a redox mechanism. Moreover, although the Ni–Zn alloy can adsorb CO intermediates in the reaction process, its ability to further convert *CO is insufficient, which is the main reason for the selective conversion of CH4 to CO. Therefore, in the development of Ni-based CO2 hydrogenation catalysts, attention should be paid to the adsorption of intermediates CO and the further hydrogenation capacity of the active sites to regulate the selectivity of the target product.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. Zhu thanks the research funding sponsored by the National Key R&D Program of China (2022YFB3805504), National Natural Science Foundation of China (22078089), Shanghai pilot Program for Basic Research (22TQ1400100-7), the Basic Research Program of Science and Technology Commission of Shanghai Municipality (22JC1400600), the Innovation Program of Shanghai Municipal Education Commission, the Shanghai Science and Technology Innovation Action Plan (22JC1403800) and SINOPEC (No. 421056).Notes and references
- S. I. Zandalinas, F. B. Fritschi and R. Mittler, Trends Plant Sci., 2021, 26, 588–599 CrossRef CAS PubMed.
- L. Al-Ghussain, Environ. Prog. Sustainable Energy, 2019, 38, 13–21 CrossRef CAS.
- W. F. Lamb, T. Wiedmann, J. Pongratz, R. Andrew, M. Crippa, J. G. J. Olivier, D. Wiedenhofer, G. Mattioli, A. Al Khourdajie, J. House, S. Pachauri, M. Figueroa, Y. Saheb, R. Slade, K. Hubacek, L. Sun, S. K. Ribeiro, S. Khennas, S. de la Rue du Can, L. Chapungu, S. J. Davis, I. Bashmakov, H. Dai, S. Dhakal, X. Tan, Y. Geng, B. Gu and J. Minx, Environ. Res. Lett., 2021, 16, 073005 CrossRef CAS.
- R. Ahmed, G. Liu, B. Yousaf, Q. Abbas, H. Ullah and M. U. Ali, J. Cleaner Prod., 2020, 242, 118409 CrossRef CAS.
- M. D. Garba, M. Usman, S. Khan, F. Shehzad, A. Galadima, M. F. Ehsan, A. S. Ghanem and M. Humayun, J. Environ. Chem. Eng., 2021, 9, 104756 CrossRef CAS.
- W. Gao, S. Liang, R. Wang, Q. Jiang, Y. Zhang, Q. Zheng, B. Xie, C. Y. Toe, X. Zhu, J. Wang, L. Huang, Y. Gao, Z. Wang, C. Jo, Q. Wang, L. Wang, Y. Liu, B. Louis, J. Scott, A.-C. Roger, R. Amal, H. He and S.-E. Park, Chem. Soc. Rev., 2020, 49, 8584–8686 RSC.
- X. Su, X. Yang, B. Zhao and Y. Huang, J. Energy Chem., 2017, 26, 854–867 CrossRef.
- Q. Zhang, J. Kang and Y. Wang, ChemCatChem, 2010, 2, 1030–1058 CrossRef CAS.
- M. Ojeda, R. Nabar, A. U. Nilekar, A. Ishikawa, M. Mavrikakis and E. Iglesia, J. Catal., 2010, 272, 287–297 CrossRef CAS.
- P. Frontera, A. Macario, M. Ferraro and P. L. Antonucci, Catalysts, 2017, 7, 59 CrossRef.
- W. Wang and J. Gong, Front. Chem. Eng. China, 2011, 5, 2–10 CAS.
- Y. Yan, Q. Wang, C. Jiang, Y. Yao, D. Lu, J. Zheng, Y. Dai, H. Wang and Y. Yang, J. Catal., 2018, 367, 194–205 CrossRef CAS.
- X. Wang, H. Shi, J. H. Kwak and J. Szanyi, ACS Catal., 2015, 5, 6337–6349 CrossRef CAS.
- J. Wang, K. Sun, X. Jia and C. Liu, Catal. Today, 2021, 365, 341–347 CrossRef CAS.
- J. Díez-Ramírez, P. Sánchez, V. Kyriakou, S. Zafeiratos, G. E. Marnellos, M. Konsolakis and F. Dorado, J. CO2 Util., 2017, 21, 562–571 CrossRef.
- C. Vogt, E. Groeneveld, G. Kamsma, M. Nachtegaal, L. Lu, C. J. Kiely, P. H. Berben, F. Meirer and B. M. Weckhuysen, Nat. Catal., 2018, 1, 127–134 CrossRef CAS.
- M. Xu, X. Liu, C. Cao, Y. Sun, C. Zhang, Z. Yang, M. Zhu, X. Ding, Y. Liu, Z. Tong and J. Xu, ACS Sustainable Chem. Eng., 2021, 9, 13818–13830 CrossRef CAS.
- S. Kattel, P. J. Ramírez, J. G. Chen, J. A. Rodriguez and P. Liu, Science, 2017, 355, 1296–1299 CrossRef CAS PubMed.
- S. Tada, T. Shimizu, H. Kameyama, T. Haneda and R. Kikuchi, Int. J. Hydrogen Energy, 2012, 37, 5527–5531 CrossRef CAS.
- H. C. Wu, Y. C. Chang, J. H. Wu, J. H. Lin, I. K. Lin and C. S. Chen, Catal. Sci. Technol., 2015, 5, 4154–4163 RSC.
- T. Pu, L. Shen, X. Liu, X. Cao, J. Xu, I. E. Wachs and M. Zhu, J. Catal., 2021, 400, 228–233 CrossRef CAS.
- X. Yuan, T. Pu, M. Gu, M. Zhu and J. Xu, ACS Catal., 2021, 11, 11966–11972 CrossRef CAS.
- L. R. Winter, E. Gomez, B. Yan, S. Yao and J. G. Chen, Appl. Catal., B, 2018, 224, 442–450 CrossRef CAS.
- W. Liao, C. Tang, H. Zheng, J. Ding, K. Zhang, H. Wang, J. Lu, W. Huang and Z. Zhang, J. Catal., 2022, 407, 126–140 CrossRef CAS.
- W. Wang, X. Li, Y. Zhang, R. Zhang, H. Ge, J. Bi and M. Tang, Catal. Sci. Technol., 2017, 7, 4413–4421 RSC.
- S. Lin, Q. Wang, M. Li, Z. Hao, Y. Pan, X. Han, X. Chang, S. Huang, Z. Li and X. Ma, ACS Catal., 2022, 12, 3346–3356 CrossRef CAS.
- Y. Wang, K. Feng, J. Tian, J. Zhang, B. Zhao, K. H. Luo and B. Yan, ChemSusChem, 2022, 15, 1–8 Search PubMed.
- Y. Pan, C. Liu and Q. Ge, J. Catal., 2010, 272, 227–234 CrossRef CAS.
- G. Garbarino, P. Riani, L. Magistri and G. Busca, Int. J. Hydrogen Energy, 2014, 39, 11557–11565 CrossRef CAS.
- W. Zhang, T. Pu, Z. Wang, L. Shen and M. Zhu, Ind. Eng. Chem. Res., 2022, 61, 9678–9685 CrossRef CAS.
- R.-P. Ye, Q. Li, W. Gong, T. Wang, J. J. Razink, L. Lin, Y.-Y. Qin, Z. Zhou, H. Adidharma, J. Tang, A. G. Russell, M. Fan and Y.-G. Yao, Appl. Catal., B, 2020, 268, 118474 CrossRef CAS.
- A. Cárdenas-Arenas, A. Quindimil, A. Davó-Quiñonero, E. Bailón-García, D. Lozano-Castelló, U. De-La-Torre, B. Pereda-Ayo, J. A. González-Marcos, J. R. González-Velasco and A. Bueno-López, Appl. Catal., B, 2020, 265, 118538 CrossRef.
- L. Zeng, Y. Wang, Z. Li, Y. Song, J. Zhang, J. Wang, X. He, C. Wang and W. Lin, ACS Appl. Mater. Interfaces, 2020, 12, 17436–17442 CrossRef CAS PubMed.
- Z. Zhang, K. Feng and B. Yan, Catal. Sci. Technol., 2022, 12, 4698–4708 RSC.
- C. Italiano, J. Llorca, L. Pino, M. Ferraro, V. Antonucci and A. Vita, Appl. Catal., B, 2020, 264, 118494 CrossRef CAS.
- R. V. Gonçalves, L. L. R. Vono, R. Wojcieszak, C. S. B. Dias, H. Wender, E. Teixeira-Neto and L. M. Rossi, Appl. Catal., B, 2017, 209, 240–246 CrossRef.
- T. A. Le, M. S. Kim, S. H. Lee, T. W. Kim and E. D. Park, Catal. Today, 2017, 293–294, 89–96 CrossRef CAS.
- H. Muroyama, Y. Tsuda, T. Asakoshi, H. Masitah, T. Okanishi, T. Matsui and K. Eguchi, J. Catal., 2016, 343, 178–184 CrossRef CAS.
- D. Li, F. Xu, X. Tang, S. Dai, T. Pu, X. Liu, P. Tian, F. Xuan, Z. Xu, I. E. Wachs and M. Zhu, Nat. Catal., 2022, 5, 99–108 CrossRef CAS.
- S. Kuld, C. Conradsen, P. G. Moses, I. Chorkendorff and J. Sehested, Angew. Chem., 2014, 126, 6051–6055 CrossRef.
- V. Schott, H. Oberhofer, A. Birkner, M. Xu, Y. Wang, M. Muhler, K. Reuter and C. Wöll, Angew. Chem., Int. Ed., 2013, 52, 11925–11929 CrossRef CAS PubMed.
- M. Behrens, F. Studt, I. Kasatkin, S. Kühl, M. Hävecker, F. Abild-Pedersen, S. Zander, F. Girgsdies, P. Kurr, B. Kniep, M. Tovar, R. W. Fischer, J. K. Nørskov and R. Schlögl, Science, 2012, 336, 893–897 CrossRef CAS PubMed.
- S. Kuld, M. Thorhauge, H. Falsig, C. F. Elkjaer, S. Helveg, I. Chorkendorff and J. Sehested, Science, 2016, 352, 969–974 CrossRef CAS PubMed.
- S. Kattel, P. Liu and J. G. Chen, J. Am. Chem. Soc., 2017, 139, 9739–9754 CrossRef CAS PubMed.
- G.-C. Wang and J. Nakamura, J. Phys. Chem. Lett., 2010, 1, 3053–3057 CrossRef CAS.
- M. Zhu, P. Tian, M. E. Ford, J. Chen, J. Xu, Y.-F. Han and I. E. Wachs, ACS Catal., 2020, 10, 7857–7863 CrossRef CAS.
- M. Gu, S. Dai, R. Qiu, M. E. Ford, C. Cao, I. E. Wachs and M. Zhu, ACS Catal., 2021, 11, 12609–12619 CrossRef CAS.
- L. F. Bobadilla, J. L. Santos, S. Ivanova, J. A. Odriozola and A. Urakawa, ACS Catal., 2018, 8, 7455–7467 CrossRef CAS.
- W. L. Vrijburg, G. Garbarino, W. Chen, A. Parastaev, A. Longo, E. A. Pidko and E. J. M. Hensen, J. Catal., 2020, 382, 358–371 CrossRef CAS.
- W. L. Vrijburg, E. Moioli, W. Chen, M. Zhang, B. J. P. Terlingen, B. Zijlstra, I. A. W. Filot, A. Züttel, E. A. Pidko and E. J. M. Hensen, ACS Catal., 2019, 9, 7823–7839 CrossRef CAS.
- E. T. Saw, U. Oemar, X. R. Tan, Y. Du, A. Borgna, K. Hidajat and S. Kawi, J. Catal., 2014, 314, 32–46 CrossRef CAS.
- C.-S. Chen, C. S. Budi, H.-C. Wu, D. Saikia and H.-M. Kao, ACS Catal., 2017, 7, 8367–8381 CrossRef CAS.
- S. Kattel, B. Yan, Y. Yang, J. G. Chen and P. Liu, J. Am. Chem. Soc., 2016, 138, 12440–12450 CrossRef CAS PubMed.
- C. Cerdá-Moreno, A. Chica, S. Keller, C. Rautenberg and U. Bentrup, Appl. Catal., B, 2020, 264, 118546 CrossRef.
- J. Zarfl, D. Ferri, T. J. Schildhauer, J. Wambach and A. Wokaun, Appl. Catal., A, 2015, 495, 104–114 CrossRef CAS.
- M. Zhu, P. Tian, X. Cao, J. Chen, T. Pu, B. Shi, J. Xu, J. Moon, Z. Wu and Y. F. Han, Appl. Catal., B, 2021, 282, 119561 CrossRef CAS.
- A. Solis-Garcia, J. F. Louvier-Hernandez, A. Almendarez-Camarillo and J. C. Fierro-Gonzalez, Appl. Catal., B, 2017, 218, 611–620 CrossRef CAS.
- N. Rui, X. Zhang, F. Zhang, Z. Liu, X. Cao, Z. Xie, R. Zou, S. D. Senanayake, Y. Yang, J. A. Rodriguez and C.-J. Liu, Appl. Catal., B, 2021, 282, 119581 CrossRef CAS.
- M. Agnelli, H. M. Swaan, C. Marquez-Alvarez, G. A. Martin and C. Mirodatos, J. Catal., 1998, 175, 117–128 CrossRef CAS.
- L. C. Wang, M. Tahvildar Khazaneh, D. Widmann and R. J. Behm, J. Catal., 2013, 302, 20–30 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ta10066e |
| This journal is © The Royal Society of Chemistry 2023 |