
Room-temperature magnetism in two-dimensional metal–organic frameworks enabled by electrostatic gating†
Qin
Yu
 abc and
Dong
Wang
*ab
abc and
Dong
Wang
*ab
aLaboratory of Flexible Electronics Technology, Tsinghua University, Beijing 100084, P. R. China
bMOE Key Laboratory of Organic OptoElectronics and Molecular Engineering, Department of Chemistry, Tsinghua University, Beijing 100084, P. R. China. E-mail: dong913@tsinghua.edu.cn
cResearch Institute of Petrolium Processing, SINOPEC, Beijing 100083, P. R. China
First published on 7th February 2023
Abstract
Two-dimensional (2D) magnetism has attracted intense attention in the area of low-dimensional materials and devices, with intriguing physics emerging and significant potential for applications in energy-efficient data storage and quantum computing. Although a robust magnetic order sustainable under room temperature is the most relevant issue for its practical applications, high temperature magnetism is rarely reported. 2D magnets obtained to date are often exfoliated from van der Waals atomic crystals with bulk magnetism. In this work, we propose a new type of 2D magnets, 2D conjugated metal–organic frameworks (MOFs), which are constructed by magnetic metal ions and conjugated ligands on a honeycomb-kagome lattice. By combining chemical modifications and an electrostatic gating approach, we attain a MOF, Mn3(THQ)2 (THQ = tetrahydroxyquinone) possessing the square-planar Mn–O coordination and minimal-sized ligand, which exhibits an ultra-high Curie temperature of 250 K in the neutral monolayer and 300 K in the hole doped one. These findings manifest both chemical and electrical means as powerful tools to tailor the magnetism of 2D MOFs on demand, and further highlights their prospect for applications in ultracompact spintronic devices.
1. Introduction
With the rapid development of two-dimensional (2D) materials, 2D magnets have attracted much attention for their many intriguing properties and potential applications in new magnetic and spintronic devices.1 The most exciting news in the field includes discovery of the intrinsic long-range magnetic order in atomically thin layers2,3 and realization of the electrical control of 2D magnetism.4–8 The giant magnetoresistance,9–11 stacking-dependent magnetic properties12,13 and pressure effects14,15 have also been observed in van der Waals layered materials and heterostructures. Representative 2D magnetic materials so far fabricated and well characterized include monolayer CrI3,2 Fe3GeTe2,8 and few-layer Cr2Ge2Te6.3 The Curie temperature has been found to decrease in the 2D limit with respect to the bulk value, and is well below 300 K which severely hampers practical applications of 2D magnets. Indeed, the room-temperature ferromagnetism in ultrathin layers is strongly pursued. Methods that can enhance the magnetic ordering temperature, such as charge doping of Cr2Ge2Te6via electrostatic gating16 and changing the Fe stoichiometry in Fe3GeTe2,17,18 have been proposed. In addition to van der Waals atomic crystals, a new type of 2D magnets, 2D metal–organic frameworks (MOFs) have caught our attention, which also show attractive properties of high porosity, chemical diversity, and superior conductivity.19–24 In 2D magnetic MOFs, transition metal ions are incorporated into lattices of various symmetry and linked by conjugated organic ligands through metal–ligand coordination. The large variety of metal ions, organic ligands, and topological structures render 2D MOFs an ideal platform for exploring the low-dimensional magnetism as well as its coupling to other intriguing properties, such as magneto-optics, magneto-electronics and multiferroics. Currently, theoretical investigations of 2D magnetic MOFs mainly focus on those with a Lieb lattice,25–29 however, the most extensively explored 2D MOFs in experiments are those with a honeycomb-kagome lattice symmetry, whose magnetic properties have not been paid much attention to,19,21,30–33 and strong magnetic interactions and high magnetic ordering temperature, Tc, have not yet been reported. The lack of fundamental understanding of the magnetic mechanism, including exchange interactions and magneto-crystalline anisotropy in 2D MOFs significantly slows down the development of new 2D magnets with desired functionalities. In the first place, how metal ions, organic ligands, and coordination atoms impact the magnetic behavior of 2D MOFs is not clear. Secondly, control of the magnetism in 2D MOFs via electric field or electrostatic gating, which is of great relevance to their device applications, has never been proposed. Thereby, tailoring magnetic properties of 2D MOFs on demand by virtue of chemical and electrical approaches is becoming a central topic of the area.In this work, we perform the first-principles electronic structure calculation and Monte Carlo simulation based on a generalized Heisenberg model to investigate the magnetic properties of 2D MOFs on a honeycomb-kagome lattice. Mn, Fe, and Co divalent ions are incorporated into these MOFs as metal nodes and triphenylene derivatives are organic linkers with O, N, and S as metal coordination atoms, respectively. Among these MOFs, Mn3(HTTP)2 (HTTP = 2,3,6,7,10,11-hexahydroxytriphenylene) with the Mn–O coordination exhibits the strongest exchange interaction and out-of-plane magneto-crystalline anisotropy. We unveil that the intralayer FM order originates from the d–π–d super-exchange interaction between metal ions on a kagome sublattice mediated by conjugated organic ligands on a honeycomb sublattice. More interestingly, either electron or hole doping above a critical value can trigger a ferromagnetic (FM) to ferrimagnetic (FIM) transition in it. By substituting HHTP with a minimal-sized organic ligand, tetrahydroxyquinone (THQ), we attain a new 2D MOF, Mn3(THQ)2, which exhibits a high Curie temperature of 250 K in the neutral monolayer and 300 K in the hole doped one. These findings manifest both chemical modifications and electrostatic gating as robust tools to control the magnetism in 2D conjugated MOFs.
2. Methods
2.1 DFT calculations
All the electronic structure calculations were performed within the framework of density functional theory (DFT) using the projector augmented-wave (PAW)34,35 method and the exchange-correlation functional of Perdew–Burke–Ernzerhof (PBE), as implemented in the Vienna ab initio simulation package (VASP 5.4).36,37 The DFT + U38 method was chosen to treat the Coulomb and exchange interactions between electrons in the localized d-orbitals of transition metals. The Coulomb (U) and exchange (J) interaction parameters were set to be U = 4 eV and J = 1 eV, which have been adopted in previous studies.21,25,26,39–41 Our test on the U and J parameters was provided in Fig. S9.† A vacuum space of 20 Å along the z direction was applied. The lattice parameters and atomic positions were fully relaxed until the force on each atom was less than 0.01 eV Å−1. The convergence criteria for the self-consistent-field energy and the energy cutoff for the plane-wave basis were 1 × 10−5 eV and 600 eV, respectively. The 3 × 3 × 1 Monkhorst–Pack k-mesh in the first Brillouin zone was used to calculate the electronic and magnetic properties of monolayer M3X2 (M = Mn, Fe, and Co; X = HHTP, HITP, and HTTP), and the 5 × 5 × 1 k-mesh was used for monolayer Mn3(THQ)2. In the calculation of DOS, the 6 × 6 × 1 k-mesh was used for the former and 15 × 15 × 1 k-mesh was used for the latter.2.2 Monte Carlo simulation
The generalized Heisenberg model was used to describe the magnetic interactions in 2D MOFs and the Curie temperature was derived from the Monte Carlo simulation based on it. For the Hamiltonian given in eqn (4), interactions were restricted to the nearest-neighbor magnetic sites, so Jij = J and Bij = B. There were three identical sites in each unit cell of the honeycomb-kagome lattice, so Ai = A too. By mapping the Heisenberg model to total energies of the FM and FIM states calculated from first-principles, we derived the expressions for J, A, and B as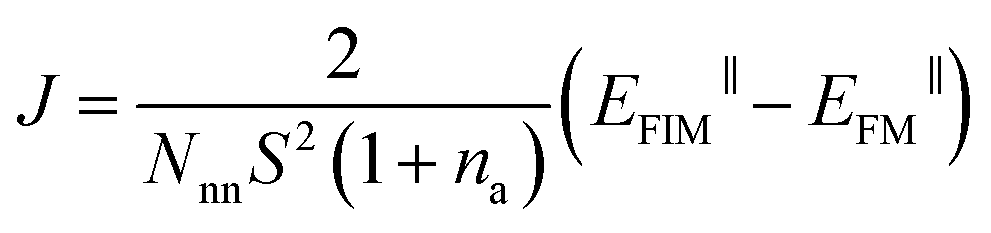 | (1) |
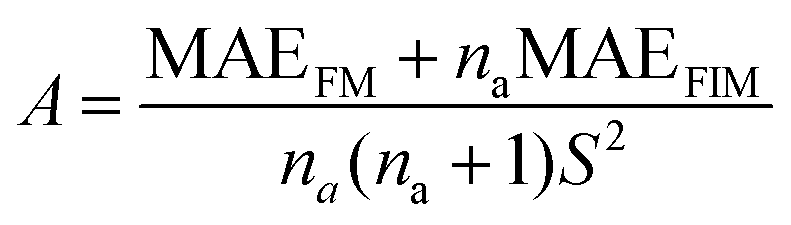 | (2) |
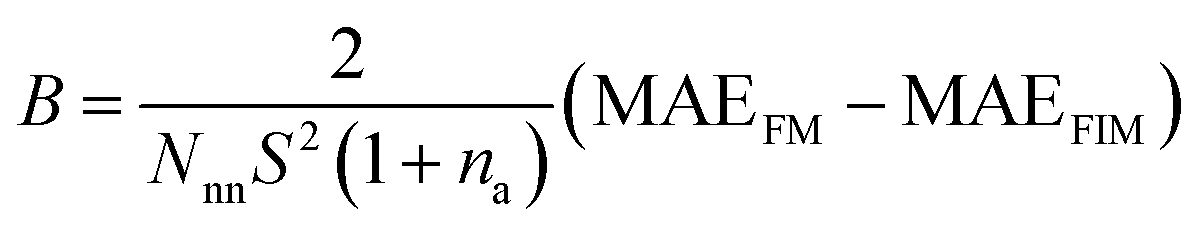 | (3) |
 . A 20 × 20 supercell was used in the Monte Carlo simulation and the adaptive Metropolis algorithm42 was chosen to determine whether a new spin configuration was accepted or rejected. At each temperature, 50 trajectories with each trajectory lasting for 15
. A 20 × 20 supercell was used in the Monte Carlo simulation and the adaptive Metropolis algorithm42 was chosen to determine whether a new spin configuration was accepted or rejected. At each temperature, 50 trajectories with each trajectory lasting for 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 steps were generated and the final data was averaged over them. The method has been applied successfully to predicting the Curie temperature of monolayer CrI3.43
000 steps were generated and the final data was averaged over them. The method has been applied successfully to predicting the Curie temperature of monolayer CrI3.43
3. Results and discussion
3.1 Magnetic ground state of 2D MOFs
We construct a series of 2D conjugated MOFs with the same formula M3X2 based on divalent transition metal ions in the third period, M = Mn, Fe, and Co and organic ligands, X = HHTP (2,3,6,7,10,11-hexahydroxytriphenylene), HITP (2,3,6,7,10,11-hexaiminotriphenylene), and HTTP (2,3,6,7,10,11-hexathiolatetriphenylene) in which the coordination atoms are O, N, and S respectively. All these MOFs share the same honeycomb-kagome lattice with organic ligands located on the honeycomb sublattice and metal ions on the kagome sublattice, as shown in Fig. 1a. Their optimized lattice parameters are provided in Table S1.† Three metal ions and two ligands exist in a rhombic unit cell of M3X2, and each metal ion is coordinated by four donor atoms (O, N, or S), forming a square-planar configuration. According to the ligand field theory, the five degenerate d-orbitals in transition metal ions will split into four sublevels,44,45 which are dxy, dxz/dyz, dz2, and dx2−y2, respectively as shown in Fig. S1.† The magnetism of these MOFs originates from the unpaired d-electrons and associated local spins residing on the transition metal ions. For Mn, Fe, and Co divalent ions, the number of unpaired electrons is 3, 2, and 1 respectively with a spin S = 3/2, 1, and 1/2, which is confirmed by our first-principles calculations. The magnetic ground state is determined by comparing total energies of two possible magnetic states, i.e., ferromagnetic (FM) and ferrimagnetic (FIM) states. We define the intralayer exchange energy as Eex = EFIM − EFM. When Eex > 0 the system is FM at 0 K and when Eex < 0 it is FIM. As shown in Table 1, all the MOFs studied are FM in their ground state, whose spin-resolved band structures are shown in Fig. S2.† Both coordination atoms and metal ions have remarkable influence on the magnetic properties of MOFs. For MOFs possessing the same metal node, such as Mn3(HHTP)2, Mn3(HITP)2, and Mn3(HTTP)2, when the coordination atom varies from O to N and to S the Eex reduces by more than one order of magnitude. A similar trend is observed in Fe3X2 and Co3X2 (X = HHTP, HITP, and HTTP). Among them, the magnetic exchange interaction in MOFs with Mn–O and Fe–O coordination is the strongest, whose Eex is 110.84 meV per f.u. and 118.76 meV per f.u., respectively. In the following, we take the Mn–O MOF, Mn3(HHTP)2 (Fig. 1b) as an example to elucidate the magnetic mechanism of this new type of 2D magnets and the role of organic ligands in establishing magnetic interactions between metal ions.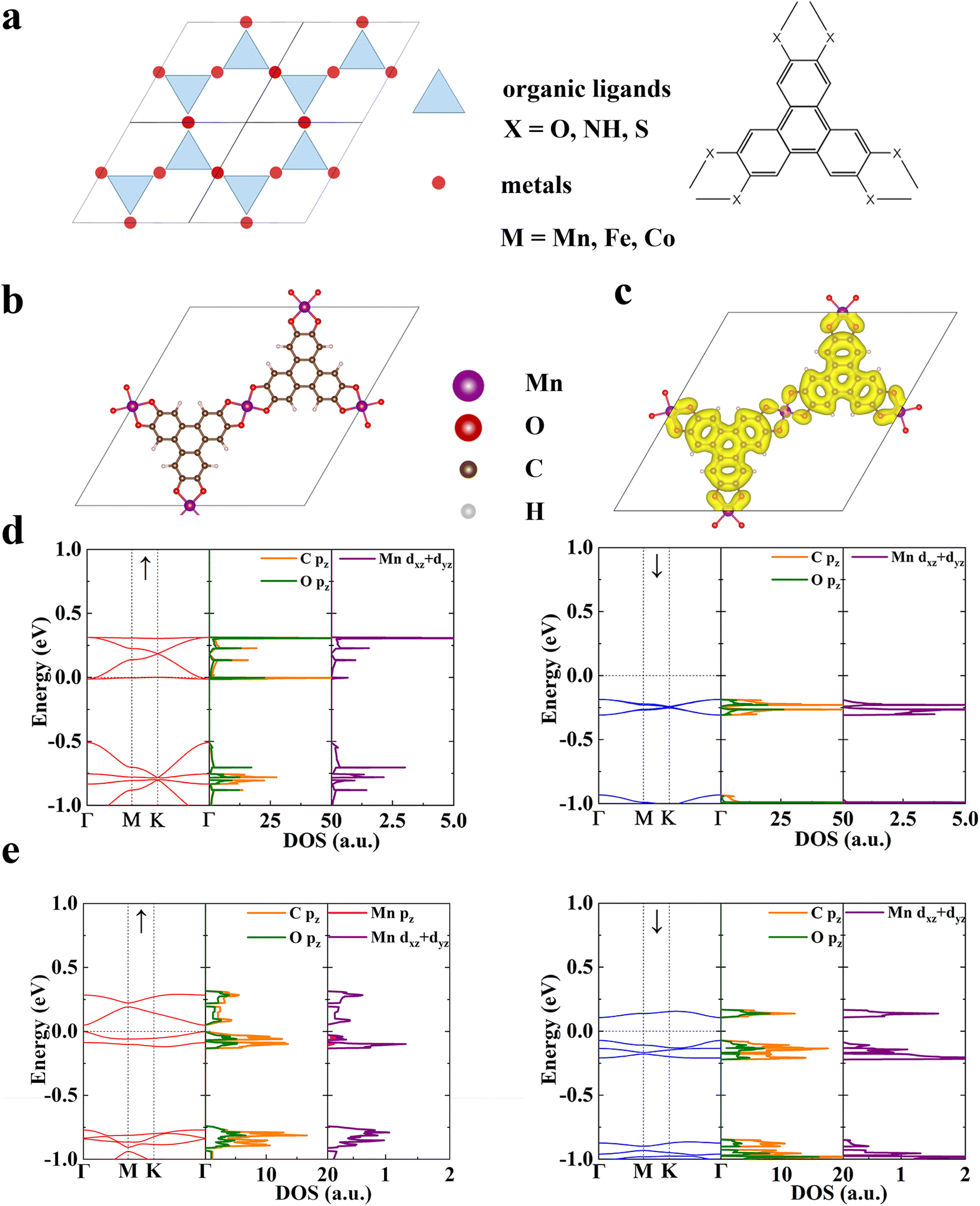 | ||
| Fig. 1 Magnetic ground state of monolayer Mn3(HHTP)2. (a) Schematics of 2D conjugated MOFs on a honeycomb-kagome lattice. Blue triangles and red circles represent organic ligands and metal ions, respectively. (b) Unit cell. (c) Total charge density distribution with an isovalue of 0.01 e/Bohr3. (d) Spin-resolved band structure and density of states (DOS) of the FM state. (e) Those of the FIM state. The red and blue lines represent majority and minority spin channels, respectively. The high symmetry k-points are Γ = (0, 0, 0), M = (1/2, 0, 0), and K = (2/3, 1/3, 0). The Fermi level has been shifted to 0. | ||
The total charge density distribution and spin-resolved band structure of Mn3(HHTP)2 in its FM ground state are shown in Fig. 1c and d, respectively. The band structure in one spin channel exhibits the typical feature of organic ligands on a honeycomb lattice: four bands sit right above the Fermi level EF, with the bottom and top ones completely dispersionless and the middle two crossing at the K point.46,47 The partial density of states (pDOS) analysis confirms that the four electronic bands arise primarily from the pz orbitals of C and O atoms in the organic ligands, with a small contribution from the dxz and dyz orbitals of Mn ions, forming the π–d conjugation. As evidenced by the partial charge density distribution at the M and K points in the Brillion zone (Fig. S3†), the bottom flat band coinciding with EF arises mainly from the π orbitals isolated on the organic ligands with destructive interference, which was previously observed in a Cu-hexaiminobenzene framework, Cu3(HAB)2 where dichotomy between local spins and conjugated electrons was demonstrated.47 The d-bands of three Mn ions on the kagome lattice show a typical feature of ‘one flat and two dispersive bands’ and are located far from the Fermi level (Fig. S4†). In the other spin channel, a bandgap of 1.80 eV shows up indicating that Mn3(HHTP)2 is a half metal. The metastable FIM state of Mn3(HHTP)2 is semiconducting with a bandgap of 0.178 eV in one spin channel while 0.051 eV in the other (Fig. 1e).
To understand the magnetic interactions in Mn3(HHTP)2, we plot the spin density isosurface in Fig. 2a. In the FM state, the spins residing on all three Mn ions are identical with S = 3/2, corresponding to the three unpaired d-electrons in each ion. And the spins on metal ions and organic ligands align in opposite directions, indicating the antiferromagnetic coupling between them. Since the distance between two nearest-neighbor Mn ions is 10.86 Å, the direct Coulomb exchange48 is impossible. Because the three unpaired d-electrons are localized on dxz/dyz and dz2 orbitals of Mn ions (Fig. S1†), we propose two super-exchange pathways responsible for the FM order in Mn3(HHTP)2. One is established between dxz/dyz orbitals of Mn ions mediated by pz orbitals of neighboring O and C atoms in the organic ligands (Fig. 2b). Here tdp,tπ, and Jπ represent the hopping integral between dxz/dyz and pz orbitals, that between two pz orbitals, and Coulomb exchange between two pz orbitals, respectively. The other pathway is established between two dz2 orbitals of Mn ions, as illustrated in Fig. 2c. In this case, the direct kinetic exchange between dz2 of Mn and pz of O is prohibited because they are orthogonal to each other, but the kinetic exchange between dz2 of Mn and px of O, together with Hund's coupling between px and pz of O, enables the super-exchange coupling between two dz2 orbitals of Mn ions. As such, the ferromagnetic interaction between metal ions will be affected significantly by the transition matrix element tdp, whose magnitude can be illustrated by the degree of π–d conjugation. We have obtained charge and spin density distributions (Fig. S5†) as well as the metal coordination bond length (Table S2†) in the secondary building units (SBUs)49 of Mn MOFs. It shows that the π–d conjugation diminishes when the coordination atom goes from O to N and to S. Accordingly, the exchange energy Eex in Mn3X2 (X = HHTP, HITP, and HTTP) decreases in the same order (Table 1). Similar results were also observed in metal-phthalocyanine-based 2D MOFs.26
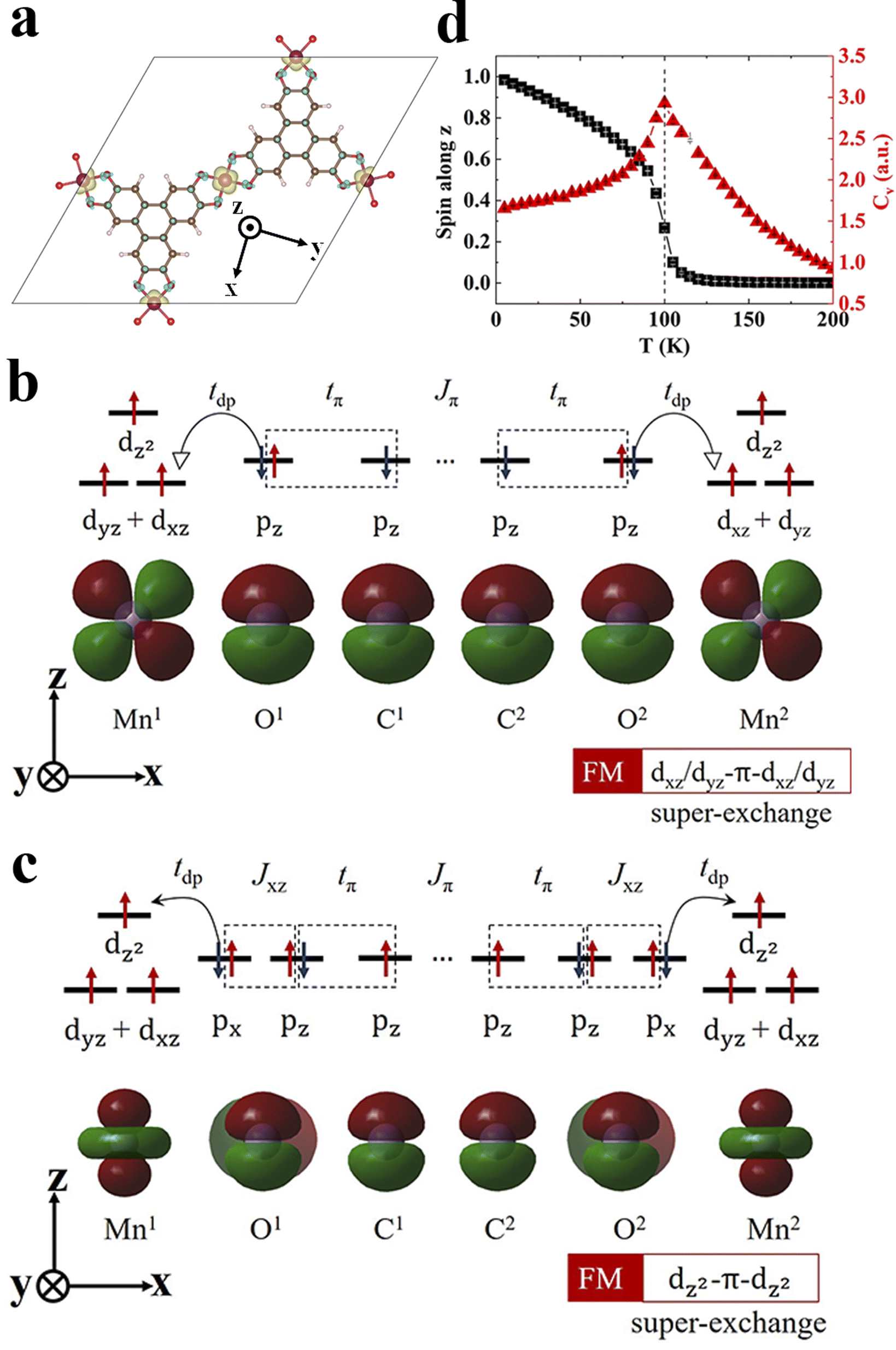 | ||
| Fig. 2 Magnetic exchange mechanism and magnetic transition in monolayer Mn3(HHTP)2. (a) Spin density isosurface with an isovalue of 0.01 e/Bohr3. The majority and minority spins are colored in yellow and cyan, respectively. (b and c) Two ferromagnetic super-exchange pathways proposed. (d) Normalized spin per Mn ion and heat capacity changing with the temperature from the Monte Carlo simulation. | ||
3.2 Curie temperature Tc of 2D MOFs
The magnetic exchange coupling tends to align neighboring spins at 0 K. As the temperature goes up, thermal fluctuations will destroy this long-range magnetic order and the system undergoes a phase transition above a critical temperature (Tc). The influence of thermal fluctuations depends on the dimensionality of a system. In a 3D system, the transition always occurs at a finite temperature. However, in a 1D system the macroscopic magnetic order cannot be retained at any finite temperature. In a 2D system, whether a phase transition occurs relies on the spin dimensionality,50 which is defined as the number of spin components involved in the magnetic interaction and also known as the magnetic anisotropy. The magnetic interactions in a system can be described by a Heisenberg model, and for a 2D magnet the Mermin–Wagner–Hohenberg theorem51 states that the magnetic anisotropy plays a key role in keeping the magnetic order from the detrimental effect of thermal fluctuations; a 2D isotropic Heisenberg model with the spin dimensionality of three predicts no long-range magnetic order at any finite temperature. Here we use a generalized Heisenberg model of the form52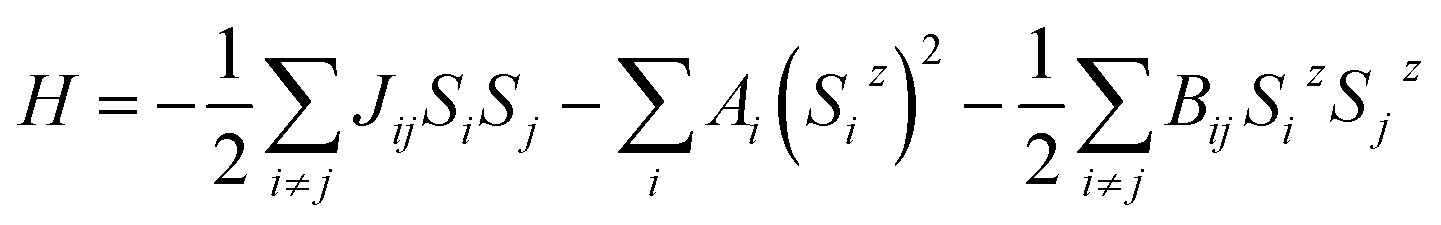 | (4) |
With all the parameters in the Heisenberg model derived, we predict Tc for 2D MOFs by performing Monte Carlo simulations. The normalized spin along the z-axis, Sz and the specific heat capacity, Cv at different temperatures are shown in Fig. 2d for Mn3(HHTP)2 whose easy axis is apparently the z-axis. The Sz drops abruptly to zero above a critical temperature Tc, and concurrently Cv diverges there. Since the magnetic couplings in Mn3(HHTP)2 and Fe3(HHTP)2 with Mn–O and Fe–O coordinations are the strongest of all, their Curie temperatures Tc are also the highest, 100 K and 105 K, respectively. The evolution of Sx and Sy with temperature is shown in Fig. S6a† for Mn3(HHTP)2. Both are nearly zero in the whole range of temperatures, which corroborates with the strong magnetocrystalline anisotropy along the z-axis. By contrast, Sx and Sy are almost identical and much larger than Sz in Fe3(HHTP)2 when T < Tc (Fig. S6b†), exhibiting the in-plane magnetic anisotropy.
3.3 Magnetic modulation by chemical modifications
The Curie temperature of above-mentioned 2D MOFs is still far below the room temperature. We have learned that when the magnetic anisotropy is similar, the exchange coupling will be the determining factor of the magnetic transition temperature, and the stronger coupling often leads to the higher Tc. For example, the exchange energy of Mn3(HHTP)2 is 110.84 meV per f.u. and Tc is 100 K, while that of Mn3(HTTP)2 is 3.57 meV per f.u. and Tc is 5 K (Table S4†). So, we should enhance the exchange coupling in 2D MOFs for its ferromagnetic order to sustain at a higher temperature, which is crucial for their practical applications. Since the ferromagnetism in monolayer Mn3(HHTP)2 originates from the super-exchange coupling between the metal d-electrons mediated by the itinerant π-electrons in organic ligand, we reasonably presume that replacement of conjugated organic ligands with small-sized ones would shorten the distance between magnetic metal ions and remarkably enhance their super-exchange interaction. MOFs constructed by small-sized organic ligands, such as hexathiolbenzene (HTB),24,54–60 hexaiminobenzene (HIB)23,61,62 and tetrahydroxyquinone (THQ)63,64 have been reported, and they show many intriguing properties ranging from topological insulators and hydrogen evolution activity to high electrical conductivity, superconducting, and photo-induced charge transport. In particular, spin arrangements on the kagome lattice lead to strong geometrical magnetic frustration in Cu3(THQ)2.64 Moreover, monolayer Mn3(HTB)2 and Mn3(HIB)2 with the square-planar Mn–S and Mn–N coordination, respectively, were predicted to be a half-metallic ferromagnet with Tc of about 212 K56 and 450 K65 based on the Ising model. Our results on M3X2 (X = HHTP, HITP, and HTTP with O, N, and S as coordination atoms, respectively) show that the Mn–O coordination possesses stronger π–d conjugation and exchange coupling than the Mn–S coordination. Therefore, we design a new MOF, Mn3(THQ)2 (Fig. 3a) with both the Mn–O coordination and the minimal-sized conjugated ligand, which should exhibit stronger magnetic coupling and higher Tc than Mn3(HHTP)2 and Mn3(HTB)2. Indeed, our calculations confirm that monolayer Mn3(THQ)2 is FM in its ground state and the exchange energy Eex is 295.31 meV per f.u., more than twice that of Mn3(HHTP)2, which unequivocally manifests chemical modifications as an effective approach to modulate the magnetism of 2D MOFs. The charge and spin density distributions of Mn3(THQ)2 and simulated scanning tunneling microscopy (STM) topographies of Mn3(THQ)2 surfaces are plotted in Fig. S7.† The STM topography with a bias of 0 eV clearly exhibits the honeycomb feature, corresponding to the itinerant electrons of organic ligands. At the bias of −2.21 eV below the Fermi level, a kagome pattern is observed, representing the d-electrons of metals ions. The subsequent Monte Carlo simulation based on the generalized Heisenberg Hamiltonian and a new set of parameters derived from the first-principles calculations predict Tc to be 250 K (Fig. 3b), substantially higher than 100 K predicted for Mn3(HHTP)2. This Tc of 250 K is even higher than the highest ferromagnetic ordering temperature of 225 K66 reported for MOFs, and the latter was observed in a mixed-valence 3D MOF Cr(tri)2(CF3SO3)0.33 (tri = 1, 2, 3-triazole). According to previous studies of van der Waals magnetic crystals including CrI3,2 Fe3GeTe2,8,67 FePS3,68 and CrGeTe3,3 the critical temperature Tc decreases as the layer thickness is reduced from 3D to 2D, so we anticipate the new MOF to show even higher Tc in its bulk form. Monolayer Mn3(THQ)2 is half metallic in its FM ground state with no bandgap in one spin channel while a bandgap of 1.51 eV in the other (Fig. 3c), and it is fully metallic in the FIM metastable state (Fig. 3d).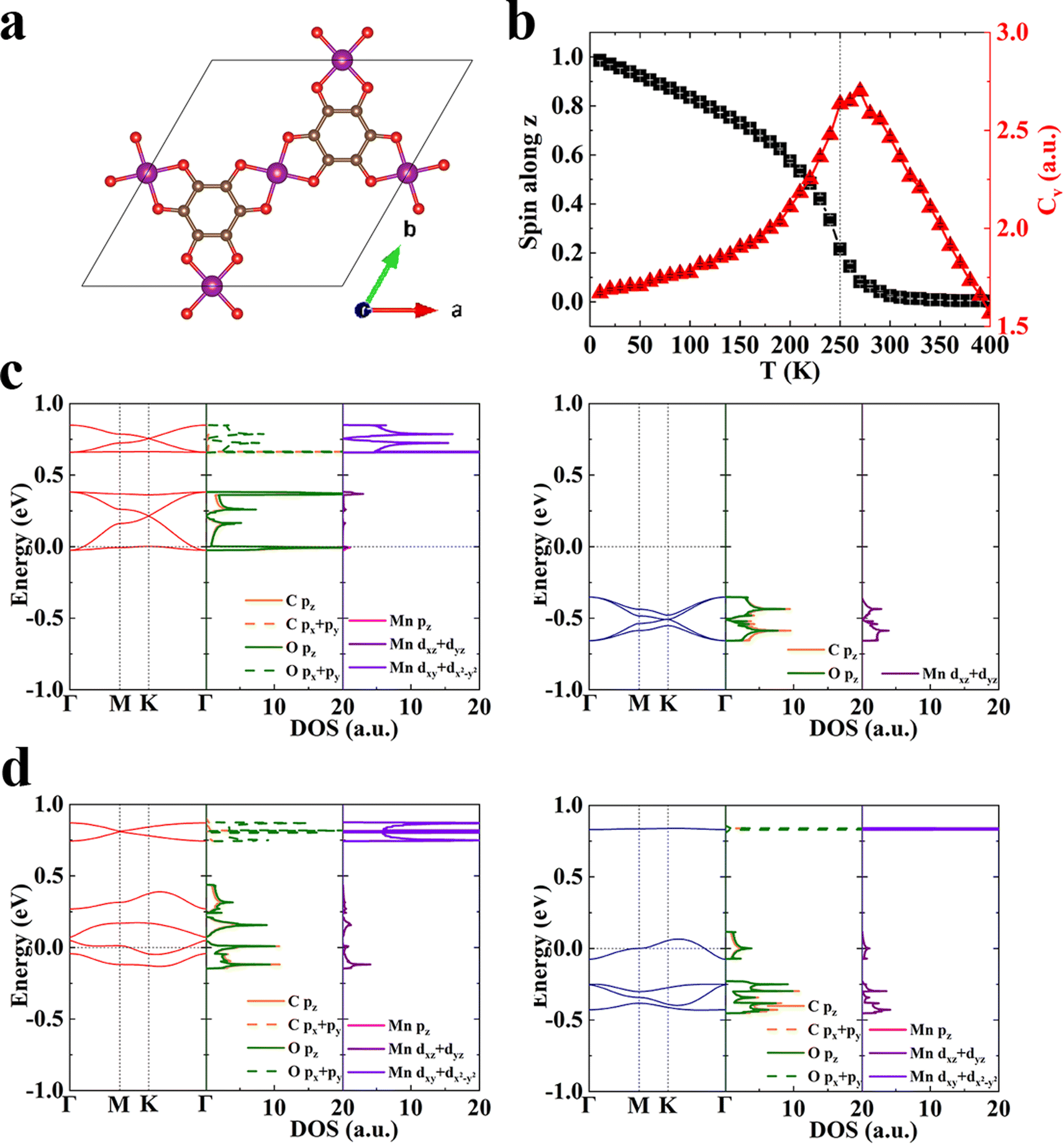 | ||
| Fig. 3 Magnetic properties of monolayer Mn3(THQ)2. (a) Unit cell. (b) Normalized spin per Mn ion and heat capacity changing with the temperature from the Monte Carlo simulation. (c) Spin-resolved band structure and pDOS of the FM state. (d) Those of the FIM state. The red and blue lines represent majority and minority spin channels, respectively. The high symmetry k-points are Γ = (0, 0, 0), M = (1/2, 0, 0), and K = (2/3, 1/3, 0). The Fermi level has been shifted to 0. | ||
3.4 Magnetic modulation by electrostatic gating
In addition to chemical modifications, which range from metal and coordination atom substitutions to the varying size of conjugated organic ligands, modulating the magnetism of 2D MOFs by physical means such as the application of an electric or magnetic field is also highly desirable and holds great promise for their device applications. Recently, the electrical control of magnetism has been realized in 2D CrI3 (ref. 4,7) and Cr2Ge2Te6.6 Both electric field and electrostatic gating approaches have been proved effective in these 2D magnets. Here we demonstrate modulation of the magnetism in monolayer Mn3(HHTP)2 and Mn3(THQ)2 by electrostatic gating and unravel the mechanism behind it.In the experimental set-up of electrostatic gating, charge carriers are injected into 2D magnets by applying a gate voltage. We calculate total energies of the charged monolayer of MOFs in their FM and FIM states, respectively, following the method proposed by Reed and co-workers in their study on the structural phase transition in 2D transition metal dichalcogenides induced by electrostatic gating.69 The energy difference between FM and FIM states, namely, the exchange energy Eex = EFIM − EFM of atomically thin Mn3(HHTP)2 has been obtained as a function of the doping charge density (Fig. 4a). It shows that either the hole or the electron doping can trigger a transition from the FM to FIM state, when a critical hole density of 1.87 e per f.u. (4.58 × 1013 e cm−2) or electron density of 1.32 e per f.u. (3.22 × 1013 e cm−2) is reached. Now we consider a capacitor structure70,71 with the monolayer of Mn3(HHTP)2 deposited on top of the dielectric layer consisting of 5 nm HfO2 and sandwiched between two parallel metal plates, on which a gate voltage is applied. The voltage required to trigger the transition is then estimated by 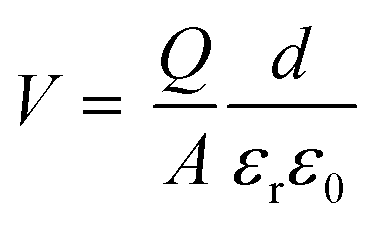 where
where 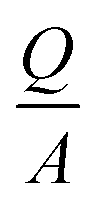 is the critical charge density, d = 5 nm is the thickness and εr = 25 is the relative permittivity of HfO2 dielectric, and ε0 is the permittivity of vacuum (8.854 × 10−12 F m−1). The critical voltage of 1.66 V and −1.16 V is obtained for hole and electron injection, respectively, which is far below the experimental breakdown voltage for HfO2 dielectric of similar thickness.69
is the critical charge density, d = 5 nm is the thickness and εr = 25 is the relative permittivity of HfO2 dielectric, and ε0 is the permittivity of vacuum (8.854 × 10−12 F m−1). The critical voltage of 1.66 V and −1.16 V is obtained for hole and electron injection, respectively, which is far below the experimental breakdown voltage for HfO2 dielectric of similar thickness.69
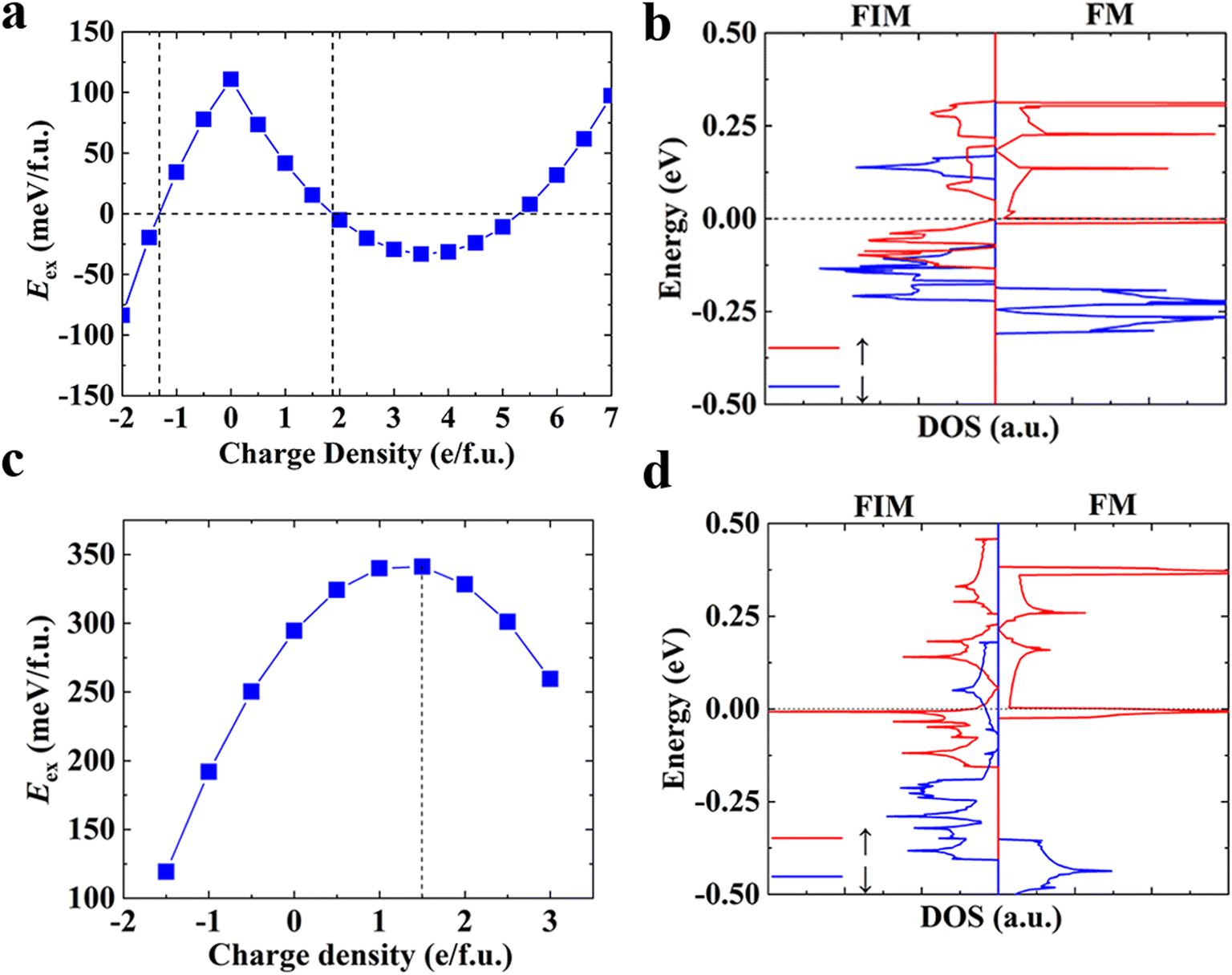 | ||
| Fig. 4 Controlling the magnetism in monolayer Mn3(HHTP)2 and Mn3(THQ)2 by electrostatic gating. (a and c) Intralayer exchange energy Eex = EFIM − EFM as a function of the doping charge density in (a) Mn3(HHTP)2 and (c) Mn3(THQ)2. The positive or negative charge density denotes extra hole or electron injected to the system. (b and d) DOS of the FIM and FM states in (b) Mn3(HHTP)2 and (d) Mn3(THQ)2. The red and blue lines represent majority and minority spin channels, respectively. The Fermi level has been shifted to 0. | ||
The neutral monolayer of Mn3(HHTP)2 is FM in its ground state, and the energy difference between FM and FIM states decreases with charge doping (Fig. 4a). When the charge density reaches a critical value, the order of their energies is reversed and the ground state becomes FIM. This result can be understood from the total DOS of neutral Mn3(HHTP)2 in Fig. 4b. When the electron is injected into it, the empty electronic states above the Fermi level will be occupied in the first place. Mn3(HHTP)2 is a half metal in the FM state, with vanishing band gap in one spin channel whereas a wide band gap in the other channel. If one electron per formula unit is injected, the Fermi level will be lifted by +0.15 eV, where a crossing of two dispersive bands leads to abruptly reduced DOS. If the electron is further injected, it will occupy higher electronic states and the energy gain of the charged monolayer will be slowed down. In contrast, although in the FIM state there exist small band gaps in both spin channels, more low-lying unoccupied electronic states are available for both channels. If the same amount of electron is injected, the energy will decrease more rapidly. As a result, the magnetic ground state turns FIM when a critical amount of electron is injected. For the positively charged monolayer, the electron is to be removed from the occupied valence bands. There are much more states available right below the Fermi level in the FIM state than the FM one. Therefore, removal of electron in the FM state costs more energy than the FIM one, leading to more energy increase in the former which will trigger the FM-to-FIM transition when the hole density reaches a critical value.
When it comes to monolayer Mn3(THQ)2 (Fig. 4c), the exchange energy decreases upon the electron doping, but the FM-to-FIM transition does not show up even after a large amount of electron is injected. While upon the hole doping, the exchange energy first increases then decreases. We can understand these results from the total DOS (Fig. 4d): there are more low-lying electronic states available above the Fermi level in the FIM than the FM state, so when the same amount of electron is injected, there will be more energy gain in the former, leading to the decreased energy difference between them. Whereas in the case of hole doping, since there exist more occupied electronic states right below the Fermi level in the FM state than the FIM one, when the electron is removed, there will be less energy cost in the former, leading to the increased energy difference between them at the start. However, an energy gap shows up below these states in the FM state, so when more electron is removed, there will be more energy cost, causing the decreased energy difference thereafter. When the hole doping density reaches 1.5 e per f.u., which amounts to 9.89 e cm−2 and can be realized by applying a gate voltage of 3.58 V based on the same capacitor model mentioned above, the energy difference between FM and FIM states, which is also known as the exchange energy, exhibits a maximum of 341.19 meV. If we ignore the effect of hole doping on the magnetic anisotropy, a Tc up to 300 K can be anticipated for the positively charged monolayer of Mn3(THQ)2 (Figure S8†).
4. Conclusions
To conclude, we have studied the magnetism of a new type of 2D materials, 2D conjugated MOFs with a honeycomb-kagome lattice symmetry, and unveiled how the magnetism is affected by the transition metal ions, coordination atoms, and the size of organic ligands. We demonstrate that the π–d conjugation between metal ions and organic linkers in 2D MOFs is essential for the long-distance magnetic interaction and communication. For 2D MOFs, high spin states of metal ions, strong couplings between the d-orbitals of metal ions and the p-orbitals of coordination atoms, and short distance between adjacent metal ions are required to facilitate efficient super-exchange interactions. We design a new MOF, Mn3(THQ)2, with the Mn–O coordination and shortest distance between adjacent metal ions to enhance the magnetic couplings between them, and it exhibits an ultra-high Curie temperature of 250 K. Other than the 2D MOFs studied here, MOFs with organic radical linkers or those combining 3d transition metal ions with high electron spins and 4d/5d transition metal ions with large spin–orbit couplings to enhance the magnetic interaction and magnetic anisotropy are also proposed to show the high temperature magnetism.72 The interlayer stacking and magnetic couplings can lead to the layer-dependent magnetism in 2D MOFs, which is of great interest for future studies. In addition to chemical modifications, we find that electrostatic gating offers an alternative route to modulate the magnetism of 2D MOFs. The charge doping above a critical value, which can be achieved by applying a gate voltage of a few volts, can either trigger a FM-to-FIM transition or enhance the magnetic couplings in 2D MOFs to attain the room-temperature ferromagnetism. As compared to the magnetic field, the voltage control of magnetism is more precise and convenient, which has technical importance for developing energy-efficient data storage. The successful modulation of magnetism in 2D MOFs by both chemical and electrical approaches, as showcased here, provides new opportunities for exploring applications of 2D magnets in ultracompact electrically controlled spintronic devices such as spin valves, magnetic tunnel junctions, and spin field-effect transistors.Author contributions
D. W. supervised this study. Q. Y. performed all the calculations. D. W. and Q. Y. discussed the results and co-wrote the paper. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no conflicts of interests.Acknowledgements
This work is supported by the National Natural Science Foundation of China (Grant No. 22073055). Computational resources are provided by the Tsinghua Supercomputing Center.Notes and references
- Q. H. Wang, A. Bedoya-Pinto, M. Blei, A. H. Dismukes, A. Hamo, S. Jenkins, M. Koperski, Y. Liu, Q. C. Sun, E. J. Telford, H. H. Kim, M. Augustin, U. Vool, J. X. Yin, L. H. Li, A. Falin, C. R. Dean, F. Casanova, R. F. L. Evans, M. Chshiev, A. Mishchenko, C. Petrovic, R. He, L. Zhao, A. W. Tsen, B. D. Gerardot, M. Brotons-Gisbert, Z. Guguchia, X. Roy, S. Tongay, Z. Wang, M. Z. Hasan, J. Wrachtrup, A. Yacoby, A. Fert, S. Parkin, K. S. Novoselov, P. Dai, L. Balicas and E. J. G. Santos, ACS Nano, 2022, 16, 6960–7079 CrossRef CAS PubMed.
- B. Huang, G. Clark, E. Navarro-Moratalla, D. R. Klein, R. Cheng, K. L. Seyler, D. Zhong, E. Schmidgall, M. A. McGuire, D. H. Cobden, W. Yao, D. Xiao, P. Jarillo-Herrero and X. Xu, Nature, 2017, 546, 270–273 CrossRef CAS PubMed.
- C. Gong, L. Li, Z. Li, H. Ji, A. Stern, Y. Xia, T. Cao, W. Bao, C. Wang, Y. Wang, Z. Q. Qiu, R. J. Cava, S. G. Louie, J. Xia and X. Zhang, Nature, 2017, 546, 265–269 CrossRef CAS PubMed.
- S. Jiang, J. Shan and K. F. Mak, Nat. Mater., 2018, 17, 406–410 CrossRef CAS PubMed.
- B. Huang, G. Clark, D. R. Klein, D. MacNeill, E. Navarro-Moratalla, K. L. Seyler, N. Wilson, M. A. McGuire, D. H. Cobden, D. Xiao, W. Yao, P. Jarillo-Herrero and X. Xu, Nat. Nanotechnol., 2018, 13, 544–548 CrossRef CAS PubMed.
- Z. Wang, T. Zhang, M. Ding, B. Dong, Y. Li, M. Chen, X. Li, J. Huang, H. Wang, X. Zhao, Y. Li, D. Li, C. Jia, L. Sun, H. Guo, Y. Ye, D. Sun, Y. Chen, T. Yang, J. Zhang, S. Ono, Z. Han and Z. Zhang, Nat. Nanotechnol., 2018, 13, 554–559 CrossRef CAS PubMed.
- S. Jiang, L. Li, Z. Wang, K. F. Mak and J. Shan, Nat. Nanotechnol., 2018, 13, 549–553 CrossRef CAS PubMed.
- Y. Deng, Y. Yu, Y. Song, J. Zhang, N. Z. Wang, Z. Sun, Y. Yi, Y. Z. Wu, S. Wu, J. Zhu, J. Wang, X. H. Chen and Y. Zhang, Nature, 2018, 563, 94–99 CrossRef CAS PubMed.
- T. Song, X. Cai, M. W. Tu, X. Zhang, B. Huang, N. P. Wilson, K. L. Seyler, L. Zhu, T. Taniguchi, K. Watanabe, M. A. McGuire, D. H. Cobden, D. Xiao, W. Yao and X. Xu, Science, 2018, 360, 1214–1218 CrossRef CAS PubMed.
- Z. Wang, I. Gutierrez-Lezama, N. Ubrig, M. Kroner, M. Gibertini, T. Taniguchi, K. Watanabe, A. Imamoglu, E. Giannini and A. F. Morpurgo, Nat. Commun., 2018, 9, 2516 CrossRef PubMed.
- D. R. Klein, D. MacNeill, J. L. Lado, D. Soriano, E. Navarro-Moratalla, K. Watanabe, T. Taniguchi, S. Mannsfeld, P. Canfield, J. Fernández-Rossier and P. Jarillo-Herrero, Science, 2018, 360, 1218–1222 CrossRef CAS PubMed.
- W. Chen, Z. Sun, Z. Wang, L. Gu, X. Xu, S. Wu and C. Gao, Science, 2019, 366, 983–987 CrossRef CAS PubMed.
- L. Thiel, Z. Wang, M. A. Tschudin, D. Rohner, I. Gutierrez-Lezama, N. Ubrig, M. Gibertini, E. Giannini, A. F. Morpurgo and P. Maletinsky, Science, 2019, 364, 973–976 CrossRef CAS PubMed.
- T. Song, Z. Fei, M. Yankowitz, Z. Lin, Q. Jiang, K. Hwangbo, Q. Zhang, B. Sun, T. Taniguchi, K. Watanabe, M. A. McGuire, D. Graf, T. Cao, J. H. Chu, D. H. Cobden, C. R. Dean, D. Xiao and X. Xu, Nat. Mater., 2019, 18, 1298–1302 CrossRef CAS PubMed.
- T. Li, S. Jiang, N. Sivadas, Z. Wang, Y. Xu, D. Weber, J. E. Goldberger, K. Watanabe, T. Taniguchi, C. J. Fennie, K. Fai Mak and J. Shan, Nat. Mater., 2019, 18, 1303–1308 CrossRef CAS PubMed.
- I. A. Verzhbitskiy, H. Kurebayashi, H. Cheng, J. Zhou, S. Khan, Y. P. Feng and G. Eda, Nat. Electron., 2020, 3, 460–465 CrossRef CAS.
- A. F. May, D. Ovchinnikov, Q. Zheng, R. Hermann, S. Calder, B. Huang, Z. Fei, Y. Liu, X. Xu and M. A. McGuire, ACS Nano, 2019, 13, 4436–4442 CrossRef CAS PubMed.
- J. Seo, D. Y. Kim, E. S. An, K. Kim, G. Y. Kim, S. Y. Hwang, D. W. Kim, B. G. Jang, H. Kim, G. Eom, S. Y. Seo, R. Stania, M. Muntwiler, J. Lee, K. Watanabe, T. Taniguchi, Y. J. Jo, J. Lee, B. I. Min, M. H. Jo, H. W. Yeom, S. Y. Choi, J. H. Shim and J. S. Kim, Sci. Adv., 2020, 6, eaay8912 CrossRef PubMed.
- A. J. Clough, J. M. Skelton, C. A. Downes, A. A. de la Rosa, J. W. Yoo, A. Walsh, B. C. Melot and S. C. Marinescu, J. Am. Chem. Soc., 2017, 139, 10863–10867 CrossRef CAS PubMed.
- D. Sheberla, J. C. Bachman, J. S. Elias, C. J. Sun, Y. Shao-Horn and M. Dinca, Nat. Mater., 2017, 16, 220–224 CrossRef CAS PubMed.
- R. Dong, P. Han, H. Arora, M. Ballabio, M. Karakus, Z. Zhang, C. Shekhar, P. Adler, P. S. Petkov, A. Erbe, S. C. B. Mannsfeld, C. Felser, T. Heine, M. Bonn, X. Feng and E. Canovas, Nat. Mater., 2018, 17, 1027–1032 CrossRef CAS PubMed.
- R. W. Day, D. K. Bediako, M. Rezaee, L. R. Parent, G. Skorupskii, M. Q. Arguilla, C. H. Hendon, I. Stassen, N. C. Gianneschi, P. Kim and M. Dinca, ACS Cent. Sci., 2019, 5, 1959–1964 CrossRef CAS PubMed.
- J. H. Dou, L. Sun, Y. Ge, W. Li, C. H. Hendon, J. Li, S. Gul, J. Yano, E. A. Stach and M. Dinca, J. Am. Chem. Soc., 2017, 139, 13608–13611 CrossRef CAS PubMed.
- X. Huang, P. Sheng, Z. Tu, F. Zhang, J. Wang, H. Geng, Y. Zou, C. A. Di, Y. Yi, Y. Sun, W. Xu and D. Zhu, Nat. Commun., 2015, 6, 7408 CrossRef CAS PubMed.
- J. Zhou and Q. Sun, J. Am. Chem. Soc., 2011, 133, 15113–15119 CrossRef CAS PubMed.
- W. Li, L. Sun, J. Qi, P. Jarillo-Herrero, M. Dinca and J. Li, Chem. Sci., 2017, 8, 2859–2867 RSC.
- X. Li, X. Li and J. Yang, J. Phys. Chem. Lett., 2020, 11, 4193–4197 CrossRef CAS PubMed.
- H. Ai, X. Liu, B. Yang, X. Zhang and M. Zhao, J. Phys. Chem. C, 2018, 122, 1846–1851 CrossRef CAS.
- X. Li and J. Yang, J. Phys. Chem. Lett., 2019, 10, 2439–2444 CrossRef CAS PubMed.
- J. A. DeGayner, I. R. Jeon, L. Sun, M. Dinca and T. D. Harris, J. Am. Chem. Soc., 2017, 139, 4175–4184 CrossRef CAS PubMed.
- L. Yang, X. He and M. Dinca, J. Am. Chem. Soc., 2019, 141, 10475–10480 CrossRef CAS PubMed.
- Y. Misumi, A. Yamaguchi, Z. Zhang, T. Matsushita, N. Wada, M. Tsuchiizu and K. Awaga, J. Am. Chem. Soc., 2020, 142, 16513–16517 CrossRef CAS PubMed.
- X. Song, X. Wang, Y. Li, C. Zheng, B. Zhang, C. A. Di, F. Li, C. Jin, W. Mi, L. Chen and W. Hu, Angew. Chem., Int. Ed., 2020, 59, 1118–1123 CrossRef CAS PubMed.
- P. E. Blochl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- V. I. Anisimov, F. Aryasetiawan and A. I. Lichtenstein, J. Phys.: Condens. Matter, 1997, 9, 767–808 CrossRef CAS.
- J. Tan, W. Li, X. He and M. Zhao, RSC Adv., 2013, 3, 7016 RSC.
- H. Wende, M. Bernien, J. Luo, C. Sorg, N. Ponpandian, J. Kurde, J. Miguel, M. Piantek, X. Xu, P. Eckhold, W. Kuch, K. Baberschke, P. M. Panchmatia, B. Sanyal, P. M. Oppeneer and O. Eriksson, Nat. Mater., 2007, 6, 516–520 CrossRef CAS PubMed.
- M. Bernien, J. Miguel, C. Weis, M. E. Ali, J. Kurde, B. Krumme, P. M. Panchmatia, B. Sanyal, M. Piantek, P. Srivastava, K. Baberschke, P. M. Oppeneer, O. Eriksson, W. Kuch and H. Wende, Phys. Rev. Lett., 2009, 102, 047202 CrossRef CAS PubMed.
- J. D. Alzate-Cardona, D. Sabogal-Suarez, R. F. L. Evans and E. Restrepo-Parra, J. Phys.: Condens. Matter, 2019, 31, 095802 CrossRef CAS PubMed.
- D. Torelli and T. Olsen, 2D Mater., 2018, 6, 015028 CrossRef.
- S. Mitra, A. K. Gregson, W. E. Hatfield and R. R. Weller, Inorg. Chem., 1983, 22, 1729–1732 CrossRef CAS.
- C. G. Barraclough, R. L. Martin, S. Mitra and R. C. Sherwood, J. Chem. Phys., 1970, 53, 1638–1642 CrossRef CAS.
- W. Jiang, X. Ni and F. Liu, Acc. Chem. Res., 2021, 54, 416–426 CrossRef CAS PubMed.
- W. Jiang, Z. Liu, J. W. Mei, B. Cui and F. Liu, Nanoscale, 2019, 11, 955–961 RSC.
- Z. Xu, Y. Li, Y. Xu and W. Duan, Chin. Sci. Bull., 2021, 66, 535–550 CrossRef.
- M. Eddaoudi, D. B. Moler, H. Li, B. Chen, T. M. Reineke, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2001, 34, 319–330 CrossRef CAS PubMed.
- M. Gibertini, M. Koperski, A. F. Morpurgo and K. S. Novoselov, Nat. Nanotechnol., 2019, 14, 408–419 CrossRef CAS PubMed.
- N. D. Mermin and H. Wagner, Phys. Rev. Lett., 1966, 17, 1133–1136 CrossRef CAS.
- D. Torelli, K. S. Thygesen and T. Olsen, 2D Mater., 2019, 6, 045018 CrossRef CAS.
- L. Onsager, Phys. Rev., 1944, 65, 117–149 CrossRef CAS.
- T. Kambe, R. Sakamoto, K. Hoshiko, K. Takada, M. Miyachi, J. H. Ryu, S. Sasaki, J. Kim, K. Nakazato, M. Takata and H. Nishihara, J. Am. Chem. Soc., 2013, 135, 2462–2465 CrossRef CAS PubMed.
- Z. F. Wang, N. Su and F. Liu, Nano Lett., 2013, 13, 2842–2845 CrossRef CAS PubMed.
- M. Zhao, A. Wang and X. Zhang, Nanoscale, 2013, 5, 10404–10408 RSC.
- T. Kambe, R. Sakamoto, T. Kusamoto, T. Pal, N. Fukui, K. Hoshiko, T. Shimojima, Z. Wang, T. Hirahara, K. Ishizaka, S. Hasegawa, F. Liu and H. Nishihara, J. Am. Chem. Soc., 2014, 136, 14357–14360 CrossRef CAS PubMed.
- A. J. Clough, J. W. Yoo, M. H. Mecklenburg and S. C. Marinescu, J. Am. Chem. Soc., 2015, 137, 118–121 CrossRef CAS PubMed.
- T. Pal, T. Kambe, T. Kusamoto, M. L. Foo, R. Matsuoka, R. Sakamoto and H. Nishihara, Chempluschem, 2015, 80, 1255–1258 CrossRef CAS PubMed.
- X. Zhang, Y. Zhou, B. Cui, M. Zhao and F. Liu, Nano Lett., 2017, 17, 6166–6170 CrossRef CAS PubMed.
- N. Lahiri, N. Lotfizadeh, R. Tsuchikawa, V. V. Deshpande and J. Louie, J. Am. Chem. Soc., 2017, 139, 19–22 CrossRef CAS PubMed.
- A. C. Hinckley, J. Park, J. Gomes, E. Carlson and Z. Bao, J. Am. Chem. Soc., 2020, 142, 11123–11130 CrossRef CAS PubMed.
- J. Nyakuchena, S. Ostresh, D. Streater, B. Pattengale, J. Neu, C. Fiankor, W. Hu, E. D. Kinigstein, J. Zhang, X. Zhang, C. A. Schmuttenmaer and J. Huang, J. Am. Chem. Soc., 2020, 142, 21050–21058 CrossRef CAS PubMed.
- Z. Meng, C. G. Jones, S. Farid, I. U. Khan, H. M. Nelson and K. A. Mirica, Angew. Chem., Int. Ed., 2022, 61, e202113569 CAS.
- J. Liu and Q. Sun, Chemphyschem, 2015, 16, 614–620 CrossRef CAS PubMed.
- J. G. Park, B. A. Collins, L. E. Darago, T. Runcevski, M. E. Ziebel, M. L. Aubrey, H. Z. H. Jiang, E. Velasquez, M. A. Green, J. D. Goodpaster and J. R. Long, Nat. Chem., 2021, 13, 594–598 CrossRef CAS PubMed.
- Z. Fei, B. Huang, P. Malinowski, W. Wang, T. Song, J. Sanchez, W. Yao, D. Xiao, X. Zhu, A. F. May, W. Wu, D. H. Cobden, J. H. Chu and X. Xu, Nat. Mater., 2018, 17, 778–782 CrossRef CAS PubMed.
- J. U. Lee, S. Lee, J. H. Ryoo, S. Kang, T. Y. Kim, P. Kim, C. H. Park, J. G. Park and H. Cheong, Nano Lett., 2016, 16, 7433–7438 CrossRef CAS PubMed.
- Y. Li, K. A. Duerloo, K. Wauson and E. J. Reed, Nat. Commun., 2016, 7, 10671 CrossRef CAS PubMed.
- Y. Sun, Z. Shuai and D. Wang, Nanoscale, 2018, 10, 21629–21633 RSC.
- C. Zhang, S. Kc, Y. Nie, C. Liang, W. G. Vandenberghe, R. C. Longo, Y. Zheng, F. Kong, S. Hong, R. M. Wallace and K. Cho, ACS Nano, 2016, 10, 7370–7375 CrossRef CAS PubMed.
- A. E. Thorarinsdottir and T. D. Harris, Chem. Rev., 2020, 120, 8716–8789 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ta08540b |
| This journal is © The Royal Society of Chemistry 2023 |