
Construction of membrane reactors coupled with conjugated network hollow microspheres for cascade production of block copolymers†
Yi Wei
Duan‡
ab,
Xi Juan
Zhang‡
ab,
Wan Lin
Guo
ab,
Min
Jian
ab,
Tao
Cai
 *ab and
Xue
Li
*ab
*ab and
Xue
Li
*ab
aKey Laboratory of Biomedical Polymers of Ministry of Education, College of Chemistry and Molecular Science, Wuhan University, Wuhan, Hubei 430072, P. R. China. E-mail: chemcaitao@whu.edu.cn; li.x@whu.edu.cn
bWuhan University Suzhou Research Institute, Suzhou, Jiangsu 215123, P. R. China
First published on 10th February 2023
Abstract
Membrane reactors have inspired a cascading and sustainable approach toward chemical production and processing, offering potential for improving reaction efficiency and addressing limitations to product purification. In this work, hollow microspheres comprising porphyrinic conjugated microporous networks (H-PPrIB-M MSs) are engineered through Sonogashira–Hagihara cross-coupling polycondensation with the assistance of silica templates. Using the photoinduced electron/energy transfer-reversible addition-fragmentation chain transfer (PET-RAFT) polymerization with H-PPrIB-M MSs as heterogeneous catalysts, polymerization was conducted under mild conditions: low-intensity household white light or far-red light, closed or open reaction vessels, ambient temperature, and ultrapure water as the solvent. Taking advantage of the robustness of H-PPrIB-M MSs, the suspended catalysts were fitted into membrane reactors to fabricate the suspended-catalyst-based membrane reactor (SCBMR) system. High polymerization kinetics, oxygen tolerance and precise control over molecular weights and polydispersities were achieved in the SCBMR systems. Separation and reutilization of the catalyst-functionalized nanocomposite was realized through facile centrifugation, which led to negligible catalyst leaching and maintenance of catalytic performance over six polymerization cycles. The purification of PHEAA-b-PNAT-b-PHPGA-CDSP triblock copolymers with purity exceeding 99% could be accomplished within 2.1 diavolumes. With the concept of such catalytic membrane reactors, this work represents a simplified and customizable synthetic tool to unlock macromolecular buildups with precisely controlled architectures and desirable properties for a variety of high-value applications.
 Xue Li | Dr Li received her PhD in Chemical Engineering under the supervision of Professor Tai-Shung Chung at the National University of Singapore. After finishing her postdoctoral training, she joined the College of Chemistry and Molecular Sciences at Wuhan University as an Associate Professor. She was recognized as a NAMS Young Membrane Scientist and received several best paper awards at chemical engineering conferences. She is also the deputy director of the Department of Applied Chemistry at Wuhan University. Her research interests include membrane separation, wastewater reclamation, polymer chemistry and material science. She has plans to embark on research relating to industrial polymer production using membranes. |
Introduction
Polymerizations are crucial industrial processes with industrialized nations consuming nearly 100 kg per person per year of polymer-relevant materials.1 With such high demand for polymers, it is of great importance to increase polymerization efficiency and reduce setup complexity for polymer production on industrial scales. In contrast to thermally initiated systems, photochemistry enables selective reactions to take place efficiently at ambient temperatures. In addition, the energy of photons supplied to these systems can be selectively accepted by chromophores at specific light wavelengths, unlike thermal energy that is transferred indiscriminately throughout entire reaction mixtures. Consequently, specific reaction pathways can be reactivated on-demand in photochemistry, in turn, allowing more precise and efficient transformations.2–5 Harnessing the unique properties of light as a synthetic tool has transformed polymer chemistry in recent years.6–8 Among these strategies, photoinduced electron/energy transfer-reversible addition-fragmentation chain transfer (PET-RAFT) polymerization is considered to be a sustainable alternative to traditional thermal-initiated polymerization, along with a wide variety of exquisite benefits including a simple experimental setup, mild reaction conditions, minimal side reactions and spatiotemporal control.9–12Reducing purification steps enhances the cost-effectiveness and environmental benignity of macromolecular syntheses to a point where production is economically attainable.13,14 Such protocols are essential for manufacturing precision polymers on an industrial scale for high-value applications. As a process that amalgamates heterogeneous catalysis with membrane separation in a single unit, membrane reactors have been demonstrated to be a promising alternative to batch reactors in small molecule and polymer synthesis with primitive adaptation utilized for thermally-triggered systems and expanded to include photochemical approaches.15–18 This actualization can be extensively ascribed to improved scalability, control over process parameters, enhanced reliability, and simplified synthetic protocols. In addition, the membrane reactor has inspired a scalable and sustainable approach toward chemical production and processing, offering unbridled potential for increasing reaction efficiency and addressing limitations in product purification.19–28 Rationally designed membrane reactors may enable the production of new materials that are prohibitively difficult to obtain by other means. Integrating PET-RAFT processes into membrane reactors has provided crucial information concerning necessary reactor design and materials, catalytic capabilities, and accessible molecular weight ranges as well as highlighting the overall ability to improve the results of polymerization relative to their batch reactor counterparts.
While homogeneous catalysts are undoubtedly useful for the majority of photopolymerizations, heterogeneous catalysts are still favorable on an industrial scale as they offer distinct advantages including straightforward separation and effective recovery.29–35 Based on the input mode of heterogeneous catalysts, membrane reactors can be categorized into suspended-catalysts-based membrane reactors (SCBMR) and catalytic active membrane reactors (CAMR). In the CAMR system, the membrane performs as a filter, while simultaneously serving as a support for active catalyst immobilization. In contrast, the heterogeneous catalysts are suspended or packed in the reaction mixture and the membrane is next to the reaction bulk in the inert type in the SCBMR system. To illustrate some of these features, we recently reported a thorough study highlighting the cascade polymerization of a range of acrylamides and acrylates using an aqueous PET-RAFT process in CAMR systems, where the polyethersulfone ultrafiltration membranes with surface-tethering eosin Y or metalloporphyrin-bound copolymers were introduced as supports.36,37 The SCBMR system offers numerous benefits due to the following reasons: (1) the concentration of the suspended catalysts can be tuned to optimize the reaction efficiency; (2) the intrinsic flexibility of suspended catalysts makes them capable of fitting in most reactor geometries and being effortlessly separated without leaving residues; (3) such systems typically employ enzymes, sacrificial substrates, and/or increased concentrations of catalysts or reducing agents to in situ consume oxygen, which could be readily eliminated in membrane filtration;5 (4) the membranes can be utilized directly without surface tethering or entrapping of catalytic active materials, which avoids exacerbating the membrane permeability and selectivity; (5) the membranes can be regenerated by backwashing, or replaced by other commercial membranes without affecting the catalytic performances.15 To the best of our knowledge, the exploration of the SCBMR system for photopolymerization is rare.
Herein, we introduce a design for the cascade production of value-added materials by exploiting the construction of membrane reactors coupled with conjugated network hollow microspheres (H-PPrIB-M MSs) as suspended heterogeneous photocatalysts to combine aqueous PET-RAFT polymerization and the membrane separation process in the SCBMR system. The H-PPrIB-M MSs were synthesized via Sonogashira–Hagihara cross-coupling polycondensation, hydrofluoric acid (HF) etching and the alkyne–azide click grafting of azido-functionalized poly(ethylene glycol) methyl ether (MPEG-N3).38–40 In particular, the H-PPrIB-M MSs were shown to be extremely effective for these transformations, allowing various acrylamides to be polymerized in batches under household white and far-red light irradiation. The resultant polymers displayed typical characteristics of RAFT polymerization with molecular weights in line with theoretical predictions and dispersities generally less than 1.15. In addition, the H-PPrIB-M MSs with large sizes and high surface areas were demonstrated to facilitate the reagent diffusion in the polymerization process, avoid membrane pore clogging during membrane filtration and improve separation efficiency. Importantly, the use of the SCBMR system eliminated the necessity for tedious deoxygenation, catalyst removal and intermittent product purification, and therefore greatly simplified the synthetic protocols. The SCBMR system provides access to multigram quantities and high purity of complex macromolecular buildups in a more straightforward and user-friendly manner.
Results and discussion
Template synthesis is a straightforward approach to the shape control of nanomaterials since the resultant shapes originating from the sacrificed templates are quite predictable.41–44 Narrowly dispersed silica microspheres (SiO2 MSs) with a diameter of 282.2 ± 24.1 nm were fabricated via an ammonia-catalyzed hydrolysis/condensation process of tetraethyl orthosilicate (TEOS) (Fig. S1a†). Polycondensation of 1 eq. 5,10,15,20-tetra(4-ethynylphenyl)-21H,23H-porphyrin (TEPP) and 2 eq. 1,4-diiodobenzene (IB) with the assistance of SiO2 MSs in a mixture of toluene and triethylamine (TEA) produced the SiO2@PPrIB core–shell MSs (Scheme 1).45–47 Field-emission scanning electron microscopy (FESEM) unveiled the formation of a uniform and coarse polymeric corona encircling an inorganic SiO2 core (Fig. 1a). The resultant solids were purified with Soxhlet extraction, and further treated with hydrofluoric acid (HF) to afford the H-PPrIB MSs as a dark brown powder in ∼65% yield, as clearly observed by FESEM and transmission electron microscopy (TEM) (Fig. 1b, c, S1 and S2†).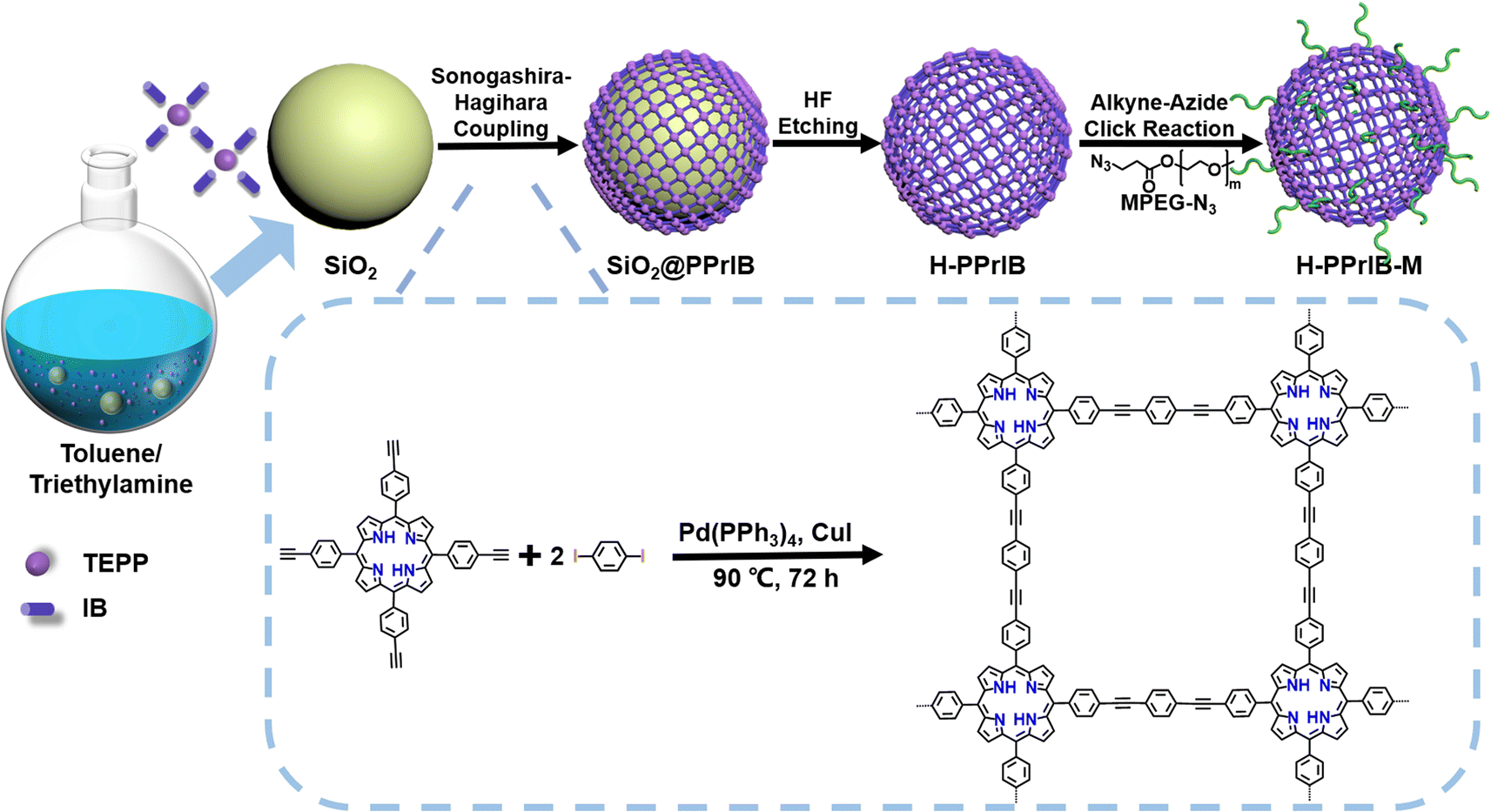 | ||
| Scheme 1 Synthetic routes for H-PPrIB-M microspheres. | ||
 | ||
| Fig. 1 Morphological observation: FESEM and TEM images of (a) SiO2@H-PPrIB-2, (b and c) H-PPrIB-2 and (d) H-PPrIB-M microspheres. | ||
The shell thickness of H-PPrIB MSs could be systematically controlled by adjusting the volume ratios of solvent systems, the quantities of monomers and SiO2 templates.41,42 Eight representative synthetic conditions were selected and the corresponding results are summarized in Table 1. In the case of the solvent system, the total volume of the solvent was fixed at 30 mL, decreasing the volume of TEA from 20 to 15 and 10 mL, accompanied by the increase in the shell thickness of H-PPrIB MSs from 19.6 ± 2.5 to 22.0 ± 1.7 and 30.5 ± 2.3 nm (Fig. 1a–c, S1c–f and S2a, b†). Remarkably, by decreasing the amount of TEPP from 85.3 to 56.9 mg or increasing the amount of SiO2 templates from 300 to 600 mg, the shell thickness of H-PPrIB MSs decreased and defective hollow structures were dominantly observed (Fig. S1g–j and S2c–e†). In contrast, by increasing the amount of TEPP from 85.3 to 113.9 mg, the resultant nanocomposites stuck together and the shell thickness became difficult to measure (Fig. S1k and l†). It is worth noting the discrete growth of PPrIB shells from SiO2 templates when TEA was replaced with viscous triethanolamine (TEOA), or there was no stirring of the reaction system (Fig. S1m and n†). Specifically, polycondensation of 1 eq. TEPP and 2 eq. IB without the addition of SiO2 templates produced the C-PPrIB conjugated microporous polymers (CMPs) (Scheme S1 and Fig. S1b†). The H-PPrIB-2 MSs were chosen for subsequent surface modification as a compromise for the well-preserved morphology and sufficient grafting anchors (alkyne groups from the TEPP component).
| Sample | SiO2 (mg) | Pd(PPh3)4 (mg) | CuI (mg) | TEPP (mg) | IB (mg) | Solvents (mL) | Reaction conditions | Stirring | PPrIB (mg) | Yieldb (%) | Shell thicknessc (nm) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Abbreviations: microspheres (MSs), conjugated microporous polymers (CMPs), tetrakis(triphenylphosphine)palladium(0) (Pd(PPh3)4), 5,10,15,20-tetra(4-ethynylphenyl)-21H,23H-porphyrin (TEPP), 1,4-diiodobenzene (IB), triethylamine (TEA), triethanolamine (TEOA). b The product yield is the experimental weight of the product over the theoretical weight of the product. The calculation of the theoretical weight of the product is based on the assumption that all the iodine is retained in the H-PPrIB MSs. c The shell thickness was determined from the TEM images in Fig. 1 and S2. | |||||||||||
| H-PPrIB-1 | 300 | 13.9 | 2.3 | 85.3 | 79.2 | Toluene(10)/TEA(20) | 90 °C, 72 h | Yes | 102.1 | 62.1 | 19.6 ± 2.5 |
| H-PPrIB-2 | 300 | 13.9 | 2.3 | 85.3 | 79.2 | Toluene(15)/TEA(15) | 90 °C, 72 h | Yes | 102.8 | 62.5 | 22.0 ± 1.7 |
| H-PPrIB-3 | 300 | 13.9 | 2.3 | 85.3 | 79.2 | Toluene(20)/TEA(10) | 90 °C, 72 h | Yes | 118.5 | 72.0 | 30.5 ± 2.3 |
| H-PPrIB-4 | 300 | 9.2 | 1.5 | 56.9 | 53.2 | Toluene(15)/TEA(15) | 90 °C, 72 h | Yes | 74.2 | 67.4 | 17.5 ± 1.4 |
| H-PPrIB-5 | 600 | 13.9 | 2.3 | 85.3 | 79.2 | Toluene(15)/TEA(15) | 90 °C, 72 h | Yes | 132.8 | 80.7 | Broken |
| H-PPrIB-6 | 300 | 18.5 | 3.0 | 113.8 | 105.7 | Toluene(15)/TEA(15) | 90 °C, 72 h | Yes | 116.2 | 52.9 | — |
| H-PPrIB-7 | 300 | 13.9 | 2.3 | 85.3 | 79.2 | Toluene(15)/TEOA(15) | 90 °C, 72 h | Yes | 104.4 | 63.5 | — |
| H-PPrIB-8 | 300 | 13.9 | 2.3 | 85.3 | 79.2 | Toluene(15)/TEA(15) | 90 °C, 72 h | No | 127.0 | 77.2 | — |
| C-PPrIB | 0 | 13.9 | 2.3 | 85.3 | 79.2 | Toluene(15)/TEA(15) | 90 °C, 72 h | No | 131.3 | 79.8 | — |
With the peripheral alkyne groups, an in situ alkyne–azide click reaction was conducted to attach azido-functionalized poly(ethylene glycol) methyl ether (MPEG-N3) via the grafting-to strategy, rendering the production of H-PPrIB-M MSs with improved aqueous dispersibility (Fig. S3a†). The surface-conjugated MPEG brushes slightly increased the shell thickness of H-PPrIB-M MSs to 25.8 ± 2.1 nm (Fig. 1d). The solid-state 13C nuclear magnetic resonance (NMR) spectrum of H-PPrIB-2 MSs revealed distinctive signals of the sp2-hybridized pyrrolic carbons and aromatic carbons in the region of δ = 112–155 ppm and signals of the alkynyl carbons in the range of δ = 86–94 ppm (Fig. 2a).45–47 The newly emerged signal for the aliphatic carbons connected to oxygen at δ = 71 ppm further corroborated the successful grafting of MPEG brushes to the H-PPrIB-M MSs (Fig. 2a). The analysis of N2 sorption isotherms based on the Brunauer–Emmett–Teller (BET) theory showed microporosity with a surface area of 630.8 m2 g−1 for the H-PPrIB-2 MSs and 600.5 m2 g−1 for the H-PPrIB-M MSs, which were much higher than that of 346.9 m2 g−1 for the C-PPrIB CMPs (Fig. 2b). Powder X-ray diffraction (XRD) studies showed the amorphous character of H-PPrIB-2, H-PPrIB-M MSs and C-PPrIB CMPs, as commonly observed for conjugated polymers prepared by Sonogashira coupling in the literature (Fig. 2c).45–47 In comparison to the TEPP and IB precursors, H-PPrIB-2, H-PPrIB-M MSs and C-PPrIB CMPs demonstrated red-shifted absorption edges with enhanced intensity over a broadened Q-band region of 530–690 nm, associated with the generation of fully π-conjugated organic frameworks (Fig. 2d).46,47 The H-PPrIB-2 MSs and C-PPrIB CMPs remained thermally inert in a nitrogen atmosphere with minor weight loss throughout the entire process, while the major weight loss for the H-PPrIB-M MSs took place at around 350 °C when gradually elevating the temperature to 800 °C, as monitored by thermogravimetric analysis (TGA) (Fig. S4†). By considering the residual weight percentage of each component at 800 °C, the surface-tethered MPEG accounted for 32.5 wt% of the H-PPrIB-M composite particles. The molar ratio of porphyrin catalytic units on the H-PPrIB-M MSs was determined to be around 0.78 × 10−6 mol mg−1.
 | ||
| Fig. 2 Structural characterization: (a) solid-state 13C NMR spectra of H-PPrIB-M and H-PPrIB-2 MSs; (b) N2 isotherms of H-PPrIB-M, H-PPrIB-2 MSs and C-PPrIB CMPs at 77 K with respective BET surface areas estimated to be 600.5, 630.8 and 346.9 m2 g−1, respectively; (c) XRD patterns and (d) UV-visible spectra of H-PPrIB-M, H-PPrIB-2 MSs and C-PPrIB CMPs; (e) XPS wide scan, (f) C 1s and (f′) N 1s core-level spectra of H-PPrIB-M MSs. | ||
The alterations in the surface composition of the H-PPrIB-2 and H-PPrIB-M MSs were substantiated by an evident attenuation of carbon (C 1s), nitrogen (N 1s) and iodine (I 3d, I 4d) signals in the X-ray photoelectron spectroscopy (XPS) measurements (Fig. 2e and S5a†). Deconvolution of the high-resolution C 1s core-level spectrum of H-PPrIB-2 MSs generated four peak components with respective binding energies (BEs) at 284.6, 285.6, 286.2 and 287.6 eV, attributable to the –![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) –C/–H–, ––N/––I, –
–C/–H–, ––N/––I, –![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and π–π* shake-up species (Fig. S5b†). Concomitantly, the ––O peak with BE at 286.2 eV, associated with the abundant ether repeat units in the MPEG chain, took a dominant position as compared to the ––C/–H– species for the H-PPrIB-M MSs (Fig. 2f). Deconvolution of the N 1s core-level spectrum of H-PPrIB-M MSs generated two peak components with respective BEs at 397.8 and 399.7 eV, assigned to the imine nitrogen ((C)
N and π–π* shake-up species (Fig. S5b†). Concomitantly, the ––O peak with BE at 286.2 eV, associated with the abundant ether repeat units in the MPEG chain, took a dominant position as compared to the ––C/–H– species for the H-PPrIB-M MSs (Fig. 2f). Deconvolution of the N 1s core-level spectrum of H-PPrIB-M MSs generated two peak components with respective BEs at 397.8 and 399.7 eV, assigned to the imine nitrogen ((C)![[N with combining low line]](https://www.rsc.org/images/entities/char_004e_0332.gif) –) and the amine nitrogen (––) in the porphyrin ring and triazole ring (Fig. 2f′). These results are consistent with the previously reported porphyrinic conjugated networks.37,48 The energy band gaps were estimated from cyclic voltammetry (CV) and were 1.16, 1.19 and 1.21 eV for H-PPrIB-M MSs, H-PPrIB-2 MSs and C-PPrIB CMPs, respectively (Fig. S6†). This result confirmed that the alteration of the size and morphology and surface functionalization of MPEG to the nanocomposites had negligible impacts on the energy band gaps of the materials. Accordingly, from the above investigations, H-PPrIB-M MSs possessed an ideal size and morphology, and improved aqueous dispersibility and photophysical properties for photocatalysis in membrane reactors.
–) and the amine nitrogen (––) in the porphyrin ring and triazole ring (Fig. 2f′). These results are consistent with the previously reported porphyrinic conjugated networks.37,48 The energy band gaps were estimated from cyclic voltammetry (CV) and were 1.16, 1.19 and 1.21 eV for H-PPrIB-M MSs, H-PPrIB-2 MSs and C-PPrIB CMPs, respectively (Fig. S6†). This result confirmed that the alteration of the size and morphology and surface functionalization of MPEG to the nanocomposites had negligible impacts on the energy band gaps of the materials. Accordingly, from the above investigations, H-PPrIB-M MSs possessed an ideal size and morphology, and improved aqueous dispersibility and photophysical properties for photocatalysis in membrane reactors.
Because of their tailorable architectures, abundant active sites, inherent porosity, chemical stability and optoelectronic properties, metal-free H-PPrIB-M MSs open a new avenue for this fundamental, yet challenging photocatalytic reaction. A model PET-RAFT system was utilized for consistency in all cases, with poly(ethylene glycol) methyl ether 4-cyano-4-[(dodecylsulfanylthiocarbonyl)sulfanyl]pentanoate (MPEG-CDSP) as the chain transfer agent (CTA), N,N-dimethylacrylamide (DMA) as the monomer and ultrapure water as the solvent, at a fixed reaction stoichiometry of [DMA]0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[MPEG-CDSP]0:[H-PPrIB-M] = 200:1:0.04 and monomer to solvent (1:1, v/v) in a closed or open system (Fig. S3b†). Nominal wavelengths of the light source were chosen according to the lower energy red-shifted Q-band of the H-PPrIB-M MSs, using household white light (1.2 mW cm−2) and far-red light (λmax = 680 nm, 4.0 mW cm−2). By analyzing the aliquots obtained during sampling at predetermined time intervals, the kinetic investigation of DMA polymerization revealed pseudo-first-order behavior for semilogarithmic plots of ln([M]0/[M]t) against exposure time t. Specifically, the polymerization performed in the closed vessel proceeded with a higher polymerization rate (kap = 0.41 h−1), whereas the polymerization performed in the open vessel occurred at a slower polymerization rate (kap = 0.35 h−1) and a prolonged induction period of 40 min under the white light (1.2 mW cm−2) irradiation (Fig. 3a). Replacement of the white light with a far-red light with higher intensity (λmax = 680 nm, 4.0 mW cm−2) resulted in a reduced kap but a concomitant increase in the induction period in both systems, which was attributed to its significantly lower extinction coefficient at this wavelength (Fig. 3a). This oxygen tolerance poses significant advantages as it eliminates the rigorous deoxygenation step that is commonly required prior to controlled radical polymerization.5 Temporal regulation was then investigated by programable “ON/OFF” experiments. Rapid chain propagation during illumination was effortlessly halted when the light was switched “OFF” and restarted as required with the next “ON” cycle with minimal inhibition and similar polymerization kinetics (Fig. 3b). Most importantly, the evolution of molecular weights (Mn,GPC) obtained from gel permeation chromatography (GPC) and the molecular weights (Mn,th) obtained from 1H NMR spectroscopy were concomitant with the narrowing of the molecular weight distribution (Đ) and in relatively good agreement with low molecular weight, whereas a more distinct deviation appeared at high monomer conversion (Fig. 3c, d and S7†). This deviation was attributed to the differences in hydrodynamic volumes between the targeted polymers and poly(methyl methacrylate) (PMMA) used as molecular weight standards. Additionally, control experiments in the absence of photocatalyst (H-PPrIB-M MSs), CTA (MPEG-CDSP) or light irradiation revealed negligible monomer conversions, further supporting the PET-RAFT mechanism (Table S1, #1–5†). Replacement of H-PPrIB-M MSs with C-PPrIB-M CMPs (C-PPrIB CMPs functionalized with MPEG-N3) in the control experiments led to the generation of polymers with comparable monomer conversions and narrow polydispersities (Table S1, #6 and 7†). In comparison to the C-PPrIB-M CMPs, the H-PPrIB-M MSs with large size and high surface area were demonstrated to facilitate the reagent diffusion in the polymerization process, avoid the membrane pore clogging during membrane filtration and improve the separation efficiency.
:[MPEG-CDSP]0:[H-PPrIB-M] = 200:1:0.04 and monomer to solvent (1:1, v/v) in a closed or open system (Fig. S3b†). Nominal wavelengths of the light source were chosen according to the lower energy red-shifted Q-band of the H-PPrIB-M MSs, using household white light (1.2 mW cm−2) and far-red light (λmax = 680 nm, 4.0 mW cm−2). By analyzing the aliquots obtained during sampling at predetermined time intervals, the kinetic investigation of DMA polymerization revealed pseudo-first-order behavior for semilogarithmic plots of ln([M]0/[M]t) against exposure time t. Specifically, the polymerization performed in the closed vessel proceeded with a higher polymerization rate (kap = 0.41 h−1), whereas the polymerization performed in the open vessel occurred at a slower polymerization rate (kap = 0.35 h−1) and a prolonged induction period of 40 min under the white light (1.2 mW cm−2) irradiation (Fig. 3a). Replacement of the white light with a far-red light with higher intensity (λmax = 680 nm, 4.0 mW cm−2) resulted in a reduced kap but a concomitant increase in the induction period in both systems, which was attributed to its significantly lower extinction coefficient at this wavelength (Fig. 3a). This oxygen tolerance poses significant advantages as it eliminates the rigorous deoxygenation step that is commonly required prior to controlled radical polymerization.5 Temporal regulation was then investigated by programable “ON/OFF” experiments. Rapid chain propagation during illumination was effortlessly halted when the light was switched “OFF” and restarted as required with the next “ON” cycle with minimal inhibition and similar polymerization kinetics (Fig. 3b). Most importantly, the evolution of molecular weights (Mn,GPC) obtained from gel permeation chromatography (GPC) and the molecular weights (Mn,th) obtained from 1H NMR spectroscopy were concomitant with the narrowing of the molecular weight distribution (Đ) and in relatively good agreement with low molecular weight, whereas a more distinct deviation appeared at high monomer conversion (Fig. 3c, d and S7†). This deviation was attributed to the differences in hydrodynamic volumes between the targeted polymers and poly(methyl methacrylate) (PMMA) used as molecular weight standards. Additionally, control experiments in the absence of photocatalyst (H-PPrIB-M MSs), CTA (MPEG-CDSP) or light irradiation revealed negligible monomer conversions, further supporting the PET-RAFT mechanism (Table S1, #1–5†). Replacement of H-PPrIB-M MSs with C-PPrIB-M CMPs (C-PPrIB CMPs functionalized with MPEG-N3) in the control experiments led to the generation of polymers with comparable monomer conversions and narrow polydispersities (Table S1, #6 and 7†). In comparison to the C-PPrIB-M CMPs, the H-PPrIB-M MSs with large size and high surface area were demonstrated to facilitate the reagent diffusion in the polymerization process, avoid the membrane pore clogging during membrane filtration and improve the separation efficiency.
 | ||
| Fig. 3 Kinetic study in batch: kinetic analyses of the H-PPrIB-M MSs-mediated PET-RAFT polymerization of DMA using a reaction stoichiometry of [DMA]0:[MPEG-CDSP]0:[H-PPrIB-M] = 200:1:0.04 in a closed or open vessel under white light (1.2 mW cm−2) or red light (λmax = 680 nm, 4.0 mW cm−2) irradiation. (a) Plots of ln([M]0/[M]t) versus exposure time t, (b) temporal control over polymerization according to the designated ON/OFF switches of light, (c) Mn,th, Mn,GPC and Đ versus monomer conversion, and (d) GPC traces of the purified PDMA polymers at different time points under red light (λmax = 680 nm, 4.0 mW cm−2) irradiation in a closed vessel. | ||
The amalgamation of PET-RAFT polymerizations and membrane separation techniques allows the simplification of the setup complexity for the cascade production of well-defined and structurally diverse macromolecules with high efficiency and minimum by-product formation. The suspended-catalysts-based membrane reactor (SCBMR, 200 mL total volume), with a residence time of 6 h, light sources of white light, 2.4 mW cm−2, and red light, λmax = 680 nm, 8.0 mW cm−2, and chemical concentration of 10 vol% monomer, with a fixed reaction stoichiometry of [DMA]0:[MPEG-CDSP]0 = 800:1, MPEG-CDSP as CTA, was adapted for the scaled-up experiments (Scheme 2 and Fig. S8†). The role of oxygen in polymerization inhibition was further probed via a method established by Boyer and coworkers, where sodium ascorbate (NaAsc) is used as a singlet-oxygen sensitizer.5 Upon establishing the role of H-PPrIB-M MSs in the SCBMR system, three H-PPrIB-M loadings of 50, 100 and 200 ppm, were investigated for the kinetic analyses. Aliquots of the reaction mixtures were taken at specific intervals via an argon-purged syringe. Due to the headspace left in the SCBMR, the polymerization kinetics without prior deoxygenation revealed an extended induction period, followed by a linear increase in ln([M]0/[M]t) with exposure time t. As expected, an increase in kap from 0.14 to 0.31 h−1 and a decrease in the induction period from 45 to 28 min by increasing the H-PPrIB-M loading from 50 to 200 ppm were identified when subjected to red light (λmax = 680 nm, 8.0 mW cm−2) irradiation (Fig. 4a). Comparatively, the acceleration in reaction kinetics (kap = 0.46 h−1) and reduction in the induction period (18 min) were expected under white light irradiation (Fig. 4a). Irrespective of these differences, the linear evolution of Mn,GPC and Mn,th with increasing monomer conversion, which coincided with decreasing dispersities, was indicative of a well-controlled polymerization with pseudo-living characteristics. The symmetrical GPC traces that shifted toward higher molecular weight revealed that the deviation between Mn,GPC and Mn,th was attributed to the difference in the hydrodynamic volumes of PDMA and PMMA references utilized (Fig. 4c, d and S9†).
 | ||
| Scheme 2 Schematic diagram of the SCBMR system designed for the cascade synthesis of block copolymers. | ||
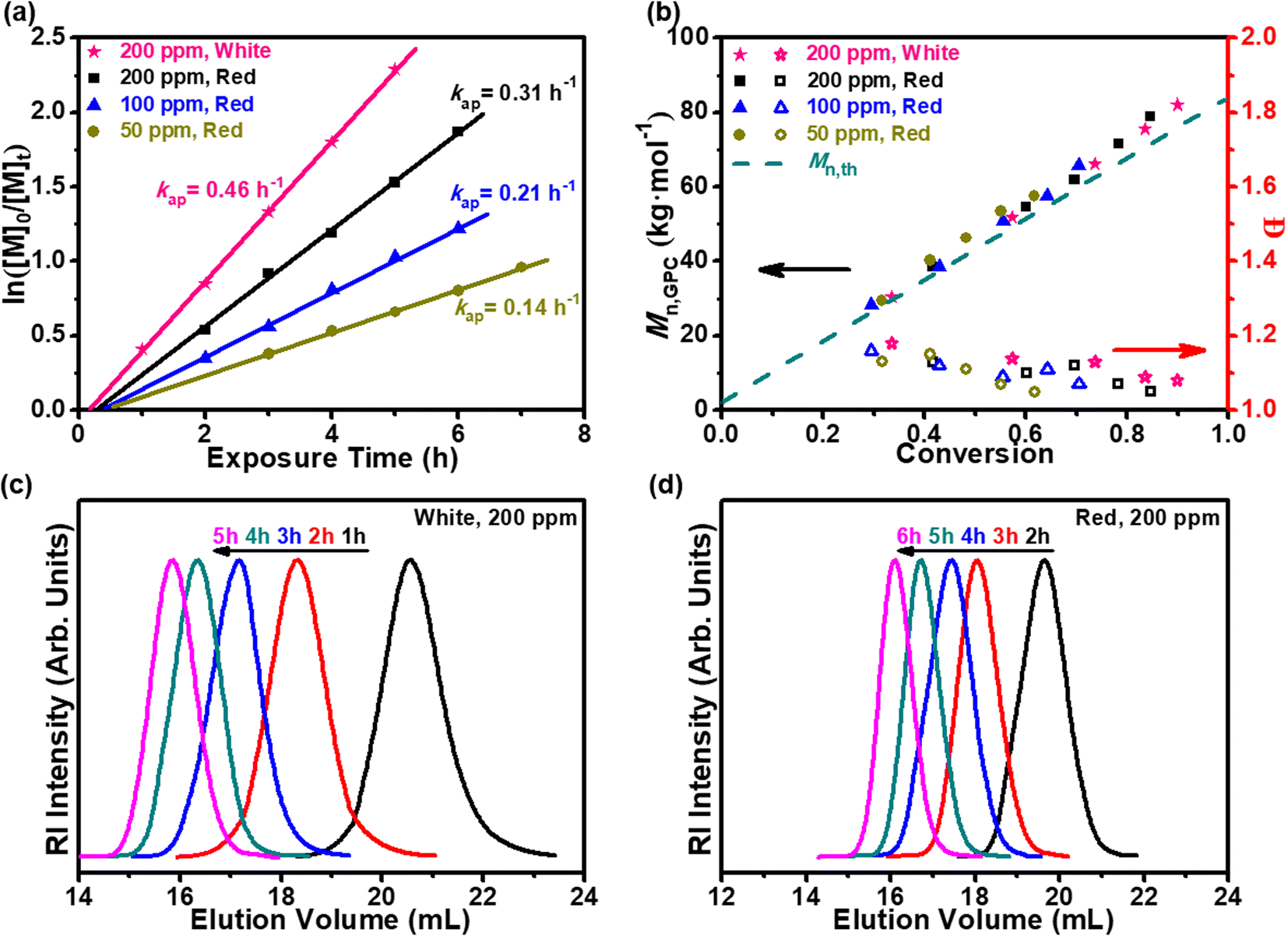 | ||
| Fig. 4 Kinetic study in the SCBMR: kinetic analyses of H-PPrIB-M-mediated PET-RAFT polymerization of DMA conducted in ultrapure water under white light (2.4 mW cm−2) or red light (λmax = 680 nm, 8.0 mW cm−2) irradiation using a reaction stoichiometry of [DMA]0:[MPEG-CDSP]0 = 800:1 without prior deoxygenation in SCBMR. (a) Plots of ln([M]0/[M]t) versus exposure time t at different catalyst concentrations with reference to monomer concentration, (b) Mn,th, Mn,GPC and Đ versus monomer conversion, (c) GPC profiles at different time points with 200 ppm of the catalyst under white light (2.4 mW cm−2) irradiation, and (d) GPC profiles at different time points with 200 ppm of the catalyst under red light (λmax = 680 nm, 8.0 mW cm−2) irradiation. | ||
To expand the monomer tolerance and the scope of accessible polymer productions, the SCBMR system was amenable to several water-soluble acrylamides (Table 2). The polymerizations were successful for N,N-diethylacrylamide [DEA (#1 and 2)], N-isopropylacrylamide [NIPAM (#3 and 4)], 4-acryloylmorpholine [AMP (#5 and 6)], N-hydroxyethyl acrylamide [HEAA (#7 and 8)] and N-[tris(hydroxymethyl)methyl] acrylamide [NAT (#9 and 10)] as water-soluble acrylamides, attaining the polydispersities of 1.06–1.16 at monomer conversions of 60.1–83.8% in 4–6 h. Despite the differences in rates, all polymerizations demonstrated excellent consistency in the experimental Mn,GPC with Mn,th. Notably, it was found that the polymerization of acrylamide-monofunctionalized hyperbranched polyglycerol (HPGA) exhibited the lowest activity, giving a conversion of 55.1% and a broader molecular weight distribution (Đ = 1.32) for the PHPGA chains after 8 h of reaction time (Table 2, #11 and 12). We attributed this to the hyperbranched structures and relatively low density of acrylamide groups in HPGA as compared to other acrylamides.49
| # | Monomer | CTAa | [Monomer]0:[CTA]0:[H-PPrIB-M] |
Time (h) | α (%) | M n,th (kg mol−1) | M n,GPC (kg mol−1) | Đ |
|---|---|---|---|---|---|---|---|---|
| a Abbreviations: CTA, chain transfer agent; DMA, N,N-dimethylacrylamide (MW 99.1 g mol−1); DEA, N,N-diethylacrylamide (MW 127.2 g mol−1); NIPAM, N-isopropylacrylamide (MW 113.2 g mol−1); AMP, 4-acryloylmorpholine (MW 141.2 g mol−1); HEAA, N-hydroxyethyl acrylamide (MW 115.1 g mol−1); NAT, N-[tris(hydroxymethyl)methyl]acrylamide (MW 175.2 g mol−1); HPGA, acrylamide-monofunctionalized hyperbranched polyglycerol (MW 1200 g mol−1); MPEG-CDSP, poly(ethylene glycol) methyl ether 4-cyano-4-[(dodecylsulfanylthiocarbonyl) sulfanyl]pentanoate (MW 2400 g mol−1). b The molecular weight was calculated using the following equation: Mn,th = [M]0/[CTA] × MWM × α + MWCTA, where [M]0, [CTA], MWM, α and MWCTA correspond to the initial monomer concentration, initial CTA concentration, molecular weight of monomer, monomer conversion derived from 1H NMR spectroscopy, and molecular weight of CTA. c Derived from GPC profiles (calibration with PMMA molecular weight standards), polydispersity index (Đ) = Mw,GPC/Mn,GPC. d The polymerizations were performed under red light (λmax = 680 nm, 8.0 mW cm−2) irradiation in ultrapure water in SCBMR. | ||||||||
| 1d | DEA | MPEG-CDSP | 800:1:0.16 |
4 | 65.4 | 69.0 | 80.6 | 1.12 |
| 2 | DEA | MPEG-CDSP | 800:1:0.16 |
6 | 80.4 | 84.2 | 99.3 | 1.09 |
| 3 | NIPAM | MPEG-CDSP | 800:1:0.16 |
4 | 69.3 | 65.2 | 73.5 | 1.09 |
| 4 | NIPAM | MPEG-CDSP | 800:1:0.16 |
6 | 83.8 | 78.3 | 88.4 | 1.06 |
| 5 | AMP | MPEG-CDSP | 800:1:0.16 |
4 | 66.5 | 77.5 | 93.2 | 1.14 |
| 6 | AMP | MPEG-CDSP | 800:1:0.16 |
6 | 81.4 | 94.3 | 110.6 | 1.08 |
| 7 | HEAA | MPEG-CDSP | 800:1:0.16 |
4 | 60.1 | 57.7 | 72.1 | 1.16 |
| 8 | HEAA | MPEG-CDSP | 800:1:0.16 |
6 | 75.6 | 72.0 | 88.5 | 1.14 |
| 9 | NAT | MPEG-CDSP | 800:1:0.16 |
4 | 68.3 | 98.1 | 116.4 | 1.12 |
| 10 | NAT | MPEG-CDSP | 800:1:0.16 |
6 | 83.0 | 118.7 | 138.0 | 1.10 |
| 11 | HPGA | MPEG-CDSP | 400:1:0.08 |
8 | 55.1 | 266.9 | 240.8 | 1.32 |
| 12 | HPGA | MPEG-CDSP | 400:1:0.08 |
12 | 78.4 | 378.7 | 338.3 | 1.28 |
| 13 | DMA | MPEG-CDSP | 400:1:0.08 |
6 | 87.8 | 37.2 | 41.2 | 1.07 |
| 14 | DMA | PDMA-CDSP | 400:1:0.08 |
6 | 83.3 | 70.2 | 77.9 | 1.08 |
| 15 | DMA | PDMA-CDSP | 400:1:0.08 |
6 | 80.2 | 102.0 | 114.1 | 1.12 |
| 16 | HEAA | MPEG-CDSP | 400:1:0.08 |
6 | 81.4 | 39.9 | 50.8 | 1.14 |
| 17 | NAT | PHEAA-CDSP | 400:1:0.08 |
6 | 84.8 | 99.3 | 123.4 | 1.18 |
| 18 | HPGA | PHEAA-b-PNAT-CDSP | 200:1:0.08 |
8 | 79.2 | 289.4 | 270.6 | 1.35 |
The expansion of membrane reactors presents polymer chemists with the opportunity to streamline multistep macromolecular syntheses; as the need for complex macromolecular products increases, so does the need for optimized reaction efficiency and reduced setup complexity. Membrane performance is typically expressed in terms of rejection and flux. The ultrafiltration membranes presented almost complete rejection of H-PPrIB-M MSs and polymers with molecular weights over 36000 Da and showed only 10% rejection to solutes with molecular weights below 1200 Da, inclusive of all the acrylamides used in this study (Fig. S10†). The large gap in the rejection of undesirable impurities as compared to the products accounts for the successful harvesting of polymers having molecular weights exceeding 36000 Da with high filtration efficiency. As the molecular weight of the solutes increased from 400 to 100000 Da, the membrane flux demonstrated a sharp decrease from 445 to 30 L m−2 h−1 (LMH), followed by a slight fluctuation in the steady-state flux (Fig. S10†). To take advantage of the rapid PET-RAFT polymerization and the SCBMR system, a one-pot chain extension was conducted, which eliminated the necessity of intermittent purification and quantitative monomer conversion for each consecutive block. A model triblock P(DMA-b-DMA-b-DMA) homopolymer was synthesized by the sequential addition of DMA with a reaction stoichiometry of [DMA]0:[MPEG-CDSP]0 = 400:1 for each block. The monomer conversions were slightly reduced for the second and third blocks, which was attributed to the increased viscosity of the polymerization solution (Fig. 5a). The GPC analysis of the polymers showed monomodal distributions with a clear shift to lower retention time after each chain extension (Fig. S11† and Table 2, #13–15). To circumvent the limitations imposed by the increased viscosity on the concentrated solutions and, therefore, the clogging of the SCBMR system, the retentate volume was kept constant by topping up the water to compensate for the volume of permeate flow during membrane filtration. In the separation process, the membrane permeability was relatively constant within the range of 85–115% of the initial value (Fig. 5c). As a time-dependent parameter, several diavolumes were employed in this study to monitor the progress of purification. The ultrafiltration membrane reached 95% removal of permeated impurities consisting of residual monomers and other reactants and obtained the PDMA-CDSP homopolymers with purity exceeding 99% in 3.3 diavolumes for the 1-1st cycle, 3.1 diavolumes for the 1-2nd cycle and 2.5 diavolumes for the 1-3rd cycle, indicative of outstanding purification efficiency (Fig. 5c).
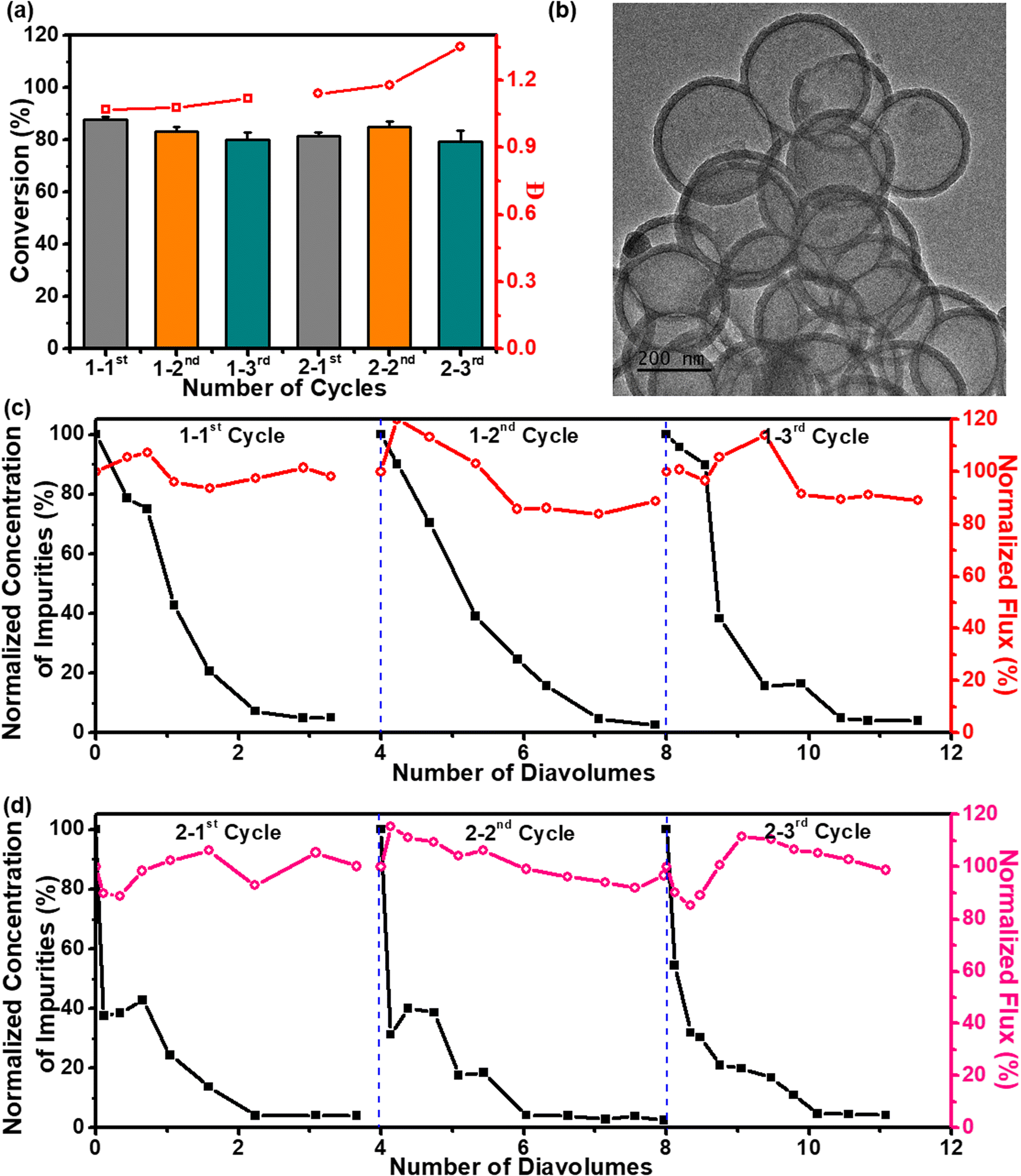 | ||
| Fig. 5 Cascade polymerization process in SCBMR: PET-RAFT polymerization was performed in ultrapure water by using H-PPrIB-M MSs as catalysts without prior deoxygenation under red light (λmax = 680 nm, 8.0 mW cm−2) irradiation in SCBMR. (a) Monomer conversion and molecular weight distribution; (b) TEM image of H-PPrIB-M MSs after six cycles of polymerization; normalized concentration of impurities in the retentate (squares) and normalized flux (circles) of the purification process as a function of the number of diavolumes for the aqueous solutions of (c) PDMA-CDSP in the first three cycles of polymerization, and (d) PHEAA-b-PNAT-b-PHPGA-CDSP in the second three cycles of polymerization. | ||
Since the chain extensions shown in the previous section were tested with success, another set of chain extensions was performed for the synthesis of polyols. The initial syntheses of PHEAA-CDSP macro-CTA in SCBMR rendered a monomer conversion of 81.4% after 6 h, with Mn,GPC of 50.8 kg mol−1 accompanied by a narrow molecular weight distribution (Đ = 1.14). Subsequently, two water-soluble acrylamides, NAT and HPGA were sequentially block copolymerized to PHEAA-CDSP macro-CTA in SCBMR without supplementary addition of the photocatalysts to give PHEAA-b-PNAT-b-PHPGA-CDSP triblock copolymers. The chain extensions were expected to achieve, and reached, stable monomer conversions (84.8% and 79.2%, respectively, for the second and third blocks), albeit with a relatively broad molecular weight distribution (Đ = 1.35) for the PHEAA-b-PNAT-b-PHPGA-CDSP copolymers (Fig. 5a, S11† and Table 2, #16–18). Owing to the intermolecular hydrogen bond attractions between polyols and water, the purification of polyols by dialysis is usually accompanied by low separation efficiency.49 To investigate this issue, the crude reaction mixture containing PHEAA-b-PNAT-b-PHPGA-CDSP copolymers was taken out and dialyzed against ultrapure water with a dialysis membrane (MWCO 10000 Da) at room temperature. The dialysate was changed using fresh ultrapure water and analyzed by a total organic carbon analyzer (TOC) at specific time intervals. Surprisingly, it took only 10 h for 72% of impurities to diffuse from the reaction mixture to the dialysate and an additional 85 h for a further 11% removal of impurities (Fig. S12†). Comparatively, membrane filtration maintained more than 85% of the initial value, and around 95% of impurities were successfully fractionated in 2.3 diavolumes for the 2-1st cycle, 2.1 diavolumes for the 2-2nd cycle and 2.1 diavolumes for the 2-3rd cycle (Fig. 5d). Apparently, the membrane filtration imposed a much higher separation efficiency than dialysis. After cascade polymerization and membrane separation in the SCBMR system, and catalyst recycling from the reaction mixture via centrifugation, the polymer products showcased a white color and the H-PPrIB-M MSs exhibited an intense brown color. The similar surface chemistry of the recycled H-PPrIB-M MSs analyzed by XPS, solid-state 13C NMR spectroscopy and UV-vis spectroscopy corroborated that negligible polymer products were entrapped in the catalysts (Fig. S13–S15 and Table S2†). The ultrafiltration membrane could be readily regenerated by backwashing with ultrapure water and subjected to another round of polymerization. Indiscernible alterations in the microstructures of the regenerated ultrafiltration membrane and recycled H-PPrIB-M MSs observed by FESEM further substantiated the robustness and integrity of the SCBMR system after six cycles of polymerization (Fig. 5b and S16†). These results demonstrate the efficiency of the SCBMR system and highlight its utility to streamline the cascade synthesis of block copolymers.
Conclusion
In this contribution, the H-PPrIB-M MSs have been synthesized via Sonogashira–Hagihara cross-coupling polycondensation, HF etching and the alkyne–azide click grafting of MPEG-N3. Using the PET-RAFT process with H-PPrIB-M MSs as heterogeneous catalysts, polymerization was conducted under extremely mild conditions: low-intensity far-red light, closed or open reaction vessels, ambient temperatures and ultrapure water as solvent. The resultant polymers displayed characteristics typical of RAFT mechanisms, with molecular weights close to theoretical predictions accompanied by narrow molecular weight distributions. The cascade production of a series of polyacrylamides has been achieved utilizing the SCBMR system, in which the H-PPrIB-M-suspended catalysts were fitted into membrane reactors. The SCBMR system provides precise and reproducible control of reaction parameters and also simplifies the synthetic process by reducing numerous tedious procedures including deoxygenation, catalyst removal and intermittent purification. Specifically, the SCBMR system could afford the purification of PHEAA-b-PNAT-b-PHPGA-CDSP triblock copolymers with purity exceeding 99% within 2.1 diavolumes, which imposed much higher separation efficiency than dialysis. Certainly, the current scope of work provides a strong foundation for the future maturation of the PET-RAFT process using membrane reactors, offering the potential for bridging the gap from an academic pursuit to the industrially relevant production of high-value materials to address the problems of modern life.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The financial support from the National Natural Science Foundation of China (51773156) and the Natural Science Foundation of Jiangsu Province (BK20211090) is acknowledged.References
- P. N. Kurek, A. J. Kloster, K. A. Weaver, R. Manahan, M. L. Allegrezza, N. D. A. Watuthanthrige, C. Boyer, J. A. Reeves and D. Konkolewicz, Ind. Eng. Chem. Res., 2018, 57, 4203–4213 CrossRef CAS.
- D. A. Corbin and G. M. Miyake, Chem. Rev., 2022, 122, 1830–1874 CrossRef CAS PubMed.
- C. Wu, N. Corrigan, C. H. Lim, W. Liu, G. Miyake and C. Boyer, Chem. Rev., 2022, 122, 5476–5518 CrossRef CAS PubMed.
- X. Pan, M. Fantin, F. Yuan and K. Matyjaszewski, Chem. Soc. Rev., 2018, 47, 5457–5490 RSC.
- J. Yeow, R. Chapman, A. J. Gormley and C. Boyer, Chem. Soc. Rev., 2018, 47, 4357–4387 RSC.
- J. C. Theriot, C.-H. Lim, H. Yang, M. D. Ryan, C. B. Musgrave and G. M. Miyake, Science, 2016, 352, 1082–1086 CrossRef CAS PubMed.
- L. Qiao, M. Zhou, G. Shi, Z. Cui, X. Zhang, P. Fu, M. Liu, X. Qiao, Y. He and X. Pang, J. Am. Chem. Soc., 2022, 144, 9817–9826 CrossRef CAS PubMed.
- J. Niu, D. J. Lunn, A. Pusuluri, J. I. Yoo, M. A. O'Malley, S. Mitragotri, H. T. Soh and C. J. Hawker, Nat. Chem., 2017, 9, 537–545 CrossRef CAS PubMed.
- Y. Zhao, Y. Ma, X. Lin and M. Chen, Angew. Chem., Int. Ed., 2020, 59, 21470–21474 CrossRef CAS PubMed.
- Z. Wu, K. Jung, C. Wu, G. Ng, L. Wang, J. Liu and C. Boyer, J. Am. Chem. Soc., 2022, 144, 995–1005 CrossRef CAS PubMed.
- C. Wu, H. Chen, N. Corrigan, K. Jung, X. Kan, Z. Li, W. Liu, J. Xu and C. Boyer, J. Am. Chem. Soc., 2019, 141, 8207–8220 CrossRef CAS PubMed.
- X. Li, S. Ye, Y. Huang, J. L. Li and T. Cai, J. Mater. Chem. A, 2019, 7, 6173–6179 RSC.
- D. J. Walsh, M. G. Hyatt, S. A. Miller and D. Guironnet, ACS Catal., 2019, 9, 11153–11188 CrossRef CAS.
- M. H. Reis, F. A. Leibfarth and L. M. Pitet, ACS Macro Lett., 2020, 9, 123–133 CrossRef CAS PubMed.
- R. van Reis and A. Zydney, J. Membr. Sci., 2007, 297, 16–50 CrossRef CAS.
- S. So, L. G. Peeva, E. W. Tate, R. J. Leatherbarrow and A. G. Livingston, Chem. Commun., 2010, 46, 2808–2810 RSC.
- I. F. J. Vankelecom, Chem. Rev., 2002, 102, 3779–3810 CrossRef CAS PubMed.
- C. Algieri, G. Coppola, D. Mukherjee, M. I. Shammas, V. Calabro, S. Curcio and S. Chakraborty, Catalysts, 2021, 11, 691 CrossRef CAS.
- X. Wang, S. Li, P. Chen, F. Li, X. Hu and T. Hua, Mater. Today Chem., 2022, 23, 100650 CrossRef CAS.
- C. Didaskalou, J. Kupai, L. Cseri, J. Barabas, E. Vass, T. Holtzl and G. Szekely, ACS Catal., 2018, 8, 7430–7438 CrossRef CAS.
- P. Kisszekelyi, A. Alammar, J. Kupai, P. Huszthy, J. Barabas, T. Holtzl, L. Szente, C. Bawn, R. Adams and G. Szekely, J. Catal., 2019, 371, 255–261 CrossRef CAS.
- Y. Wang, R. Z. Liang, T. Z. Jia, X. L. Cao, Q. Wang, J. R. Cao, S. Li, Q. Shi, L. Isaacs and S. P. Sun, J. Am. Chem. Soc., 2022, 144, 6483–6492 CrossRef CAS PubMed.
- A. Yao, D. Hua, Z. F. Gao, J. Y. Pan, A. R. Ibrahim, D. Y. Zheng, Y. P. Hong, Y. Liu and G. W. Zhan, J. Membr. Sci., 2022, 652, 120465 CrossRef CAS.
- Q. Ding, Z. Zhang, Y. Liu, K. Chai, R. Krishna and S. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202208134 CAS.
- J. L. Li, Y. Zhang, S. Zhang, M. Liu, X. Li and T. Cai, J. Mater. Chem. A, 2019, 7, 8167–8176 RSC.
- H. Jiang, Y. Liu, W. Xing and R. Chen, Ind. Eng. Chem. Res., 2021, 60, 8969–8990 CrossRef CAS.
- Y. Lu, W. Liu, K. Wang and S. Zhang, J. Mater. Chem. A, 2022, 10, 20101–20110 RSC.
- Y. Lu, L. Zhang, L. Shen, W. Liu, R. Karnik and S. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2111360118 CrossRef CAS PubMed.
- S. Allison-Logan, Q. Fu, Y. Sun, M. Liu, J. Xie, J. Tang and G. G. Qiao, Angew. Chem., Int. Ed., 2020, 59, 21392–21396 CrossRef CAS PubMed.
- Z. Xia, B. Shi, W. Zhu, Y. Xiao and C. Lu, Adv. Funct. Mater., 2022, 32, 2207655 CrossRef CAS.
- Y. Zhu, D. Zhu, Y. Chen, Q. Yan, C.-Y. Liu, K. Ling, Y. Liu, D. Lee, X. Wu, T. P. Senftle and R. Verduzco, Chem. Sci., 2021, 12, 16092–16099 RSC.
- S. Dadashi-Silab, F. Lorandi, M. J. Ditucci, M. Sun, G. Szczepaniak, T. Liu and K. Matyjaszewski, J. Am. Chem. Soc., 2021, 143, 9630–9638 CrossRef CAS PubMed.
- W. L. Guo, Y. Zhou, B. Duan, W. F. Wei, C. Chen, X. Li and T. Cai, Chem. Eng. J., 2022, 429, 132120 CrossRef CAS.
- Y. Huang, X. Li, J. L. Li, B. Zhang and T. Cai, Macromolecules, 2018, 51, 7974–7982 CrossRef CAS.
- X. Li, Y. C. Zhang, S. Ye, X. R. Zhang and T. Cai, J. Mater. Chem. A, 2020, 8, 25363–25370 RSC.
- Y. Zhao, S. Shao, J. Xia, Y. Huang, Y. C. Zhang, X. Li and T. Cai, J. Mater. Chem. A, 2020, 8, 9825–9831 RSC.
- C. Chen, G. A. Zhou, H. R. Zhang, X. Tang, J. N. Cheng, Y. H. Zhao, X. Li and T. Cai, Chem. Eng. J., 2021, 424, 130395 CrossRef CAS.
- S. Luo, Z. Zeng, H. Wang, W. Xiong, B. Song, C. Zhou, A. Duan, X. Tan, Q. He, G. Zeng, Z. Liu and R. Xiao, Prog. Polym. Sci., 2021, 115, 101374 CrossRef CAS.
- W. Zhang, H. Zuo, Z. Cheng, Y. Shi, Z. Guo, N. Meng, A. Thomas and Y. Liao, Adv. Mater., 2022, 34, 2104952 CrossRef CAS PubMed.
- D. Taylor, S. J. Dalgarno, Z. Xu and F. Vilela, Chem. Soc. Rev., 2020, 49, 3981–4042 RSC.
- N. Kang, J. H. Park, M. Jin, N. Park, S. M. Lee, H. J. Kim, J. M. Kim and S. U. Son, J. Am. Chem. Soc., 2013, 135, 19115–19118 CrossRef CAS PubMed.
- N. Park, K. C. Ko, H. W. Shin, S. M. Lee, H. J. Kim, J. Y. Lee and S. U. Son, J. Mater. Chem. A, 2016, 4, 8010–8014 RSC.
- W. Huang, B. C. Ma, D. Wang, Z. J. Wang, R. Li, L. Wang, K. Landfester and K. A. I. Zhang, J. Mater. Chem. A, 2017, 5, 3792–3797 RSC.
- X. Zhuang, D. Gehrig, N. Forler, H. Liang, M. Wagner, M. R. Hansen, F. Laquai, F. Zhang and X. Feng, Adv. Mater., 2015, 27, 3789–3796 CrossRef CAS PubMed.
- R. Chinchilla and C. Najera, Chem. Rev., 2007, 107, 874–922 CrossRef CAS PubMed.
- X. Yu, Z. Yang, B. Qiu, S. Guo, P. Yang, B. Yu, H. Zhang, Y. Zhao, X. Yang, B. Han and Z. Liu, Angew. Chem., Int. Ed., 2019, 58, 632–636 CrossRef CAS PubMed.
- X. Li, Y. C. Zhang, Y. Zhao, H. P. Zhao, B. Zhang and T. Cai, Macromolecules, 2020, 53, 1550–1556 CrossRef CAS.
- X. Li, Y. Huang, W. F. Wei, W. L. Guo, Z. Luo, J. Xu and T. Cai, Chem. Eng. J., 2022, 434, 134692 CrossRef CAS.
- G. Kasza, G. Kali, A. Domjan, L. Petho, G. Szarka and B. Ivan, Macromolecules, 2017, 50, 3078–3088 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ta08634d |
| ‡ These authors contribute equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |