
A binder-driven cathode–electrolyte interphase via a displacement reaction for high voltage Na3V2(PO4)2F3 cathodes in sodium-ion batteries†
Dae Hui
Yun‡
a,
Jinju
Song‡
a,
Jiseong
Kim
a,
Joon Kyo
Seo
a,
Joonhee
Kang
b,
Sohyun
Park
c,
Jaekook
Kim
 c,
Dong-Joo
Yoo
*d and
Sunghun
Choi
*a
c,
Dong-Joo
Yoo
*d and
Sunghun
Choi
*a
aGwangju Clean Energy Research Center, Korea Institute of Energy Research (KIER), 270-25 Samso-ro, Buk-gu, Gwangju 61003, Republic of Korea. E-mail: s.h.choi@kier.re.kr
bComputational Science & Engineering Laboratory, Korea Institute of Energy Research (KIER), 152 Gajeong-ro, Yuseong-gu, Daejeon 34129, Republic of Korea
cDepartment of Materials Science and Engineering, Chonnam National University, Gwangju 61186, Republic of Korea
dSchool of Mechanical Engineering, Korea University, 145 Anam-ro, Seongbuk-gu, Seoul 02841, Republic of Korea. E-mail: djyoo@korea.ac.kr
First published on 10th February 2023
Abstract
Sodium super ionic conductor (NASICON)-structured Na3V2(PO4)2F3 (NVPF) is a promising cathode for application in sodium-ion batteries (SIBs) because of its high working potential (3.7 V and 4.2 V vs. Na/Na+) and structural stability. Nonetheless, interfacial instability deteriorates its electrochemical performance. Therefore, to overcome this limitation, we introduced a sodium polyacrylate (NaPAA) binder for NVPF cathodes. The NaPAA binder effectively suppresses electrolyte decomposition by uniformly covering NVPF particles. Furthermore, the sodium carboxylate group of R–COONa in the NaPAA binder can react with the HPO2F2 intermediate generated by the hydrolysis of NaPF6 and be converted into R–COOH and NaPO2F2via the displacement of Na+ by H+. This results in the formation of a stable and Na-ion conductive NaPO2F2-rich cathode–electrolyte interphase (CEI) layer. In addition, the NaPAA-based electrode exhibits desirable cycling and rate performances compared to those of conventional poly(vinylidene difluoride)-based electrodes. This study provides new insights into the design of CEI layers by introducing chemical functional groups in the binder for high-performance SIB cathodes.
Introduction
With the rapid commercialization of electric vehicles (EVs) and energy storage systems (ESSs), the demand for secondary batteries has increased significantly. Although lithium-ion batteries (LIBs) can meet the demand of the rechargeable battery market, concerns related to the limited resources of expensive metals such as cobalt, nickel, and lithium have shifted research interest to sodium-ion batteries (SIBs).1,2 Sodium is an abundant element and comprises 2.64% of the earth's crust, that is 440 times higher than the lithium content.3 The inertness of sodium toward aluminum possibly lowers the cost of SIBs, as expensive copper foil can be replaced with aluminum foil for anode current collectors.2 Moreover, the similarity of the physicochemical properties between lithium and sodium has accelerated the development of active materials for SIBs.1,3 However, to capture the market for LIBs, considerable progress must be made toward improving the stability and kinetics of cathode materials used for SIBs.Various materials, such as layered oxides,4,5 polyanions,6–8 and Prussian blue9–11 analogs, have been explored as cathodes for SIBs. Among them, Na3V2(PO4)2F3 (NVPF) is regarded a promising candidate owing to its high operating potential (3.7 V and 4.2 V vs. Na/Na+) resulting from the strong ionicity of the F–V bond, rapid sodium ion conduction with a large interstitial space, and high structural stability.6,12 However, NVPF, which has a NASICON structure, exhibits intrinsically low electrical conductivity and sluggish electrochemical kinetics.13,14 Accordingly, several approaches, including morphological engineering15 and the use of carbon composites16–20 with graphene, carbon nanotubes, or porous carbon, have been adopted to achieve improved electrical conductivity. Although these approaches are effective, undesirable degradation reactions at the cathode–electrolyte interface limit their practical application.21 Specifically, sodium hexafluorophosphate (NaPF6), widely used as a salt in carbonate electrolytes, can react with trace water to generate hydrofluoric acid (HF) via HxPOyFz intermediates.22 HF causes the dissolution of transition metal ions, which adversely affects both the cathode and anode interfaces. Furthermore, the decomposition of conventional carbonate solvents accelerates the formation of an unstable cathode–electrolyte interphase (CEI) layer.23 Considering that electrochemical stability and kinetics are closely related to the CEI layer, a binder is expected to have a significant impact on battery performance because it initially covers the cathode surface and actively participates in the formation of the CEI layer.
A binder plays a role in holding active particles during operation via adhesive and cohesive forces. For example, sodium carboxylate-based binders13,23–26 can strongly interact with the surface of metal-oxide-based NVPF cathodes via ion–dipole interactions, which tightly bind the electrode components. Thus, these sodium carboxylate-based binders exhibit improved mechanical properties, such as robust adhesion and uniform coverage of the active particles, compared to conventional poly(vinylidene difluoride) (PVDF) binders, which exhibit weak adhesion via van der Waals forces. Although these enhanced mechanical properties contribute to the improvement in the performance of alloyed anode materials27–29 that undergo high volume expansion, they cannot explain the behavior of NVPF cathodes that undergo significantly low volume changes (<4%) during cycling.6 In addition, the role of binders in imparting excellent rate capabilities is debatable because the ionic conductivities of carboxylate-based binders are insufficient to attain the required current densities.
In this study, we clarified the binder-driven formation of a stable Na-ion-conductive CEI layer using a sodium polyacrylate (NaPAA) binder. The sodium carboxylate group of R–COONa in the NaPAA binder enables uniform coverage of the particle surface and effectively suppresses electrolyte decomposition during cycling. Notably, these functional groups are highly involved in the formation of a stable Na-ion conductive CEI layer by reacting with the HPO2F2 intermediate in NaPF6 hydrolysis, resulting in a stable cycling performance and excellent rate capability. To the best of our knowledge, this is the first study clarifying the mechanism by which specific functional groups in the binder develop multifunctional CEI layers in SIB cathodes. This study provides useful insights on how to design binders for high-performance SIBs.
Experimental
Synthesis of the NVPF cathode
Na3V2(PO4)2F3 was prepared by a refluxing method using sodium acetate, vanadium acetylacetonate, phosphoric acid, and ammonium fluoride in a stoichiometric molar ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:2:3, respectively. All precursors were dissolved in diethylene glycol for 24 h. The solution was refluxed at 260 °C for 6 h in a round-bottom flask equipped with a condenser. The resulting powder was washed several times with acetone to remove organic solvents and compounds. The dried powder was heated at 650 °C for 8 h under an argon atmosphere.
:2:2:3, respectively. All precursors were dissolved in diethylene glycol for 24 h. The solution was refluxed at 260 °C for 6 h in a round-bottom flask equipped with a condenser. The resulting powder was washed several times with acetone to remove organic solvents and compounds. The dried powder was heated at 650 °C for 8 h under an argon atmosphere.
Synthesis of the NaPAA binder
Poly(acrylic acid) (Mw = 450000, Sigma Aldrich, USA) was dissolved in distilled water and then 0.1 N sodium hydroxide (NaOH, Sigma Aldrich, USA) solution was added dropwise while stirring until the pH reached 7. The prepared solution was lyophilized to obtain a white powder.
Preparation of NVPF electrodes
Electrodes were fabricated using the synthesized NVPF powder, Super P (Timcal), and binder. The synthesized NaPAA and PVDF (Mw = 1000000, Kureha, Japan) were dissolved in distilled water and N-methyl-2-pyrrolidone (NMP, SAM CHEON, South Korea), respectively. For the working electrodes, a slurry was prepared by dispersing NVPF, Super P, and each binder at a weight ratio of 8:1:1, respectively. The slurry was cast onto an Al current collector using a doctor blade, dried at 80 °C in air for 1 h, and subsequently dried at 80 °C in a vacuum oven overnight. The areal loading of NVPF was 1.5 mg cm−2. CR2032 coin-type half-cells were assembled in an Ar-filled glove box by sandwiching a glass fiber membrane (Whatman®, England) with sodium metal (Sigma Aldrich, USA) as the counter electrode. 1.0 M sodium hexafluorophosphate (NaPF6) in 1:1 (v/v) ethylene carbonate (EC)/propylene carbonate (PC) containing (5 wt%) fluoroethylene carbonate (FEC) (Enchem, South Korea) was used as an electrolyte.
Characterization
The chemical structures of the PVDF and NaPAA binders were analyzed using Fourier transform infrared spectroscopy (FT-IR, Spectrum 400, PerkinElmer, USA). The adhesive force of the electrode was obtained via 180° peeling tests using a universal testing machine (UTM, AG-X plus, Shimadzu, Japan). Using 3M double-sided tape, one side was attached to the electrode sheet and the other to a slide glass. The upper and lower grips of the UTM held the end of the slide glass and electrode sheet, respectively, and then peeled off at a speed of 25 mm min−1. During the test, the load was continuously recorded and the adhesive force was calculated using the average load. The powder and ex situ XRD patterns of NVPF were recorded using a PANalytical X'Pert PRO multipurpose X-ray diffractometer with Ni-filtered Cu Kα radiation (λ = 1.5406 Å) operating at 40 kV and 30 mA at 2θ values ranging from 10° to 80° in steps of 0.02°. The carbon content in the NVPF cathode was characterized by thermogravimetric analysis (TGA2, Mettler Toledo). The morphology, size, and binder coverage of NVPF were observed using high-resolution transmission electron microscopy (HR-TEM, FEI Tecnai F20, USA). The electrode morphology was visualized using a field-emission scanning electron microscope (FE-SEM, JSM-7500F, JEOL and Verios 5 UC, Thermo Fisher Scientific, USA). The chemical bonds of each electrode were confirmed using X-ray photoelectron spectroscopy (XPS, K-ALPHA+, Thermo Fisher Scientific, USA). The mass spectra of the CEI components of each electrode were characterized by time-of-flight secondary ion mass spectroscopy (ToF-SIMS, ION-TOF GmbH, Germany). The electrical resistance of each electrode was measured using a four-point probe system (CMT-100S; AIT, South Korea).Electrochemical measurements
In galvanostatic battery tests, three formation cycles were assessed at 0.1C (12.8 mA g−1) to stabilize the CEI layers for all electrodes, and following cycles were assessed at 1C (128 mA g−1) or 10C (1280 mA g−1) in the potential range of 2.0–4.5 V vs. Na/Na+ using a battery cycler (WBCS 300 L, WonAtech, South Korea). Rate performance was measured at various C-rates from 0.5C to 30C. Cyclic voltammetry (CV) in the 2.0–4.5 V potential window at a scan rate of 0.05 mV s−1 was conducted to check the electrochemical stability of the binder-super P composite in a weight ratio of 3:1 by using a potentiostat (VMP3, Bio-logic, France). Electrochemical impedance spectroscopy (EIS) analysis was performed using a potentiostat (VMP3, Bio-logic) over the frequency range of 0.01 Hz to 1 MHz and an amplitude of 10 mV. The self-discharge rate was observed by monitoring the voltage drop for 500 h after being fully charged to 4.5 V.
DFT calculations
Geometric optimization and single-point energy calculations to obtain binding energies of sodium ions in molecules and energies of reactants/products were conducted based on the three-parameter Becke model with the Lee–Yang–Par modification (B3LYP) in the GAUSSIAN16 software package. We used the 6-311 + + G (3df, 3pd) basis set containing diffusive functions to address the charge in molecular systems.30,31The binding energy of sodium ions in a molecular system was obtained starting with geometry optimization for a molecule, followed by single-point energy calculations. The optimized molecular structure was then divided into a sodium ion and single-anion model for geometry optimization and single-point energy calculations. The single-point energies for the molecule, sodium ion, and single anion were used to calculate the binding energy.
Results and discussion
The chemical structures of NaPAA and PVDF binders are shown in Fig. 1a. The NaPAA binder was synthesized via the neutralization reaction of poly(acrylic acid) with sodium hydroxide and characterized by Fourier-transform infrared (FT-IR) spectroscopy (Fig. S1a†), with peaks in the range of 1510–1650 cm−1 and 1280–1400 cm−1 corresponding to asymmetric and symmetric vibrations, respectively. The NVPF cathode was synthesized via a facile polyol-assisted synthesis, and the structure was characterized by powder X-ray diffraction (XRD) analysis indexing the data to the tetragonal structure with the P42/mmm space group (JCPDS #01-089-8485; Fig. S1b†).32 NVPF was coated with 4.1 wt% carbon confirmed by thermogravimetric analysis (Fig. S2†). Scanning electron microscopy (SEM) analysis was conducted to compare the morphologies of the NaPAA- and PVDF-based electrodes. The NaPAA-based electrode showed a highly uniform distribution of active particles (Fig. 1b). In contrast, the PVDF-based electrode has macropores (circled in red in Fig. 1c) and an uneven electrode morphology. In the peeling-off test (Fig. 1d), the average peeling force of the NaPAA-based electrode was estimated to be 0.25 N mm−1, whereas the PVDF-based electrode showed negligible adhesion due to weak van der Waals interactions. This is because the sodium carboxylate group in the NaPAA binder strongly interacts with the surface of the NVPF cathode via ion–dipole interactions. According to the four-point analysis, the NaPAA-based electrode with high integrity exhibited a lower electrical resistance of 3.5 mΩ cm−1 compared with 6.2 mΩ cm−1 of the PVDF-based electrode (Fig. S3†).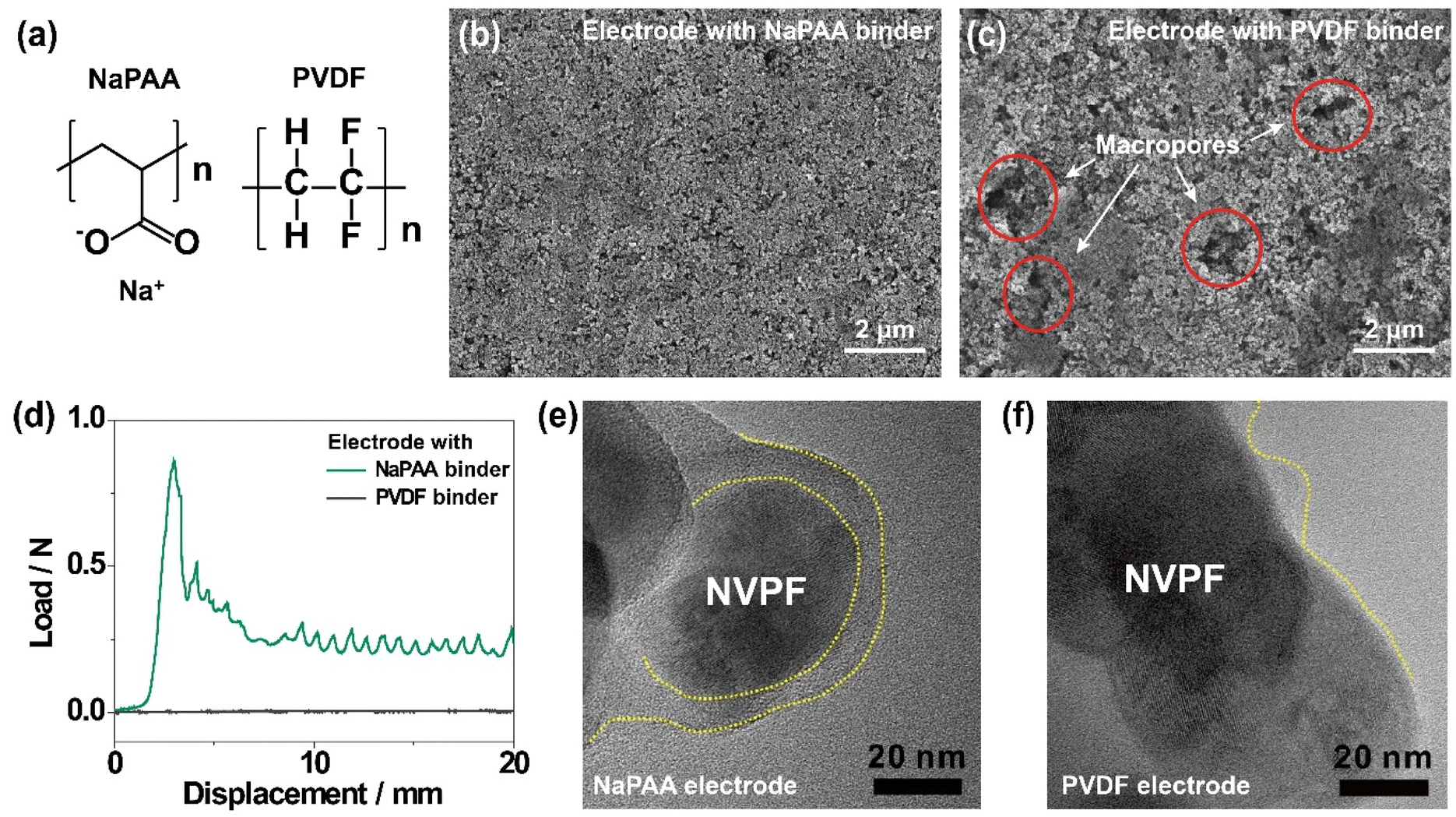 | ||
| Fig. 1 (a) Chemical structures of NaPAA and PVDF binders. SEM images of electrodes with (b) NaPAA and (c) PVDF binders. (d) Peeling force versus displacement profiles of electrodes with NaPAA and PVDF binders. TEM images of NVPF particles with (e) NaPAA and (f) PVDF binders. | ||
Transmission electron microscopy (TEM) images showed that the surface of the NVPF particles was covered by the NaPAA binder with 10 nm thickness (Fig. 1e). In contrast, the coverage of the PVDF binder was irregular, and the exposed surfaces of the NVPF particles were observed (Fig. 1f). Poor binder coverage possibly accelerated electrolyte decomposition by directly exposing the cathode surface to the electrolyte, mainly when operating at high voltages (>4.2 V vs. Na/Na+),33 resulting in capacity decay. The uniform coverage of the NaPAA binder on the NVPF cathodes was also observed at a lower magnification (Fig. S4†).
Fig. 2a shows the rate performance of the NaPAA- and PVDF-based electrodes in the potential range of 2.0–4.5 V vs. Na/Na+. When the C-rate decreased stepwise from 0.5C to 30C, the NaPAA- and PVDF-based electrodes showed capacity retentions of 62% and 13%, respectively. The high performance of the NaPAA-based electrode can be attributed to the low electrical resistance and the formation of a Na-ion conductive CEI layer induced by the NaPAA binder. In addition, the binder significantly affects the cycling performance. When cycled at 1C (1C = 128 mA g−1) (Fig. 2b), the NaPAA- and PVDF-based electrodes delivered initial capacities of 110 mA h g−1 and 101 mA h g−1, respectively, and retained 87% and 67% of their capacities, respectively, after 200 cycles. The average coulombic efficiencies (CEs) were 99.5% and 99.2%, respectively, implying that the uniform coverage of the NaPAA binder on the NVPF surface effectively suppressed electrolyte decomposition. The voltage profiles of the NaPAA-based electrode overlap above 200 cycles compared to that of the PVDF-based electrode (Fig. S5†). The high performance of the NaPAA-based electrode was maintained at a mass loading of 4.3 mg cm−2 (Fig. S6†). Trends in the cycling performance and discharge capacity were retained even at 10C (Fig. 2c). The NaPAA-based electrode maintained 70% of its initial capacity, whereas the PVDF-based electrode retained only 50% of its initial capacity after 2000 cycles.
 | ||
| Fig. 2 (a) Rate capability measurement of NaPAA and PVDF electrodes. Cycling performances of NaPAA and PVDF electrodes at (b) 1C and (c) 10C. CV profiles of (d) NaPAA and (e) PVDF electrodes at various scan rates. (f) B-values based on peak oxidation and reduction currents extracted from (d and e). | ||
The ex situ XRD patterns of the NaPAA- and PVDF-based electrodes after 200 cycles were similar to those of the pristine state (Fig. S7†), indicating that the capacity decay originated from interfacial instability and not structural collapse of the NVPF cathode. To verify the dependence of the interfacial stability on the binder, we performed cyclic voltammetry (CV) on the carbon electrodes. The NaPAA binder showed a lower current intensity than that of the PVDF binder, specifically in the high-voltage range over 4.0 V (Fig. S8a†). In addition, self-discharge measurements of NaPAA- and PVDF-based electrodes charged to 4.5 V were conducted by observing the voltage drop over time (Fig. S8b†). The NaPAA-based electrode retained a higher voltage (3.98 V) after 500 h than that of the PVDF-based electrode (3.79 V). These results indicate that the NaPAA binder enables the stable CEI layer formation compared to the PVDF binder.
To clarify the improved electrochemical kinetics of the NaPAA-based electrode, we conducted CV tests at various scan rates from 0.05 mV s−1 to 2 mV s−1 (Fig. 2d and e). The NaPAA-based electrode exhibited sharp reduction/oxidation current peaks and a small voltage peak shift of 0.14 V. In contrast, the PVDF-based electrode exhibited broad current peaks and a relatively large voltage peak shift of 0.22 V, implying a high overpotential due to slow kinetics. In addition, the b-values of the reduction and oxidation peaks were calculated using the following relationship between the peak current (Ip) and the scan rate (v):
| Ip = avb |
The b-value was extracted from the slope of the log–log plot of v vs. Ip (Fig. 2f). Generally, a b-value of 0.5 indicates a diffusion-controlled reaction, while a b-value of 1 indicates a capacitive reaction with negligible charge transfer resistance.34,35 For all peaks, the b value of the NaPAA-based electrode was higher than that of the PVDF-based electrode. These results are consistent with the improved rate capability of the NaPAA-based electrode.
To investigate the effect of the binder on the CEI layer, we conducted X-ray photoelectron spectroscopy (XPS) of the NaPAA- and PVDF-based electrodes in the pristine state, after three cycles, and after 200 cycles (Fig. 3). In the F 1s spectra of the pristine state, both NaPAA- and PVDF-based electrodes exhibited a strong V–F peak (684.1 eV) originating from the NVPF cathode, and the peak at 687.8 eV assigned to the C–F bond from the PVDF binder was observed in the PVDF-based electrode. After 3 cycles, NaF and NaPO2F2 peaks appeared and were assigned to 684.8 eV and 686.5 eV, respectively, due to the decomposition of the PF6− anion in the electrolytes. It is noteworthy that PO2F2− can be produced by two distinct pathways; one is hydrolysis with trace water in electrolytes and the other is an electrochemical reaction with carbonates.36,37 While the hydrolysis rapidly occurs due to the high reactivity of water, the electrochemical reaction occurs slowly upon cycling. Interestingly, the peak intensity of NaPO2F2 for the NaPAA electrode was more prominent than that for the PVDF electrode, while the NaF peak was almost absent. It can be concluded that the NaPAA binder played a key role in the selective formation of NaPO2F2 during initial cycles by rapid hydrolysis. In contrast, both NaF and NaPO2F2 simultaneously increased in the PVDF-based electrode by electrochemical oxidation. After 200 cycles, the NaPAA-based electrode showed a small decrease in NaPO2F2 and an increase in the other peaks of NaF and C–F, while the PVDF-based electrode exhibited a significant increase in both NaF and NaPO2F2. Overall, it was confirmed that the NaPO2F2 in the NaPAA-based electrode was positioned at the inner CEI layer, while the NaPO2F2 was distributed throughout the CEI layer in the PVDF-based electrode.
 | ||
| Fig. 3 XPS F 1s profiles of NVPF electrodes with (a) NaPAA and (b) PVDF binders in the pristine state, after 3 cycles, and after 200 cycles. | ||
Furthermore, we characterized the relative quantity of the components in the CEI layers by conducting time-of-flight secondary ion mass spectrometry (ToF-SIMS) in the negative ion data collection mode of the pristine NaPAA- and PVDF-based electrodes and after three cycles (Fig. S9†). Consistent with the F 1s XPS results, the peak for the PO2F2− anion fragment (m/z = 100.96) related to NaPO2F2 was more prominent in the NaPAA-based electrode than that in the PVDF-based electrode after three cycles, while the peak intensity of the NaF− anion fragment (m/z = 42.0) was almost the same as that of the pristine state. In addition, the intensity of the C3H3O− anion fragment (m/z = 55.02), which is an indicator of the organic components originating from the decomposition of the electrolyte solvent,38 was lower in the NaPAA-based electrode than that in the PVDF-based electrode. These combined XPS and ToF-SIMS analyses confirmed that the NaPAA binder enabled the stable NaPO2F2-rich CEI layer formation, that is consistent with the superior electrochemical performances.
To understand the distinct mechanism underlying the selective formation of the NaPO2F2-rich CEI layer by the NaPAA binder, we conducted ex situ FT-IR spectroscopy of both the NaPAA- and PVDF-based electrodes in the pristine state and after three cycles (Fig. 4a and b). In the case of the pristine NaPAA-based electrode, the sodium carboxylate group of R–COONa could be clearly identified from the peak at 1550 cm−1 for the asymmetric vibration, which is consistent with that of the NaPAA binder. After three cycles, the peak intensity of R–COONa decreased, but the carboxylic group of R–COOH appeared noticeably with the peak at 1700 cm−1 for the stretching vibration, implying a displacement reaction of Na+ to H+. In contrast, there was no change observed in the peaks of the PVDF-based electrode during the cycling (Fig. 4b).
 | ||
| Fig. 4 FT-IR absorbance spectra of (a) NaPAA- and (b) PVDF-based electrodes. For comparison, binder, pristine, and cycled electrodes were measured. (c) Energy difference between the NaPAA binder and HPO2F2. (d) Binding energies of NaF and NaPO2F2 from DFT calculations. | ||
It is widely known that trace water in electrolytes triggers hydrofluoric acid (HF) formation by hydrolysis of NaPF6.22 HF is formed through the following steps:
| NaPF6 + H2O ↔ NaF + POF3 + 2HF | (1) |
| POF3 + H2O ↔ HPO2F2 + HF | (2) |
| HPO2F2 + H2O ↔ H2PO3F + HF | (3) |
We assumed that the selective formation of NaPO2F2 in the CEI layer of the NaPAA-based electrode occurred during the initial cycles, because the R–COONa in the NaPAA binder could react with the HPO2F2 intermediate generated by the hydrolysis of NaPF6 and is converted into NaPO2F2 and R–COOH via the following displacement reaction:
| RCOONa + HPO2F2 → RCOOH + NaPO2F2 (ΔE = −0.69 eV) |
The displacement reaction was thermodynamically favorable owing to the lower Gibbs free energy of the products obtained using density functional theory (DFT) calculations (Fig. 4c).
Notably, the NaPO2F2-rich CEI layer in the NaPAA-based electrode is more beneficial in terms of Na-ion conductivity, which is related to the excellent rate performance of the NaPAA-based electrode. To understand the fast kinetics of the NaPAA-based electrode, we compared the binding energy between Na ions and their counter anions (F− and PO2F2−) in the inorganic components because it is acceptable that a lower binding energy induces a higher Na ion conductivity in the components (Fig. 4d).39,40 In the DFT results, NaPO2F2 showed a lower binding energy than that of NaF, indicating a higher Na-ion conductivity in NaPO2F2. From the combined analysis of FT-IR and DFT calculations, it was demonstrated that the NaPAA binder enabled the formation of the Na-ion conductive NaPO2F2-rich CEI layer and simultaneously prevented additional detrimental HF formation by the displacement reaction (Fig. 5).
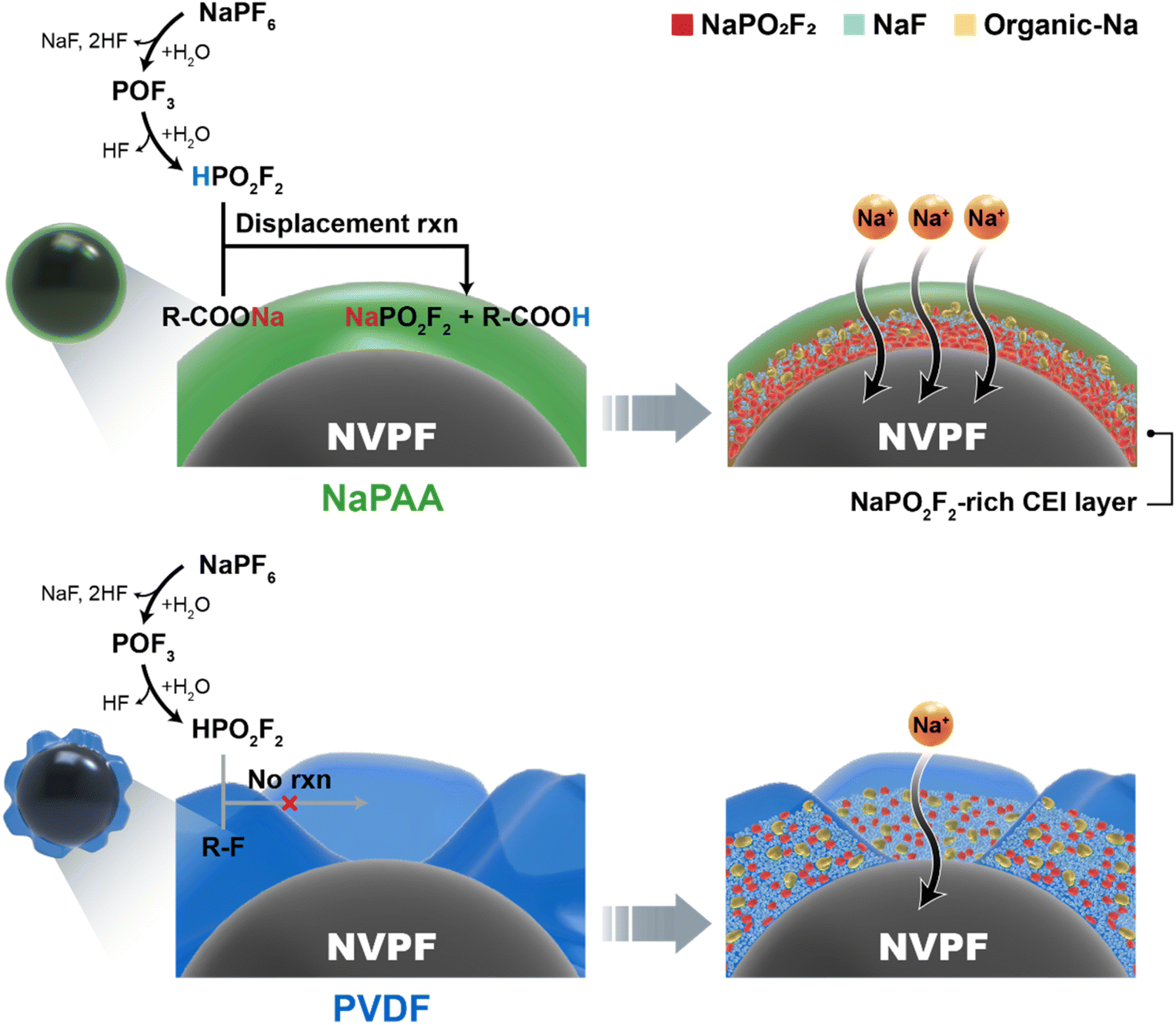 | ||
| Fig. 5 Schematic illustration of the effect of NaPAA and PVDF binders on CEI layer formation. | ||
Conclusions
In conclusion, we have developed a stable and Na-ion-conductive NaPO2F2-rich CEI layer by adopting a NaPAA binder for NVPF cathodes. The sodium carboxylate group of R–COONa in the NaPAA binder induced strong ion–dipole interactions with the NVPF cathodes, which resulted in the uniform surface coverage and improved the distribution of active materials on the electrode scale. By reacting with the HPO2F2 intermediate generated from the NaPF6 hydrolysis, the NaPAA binder contributed to the stable and ion-conductive NaPO2F2-rich CEI layer formation via a displacement reaction. Thus, the NaPAA-based electrode delivered a high-rate capability of 62% capacity retention at 30C and an excellent cyclability of 70% capacity retention after 2000 cycles at 10C. This study presents a new concept of binder-driven CEI layers for high-performance SIB cathodes.Author contributions
D. H. Y., J. S., D.-J. Y. and S. C. designed the research. D. H. Y., J. S., S. P., J. K. performed the experimental work. J. K. S. and J. K. performed the DFT calculations. D. H. Y., J. S., D.-J. Y. and S. C. wrote the manuscript. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. K. acknowledges the financial support from the National Research Foundation of Korea (NRF) grant funded by the Korea government (MIST) (NRF-2021R1A4A1052051). D. Y. acknowledges the financial support from the Korea University Grant (K2204461 and K2206311). S. C. acknowledges the financial support from the Technology Innovation Program (Grant no. 20009985) funded by the Ministry of Trade, Industry & Energy (MOTIE, Korea) and the research development program of the Korea Institute of Energy Research (C2-2472).References
- C. Vaalma, D. Buchholz, M. Weil and S. Passerini, Nat. Rev. Mater., 2018, 3, 18013 CrossRef.
- P. K. Nayak, L. Yang, W. Brehm and P. Adelhelm, Angew. Chem., Int. Ed., 2018, 57, 102–120 CrossRef CAS PubMed.
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Chem. Rev., 2014, 114, 11636–11682 CrossRef CAS PubMed.
- Y. Xiao, N. M. Abbasi, Y. F. Zhu, S. Li, S. J. Tan, W. Ling, L. Peng, T. Yang, L. Wang, X. D. Guo, Y. X. Yin, H. Zhang and Y. G. Guo, Adv. Funct. Mater., 2020, 30, 2001334 CrossRef CAS.
- N. Ortiz-Vitoriano, N. E. Drewett, E. Gonzalo and T. Rojo, Energy Environ. Sci., 2017, 10, 1051–1074 RSC.
- Q. Ni, Y. Bai, F. Wu and C. Wu, Adv. Sci., 2017, 4, 1600275 CrossRef PubMed.
- P. Barpanda, L. Lander, S.-i. Nishimura and A. Yamada, Adv. Energy Mater., 2018, 8, 1703055 CrossRef.
- Y. Cao, C. Yang, Y. Liu, X. Xia, D. Zhao, Y. Cao, H. Yang, J. Zhang, J. Lu and Y. Xia, ACS Energy Lett., 2020, 5, 3788–3796 CrossRef CAS.
- J. Qian, C. Wu, Y. Cao, Z. Ma, Y. Huang, X. Ai and H. Yang, Adv. Energy Mater., 2018, 8, 1702619 CrossRef.
- W. Wang, Y. Gang, Z. Hu, Z. Yan, W. Li, Y. Li, Q. F. Gu, Z. Wang, S. L. Chou, H. K. Liu and S. X. Dou, Nat. Commun., 2020, 11, 980 CrossRef CAS PubMed.
- P. Hu, W. Peng, B. Wang, D. Xiao, U. Ahuja, J. Réthoré and K. E. Aifantis, ACS Energy Lett., 2019, 5, 100–108 CrossRef.
- L. Zhu, H. Wang, D. Sun, Y. Tang and H. Wang, J. Mater. Chem. A, 2020, 8, 21387–21407 RSC.
- Y. Subramanian, W. Oh, W. Choi, H. Lee, M. Jeong, R. Thangavel and W.-S. Yoon, Chem. Eng. J., 2021, 403, 126291 CrossRef CAS.
- T. Jiang, G. Chen, A. Li, C. Wang and Y. Wei, J. Alloys Compd., 2009, 478, 604–607 CrossRef CAS.
- Y. Qi, L. Mu, J. Zhao, Y.-S. Hu, H. Liu and S. Dai, J. Mater. Chem. A, 2016, 4, 7178–7184 RSC.
- L. Deng, G. Sun, K. Goh, L.-L. Zheng, F.-D. Yu, X.-L. Sui, L. Zhao and Z.-B. Wang, Electrochim. Acta, 2019, 298, 459–467 CrossRef CAS.
- M. Wang, X. Huang, H. Wang, T. Zhou, H. Xie and Y. Ren, RSC Adv., 2019, 9, 30628–30636 RSC.
- Q. Liu, D. Wang, X. Yang, N. Chen, C. Wang, X. Bie, Y. Wei, G. Chen and F. Du, J. Mater. Chem. A, 2015, 3, 21478–21485 RSC.
- Q. Liu, X. Meng, Z. Wei, D. Wang, Y. Gao, Y. Wei, F. Du and G. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 31709–31715 CrossRef CAS PubMed.
- T. Broux, F. Fauth, N. Hall, Y. Chatillon, M. Bianchini, T. Bamine, J.-B. Leriche, E. Suard, D. Carlier, Y. Reynier, L. Simonin, C. Masquelier and L. Croguennec, Small Methods, 2019, 3, 1800215 CrossRef CAS.
- Y. You, S. Xin, H. Y. Asl, W. Li, P.-F. Wang, Y.-G. Guo and A. Manthiram, Chem, 2018, 4, 2124–2139 CAS.
- P. Barnes, K. Smith, R. Parrish, C. Jones, P. Skinner, E. Storch, Q. White, C. Deng, D. Karsann, M. L. Lau, J. J. Dumais, E. J. Dufek and H. Xiong, J. Power Sources, 2020, 447, 227363 CrossRef CAS.
- H. Li, C. Guan, J. Zhang, K. Cheng, Q. Chen, L. He, X. Ge, Y. Lai, H. Sun and Z. Zhang, Adv. Mater., 2022, 34, 2202624 CrossRef CAS PubMed.
- Z. Y. Gu, Z. H. Sun, J. Z. Guo, X. X. Zhao, C. D. Zhao, S. F. Li, X. T. Wang, W. H. Li, Y. L. Heng and X. L. Wu, ACS Appl. Mater. Interfaces, 2020, 12, 47580–47589 CrossRef CAS PubMed.
- Y.-Y. Zhang, S.-J. Zhang, J.-T. Li, K. Wang, Y.-C. Zhang, Q. Liu, R.-S. Xie, Y.-R. Pei, L. Huang and S.-G. Sun, Electrochim. Acta, 2019, 298, 496–504 CrossRef CAS.
- H. Xu, K. Jiang, X. Zhang, X. Zhang, S. Guo and H. Zhou, ACS Appl. Mater. Interfaces, 2019, 11, 26817–26823 CrossRef CAS PubMed.
- C. Bommier and X. Ji, Small, 2018, 14, 1703576 CrossRef PubMed.
- H. Gao, W. Zhou, J.-H. Jang and J. B. Goodenough, Adv. Energy Mater., 2016, 6, 1502130 CrossRef.
- P.-F. Cao, G. Yang, B. Li, Y. Zhang, S. Zhao, S. Zhang, A. Erwin, Z. Zhang, A. P. Sokolov, J. Nanda and T. Saito, ACS Energy Lett., 2019, 4, 1171–1180 CrossRef CAS.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- S. Park, J. Song, S. Kim, B. Sambandam, V. Mathew, S. Kim, J. Jo, S. Kim and J. Kim, Nano Res., 2019, 12, 911–917 CrossRef CAS.
- Y. You and A. Manthiram, Adv. Energy Mater., 2017, 8, 1701785 CrossRef.
- X. Yang and A. L. Rogach, Adv. Energy Mater., 2019, 9, 1900747 CrossRef.
- V. Augustyn, J. Come, M. A. Lowe, J. W. Kim, P. L. Taberna, S. H. Tolbert, H. D. Abruna, P. Simon and B. Dunn, Nat. Mater., 2013, 12, 518–522 CrossRef CAS PubMed.
- B. S. Parimalam, A. D. MacIntosh, R. Kadam and B. L. Lucht, J. Phys. Chem. C, 2017, 121, 22733–22738 CrossRef CAS.
- Y. Yu, P. Karayaylali, Y. Katayama, L. Giordano, M. Gauthier, F. Maglia, R. Jung, I. Lund and Y. Shao-Horn, J. Phys. Chem. C, 2018, 122, 27368–27382 CrossRef CAS.
- W. Li, A. Dolocan, P. Oh, H. Celio, S. Park, J. Cho and A. Manthiram, Nat. Commun., 2017, 8, 14589 CrossRef PubMed.
- H. Zhang, C. Li, M. Piszcz, E. Coya, T. Rojo, L. M. Rodriguez-Martinez, M. Armand and Z. Zhou, Chem. Soc. Rev., 2017, 46, 797–815 RSC.
- H. Huo, B. Wu, T. Zhang, X. Zheng, L. Ge, T. Xu, X. Guo and X. Sun, Energy Storage Mater., 2019, 18, 59–67 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: FT-IR, XRD, electrode resistances, TEM, and voltage, CV, and ToF-SIMS profiles. See DOI: https://doi.org/10.1039/d2ta07990a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |