
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Characterisation of direct ammonia proton conducting tubular ceramic fuel cells for maritime applications†
Kamil M.
Nowicki
 ab,
George
Carins
ab,
John
Bayne
ac,
Chayopas
Tupberg
ab,
Gavin J.
Irvine
a and
John T. S.
Irvine
*ab
ab,
George
Carins
ab,
John
Bayne
ac,
Chayopas
Tupberg
ab,
Gavin J.
Irvine
a and
John T. S.
Irvine
*ab
aSchool of Chemistry, University of St Andrews, Fife KY16 9ST, St Andrews, UK. E-mail: jtsi@st-andrews.ac.uk
bUniversity of St Andrews, ZEM Fuel Systems, c/o Chemistry Building, St Andrews, Fife KY16 9ST, UK
cLow Emissions Resources Global, Block 2 Unit A, Mid-Craigie Road, Dundee DD4 7RH, UK
First published on 28th November 2022
Abstract
Ammonia is now being widely considered as a carrier for low carbon hydrogen due to its favourable physical properties and the existing infrastructure for its transport, storage and distribution. The direct utilisation of ammonia in Protonic Ceramic Fuel Cells (PCFCs) has the advantages of ammonia cracking activity and avoiding mixing of ammonia with generated steam. Here we seek to demonstrate a low-carbon electrical power generation system based on a PCFC fueled by ammonia for port and maritime applications. The performance of a 36 cm2 tubular cell with a proton conductive BaCe0.7Zr0.1Y0.16Zn0.04O3−δ (BCZYZ) electrolyte, composite Ni/BaCe0.7Zr0.1Y0.16Zn0.04O3−δ (Ni/BCZYZ) fuel electrode and La0.8Sr0.2Co0.5Fe0.5O3−δ/BaCe0.7Zr0.1Y0.16Zn0.04O3−δ LSCF/BCZYZ air electrode has been investigated using pure ammonia fuel. The tested cell generated up to 8.5 W with a maximum power density of 0.236 W cm−2 at 750 °C. The cell was operated in pure NH3, repeatedly characterised using I–V and EIS techniques, and tested under load to generate current for more than 140 h, with a fairly stable performance at a current above 8 A.
Introduction
Shipping is a central mode of the transport industry, and decarbonisation of the sector is required to meet net zero targets. Currently, the shipping industry generates about 3% of global CO2 emissions and 17% of atmospheric pollution. As mandated by the international treaty,1 large vessels with a power consumption of more than 130 kW or modified since 2000 must control emissions, including SOx, NOx and CO2, in so-called emission control areas, where the vessels approach ports and are close to the land. As outlined by the International Marine Organisation, the targets are to cut at least 50% of GHG emissions from shipping by 2050 and eliminate them as soon as possible before the end of the century.Emission control could be achieved by implementing clean and alternative fuels and technology, e.g., moving from fossil fuels to renewable energy. Fuel cells became an attractive solution for low-carbon energy generation, where green hydrogen is often considered the likely energy carrier.2,3 However, hydrogen is problematic for long-distance and heavy-duty applications due to its low energy density. Recently, a new concept has emerged to use ammonia (NH3) produced from renewable energy for fueling the shipping industry.4 Already, Japan and several large international shipping companies have committed to developing ships using ammonia as a primary fuel.5–7 Moreover, several governments have highlighted ammonia as a major part of their decarbonisation goals.8 For example, Japan has highlighted ammonia for use in co-fire power plants, with a goal of 20% ammonia demonstration by 2023.9 Saudi Arabia has committed 5 billion USD to build a solar-powered electrolysis plant that will be used to produce ammonia.10
Various green fuels have been considered for maritime, including ammonia, liquid hydrogen, synthetic Liquid Natural Gas (LNG), methanol and diesel.4,11 Among these, NH3 could be the most promising, thanks to the high energy density (4 kW h kg−1), high hydrogen content (17.7 wt%) and lack of CO2 emissions. Also, the costs related to the liquefaction of hydrogen could be avoided, lightly pressurised ammonia at room temperature is in the liquid phase (boiling point of −33.4 °C at atmospheric pressure) and ammonia has already existing infrastructure for transport, storage and distribution.12,13 Issues remain for implementing ammonia as a carbon-free energy vector, such as green production – 80% of the 90 Mt H2 used in 2020 was from fossil fuel sources, with 30 Mt going to the production of ammonia alone.8 However, so-called green production is seen as a feasible alternative in which ammonia is synthesised via the Haber–Bosch process from hydrogen produced by electrolysis driven by renewable energy14–16 or even through an electrochemical synthesis from water and nitrogen through an electrolysis cell.12,17,18
Research is ongoing into the use of ammonia as fuel both in combustion engines and in fuel cells.19,20 Ammonia has several issues as a direct fuel for combustion: low flammability, high NOx emissions, and low flame speeds.21 Alternatively, ammonia can be electrochemically oxidised using a fuel cell. Fuel cells directly convert chemical energy into electrical energy, giving them a significantly higher theoretical efficiency, and generation of NOx could be avoided using fuel cells, with H2O and N2 being the only emissions.
Solid Oxide Fuel Cells (SOFCs) are high-temperature fuel cells where the electrolyte is made of ion-conductive solid ceramics. Traditional SOFCs conduct oxide ions (O2−) across a solid electrolyte to the fuel side, where H2 is electrochemically oxidised, generating current. In state-of-the-art devices, 8% doped yttria-stabilised zirconia (Zr0.84Y0.16O2.08, YSZ) is used as the electrolyte, the Ni/YSZ composite as the fuel electrode22 and the LSM/YSZ composite (LaxSr1−xMnO3–Zr0.84Y0.16O1.92) as the oxygen electrode.23,24
Another type of SOFC is a proton conductive ceramic fuel cell (PCFC), where hydrogen protons are conducted to the oxygen side. The most promising electrolytes for PCFCs are BaCe1−x−yZrxYyO3−δ (BCZY), known for their high protonic conductivity and stability.25–27 The higher Ce content gives better protonic conductivity, but BCZY materials with the higher Ce content have the worst chemical stability in the atmosphere of CO2 or steam.28,29 The addition of Zr on the B-site improves the chemical stability of barium cerate-based materials but at the expense of the material conductivity.28 A composition, with 70% of Ce on the B-site, appears to give an efficient compromise between those two factors.30
Doping Zr into the B-site of barium cerates provides the advantage of chemical stability. However, due to the poor grain-boundary conductivity of barium zirconates, such materials require very high temperatures for complete densification.31–33 The high sintering temperature causes production and barium evaporation issues.34 Therefore, there is a concern about the application of PCFCs on a large scale, and only a few researchers have managed to produce a cell with a large surface area delivering sizable power.35,36 Used here, 4% Zn doped BCZY (BaCe0.7Zr0.1Y0.16Zn0.04O3−δ, BCZYZ) can diminish some manufacturing problems. A low level of ZnO doping into barium zirconate (Ba0.97Zr0.77Y0.19Zn0.04O3−δ) significantly reduced the sintering temperature without a noticeable impact on material conductivity.31 Moreover, doping ZnO into (Ba–Ce0.5Zr0.3Y0.16Zn0.04O3−δ) greatly improved its stability against carbonation.31 Other aids also were investigated as the sintering aids, such as 1% doping of metal oxide of Cu, Ni, Zn, Fe, Co, Ti in BaCe0.9Gd0.1O3−δ. They promoted grain agglomeration and decreased sintering temperature for up to 150 °C. Materials with Zn and Cu doping produced the highest conductivity; however, there is a concern about Cu doping due to the formation of an impurity phase and its reduction to the metallic phase;37 hence doping with Zn seems to be a preferable option.
Thanks to the high operating temperature and catalytic activity of Ni composite electrodes for ammonia cracking,15,32 high-temperature fuel cells seem to be a preferable option for ammonia fuel. Ammonia can be split in high-temperature fuel cells into H2 and N2, with much higher efficiency than, for example, in low-temperature fuel cells, where auxiliary units are required and typically a high sensitivity to poisoning by ammonia yields significant challenges. Also, the produced heat from power generation could be utilised for the endothermic reaction of NH3 splitting, improving the theoretical efficiency of the process.38
In terms of using ammonia as a fuel in oxide ion conducting SOFCs some important advances have been achieved;15,32 however, there are some important advantages when utilising protonic conducting oxides. PCFCs have the advantage of higher catalytic activity for the NH3 cracking reaction.15,38 Also, in the protonic type of SOFC, water is produced at the air electrode, minimising corrosion issues and generation of NOx from electrochemical oxidation when oxide ions diffuse to the fuel electrode in classic oxide conducting SOFCs.38,39 If the water is generated on the fuel side, the exhaust gas would contain a mixture of a small amount of unconverted ammonia and steam, which, when cooled in the exhaust, yields a very corrosive liquid solution or yields NOx when combusted.
PCFCs have been demonstrated for use with ammonia fuel in planar button cells26,39–50 and micro tubular cells.51 Whilst early results demonstrated very low power they did show effective cracking of ammonia even at 1%Ni loading.46 Yang et al. developed a cell with a Ni/BaCe0.75Y0.25O3−δ (Ni/BCY25) fuel electrode, BaCe0.9Y0.1O3−δ (BCY10) electrolyte and Sm0.5Sr0.5CoO3−δ (SSC) oxygen electrode.39 The author showed that the activity of the Ni/BaCe0.8Y0.2O3−δ Ni/BCZ20 composite for ammonia decomposition is much higher than in the case of Ni–YSZ and Ni–CGO composites. They also studied the effect of water in fuel stream, which although initially improved the cell performance eventually has deteriorating influence. One of the highest power densities on ammonia PCFCs was achieved by Aoki et al.48 through depositing a thin 1 μm layer of BaCe0.7Zr0.1Y0.2O3−δ (BCZY) electrolyte on a Pd solid anode film and La0.6Sr0.4Co0.2Fe0.8O3−δ (LSCF) oxygen electrode. They achieved 0.58 W cm−2 power density at 600 °C, and the cell was operated for about 3 h at 600 °C at about 0.6 A cm−2 current density. A similar power density, 0.6 W cm−2, was achieved by Duan et al. who tested a set of PCFC button cells with various fuels, including ammonia.49 Zhu et al. reported ammonia-fed reversible protonic ceramic fuel cells with a Ru-based catalyst.52 operating for 1250 h; however, ammonia was cracked in a separate reactor. When the button cell was operated under direct ammonia, the voltage dropped in 15 h. He et al. utilised a Pd added electrolyte to produce 0.724 W cm−2 at 650 °C with direct ammonia feed.50 A stability test was performed at 550 °C for about 200 h in pure H2 and 130 h in ammonia fuel on a small laboratory scale fuel cell. 1 W cm−2 at 700 °C in a PCFC working directly on NH3 was reported in a micro tubular cell with the BaZr0.1Ce0.7Y0.1Yb0.1O3−δ (BZCYYb) electrolyte, Ni/BZCYYb composite fuel electrode and PrBa0.5Sr0.5Co1.5Fe0.5O5+δ (PBSCF) oxygen electrode.51 The low durability of the cell was improved by adding an iron layer on the outer layer of the fuel electrode.
Although large PCFC cells (planar) have been demonstrated on continuous H2,36 this has not yet been achieved on ammonia. Here, we present a large Ni/BCZYZ supported tubular cell with a thin BCZYZ electrolyte and La0.8Sr0.2Co0.5Fe0.5O3−δ/BaCe0.7Zr0.1Y0.16Zn0.04O3−δ (LSCF/BCZY) oxygen electrode, with a 36 cm2 surface area and peak power of 8.5 W at 750 °C, working durably under NH3, which is a breakthrough in the scale and applicability of proton conductive PCFCs.
Results and discussion
XRD analysis
X-ray diffraction patterns of the used BCZYZ and LSCF are shown in ESI Fig. S1(a).† Unit cell parameters from Rietveld refinement are listed in ESI Table S1.† BaCe0.7Zr0.1Y0.16Zn0.04O3−δ (BCZYZ) possessed a single phase of orthorhombic symmetry (Pbnm space group) in accord with prior studies.37 For LSCF, a single perovskite phase was observed with the R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group. LSCF with a Co/Fe ratio of 50/50 provided a pure phase in rhombohedral symmetry which was more stable than in orthorhombic symmetry.53
m space group. LSCF with a Co/Fe ratio of 50/50 provided a pure phase in rhombohedral symmetry which was more stable than in orthorhombic symmetry.53
Characterisation of cell structure
A tubular cell was produced by extrusion of the NiO/BCZYZ support and the multistep co-firing process with a dip-coated BCZYZ electrolyte and LSCF/BCZYZ oxygen electrode.54Fig. 1(a) shows the BSE/SEM analysis of the cell microstructure after reduction with H2. The SEM analysis of the cross-section of the cell shows a dense ∼40 μm BCZYZ electrolyte, porous ∼1100 μm Ni/BCZYZ fuel electrode support and ∼5 μm LSCF/BCZYZ oxygen electrode. | ||
| Fig. 1 SEM micrographs of the tubular cell structure after reduction. (a) The cross-section of the cell (BSE). (b) The cross-section of the cell (SEI). (c) Ni/BCZYZ fuel electrode. (d) LSCF/BCZYZ oxygen electrode. | ||
The image in Fig. 1(a) was obtained with a Back Scattering Electron probe (BSE) and was used for porosity measurement of the fuel electrode with ImageJ software,55 thanks to the high contrast between the resin, ceramic and metal. Two phases visible on the fuel electrode side are Ni with a darker colour, and a brighter phase, BCZYZ. The fuel electrode shows around 15% total porosity, made of large pores formed between the particles and smaller pores at the BCZYZ/Ni interface and in the Ni itself, due to NiO reduction, Fig. 1(c) and (d) show the 5 μm LSCF/BCZYZ oxygen electrode and its porosity which is about 50%. The thickness of the oxygen electrode is quite low placing some constraints on current collection to avoid some additional sheet resistance; therefore, it should be optimised in future studies. The electrode was sintered as the composite to ensure a good interface with the BCZYZ electrolyte. Both electrodes have good adhesion with the BCZYZ electrolyte and no cracking or delamination is observed in the cell structure.
Electrochemical characterisation
The cell performance was characterised by current–voltage (I–V) measurements and impedance analysis (EIS) under various conditions, such as varying temperature, flow of ammonia, applied potential and fuel gas mixture.Impedance spectroscopy (EIS) is a transient-response technique used to test cells electrochemically.56–58 EIS can be used to separate processes occurring at various speeds (frequency), giving insight into their kinetics. The Nyquist plot of impedance spectra illustrates ohmic resistance, indicated by the intersection point of the plot with the x-axis at high frequency and polarisation in the upper part of the plot above the x-axis. For the well-optimised cells, the ohmic resistance Rs mainly comes from the electrolyte, as electrodes should have a high electronic conductivity. The frequency-dependent polarisation responses are related to various processes from fuel and oxygen electrodes. Usually, they are related to the diffusion of gases, their conversion on the electrode, transport of species and charge transfer reaction.59
The individual processes could not always be easily separated from the Nyquist plot. Those at close frequencies, either from the same electrode or the opposite ones, tend to merge; in our case, only two arcs are evident (Fig. 3(b)). The equivalent circuit could be constructed using complex non-linear least squares (CNLS) fits to distinguish and characterise ohmic resistance and polarisation.60 However, the fitting becomes challenging without previous identification of some of their parameters, as the method requires a pre-defined model with a specific number and type of elements. Often, the equivalent circuit could be misinterpreted, and even a substantially different system could respond similarly. Alternatively, the number, size, and frequency of those responses could be pre-determined with DRT61,62 constructed from the real or imaginary part of impedance spectra. In the usual approach, their dependency on various conditions is measured, e.g. how processes change with gas composition on one of the electrodes, applied voltage or when working at various temperatures.61 Theoretically, processes could be identified by the value of their characteristic frequencies, capacitance and activation energy. However, one should keep in mind that processes on porous electrodes and interface strongly depend on the cell structure and characteristics, and direct comparison between the same type of system but different cells is not straightforward;63 also, obtaining such data for PCFC is relatively rare, as most of the work has been done on the Ni/YSZ system.
 | ||
| Fig. 2 Scheme of the PCFC operating on ammonia, with the LSCF/BCZYZ oxygen electrode (top) and Ni/BCZYZ fuel electrode (bottom). | ||
Several steps could be considered for NH3 utilisation in the PCFC system (Fig. 2). Processes on the fuel electrode include gas diffusion, ammonia decomposition to N2 and H2 and splitting of H2 to produce electrons and protons.15,47,48,64 At the oxygen electrode, oxygen air is reduced to oxygen ions which join with hydrogen protons to produce water.65
The DRT characteristics of data measured at various ammonia flows and applied potentials helped us to separate most resistive contributions taking place during the cell operation, Fig. 3. Although a more detailed analysis needs to be done to assign them to specific processes or electrodes unambiguously, we can still observe their behaviour and determine their fundamental parameters. The DRT shows up to five processes, with a dominance at middle to low frequency, Fig. 3(c). Under a lower flow of ammonia, along the ohmic resistance, the polarisation processes P2, P3 and P5 are affected, indicating the fuel electrode responses. Other processes, P1 and P4, are not much affected by the fuel flow and are believed to come from the oxygen side, Fig. 3(c).
 | ||
| Fig. 3 The electrochemical performance of the tubular cell at various flow rates of NH3. (a) I–V curve of the cell operating at 66 and 133 ml min−1 flow of NH3. (b and c) Corresponding EIS and DRT under OCV conditions. (d and e) EIS and DRT at 66 ml min−1 flow of NH3 at various applied DC potentials in fuel cell mode. | ||
Assigning P5 to a specific process does not raise much doubt. The low-frequency process most likely shows a concentration loss of the gas phase on the fuel electrode. P5 emerges at lower ammonia flow and gradually increases under an applied potential, Fig. 3(d) and (e), and eventually becomes dominant at the fuel starvation point. Gas diffusion processes usually occur at a low frequency and have low activation energy, as they do not depend on material properties, but only on microstructure. They mainly depend on the porosity of the electrode and fuel concentration, which, if too low, may lead to a limitation at high current density.
P2 and P3 likely are related to the H2 protonation process in the active region of the fuel electrode. It is generally assumed that the electrochemical oxidation of H2 could take place up to 20 μm from the electrolyte, while regions further from the interface could show the contribution of gas diffusion. The process in an active region would include diffusion of H2 gas to the vicinity of the Ni surface and its adsorption/dissociation on Ni.63 In the next step, H+ species would migrate to the interface with BCZYZ, through the surface or bulk from where it will be conducted through the electrolyte to the oxygen electrode. Aoki48 evaluated the mechanism on the BaCe0.7Zr0.1Y0.2O3−δ/Pd (BCZY/Pd) metal interface with impedance spectroscopy and concluded that the monoatomic hydrogen dissolves at the BCZYZ/Pd interface and a proton is transferred from metal to the interface. In work on the Pd/BaCe0.8Y0.2O3−δ/L0.6Sr0.4Co0.2Fe0.8O3 cell,64 the equivalent circuit was made of two elements, charge transfer resistance on the oxygen electrode at 104–102 Hz and mass transfer on the fuel side 101–10−1 Hz. The authors also recognised contribution in the middle-frequency range, 102 Hz.
Polarisation was not evident as it merged with other elements. However, increased contribution at lower fuel concentration indicated fuel concentration dependence, and the processes on the fuel electrode were relatively small compared to processes on the oxygen side and ohmic resistance.
P2 and P3 decrease with a lower flow of ammonia, which somehow is counterintuitive. The slope of the curve is slightly lower at 66 ml min−1, which also suggests better kinetics. One possible explanation is the higher temperature inside the cell due to the smaller concentration of ammonia and the extent of the endothermic cracking reaction. However, the measured temperature was only around 6 °C higher for the test with 66 ml min−1 of ammonia, which cannot explain the significant change in ohmic and polarisation resistance. Similarly, Miyazaki47 recognised a decrease in total polarisation resistance at the fuel electrode and a slight change in ohmic resistance at a lower flow and concentration of ammonia. The authors studied the catalytic activity of the Ni/BCZY composite and concluded that the ammonia decomposition rate was significantly reduced at higher flow rates due to the short contact of the gas with the catalyst. They concluded that at higher flow rates, the fuel electrode polarisation was affected by the insufficient conversion of ammonia, while the increase of ohmic resistance was believed to be related to the decrease in temperature due to the endothermic effect.
The LSCF used in our study for the oxygen electrode is a mixed oxygen and electronic conductor, responsible for oxygen gas reduction and oxygen ion conduction in such applications. The possible elementary steps on the LSCF-based oxygen electrode in the PCFC would include diffusion of the oxygen gas to the electrode surface, adsorption of oxygen on the LSCF surface, reduction of oxygen to oxygen ions, diffusion of oxygen ions through the LSCF to the interface and finally, water production at the triple phase boundary (TPB) with BCZYZ (Fig. 2).65
Similar values of frequency and capacitance to those for the processes associated with the LSCF electrode in PCFCs44,66 let us assign P1 to interfacial resistance at the oxygen electrode, which includes water formation in charge transfer between oxygen species and protons. The P4 process is most likely related to concentration losses due to its low frequency and response under an applied potential. Under applied potential, P4 slightly increases at 0.1 V voltage bias and moves to a lower frequency. At higher overpotential, the process cannot be recognised as it most likely merged with fuel electrode process P5.
The cell performance tested at 133 ml min−1 of NH3 at 650–750 °C is shown in Fig. 4. The generated power increased significantly with rising temperature, and up to 7.7 W was generated at 750 °C from a single cell with 36 cm2 of surface area when tested in the 0–10 A range. Due to the limitation of power load, the peak power was determined by connecting a series of low resistance, high power resistors as the load for the cell and was equal to 8.5 W, reaching 0.236 W cm−2 maximum power density, Fig. S2.†
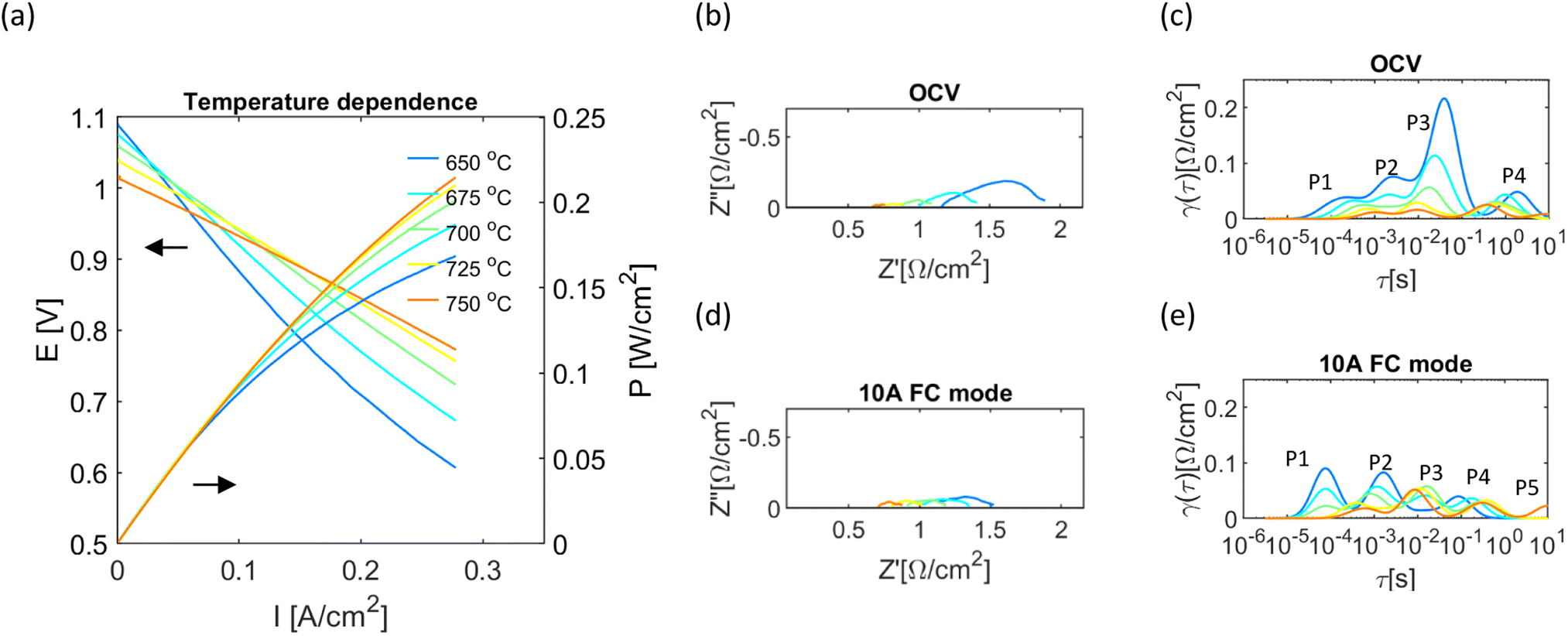 | ||
| Fig. 4 The electrochemical performance of the tubular cell operating at 650–750 °C with 133 ml min−1 of NH3. (a) I–V curve of the cell (b and c) EIS and DRT at 650–750 °C under OCV conditions. (d and e) EIS and DRT at 650–750 °C at 10 A in fuel cell mode. | ||
When changing current, the slope of the line seems to decrease, this is especially visible for lower test temperature, which could be related to cell temperature increasing through Joule heating – according to the temperature measurement inside a cell. The slope change is lower at higher temperatures and even increases at 750 °C. Such behaviour could be related to increased concentration losses, leading to competition between temperature and concentration factors as well as lessening of Joule heating with higher electrolyte conductivity at higher temperature.
The OCV was between 1.08 and 1.01, decreasing with rising temperature, which agrees with the thermodynamics of H2 containing gas rather than the direct oxidation of NH3. For the direct conversion of ammonia, the theoretical OCV value increases with rising temperature and is much higher than for H2 (ref. 38) (Table S2†). Nevertheless, the relatively high value of OCV indicates good gas-tightness of the cell, sufficiently dense electrolyte and robust structure.
For the OCV conditions in the investigated temperature range, the DRT shows at least four processes, with a dominant contribution from P2 and P3 processes at middle to low frequency, previously assigned to the fuel electrode (Fig. 3(c)). Similarly, DRT at 10 A (Fig. 3(e)) shows up to four processes, but the size and frequency changed under applied anodic polarisation. Apart from the number and approximation of characteristic frequency of circuit elements, DRT gives the possibility to find their resistance, as the area under the peak corresponds to the ohmic resistance of the dynamic process;61 this was achieved by fitting Gaussian peaks into DRT (Fig. S3(a) and (b)†).
The polarisation and ohmic resistance of the cell, obtained from Gaussian fitting, were plotted based on the Arrhenius dependence for OCV and fuel cell operation, Fig. 5. The linearity of the plots confirms the correctness of the Gaussian estimates.
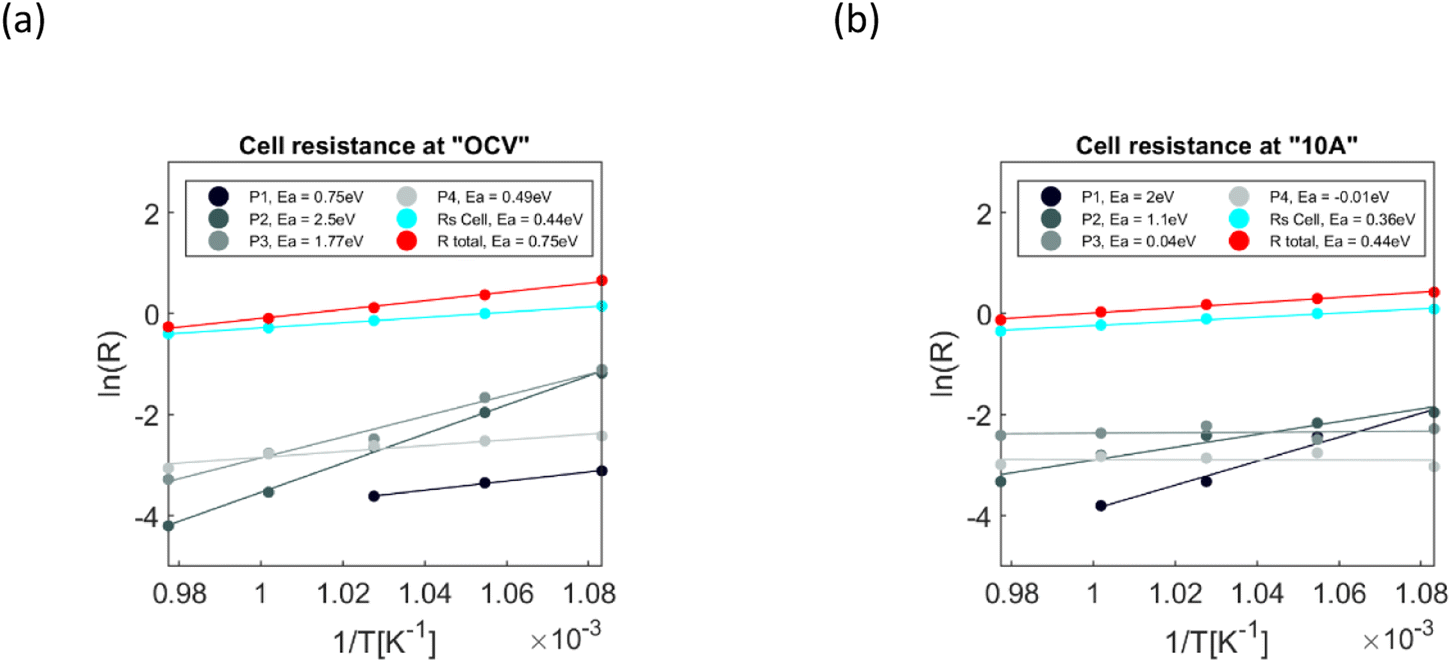 | ||
| Fig. 5 Arrhenius plot of ohmic and polarization resistance at 650–750 °C, where “R total” is the sum of all resistive contributions. (a) Under OCV conditions (b) at 10 A in the fuel cell mode. | ||
R s, the ohmic resistance is dominant and is responsible for the highest voltage drop during the cell operation; under OCV conditions, values of activation energy and conductivity are close to what were reported for 5% Zn doped BCZYZ under dry conditions.37 The conductivity extracted from Rs was equal to 0.0103 S cm−1 in dry NH3 at 700 °C, while Hossain et al. achieved 0.00958 S cm−1 in dry 5% H2 in Ar at 700 °C. Those results indicate that the ohmic resistance mainly comes from the electrolyte.37 The ohmic resistance dropped under an applied current, which could be related to the increased temperature inside a cell during the operation, but other factors than the temperature could influence it. The decrease in ohmic resistance could be related to an increase in proton carrier concentration due to a higher rate of H2O production on the oxygen electrode side, thus increasing the concentration of hydroxyl ions in the vicinity of the electrolyte/oxygen electrode interface.64 The resistance of the high-frequency P1 process, assigned to the interfacial process at the oxygen electrode, increases at an applied current, while its activation energy (Ea) rises from 0.75 eV at OCV to 2 eV at 10 A. The fuel electrode P2 process is thermally activated; its Ea changes from 2.5 eV at OCV to 1.1 eV under fuel cell operating conditions. The fuel electrode process P3, from being very high-temperature dependent at OCV, changes to being almost no temperature dependent at 10 A; this may indicate some dissociative-adsorption processes coupled with gas diffusion, which becomes a dominant factor at higher overvoltage. Similar behaviour could be observed for P4, previously assigned to concentration losses on the oxygen electrode. Very low activation energy at an applied voltage confirms coupling with gas diffusion. One more process, P5, becomes visible at 10 A (Fig. 4(e)), which also becomes visible in a test we conducted at lower NH3 flow, Fig. 3(c), suggesting a fuel electrode concentration loss.
With the number and polarisation resistance of elements known, also the equivalent circuit (EC) was constructed,67 Fig. S3(c) and (d),† and fitting to EC gives very close results in terms of ohmic and polarisation resistance to Gaussian fitting into DRT spectra. EC consists of an ohmic resistance (Rs) and four semicircles; the inductance (3.7 × 10−7) was fitted to the impedance at 650 °C and extracted from the data. The semicircles were modelled by resistance, and constant phase elements (CPEs) connected in parallel (R/CPE). R represents the polarisation process's resistance, while CPEs represent its capacitive behaviour and are modelled by two parameters. With the R and CPE known, the values of capacitance and characteristic frequency were calculated, Table 1. Due to the diminishing contribution of polarisation at a higher temperature, the fitting was done at 650–700 °C, which offers a better estimation.
| No. | Description | Char. freq. [Hz] at 650 °C OCV/10 A | E a [eV] at 650–700 °C OCV/10 A | Capacitance [F cm−2] at 650 °C OCV/10 A |
|---|---|---|---|---|
| R s | The ohmic resistance of the cell | — | 0.44/0.36 | — |
| P1 | Oxygen electrode | 1310/7584 | 0.78/2 | 2 × 10−3/1.1 × 10−4 |
| P2 | Fuel electrode | 35/256 | 2.5/1.1 | 1.3 × 10−2/4.1 × 10−3 |
| P3 | Fuel electrode | 3.6/29 | 1.77/0.04 | 1.5 × 10−1/7.7 × 10−2 |
| P4 | Oxygen electrode | 0.18/1.9 | 0.49/−0.01 | 12.7/1.64 |
Considering the high operating temperature and excellent catalytic properties of Ni and barium cerate-based materials15,38,39 for ammonia decomposition, we expect that ammonia would be almost entirely cracked to H2 and N2 on the fuel electrode; and rather than direct oxidation of ammonia, the hydrogen will be used in the electrochemical reaction. Yang et al.39 compared the catalytic activity of Ni/BCe0.75Y0.25O3−δ with those of Ni/8 mol% YSZ (8YSZ) and Ni/Ce0.9Gd0.1O1.95 (CGO) composites for the ammonia breaking reaction. The reaction rate increased with temperature; for Ni/BaCe0.75Y0.25O3−δ, a 98.6% gas conversion was achieved at 600 °C, whereas for Ni/YSZ it was only 50%, while Ni/CGO showed 60% conversion efficiency. Similar results were achieved in the further work of the group on Ni/BCe0.4Zr0.4Y0.2O3−δ, where 100% ammonia decomposition was achieved below 600 °C.47
These assumptions are also confirmed in our experiment (Fig. 6), which compares the current–voltage and impedance characteristics of the cell performance at 133 ml min−1 flow of NH3 and in the mixture of H2/N2 (200/66 ml min−1), which have the same flow and stoichiometry assuming complete ammonia decomposition.
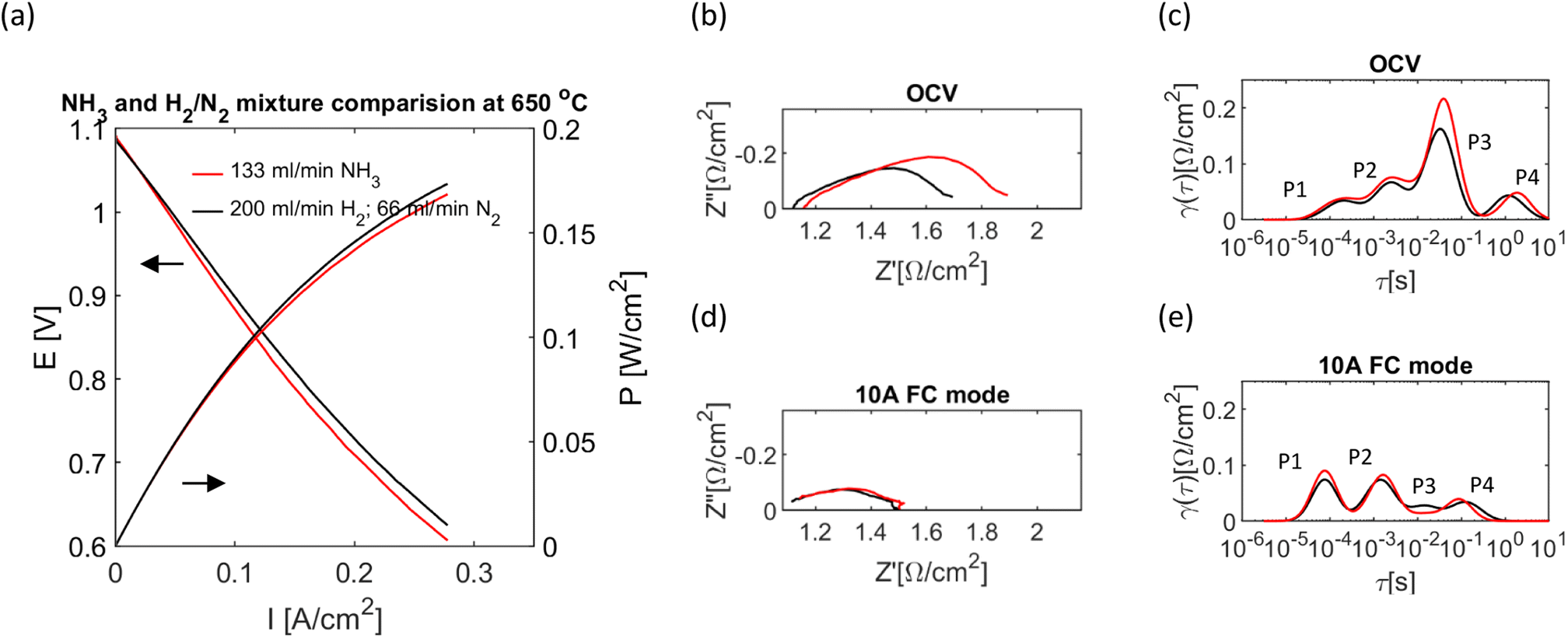 | ||
| Fig. 6 The comparison of cell operating with the H2/N2 mixture and NH3 at 650 °C. (a) I–V characteristics. (b and c) Corresponding EIS and DRT under OCV conditions. (d and e) Corresponding EIS and DRT at 10 A in fuel cell mode. | ||
The same shape of the I–V curves suggests similar electrode kinetics in both cases, further confirmed by impedance data showing no difference in the shape of the EIS or DRT curve for the whole frequency range (Fig. 6(b)–(e)). The slightly lower performance of ammonia could be explained by the lower temperature inside the cell measured by the thermocouple (the 650 °C reported in the plot is furnace temperature). A thermocouple was placed in the top part of the cell; the measured temperature was 661 °C for the H2/N2 mixture and 653 °C for NH3 fuel while at the maximum current (10 A) the measured temperature was 694 °C and 679 °C, respectively. Under OCV conditions (Fig. 6(c)), the P2 & P3 processes increase at a higher rate than P1 and P4 for ammonia fuel, which could be due to the higher activation energy of those as indicated in Fig. 5(a). Respectively, the higher increase of P1 and P2 in comparison to P3 and P4 when current was applied (Fig. 6(e)) could be related to the change of their activation energy during fuel cell operation, Fig. 5(b). Higher temperature inside a cell than in the furnace may be due to small leaks in the cell; thus, burning of H2 and heating the cell from outside; we do not expect that O2 would diffuse into the cell through the eventual leaks due to slight overpressure (8 mbar) on the fuel side. The higher temperature during the fuel cell operation comes from the exothermic reaction of H2 oxidation and Joule heating. The smaller increase in temperature for the ammonia fed of the cell is due to the endothermic reaction of ammonia cracking, partially compensating for the exothermic fuel cell processes. The ammonia cracking was further confirmed by outlet gas analysis at 750 °C with gas chromatography, where no ammonia in the gas stream was detected.
Cell operation and stability
Cell operation for a total time of about 170 h is shown in Fig. 7. The stability test was conducted at a constant potential (−0.25 V relative to OCV) for 4 cycles under 133 ml min−1 flow of pure NH3 for more than 140 h. Between the discharging cycles, the cell was characterised by I–V and EIS characteristics (indicated in the figure by voltage fluctuations); their comparison gives a more transparent overview of how individual processes are affected by stability loss, Fig. 8. | ||
| Fig. 7 Performance of the cell during the operation time. | ||
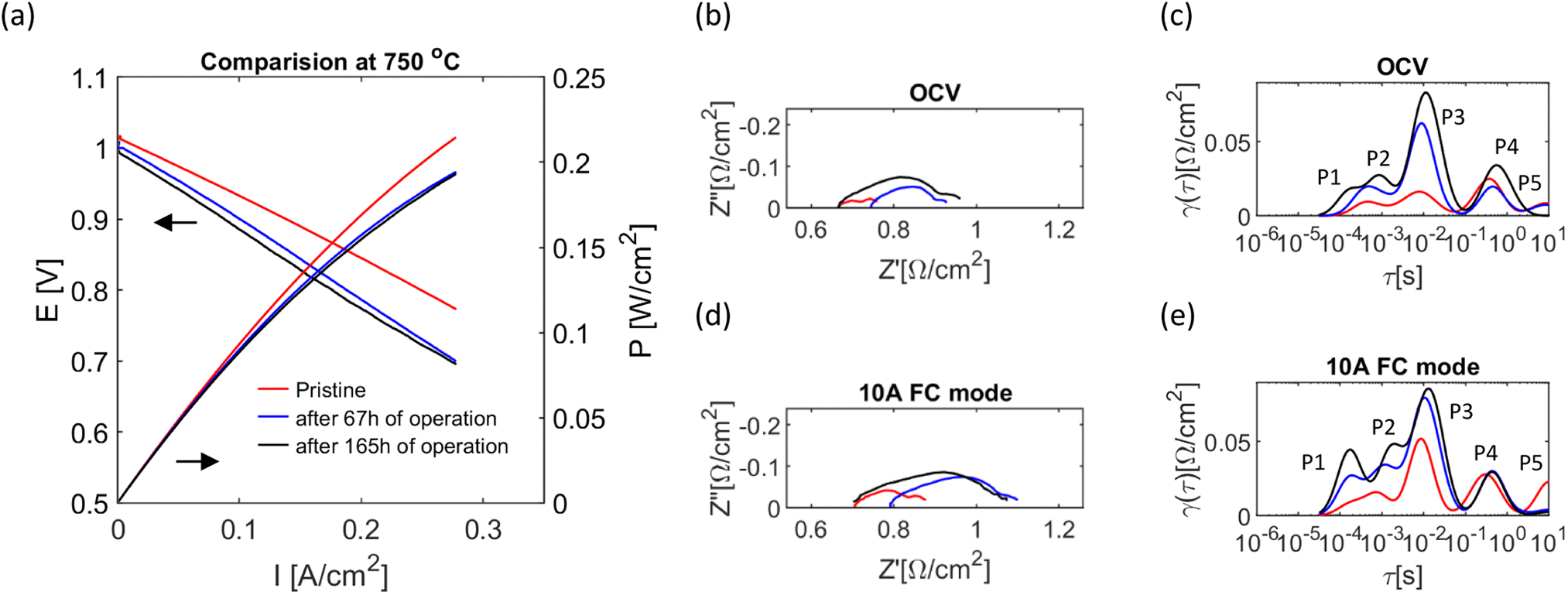 | ||
| Fig. 8 The electrochemical performance of the tubular cell at various operation periods at 750 °C. (a) I–V curve of the cell operating in fuel cell mode. (b and c) Corresponding EIS and DRT under OCV conditions. (d and e) Corresponding EIS and DRT at 10 A in fuel cell mode. | ||
The first stability cycle was conducted at 650 °C under 133 ml min−1 flow of pure NH3 for ∼20 h. At this point, the cell had the highest degradation rate and lost up to 12% of the current. The temperature inside the cell increased from 653 to 656 °C due to the Joule heating and exothermic reaction of H2 oxidation, Fig. S4(a).† Following changes in generated current, the temperature gradually decreased, likely due to the drop in electrochemical activity; hence, less heat was produced. Also, with the deterioration in generated current, the exhaust flow went up, presumably due to the drop in fuel utilization. Next, the temperature was increased to 750 °C, the cell was electrochemically characterised, and another stability run with the same ammonia flow was conducted for ∼ 20 h. A total 7.5% current loss was registered in this cycle, mainly during the first 10 h, and then the degradation rate started to decrease. As mentioned previously, the exhaust flow increased, and the temperature inside the cell dropped following trends in generated current, Fig. S4(b).† The I–V test after the second cycle (“after 67 hours of operation” in Fig. 8) has slightly lower OCV and a higher slope than that of a “pristine cell”. Nyquist and DRT plots show an increase in ohmic resistance and all polarisation processes. Presumably, the change in OCV is associated with an increasing leaking rate. The drop in polarisation could be caused by the changes in the microstructure or stoichiometry of electrodes. An increase in ohmic resistance could be caused by the electrolyte. As virtually all processes have increased their resistance, it is also possible that the current collector lost its initial efficiency, either on oxygen or a fuel electrode side.
The third stability run was made for ∼20 h under the same conditions as the previous cycle. The degradation rate further decreased, leading to a 2.5% performance drop. The behaviour of the exhaust gas flow signal was different than in the previous stability runs, Fig. S4(c).† The flow of exhaust gases slightly decreased during the cell operation, indicating an increase in leakage rate. The fourth cycle was conducted for 75 h. Some fluctuations in current and flow were registered, but no power was lost during the last discharge. The I–V curve and EIS after the fourth cycle are shown in Fig. 8 as “after 165 h of operation”. The OCV slightly decreases, the cause of which could be deterioration of leaks. The polarisation resistance further increased for all mentioned processes; while, the ohmic resistance significantly decreased.
Summing up, the stability test shows a higher degradation rate at the start than at the end, which could lead to complete stabilization of the current for long-term operation. A higher degradation rate at the initial time is typical for SOFCs. Unfortunately, the increased leaking rate in our cells made it impossible to continue the test for a longer time. Leak, between the metal cap and cell at the top, has led to a quick decline of generated current, in further (not shown) part of the test.
Leaks will significantly reduce the cell performance and accelerate its degradation, which likely is the case in our cell. Besides fuel draining, they could create unfavourable gas conditions on both electrodes, thus, decreasing their functionality. Moreover, the mixture of air and H2 would burn, creating local hot spots in the cell. Therefore, improving cell seals is our priority in further developing our ammonia-powered fuel cells.
Experimental
Cell preparation
The tubular cell evaluated in this work was fabricated by extrusion of the NiO/BaCe0.7Zr0.1Y0.16Zn0.04O3−δ (NiO/BCZYZ) tubular support and dip-coating of the BCZYZ electrolyte and the La0.8Sr0.2Co0.5Fe0.5O3−δ/BaCe0.7Zr0.1Y0.16Zn0.04O3−δ (LSCF/BCZYZ) oxygen electrode. The process parameters were optimised to achieve thin and uniform functional layers; the manufacturing method of the cells is given elsewhere.54Cell assembly
100 μm Inconel 625, 1 mm nickel wire and 1 mm nickel sheet were formed into collars and brazed onto both ends of the cell using a CuO/Ag braze material. The assembly was then heated to 1030 °C at 10 °C min−1, allowed to dwell for 12 minutes and cooled to room temperature at 10 °C min−1. This burned off binders and solvent vehicles and melted the braze material, ensuring a hermetic seal between the collars and the cell.One open and one closed end cap were formed from Inconel 625 and nickel and were brazed to each end of the cell using proprietary braze materials and methods. The open cap was brazed to an alumina tube for the support and interface with the test infrastructure. Ni wires, welded to the fuel cell's cap were woven and placed into the cell for the fuel electrode current collection.
Ferro 903B AG LTCC conductor paste was applied to the air side electrode to ensure optimal current collection. A 2 mm silver (99.9%) wire was formed into an air side electrode current bus, and this was affixed to the electrode by wrapping 1 mm silver wire at regular intervals. Additional conductor paste was then applied to the wires to enhance the contact between the silver wires and the conductor (Fig. 9).
 | ||
| Fig. 9 Cell assembly and working principle. (a) The image of the cell mounted on the testing station. (b) The principle of cell operation. | ||
Electrochemical test/data collection
Electrochemical tests were carried out using a Biologic (VSP-300) and EZ lab software. The cell was characterised for about seven days by performing a series of tests, including reduction, cycling under OCV conditions, electrochemical characterisation (I–V and EIS), and about 140 h of stability test at a constant voltage and 133 ml min−1 of pure NH3.Besides the furnace temperature, the temperature inside the cell was monitored using a thermocouple placed inside a cell in its top part. The anode exhaust flow was measured using a mass flow controller. The mass flow controller generated a voltage signal equivalent to the exhaust mass flow, assuming pure H2. As the composition of gases changes during the operation (ratio of H2 to N2), it is not possible to measure an exact flow with this method; however, it gives a good indication of its relative change.
Before electrochemical characterisation, the fuel electrode was reduced in 100 ml min−1 of H2 and 100 ml min−1 of N2 mixture at 650 °C overnight. Current–voltage (I–V) measurements and electrochemical impedance spectroscopy (EIS) were used to characterise the cell performance at 25 °C intervals between 650 °C and 750 °C after reduction. The gas flow rate used for the analysis was 133 ml min−1 of anhydrous ammonia, or a synthetic mixture of hydrogen/nitrogen was used to replicate the equivalent molar value of gas (H2 (200 ml min−1)/N2 (66 ml min−1)) assuming total decomposition of ammonia. The cathode side was open to the air atmosphere of the furnace. Additionally, 500 ml min−1 of air was delivered to the furnace cavity to ensure faster air exchange with the environment. For the stability test, the cell was discharged at a constant potential (−0.25 V relative to OCV).
The EIS was taken in the 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–0.1 Hz frequency range with an excitation voltage amplitude of 20 mV. The free Matlab-based DRTtool by Wan et al.68 was used to construct DRT. Before conducting analysis, the quality of impedance spectra was investigated with Kramers–Kroning (K.K.) transforms.69,70 As the real and imaginary parts are connected through Kramers–Kroning transformations,64 only the imaginary part of impedance could be considered. Good quality data should not show a more significant residual than 0.5%.61
000–0.1 Hz frequency range with an excitation voltage amplitude of 20 mV. The free Matlab-based DRTtool by Wan et al.68 was used to construct DRT. Before conducting analysis, the quality of impedance spectra was investigated with Kramers–Kroning (K.K.) transforms.69,70 As the real and imaginary parts are connected through Kramers–Kroning transformations,64 only the imaginary part of impedance could be considered. Good quality data should not show a more significant residual than 0.5%.61
Complex non-linear least-squares (CNLS) approximation was used for EIS analysis with the equivalent circuit,53 consisting of elements connected in series or parallel that correspond to individual electrode processes. The impedance spectra were fitted to the equivalent circuit using Z-view software.71
Using an Agilent micro GC, the anode exhaust gas was analysed at 100 second intervals for hydrogen, nitrogen and residual ammonia. Using Soprane software, the chromatograms were analysed to give percent gas composition. Maximum power was determined by connecting a series of low resistance, high power resistors as the load across the cell.
XRD and SEM characterisation
XRD analysis was carried out using a PANalytical X-ray diffractometer with CuKα1 radiation (λ = 1.54056 Å) in the range 2θ = 10–90. Scanning electron microscopy was carried out using an FEI Scios to image fractured cross-sections of the tubular cell.The cell described as “after reduction” in Fig. 1 was reduced with pure H2 and characterised by I–V and EIS for one day in pure ammonia fuel. The cell was from the same manufacturing batch as the cell whose performance was evaluated in this paper.
Conclusions
A large tubular cell with 36 cm2 of surface area based on a BCZYZ proton conductive electrolyte was successfully fabricated by the extrusion and dip-coating method.With the help of DRT, polarisation processes in the cell could be identified and characterised; the presented method also helped develop the equivalent circuit by CNLS approximation. The cell performance was mainly affected by the ohmic resistance of the electrolyte and processes on the fuel electrode.
The cell was characterised for an extended operation time of 170 hours in pure ammonia fuel, showing exceptional stability and potential for direct ammonia fuel cell applications. The tested cell generated up to 8.5 W with a maximum power density of 0.236 W cm−2 at 750 °C. A satisfying peak power allows for relatively high fuel utilisation, reaching 30% at 133 ml min−1 flow of ammonia.
Conflicts of interest
There are no conflicts to declare.Notes and references
- IMO, Initial Imo Strategy on Reduction of GHG Emission From Ships, Resolution MEPC.304(72), 2018 Search PubMed.
- C. Graves, S. D. Ebbesen, M. Mogensen and K. S. Lackner, Renewable Sustainable Energy Rev., 2011, 15, 1–23 CrossRef CAS.
- European Commission, A Hydrogen Strategy for a Climate-Neutral Europe, 2020 Search PubMed.
- J. Hansson, S. Brynolf, E. Fridell and M. Lehtveer, Sustainability, 2020, 12, 10–14 CrossRef.
- NYK Line, https://www.nyk.com/english/news/2020/20200812_01.html, accessed July 2022.
- Public, http://www.publicnow.com/view/342623DC5C5DFB326C4D4244D0CA15A2E597ED72?1633690161, accessed July 2022.
- Warstila, http://www.publicnow.com/view/342623DC5C5DFB326C4D4244D0CA15A2E597ED72?1633690161, accessed July 2022.
- IEA, Global Hydrogen Review, 2021 Search PubMed.
- Ammonia Energy Association, https://www.ammoniaenergy.org/articles/japans-road-map-for-fuel-ammonia/, accessed July 2022.
- A. Tullo, C&EN Glob. Enterp., 2021, 99(8), 20–22 Search PubMed.
- J. Hansson, S. Månsson, S. Brynolf and M. Grahn, Biomass Bioenergy, 2019, 126, 159–173 CrossRef.
- R. Lan, J. T. S. Irvine and S. Tao, Int. J. Hydrogen Energy, 2012, 37, 1482–1494 CrossRef CAS.
- A. Klerke, C. H. Christensen, J. K. Nørskov and T. Vegge, J. Mater. Chem., 2008, 18, 2304–2310 RSC.
- S. Giddey, S. P. S. Badwal, C. Munnings and M. Dolan, ACS Sustainable Chem. Eng., 2017, 5, 10231–10239 CrossRef CAS.
- J. Yang, A. F. S. Molouk, T. Okanishi, H. Muroyama, T. Matsui and K. Eguchi, ACS Appl. Mater. Interfaces, 2015, 7, 28701–28707 CrossRef CAS PubMed.
- S. Chatterjee, R. K. Parsapur and K. W. Huang, ACS Energy Lett., 2021, 6, 4390–4394 CrossRef CAS.
- M. A. Shipman and M. D. Symes, Catal. Today, 2017, 286, 57–68 CrossRef CAS.
- G. Soloveichik, Nat. Catal., 2019, 2, 377–380 CrossRef CAS.
- C. Zamfirescu and I. Dincer, J. Power Sources, 2008, 185, 459–465 CrossRef CAS.
- D. Miura and T. Tezuka, Energy, 2014, 68, 428–436 CrossRef CAS.
- H. Kobayashi, A. Hayakawa, K. D. K. A. Somarathne and E. C. Okafor, Proc. Combust. Inst., 2019, 37, 109–133 CrossRef CAS.
- H. S. Spacil, US Pat., US 3558360, 1970.
- N. Q. Minh and T. Takahashi, Science and Technology of Ceramic Fuel Cells, Elsevier, 1995 Search PubMed.
- S. B. Adler, Chem. Rev., 2004, 104, 4791–4843 CrossRef CAS PubMed.
- C. Zuo, S. Zha, M. Liu, M. Hatano and M. Uchiyama, Adv. Mater., 2006, 18, 3318–3320 CrossRef CAS.
- Y. Lin, R. Ran, Y. Guo, W. Zhou, R. Cai, J. Wang and Z. Shao, Int. J. Hydrogen Energy, 2010, 35, 2637–2642 CrossRef CAS.
- Y. Meng, J. Gao, H. Huang, M. Zou, J. Duffy, J. Tong and K. S. Brinkman, J. Power Sources, 2019, 439, 227093 CrossRef CAS.
- F. Chen, O. T. Sørensen, G. Menga and D. Penga, J. Mater. Chem., 1997, 7(3), 481–485 RSC.
- S. V. Bhide and A. V. Virkar, J. Electrochem. Soc., 1999, 146, 2038–2044 CrossRef CAS.
- K. H. Ryu and S. M. Haile, Solid State Ionics, 1999, 125, 355–367 CrossRef CAS.
- S. Tao and J. T. S. Irvine, Adv. Mater., 2006, 18, 1581–1584 CrossRef CAS.
- H. G. Bohn and T. Schober, J. Am. Ceram. Soc., 2000, 83, 768 CrossRef CAS.
- K. D. Kreuer, S. Adams, W. Munch, A. Fuchs, U. Klock and J. Maier, Solid State Ionics, 2001, 145, 295 CrossRef CAS.
- B. Beyribey, H. Kim and J. Persky, Ceram. Int., 2021, 47, 1976–1979 CrossRef CAS.
- H. An, H. W. Lee, B. K. Kim, J. W. Son, K. J. Yoon, H. Kim, D. Shin, H. I. Ji and J. H. Lee, Nat. Energy., 2018, 3, 870–875 CrossRef CAS.
- R. J. Braun, A. Dubois, K. Ferguson, C. Duan, C. Karakaya, R. J. Kee, H. Zhu, N. Sullivan, E. Tang, M. Pastula, A. Wood, T. Joia and R. O'Hayre, ECS Trans., 2019, 91, 977–1008 Search PubMed.
- S. Hossain, et al. , Int. J. Hydrogen Energy, 2018, 43, 894–907 CrossRef CAS.
- J. Yang, T. Akagi, T. Okanishi, H. Muroyama, T. Matsui and K. Eguchi, Fuel Cells, 2015, 15, 390–397 CrossRef CAS.
- J. Yang, A. F. S. Molouk, T. Okanishi, H. Muroyama, T. Matsui and K. Eguchi, ACS Appl. Mater. Interfaces, 2015, 7, 7406–7412 CrossRef CAS PubMed.
- L. Pelletier, N. Maffei and A. McFarlan, J. Power Sources, 2005, 145, 262–265 CrossRef CAS.
- Q. Ma, J. Ma, S. Zhou, R. Yan, J. Gao and G. Meng, J. Power Sources, 2007, 164, 86–89 CrossRef CAS.
- G. G. M. Fournier, I. W. Cumming and K. Hellgardt, J. Power Sources, 2006, 162, 198–206 CrossRef CAS.
- G. Meng, C. Jiang, J. Ma, Q. Ma and X. Liu, J. Power Sources, 2007, 173, 189–193 CrossRef CAS.
- K. Xie, R. Yan, D. Dong, S. Wang, X. Chen, T. Jiang, B. Lin, M. Wei, X. Liu and G. Meng, J. Power Sources, 2008, 179, 576–583 CrossRef CAS.
- L. Zhang and W. Yang, J. Power Sources, 2008, 179, 92–95 CrossRef CAS.
- N. Maffei, L. Pelletier and A. McFarlan, J. Power Sources, 2008, 175, 221–225 CrossRef CAS.
- K. Miyazaki, H. Muroyama, T. Matsui and K. Eguchi, Sustainable Energy Fuels, 2020, 4, 5238–5246 RSC.
- Y. Aoki, T. Yamaguchi, S. Kobayashi, D. Kowalski, C. Zhu and H. Habazaki, Met. Sustainability, 2018, 2, 1700088 Search PubMed.
- C. Duan, R. J. Kee, H. Zhu, C. Karakaya, Y. Chen, S. Ricote, A. Jarry, E. J. Crumlin, D. Hook, R. Braun, N. P. Sullivan and R. O’Hayre, Nature, 2018, 557, 217–225 CrossRef CAS PubMed.
- F. He, Q. Gao, Z. Liu, M. Yang, R. Ran, G. Yang, W. Wang, W. Zhou and Z. Shao, Adv. Energy Mater., 2021, 11, 2003916 CrossRef CAS.
- Y. Pan, H. Zhang, K. Xu, Y. Zhou, B. Zhao, W. Yuan, K. Sasaki, Y. Choi, Y. Chen and M. Liu, Appl. Catal., B, 2022, 306, 121071 CrossRef CAS.
- L. Zhu, C. Cadigan, C. Duan, J. Huang, L. Bian, L. Le, C. H. Hernandez, V. Avance, R. O'Hayre and N. P. Sullivan, Commun. Chem., 2021, 4, 1–10 CrossRef.
- L. W. Tai, M. M. Nasrallah, H. U. Anderson, D. M. Sparlin and S. R. Sehlin, Solid State Ionics, 1995, 76, 259–271 CrossRef CAS.
- B. Beyribey, J. Bayne and J. Persky, Ceram. Int., 2022, 48, 6046–6051 CrossRef CAS.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
- J. E. Bauerle, J. Phys. Chem. Solids, 1969, 30, 2657–2670 CrossRef CAS.
- J. T. S. Irvine, D. C. Sinclair and A. R. West, Adv. Mater., 1990, 2, 132–138 CrossRef CAS.
- A. Lasia, Electrochemical Impedance Spectroscopy and its Applications, Springer, 2014 Search PubMed.
- H. Zhu, R. J. Kee, V. M. Janardhanan, O. Deutschmann and D. G. Goodwin, J. Electrochem. Soc., 2005, 152, A2427 CrossRef CAS.
- B. A. Boukamp, Solid State Ionics, 1986, 20, 31–44 CrossRef CAS.
- A. Leonide, SOFC Modelling and Parameter Identificationby means of Impedance Spectroscopy, Karlsruher Institut für Technologie (KIT), 2010 Search PubMed.
- H. Schichlein, A. C. Müller, M. Voigts, A. Krügel and E. Ivers-Tiffée, J. Appl. Electrochem., 2002, 32, 875–882 CrossRef CAS.
- M. Mogensen, J. Høgh, K. V. Hansen and T. Jacobsen, ECS Trans., 2007, 7, 1329–1338 CrossRef CAS.
- Y. Aoki, et al. , J. Phys. Chem. C, 2016, 120, 15976–15985 CrossRef CAS.
- R. Strandbakke, V. A. Cherepanov, A. Y. Zuev, D. S. Tsvetkov, C. Argirusis, G. Sourkouni, S. Prünte and T. Norby, Solid State Ionics, 2015, 278, 120–132 CrossRef CAS.
- A. Grimaud, et al. , J. Electrochem. Soc., 2012, 159, B683–B694 CrossRef CAS.
- B. A. Boukamp and J. R. Macdonald, Solid State Ionics, 1994, 74, 85–101 CrossRef CAS.
- T. H. Wan, M. Saccoccio, C. Chen and F. Ciucci, Electrochim. Acta, 2015, 184, 483–499 CrossRef CAS.
- B. A. Boukamp, J. Electrochem. Soc., 1995, 142, 1885–1894 CrossRef CAS.
- B. A. Boukamp, Solid State Ionics, 2004, 169, 65–73 CrossRef CAS.
- P. Johnson, ZPlot, ZView Electrochemical Impedance Software, Scribner Associates Inc., 2000 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ta07310b |
| This journal is © The Royal Society of Chemistry 2023 |