
Roles of the self-reconstruction layer in the catalytic stability of a NiFeP catalyst during the oxygen evolution reaction†
Fuzhen
Zhao
a,
Xinyu
Mao
a,
Xin
Zheng
b,
Huicong
Liu
 a,
Liqun
Zhu
a,
Weiping
Li
a,
Ze
Wang
a and
Haining
Chen
*a
a,
Liqun
Zhu
a,
Weiping
Li
a,
Ze
Wang
a and
Haining
Chen
*a
aSchool of Materials Science and Engineering, Beihang University, No. 37 Xueyuan Road, Haidian District, Beijing 100191, China. E-mail: chenhaining@buaa.edu.cn
bState Key Laboratory of Heavy Oil Processing, China University of Petroleum, Beijing 102249, China
First published on 22nd November 2022
Abstract
The roles of the self-reconstruction NiFe–OOH–P layer in the catalytic stability of the NiFeP catalyst during the oxygen evolution reaction (OER), especially in terms of the atomic dissolution and the adsorption behavior of formed bubbles, are seldom studied. Herein, through a systematic study on crystalline (Ni1−xFex)3P, a NiFeP catalyst, it was revealed that although Fe & P dissolve rapidly from the self-reconstruction layer, the self-reconstruction layer could suppress Fe & P dissolution from (Ni1−xFex)3P, tending to form a stable (Ni1−xFex)3P–O/NiOOH heterostructure. DFT simulations demonstrate that the synergistic effect between Fe–O of (Ni1−xFex)3P–O and NiOOH could still efficiently catalyze the OER. Thus, atomic dissolution does not obviously lower the activity. However, the self-reconstruction layer would change the original interface and increase the surface adsorbed bubbles, which determines the degradation of OER performance. Eventually, a superaerophobic heterostructure of (Ni1−xFex)3P/NiFe–OH–P is prepared by changing reconstruction conditions. The catalyst not only exhibits excellent activity with an overpotential of 233 mV at 10 mA cm−2, but also obtains dramatically enhanced stability with a decrease in current density from 120 to 100 mA cm−2 after a 120 h potentiostatic test.
Introduction
Hydrogen is one of the most promising clean energies to replace fossil fuels.1,2 Electrochemical water splitting is an effective approach to produce hydrogen, including two half-reactions the hydrogen evolution reaction (HER) and the oxygen evolution reaction (OER). Compared with the two-electron HER process, the four-electron OER is a more sluggish kinetics process, determining the reaction rate of electrochemical water splitting.3–5 Therefore, many studies have been carried out to synthesize catalysts for activity enhancement of the OER.6–11Among various OER catalysts, nickel–iron phosphide (NiFeP) is an important catalyst, which would generate P-doped (NiFe)OOH ((NiFe)OOH–P) to catalyze the reaction.12–17 More importantly, NiFeP has exhibited state-of-the-art OER catalytic activities,18–21 which is attributed to the modulated binding energy of O-containing intermediates and high conductibility.22 However, the practical design of OER catalysts requires not only excellent activity but also outstanding stability.23,24 These studies usually focused on understanding the OER active sites of NiFeP catalysts. The stability mechanisms and the strategy to improve stability are seldom studied.
The decrease in the stability of OER catalysts, especially at a high current density (≥100 mA cm−2), is usually attributed to the following reasons: (i) the adhesion of bubbles on the electrode, which could block electrolyte diffusion and inhibit the mass transfer process, and (ii) phase transition induced by active site dissolution.25–30 So far, almost no insightful study on the effect of atomic dissolution (especially for Fe & P) on the catalytic stability of NiFeP catalysts has been conducted. In addition, it remains unclear how bubble absorption behaves on the electrode due to the generation of the self-reconstruction layer during the OER, particularly at a high current density (≥100 mA cm−2).
Herein, we conducted a systematic study on the effects of the self-reconstruction NiFe–OOH–P layer on the catalytic stability of crystalline (Ni1−xFex)3P, a NiFeP catalyst. At a high current density (100 mA cm−2) in electrolyte, although Fe & P dissolved rapidly from the self-reconstruction layer, the self-reconstruction layer could suppress Fe & P dissolution from (Ni1−xFex)3P, tending to form a stable (Ni1−xFex)3P–O/NiOOH heterostructure eventually (Fig. 1a). DFT simulations indicate that the synergistic effect between Fe–O of (Ni1−xFex)3P–O and NiOOH would endow the heterostructure with excellent activity. Thus, atomic dissolution tenuously affects the stability. However, the self-reconstruction layer would change the electrode morphology for increasing the amount of adsorbed bubbles, which would lower the OER performance. By constructing a superaerophobic heterostructure of (Ni1−xFex)3P/NiFe–OH–P, the absorption of bubbles on the electrode was mitigated, which not only achieved excellent activity but also exhibited dramatically enhanced stability with a slight decrease in current density from 120 to 100 mA cm−2 within a 120 h potentiostatic test.
 | ||
| Fig. 1 Changes in the morphology and composition after 30 h OER measurements. (a) Schematic diagram of surface reconstruction. (b) Cross-sectional and (c) top-view SEM images of the as-deposted catalyst. (d) Cross-sectional and (e) top-view SEM images of the catalyst after 30 h stability measurements. (f) XRD patterns, (g) Ni 2p XPS spectra and (h) Fe 2p XPS spectra of the catalyst before and after 30 h stability measurements. | ||
Results and discussion
Changes in the morphology and structure after the OER with self-reconstruction
Scanning electron microscopy (SEM) images were taken to show the change in the morphology of the catalyst before and after 30 h OER measurements. As shown in Fig. 1b, the as-deposited coating is ∼500 nm thick. Fig. 1c presents that the coating uniformly grows on the nickel foam with a rough surface, which might be attributed to H2 etching during annealing. After the 30 h OER measurements, a self-construction layer (∼100 nm) forms on the coating (Fig. 1d) with the surface becoming flat (Fig. 1e). According to the literature, the change could be attributed to the generation of (NiFe)OOH–P during OER measurements.17,31–33To evaluate the change in the structure and composition, more characterization studies were conducted. As shown in Fig. 1f, the X-ray diffraction (XRD) pattern of the powder sample scratched from the nickel foam exhibits diffraction peaks at 41.8°, 42.8°, 43.6°, 45.2°, 46°, 46.6°, 50.5° and 52.7°, which could be indexed to the (231), (330), (112), (240), (202), (141), (222) and (132) planes of Ni3P, respectively. No Fe-related diffraction peaks are detected, indicating that Fe is more likely to be doped into Ni3P. After 30 h OER measurements, no obvious change in the XRD pattern is found, implying the amorphous nature of the reconstructed layer. The FTIR spectra in Fig. S2a† present that the peak of –OH groups is greatly enhanced after the measurements, which could be attributed to the generation of (NiFe)OOH–P during the OER.17,31–33
As shown in X-ray photoelectron spectroscopy (XPS) spectra in Fig. 1g and h, a peak at 852.57 eV in the Ni XPS spectrum of (Ni1−xFex)3P could be indexed to the 2p3/2 region of metallic Ni, while the other two peaks at 855.66 eV and 860.63 eV in the 2p3/2 region should correspond to the Nix+ and the shakeup satellite (sat.) peaks, respectively. The generation of Nix+ is attributed to oxidation in an air atmosphere. After 30 h measurements, the intensity of the metallic Ni (852.7 eV) greatly decreases, and the Nix+ peak and the satellite peak become intense, which is attributed to the surface reconstruction for forming the heterostructure. In addition, the obvious positive shift of the Nix+ peak and the satellite peak is caused by the weakening of the nuclear shielding, which could be contributed to the generation of (NiFe)OOH–P during the OER. The Fe XPS spectrum of (Ni1−xFex)3P in the 2p3/2 region (Fig. 1h) shows two peaks at 706.8 eV and 712.6 eV, which correspond to the metallic Fe and Fe3+, respectively. The existence of Fe3+ is attributed to air oxidation. After OER measurements, the catalyst with the reconstructed heterostructure exhibits a low intensity metallic Fe peak and a more intense Fe3+ peak, which also indicates the generation of (NiFe)OOH–P on the surface during the OER.
Protection effect of the self-construction layer
Transmission electron microscopy (TEM) images were taken to show the change in the morphology and structure of the catalyst after 30 h OER measurements. The TEM image in Fig. 2a and the corresponding selected-area electron diffraction (SAED) pattern in Fig. 2c exhibit an obvious single-crystal feature of (Ni1−xFex)3P with a grain size of ∼500 nm. The high-resolution TEM (HRTEM) image in Fig. 2b shows a lattice distance of 0.217 nm, which could be indexed to the (231) plane of (Ni1−xFex)3P. Furthermore, the corresponding energy dispersive spectra (EDS) in Fig. 2d–f show that the Ni, Fe and P elements are uniformly distributed, and the Fe/Ni ratio is 12.99%, while the P/Ni ratio is 37.96% (Table S2†). After 30 h OER measurements, a self-construction layer is found on the surface (Fig. 2g) and the HRTEM image (Fig. 2h) presents two clearly divided parts. The part with lattice fringes is indexed to the (231) plane of (Ni1−xFex)3P, the other part without lattice fringes corresponds to the amorphous self-construction layer. The SAED pattern in Fig. 2i still shows an obvious single-crystal feature of (Ni1−xFex)3P, further proving the amorphous nature of the self-construction layer. The EDS mapping images (Fig. 2j–l) exhibit a clear boundary between the (Ni1−xFex)3P and the self-reconstruction layer. The Fe/Ni ratio decreases to 8.83%, while the P/Ni ratio decreases to 35.83% (Table S2†), which indicates that Fe & P are easier to dissolve into the electrolyte than Ni, consistent with previous reports.23 Meanwhile, large amounts of Ni, Fe and P are still maintained in (Ni1−xFex)3P. For the self-construction layer, only intense Ni signals are detected through the whole structure, while the Fe and P elements are hardly detected. These results suggested that Fe and P elements would rapidly dissolve from the self-construction layer, while the self-construction layer could suppress the Fe and P dissolution from (Ni1−xFex)3P. | ||
| Fig. 2 Changes in the morphology and composition after OER measurements. (a and g) TEM images, (b and h) HRTEM images, (c and i) SAED patterns, (d and j) Ni EDS elemental mapping images, (e and k) Fe EDS elemental mapping images and (f and l) P EDS elemental mapping images of the catalyst (a–f) before and (g–l) after stability measurements. (m) TEM image and (n–p) EDS elemental mapping images of the exfoliated self-reconstruction layer after 10 h measurements. (q) TEM image, and (r–t) EDS elemental mapping images of the exfoliated self-reconstruction layer after 30 h measurements. | ||
For the exfoliated self-construction layer after 10 h measurements (Fig. 2m and S4a†), EDS mapping images in Fig. 2n–p present uniformly distributed Ni, Fe and P elements, indicating the formation of (NiFe)OOH–P during the OER. After 30 h measurements (Fig. 2q and S4b†), most of the Fe and P dissolved into the electrolyte, and Ni was left (Fig. 2r–t), which indicates that the (NiFe)OOH–P tends to become NiOOH with the OER proceeding. Meanwhile, the Fe/Ni ratios after 10 h and 30 h measurements are 13.79% and 1.54%, respectively. By comparing the Fe/Ni ratio in Fig. 2k and l (8.83%), it could be concluded that though most Fe in (NiFe)OOH–P rapidly dissolves into the electrolyte, the Fe dissolution from (Ni1−xFex)3P is very slow due to the protection of the self-reconstruction layer. Meanwhile, it could also be inferred that the P dissolution from the (Ni1−xFex)3P is very slow (Table S2†). For further comparing the change in the structure of the self-construction layer, Raman spectra were collected. As shown in Fig. S17,† (NiFe)OOH–P and NiOOH present obviously different results. For the sample after the 30 h test, the Raman peaks at 463.1 and 559.7 cm−1 are attributed to the eg bending vibration and the A1g stretching vibration of Ni–O in NiOOH. For the sample with a large amount of Fe retained in the self-construction layer, the intensity ratio (I463.1/I559.7) obviously decreases in the (NiFe)OOH–P. Therefore, it could be concluded that the sample tends to become the (Ni1−xFex)3P/NiOOH heterostructure after long-term OER measurements.
For the sample after 30 h measurements, the EDS mapping images indicate that Fe & P in the self-reconstruction layer almost completely dissolve in the solution, and the Fe & P dissolution from the inner (Ni1−xFex)3P is suppressed by this layer (see the first and second parts of Fig. 3a). Therefore, only small amounts of Fe & P dissolve into the electrolyte (green lines in Fig. 3c and d). After etching part of self-reconstruction layer (see the third part of Fig. 3a), the dissolution of Fe & P from the inner (Ni1−xFex)3P is unable to be suppressed. The etched sample (Fig. 3b) leached more Fe & P in the electrolyte compared to the sample after 30 h measurements (blue lines in Fig. 3c and d). Therefore, it could be concluded that the self-construction layer could act as a protective layer to suppress the Fe & P dissolution from (Ni1−xFex)3P.
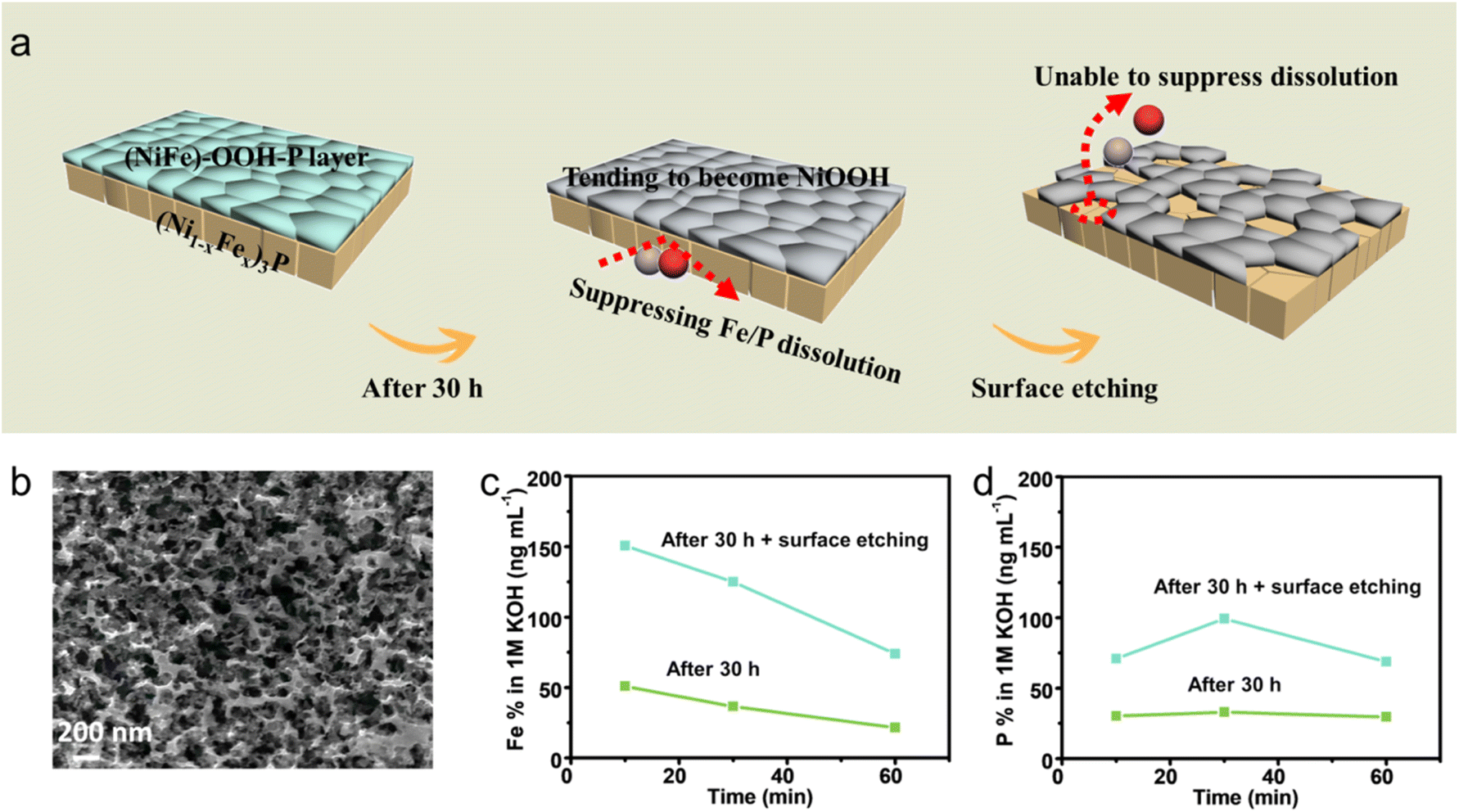 | ||
| Fig. 3 ICP-MS for proving the protection effect of the self-construction layer. (a) Schematic diagram demonstrating the protection effect of the self-reconstruction layer. (b) SEM image of the tested sample after surface etching. ICP-MS measurements for (c) Fe and (d) P concentrations in the electrolyte during the OER test at the current density of 100 mA cm−2. | ||
Effect of the self-construction layer on the behavior of bubble adhesion
In order to evaluate the effect of the self-reconstruction layer on the behavior of bubble adhesion, bubble contact angle measurements were conducted before and after 30 h OER measurements. The contact angle of the untested electrode surface is 135.2° (Fig. 4a), and the surface contact angle decreases to 121.1° after 30 h (Fig. 4b), which could be attributed to the change in surface roughness. Thus, bubble adhesion was not obvious at the beginning of the test (Fig. 4c), but the generation of the self-reconstruction layer results in more and more bubble adsorption after 30 h (Fig. 4d). The adsorbed bubbles could block the electrolyte diffusion and lower the OER performance.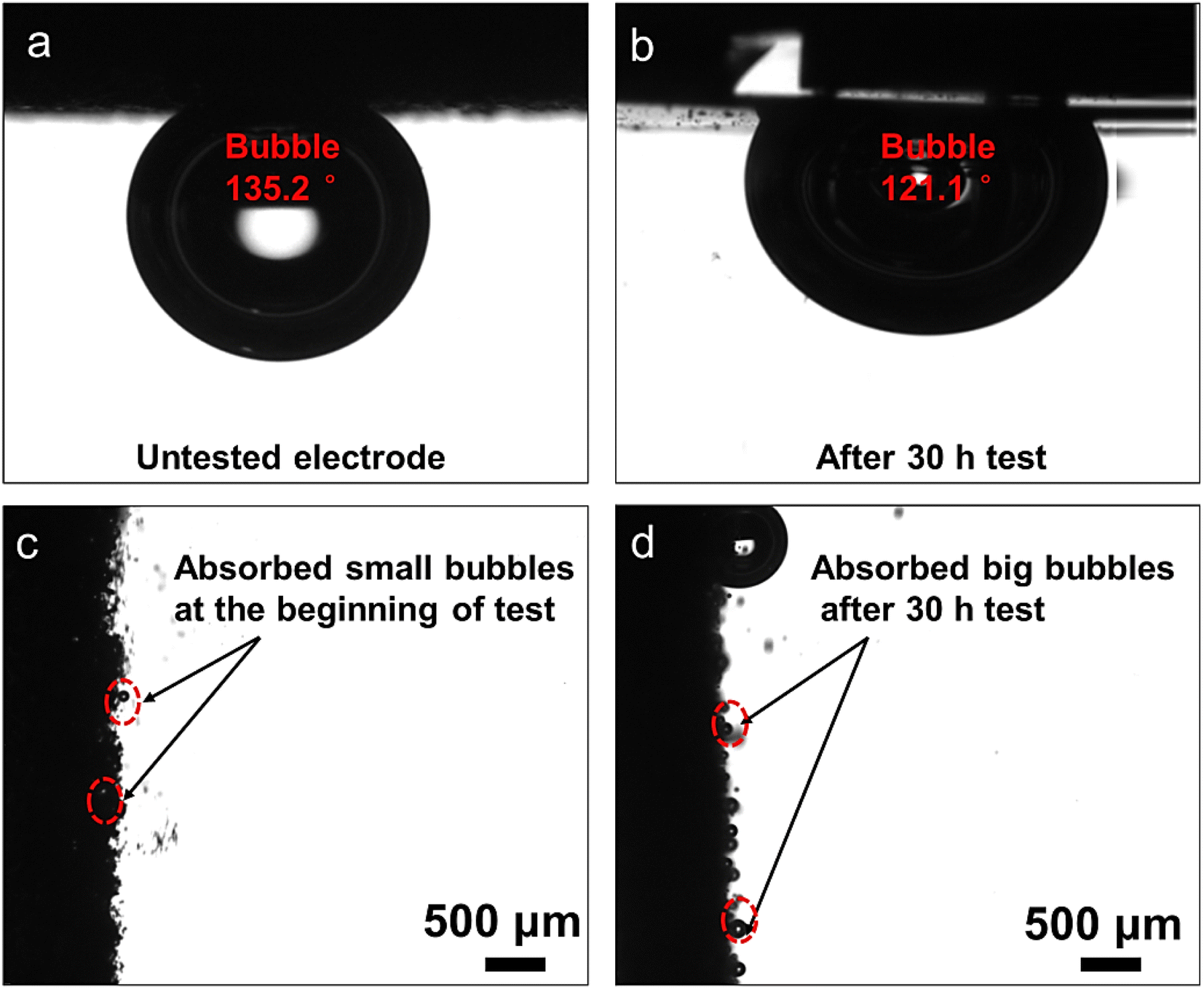 | ||
| Fig. 4 Bubble contact angle measurements of the electrodes (a) before and (b) after 30 h OER measurements. The behavior of bubble adhesion during the OER of the electrodes (c) before and (d) after 30 h OER measurements. | ||
Effects of atomic dissolution and bubble adhesion on OER performances
The above discussion suggests that the changes in OER performances could be attributed to the atomic dissolution (especially for Fe and P) and bubble adhesion. However, it is unclear which reason is the predominant one during the OER. Thus, a simple strategy was performed every 10 h (Fig. 5a), by which the tested catalyst was taken out from the electrolyte and placed in a H2 and Ar atmosphere for 20 min to remove surface O2, and then re-tested. In this way, the effect of the two negative factors on stability could be separately evaluated.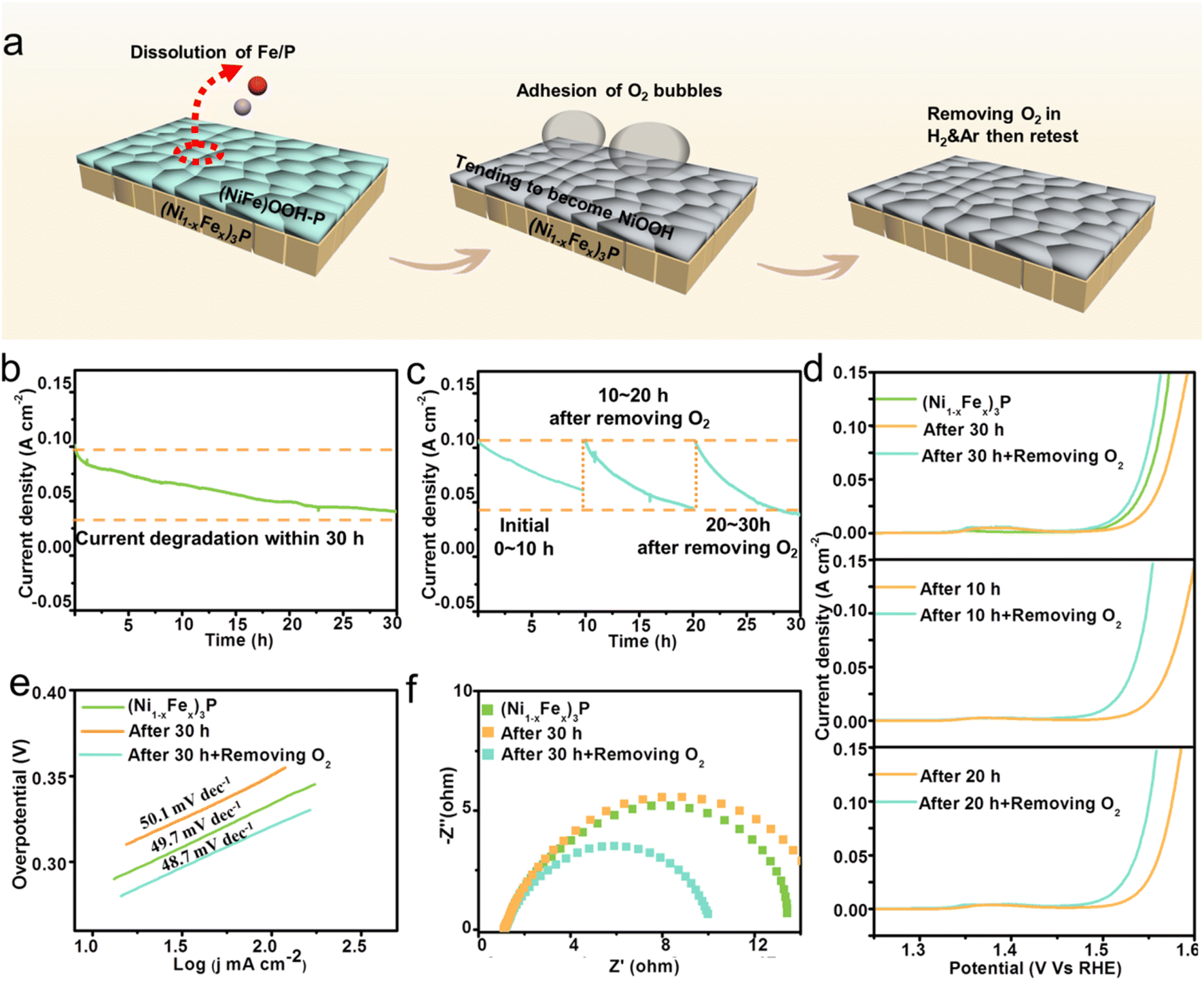 | ||
| Fig. 5 Effects of atomic dissolution and bubbles adhesion on OER performances. (a) The schematic diagram of the experiment. (b) The i–t curve within 30 h measurements of (Ni1−xFex)3P. (c) The i–t curves every 10 h after removing O2 for the stability measurements of (Ni1−xFex)3P. (d) LSV curves. (e) Tafel plots. (f) EIS spectra. | ||
For stability measurements, i–t curves were obtained in a three electrode system in 1 M KOH with a constant potential. As shown in Fig. 5b, the current density drops drastically from 101 mA cm−2 to 40.7 mA cm−2 after 30 h measurements. The degradation could be attributed to the atomic dissolution and the adhesion of O2 bubbles. In order to investigate which one is the determining factor, i–t measurements were re-conducted after the operation of removing O2 every 10 hours. The performance at the beginning of re-test curves could be regarded as the result affected by atomic dissolution rather than bubble adhesion, and the corresponding i–t curves are shown in Fig. 5c. Interestingly, the curves exhibit periodic-like degradation, with nearly unchanged initial current densities after O2 removal every 10 h, then decreasing as the measurement progresses. The almost unchanged initial current densities at the beginning of re-test curves imply that atomic dissolution does not obviously affect the stability, and the current degradation in the periodic-like curves is attributed to a repeated process from an increased electrochemical surface area after O2 removal to a gradually decreased surface area as O2 bubbles adsorption. Furthermore, the i–t curves of 10–20 h and 20–30 h decrease faster than that of 0–10 h, indicating that the generation of the flat self-reconstruction layer could accelerate the process of O2 bubble adhesion. Therefore, the adhesion of O2 bubbles is the determining factor for catalytic stability, and it could be concluded that the current degradation in Fig. 5b is mainly caused by the adsorbed O2 bubbles instead of atomic dissolution.
The IR-corrected LSV curves in 1 M KOH electrolyte were further measured to evaluate the change in the catalytic activity for the (Ni1−xFex)3P catalyst. As shown in Fig. 5d, the as-synthesized (Ni1−xFex)3P exhibits an overpotential of 284 mV at the current density of 10 mA cm−2. After 30 h measurements, the activity of the catalyst declines, with an increased overpotential of 301 mV at the current density of 10 mA cm−2. However, after the operation of removing surface O2 bubbles, the catalytic activity is greatly enhanced with an overpotential of 275 mV, even higher than that of the as-synthesized (Ni1−xFex)3P. In general, the catalytic activity would degrade when the surface atoms are dissolved into the electrolyte, but our result is opposite for the O2-removed catalyst. The atomic dissolution from the catalyst does not lower the OER performance. The recovery of the OER performance after the removal of O2 bubbles indicates the important role of the absorbed O2 in determining the catalytic activity, which would isolate the catalyst from the electrolyte and hence inhibit the OER. Similarly, the recovery of the OER performance after removing O2 bubbles is also found for catalysts after 10 h or 20 h measurements, which further confirms our understanding.
In order to compare the kinetics, Tafel plots were transformed from the LSV curves. As shown in Fig. 5e, the Tafel slope for (Ni1−xFex)3P is 49.7 mV dec−1. After 30 h measurements, the Tafel slope increases to 50.1 mV dec−1, indicating more sluggish kinetics. After removing O2, the slope decreases to 48.7 mV dec−1, suggesting faster reaction kinetics than the original (Ni1−xFex)3P. Electrochemical impedance spectroscopy (EIS) was conducted to study the charge transfer resistance at the electrode/electrolyte interface. As presented in Fig. 5f, the transfer resistance (Rct) of (Ni1−xFex)3P obviously increases from 12.46 Ω to 14.27 Ω after 30 h measurements, which should be attributed to the absorption of O2 bubbles on the surface. After removing the O2 bubbles, Rct decreases to 9.12 Ω, indicating faster charge transfer than the original (Ni1−xFex)3P. In addition, the Tafel plots and EIS spectra of the catalyst after 10 and 20 h (Fig. S7 and S8†) measurements also present similar changes before and after removing O2 bubbles.
DFT calculations for explaining the insignificant effect of atomic dissolution on OER stability
As mentioned above, the (Ni1−xFex)3P/(NiFe)OOH–P heterostructure would tend to become (Ni1−xFex)3P/NiOOH, with Fe & P dissolving rapidly from (NiFe)OOH–P but slowly from (Ni1−xFex)3P due to the protection of the (NiFe)OOH–P layer. However, the Fe & P dissolution does not obviously affect the catalytic activity, suggesting that the (Ni1−xFex)3P/NiOOH heterostructure has excellent activity. To confirm this, DFT calculations were conducted. The calculated OER reactions include four key steps in alkaline electrolyte:| 4OH− + surface (sur) − e− → sur-OH* + 3OH− |
| sur-OH* + 3OH− − e− → sur-O* + 2OH− + H2O |
| sur-O* + 2OH− + H2O − e− → sur-OOH* + OH− + H2O |
| sur-OOH* + OH− + H2O − e− → sur + 2H2O + O2 |
The (Ni1−xFex)3P/NiOOH heterostructure is shown in Fig. 6a. After comparing the adsorption reactions on (Ni1−xFex)3P and NiOOH, the Fe atoms in (Ni1−xFex)3P tend to form Fe–O*. The corresponding detailed Gibbs free energy diagrams of the reactions on the Fe atom are shown in Fig. 6d. Under standard conditions (U = 0 V), the first step presents a ΔG of −0.51 eV, but the other steps present rather high energy barriers (ΔG > 0 eV), indicating that the formation of Fe–OH* would be a spontaneous reaction. At the ideal OER potential (U = 1.23 V), the first and second steps show a ΔG of −1.74 eV and −0.7 eV, respectively, indicating an exothermic process to form Fe–OH* and then Fe–O*. However, there is a very high energy barrier to form Fe–OOH* (ΔG = 1.72 eV), implying that the first and second steps to generate Fe–O* from the (Ni1−xFex)3P part are simple but the other two-electron OER steps are difficult to conduct. Therefore, it is reasonable that the heterostructure tends to transform into (Ni1−xFex)3P–O/NiOOH instead of (Ni1−xFex)3P/NiOOH after long-term OER, and the model with doped O atoms should be used for DFT calculations.
 | ||
| Fig. 6 DFT calculations to investigate the OER mechanism. Models of (a) (Ni1−xFex)3P/NiOOH, (b) NiOOH and (c) (Ni1−xFex)3P–O/NiOOH. The calculated Gibbs free energy of (d) (Ni1−xFex)3P/NiOOH, (e) NiOOH and (f) (Ni1−xFex)3P–O/NiOOH. | ||
For the individual NiOOH (Fig. 6b), as a control model, the corresponding Gibbs free energy diagrams (Fig. 6e) indicate that the conversion of OH* to O* is the determining step with a ΔG of 1.84 eV (U = 0 V). Therefore, at 1.23 V the diagrams present a rather high overpotential of 0.61 V indicating that the OER of NiOOH suffers from the supply of O*, consistent with previous reports.34,35
For the (Ni1−xFex)3P–O/NiOOH heterostructure (Fig. 6c), the corresponding Gibbs free energy diagrams in Fig. 6f indicate that the determining step of forming O* at the Fe–O in (Ni1−xFex)3P–O shows a ΔG of 1.62 eV (U = 0 V). Therefore, (Ni1−xFex)3P–O/NiOOH presents an overpotential of 0.39 V according to the diagrams at 1.23 V, which is much lower than that of the individual NiOOH (0.61 V). Due to the low energy barrier, the remaining two-electron OER steps performed on the heterostructure's NiOOH ports would be provided with an adequate amount of O*. Therefore, the synergistic effect between NiOOH and (Ni1−xFex)3P–O could efficiently catalyze the OER.
Constructing a superaerophobic structure for long-term OER stability
Until now, several key points have been confirmed: (i) the dissolution of the surface Fe during OER measurement does not obviously affect the activity of the catalyst. (ii) The Fe dissolution from (Ni1−xFex)3P is very slow during the OER and the (Ni1−xFex)3P–O/NiOOH heterostructure tends to finally form, which could maintain excellent activity. (iii) The change in OER performance is mainly caused by the absorption of O2 bubbles.Therefore, it is important to remove O2 bubbles to improve long-term OER stability. As previously discussed, constructing a superaerophobic surface is a good strategy, which could be achieved by forming rough micro/nano structures.25,26,36 To construct this structure, we changed the formation condition of the self-construction layer by immersing (Ni1−xFex)3P in a KOH solution with a high concentration (2.5 M) at 80 °C, and a NiFe–OH–P layer was generated. Under high concentration and high temperature, the reactions rate would be extremely high, which would boost growth rate of the NiFe–OH–P to form a superaerophobic surface (Fig. 7a). The superaerophobic surface could not only rapidly release the formed bubbles but also inhibit the generation of a flat self-reconstruction layer. As shown in Fig. 7b, the catalyst presents a rougher surface than the original self-reconstruction layer (Fig. 1e), and the width of the nanosheets is about 200 nm.
 | ||
| Fig. 7 (a) Schematic diagram of the construction of the superaerophobic heterostructure. (b) SEM image of the superaerophobic heterostructure. (c) Bubble contact angle measurements of the superaerophobic heterostructure. (d) The behavior of bubble adhesion of the superaerophobic heterostructure. (e) TEM image and (f–h) EDS elemental mapping images of the superaerophobic heterostructure. (i) TEM image and (j–l) EDS elemental mapping images of the superaerophobic heterostructure after stability measurements. | ||
As expected, compared with the original self-reconstruction layer (121.1°), the bubble contact angle for the rougher surface increases to 161.5° (Fig. 7c). Therefore, a superaerophobic electrode is obtained, which could favor the release of bubbles during the OER (Fig. 7d). The TEM image in Fig. 7e exhibits a clear heterostructure, in which a sheet-like structure is formed on the single-crystal (Ni1−xFex)3P (Fig. S10a†). By combining with the surface Raman spectrum (Fig. S17†), the result presents two peaks at 447.9 and 587.5 cm−1, which are assigned to the A1g stretching modes of Ni–OH and Ni–O in NiFe–OH–P. Thus, the sheet-like structure should be NiFe–OH–P. The HRTEM image in Fig. S10b† presents a lattice distance of 0.190 nm, corresponding to the (141) plane of (Ni1−xFex)3P, and NiFe–OH–P shows amorphous nature. Moreover, the EDS mapping images in Fig. 7g and h show considerable amounts of Fe and P in the nanosheet, which would be significantly reduced after stability measurements (Fig. 7k and l). In contrast, Fe and P in (Ni1−xFex)3P could remain after stability measurements. Therefore, it also demonstrates that the (Ni1−xFex)3P–O/NiOOH heterostructure tends to form after long-term OER.
OER performance of the superaerophobic heterostructure
Electrochemical measurements were conducted to evaluate the OER performance of the superaerophobic heterostructure. The reverse scan results of IR-corrected cyclic voltammetry (CV) curves (Fig. 8a) obtained in 1 M KOH solution show that the superaerophobic heterostructure presents an overpotential of 233 mV at the current density of 10 mA cm−2, much lower than that of the (Ni1−xFex)3P-induced catalysts (Fig. 5d). The i–t curve in Fig. 8b indicates that after 120 h measurements, the current density only exhibits a slight decrease from 120 to 100 mA cm−2, which is much better than that of the original (Ni1−xFex)3P (101 to 40.7 mA cm−2 within 30 h). The dramatically enhanced stability should be attributed to the superaerophobic heterostructure, which would accelerate the release of the absorbed O2 bubbles during the OER. The inset in Fig. 8b shows a zoomed region of the i–t curve. As shown, the zoomed curve exhibits regular fluctuations, which is induced by the formation and release of O2 bubbles during the OER. As presented in Fig. 8a, an overpotential of 246 mV at the current density of 10 mA cm−2 from the reverse scan result could be obtained after 120 h measurements, further confirming the outstanding stability of the superaerophobic heterostructure. The SEM image in Fig. S13a† presents that the rough surface with nanosheet edges could still be retained after a 120 h OER, and the generation of the flat self-reconstruction layer is totally suppressed. Therefore, the superaerophobic electrode could still release the formed bubbles after long-term OER with the bubble contact angle slightly reduced to 154.2° (Fig. S16†), which well improved the OER stability. | ||
| Fig. 8 Electrochemical measurements of the superaerophobic heterostructure. (a) CV curves before and after 120 h measurements, (b) i–t curve for stability evaluation, (c) Tafel slope, (d) EIS spectrum, (e) CV curves for the ECSA and (f) ECSA results. | ||
The Tafel plot (Fig. 8c) and EIS spectrum (Fig. 8d) present a slope of 41.3 mV dec−1 and a transfer resistance of 3.33 Ω, respectively. Both parameters are better than those of the (Ni1−xFex)3P-induced catalysts (Fig. 5e and f), indicating an improvement in reaction kinetics. Meanwhile, the electrochemical active surface area (ECSA) result of the superaerophobic electrode (Fig. 8f) shows 3 times higher Cdl (4.675 mF cm−2) than the original (Ni1−xFex)3P (1.41 mF cm−2), further confirming the rougher surface of the superaerophobic heterostructure.
Conclusion
We have conducted a systematic study on the effect of the self-reconstruction layer on the stability of crystalline (Ni1−xFex)3P, a NiFeP catalyst. During the OER, (Ni1−xFex)3P could initially form a (Ni1−xFex)3P–O/(NiFe)OOH–P heterostructure, which tended to become (Ni1−xFex)3P–O/NiOOH because Fe & P dissolved rapidly from the (NiFe)OOH–P but slowly from the (Ni1−xFex)3P–O under the protection of the (NiFe)OOH–P layer. As proved, the atomic dissolution almost hardly affected the catalytic stability, because the synergistic effect between (Ni1−xFex)3P–O and NiOOH could still efficiently catalyze the OER. However, the increase of adsorbed O2 bubbles on the surface would significantly lower the OER performance, which should be released to achieve long-term OER stability. By constructing a superaerophobic heterostructure of (Ni1−xFex)3P/NiFe–OH–P in the KOH solution with a high concentration, the OER catalyst exhibited excellent stability.Experimental section
Preparation of materials
Ferrous sulfate (FeSO4·7H2O), sodium citrate dihydrate (Na3C6H5O7·2H2O), sodium sulfate (Na2SO4), potassium hydroxide (KOH), boric acid (H3BO3), potassium carbonate (K2CO3) muriatic acid (HCl) and sulfuric acid (H2SO4) were purchased from the Beijing Chemical Works (Beijing, China). Sodium hypophosphite (NaH2PO2·H2O) was purchased from Innochem-Beijing. Nickel sulfate (NiSO4·6H2O) and sodium hydroxide (NaOH) were purchased from Shanghai Aladdin Bio-Chem Technology Co., LTD. Trisodium phosphate anhydrous (Na3PO4) were purchased from Shanghai Macklin Biochemical Co., LTD. Nickel foam was purchased from the Tianyu factory of Shandong province.Synthesis steps
The synthesis process of (Ni1−xFex)3P included the following steps: (1) the operations of oil removal and oxide layer removal were conducted, as presented in previous work.37 (2) The chemical plating solution containing 15 g per L FeSO4·7H2O, 15 g per L NiSO4·6H2O, 150 g per L NaH2PO2·H2O, 20 g per L NaSO4, 30 g per L H3BO3 and 60 g per L Na3C6H5O7·2H2O was used. NaOH was used to modulate pH to 10. (3) Ni foam was immersed in the chemical plating solution at 80 °C for 1 h. (4) To obtain clean crystalline (Ni1−xFex)3P, the product of chemical plating was annealed at 400 °C for 4 h in an Ar environment and then at 600 °C for 35 min in H2 and Ar environments. (5) The loading content of the achieved (Ni1−xFex)3P on the Ni foam (1 cm × 1 cm) is 2.7 mg.In order to achieve the surface etching sample, the tested electrode was immersed in 50 g per L citric acid solution for 10 min at 80 °C. Ammonia water was used to modulate pH to 3.5.
The superaerophobic heterostructure of (Ni1−xFex)3P/NiFe–OH was synthesized by etching (Ni1−xFex)3P in 2.5 M KOH at 80 °C for 5 h.
Materials characterization
SEM measurements were conducted on a ZEISS SUPRA55 field-emission electron microscope. FET Tecnai F20 and JEM-2100F (operating at 200 keV) were used to perform TEM and EDS measurements. XRD patterns were collected on a Rigaku D/max 2200pc powder diffractometer (Cu Kα λ = 1.5406 Å). FTIR (Thermo Fisher Nicolet is50) and XPS (Thermo Scientific Escalab 250Xi, USA) were used to characterize the chemical compositions. An Agilent 7800 ICP-MS was used to measure the ionic concentration of electrolyte. The EQCM results were collected on a CHI 440E electrochemical workstation.Measurement of OER performance
For measuring the OER performance, a CHI 760E electrochemical workstation was used. During the measurements, a three-electrode system in 1 M KOH (pH = 13.8) was used. Ni foam (1 cm × 1 cm) with the catalyst, Hg/HgO electrode and Pt sheet (1 cm × 1 cm) were used as the working electrode, reference electrode and counter electrode, respectively.The equation of E(RHE) = E(Hg/HgO) + 0.098 + 0.059pH is used to calculate the RHE potential. For activity measurements, the CV and LSV measurements were conducted at a scan rate of 5 mV s−1 (95% IR-correction).
For kinetics comparisons, the Tafel plots were obtained from CV or LSV curves. The EIS patterns were measured at an overpotential of 250 mV vs. the RHE potential. The frequency range and AC amplitude were set at 100 kHz to 0.1 Hz and 5 mV, respectively.
CV curves with scan rates from 50 mV s−1 to 100 mV s−1 were collected to calculate the ECSA. The electrochemical double layer capacitance (Cdl) was calculated based on the current change of the increasing scan rates from 50 mV s−1 to 100 mV s−1.
DFT calculations
The DFT calculations of this work are conducted by using the Vienna Ab initio Simulation Package (VASP) with the Projector Augmented Wave (PAW) method. The exchange–correlation energy was calculated by the generalized gradient approximation (GGA) of the Perdew–Burke–Ernzerhof (PBE) approach. The kinetic energy cut-off of electron wave functions is set as 400 eV and 450 eV for coarse and medium quality cell optimization, respectively. The conjugated gradient method was used to conduct the geometry optimizations. During every geometry optimization, the convergence threshold is set as 10−5 eV in energy. The break condition for the ionic relaxation loop is 0.05 eV Å−1 in force. The DFT-D3 method was used to describe the van der Waals interactions. All the calculations were spin polarized. U-Values applied to the d-orbitals of Fe and Ni are set as 2.56 eV and 5.20 eV, respectively.The Gibbs free energy of adsorption species was obtained according to equation G = E + ZPE − TS. The E, ZPE, and TS are the total electronic-energy at 300 K, the zero point energy correction, and the entropy correction (300 K), respectively. The values of ZPE − TS were calculated using vaspkit code presented in Table S1.†38
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (51971012).References
- X. Long, H. Lin, D. Zhou, Y. An and S. Yang, ACS Energy Lett., 2018, 3, 290–296 CrossRef CAS.
- M. Tahir, L. Pan, F. Idrees, X. Zhang, L. Wang, J.-J. Zou and Z. L. Wang, Nano Energy, 2017, 37, 136–157 CrossRef CAS.
- M. Ju, X. Wang, X. Long and S. Yang, CrystEngComm, 2020, 22, 1531–1540 RSC.
- M. Huynh, C. Shi, S. J. Billinge and D. G. Nocera, J. Am. Chem. Soc., 2015, 137, 14887–14904 CrossRef CAS.
- J. Mohammed-Ibrahim, J. Power Sources, 2020, 448, 227375 CrossRef CAS.
- B. Kang, X. Jin, S. M. Oh, S. B. Patil, M. G. Kim, S. H. Kim and S.-J. Hwang, Appl. Catal., B, 2018, 236, 107–116 CrossRef CAS.
- L. Wang, Y. Zhang, X. Li, Y. Xie, J. He, J. Yu and Y. Song, Sci. Rep., 2015, 5, 1–12 CAS.
- J. Jiang, F. Sun, S. Zhou, W. Hu, H. Zhang, J. Dong, Z. Jiang, J. Zhao, J. Li and W. Yan, Nat. Commun., 2018, 9, 1–12 CrossRef.
- Y. Liu, X. Liang, L. Gu, Y. Zhang, G.-D. Li, X. Zou and J.-S. Chen, Nat. Commun., 2018, 9, 1–10 CrossRef.
- A. T. Swesi, J. Masud and M. Nath, Energy Environ. Sci., 2016, 9, 1771–1782 RSC.
- K. Wan, J. Luo, C. Zhou, T. Zhang, J. Arbiol, X. Lu, B. W. Mao, X. Zhang and J. Fransaer, Adv. Funct. Mater., 2019, 29, 1900315 CrossRef.
- J. Chen, Y. Li, G. Sheng, L. Xu, H. Ye, X.-Z. Fu, R. Sun and C. P. Wong, ChemCatChem, 2018, 10, 2248–2253 CrossRef CAS.
- F. Hu, S. Zhu, S. Chen, Y. Li, L. Ma, T. Wu, Y. Zhang, C. Wang, C. Liu, X. Yang, L. Song, X. Yang and Y. Xiong, Adv. Mater., 2017, 29, 1606570 CrossRef.
- J. Lian, Y. Wu, H. Zhang, S. Gu, Z. Zeng and X. Ye, Int. J. Hydrogen Energy, 2018, 43, 12929–12938 CrossRef CAS.
- J. Liu, X. Liu, H. Shi, J. Luo, L. Wang, J. Liang, S. Li, L.-M. Yang, T. Wang, Y. Huang and Q. Li, Appl. Catal., B, 2022, 302, 120862 CrossRef CAS.
- X. Luo, P. Ji, P. Wang, X. Tan, L. Chen and S. Mu, Adv. Sci., 2022, 9, 2104846 CrossRef CAS.
- T. Kou, S. Wang and Y. Li, ACS Mater. Lett., 2021, 3, 224–234 CrossRef CAS.
- B. Yang, D. Bin, A. G. Tamirat, Y. Liu, L. Liu and B. Liu, Electrochim. Acta, 2020, 331, 135360 CrossRef CAS.
- H. Liang, A. N. Gandi, C. Xia, M. N. Hedhili, D. H. Anjum, U. Schwingenschlögl and H. N. Alshareef, ACS Energy Lett., 2017, 2, 1035–1042 CrossRef CAS.
- K. Liu, F. Wang, P. He, T. A. Shifa, Z. Wang, Z. Cheng, X. Zhan and J. He, Adv. Energy Mater., 2018, 8, 1703290 CrossRef.
- S. Sun, X. Zhou, B. Cong, W. Hong and G. Chen, ACS Catal., 2020, 10, 9086–9097 CrossRef CAS.
- Y. Li, R. Li, D. Wang, H. Xu, F. Meng, D. Dong, J. Jiang, J. Zhang, M. An and P. Yang, Int. J. Hydrogen Energy, 2021, 46, 5131–5149 CrossRef CAS.
- D. Y. Chung, P. P. Lopes, P. F. B. D. Martins, H. He, T. Kawaguchi, P. Zapol, H. You, D. Tripkovic, D. Strmcnik and Y. Zhu, Nat. Energy, 2020, 5, 222–230 CrossRef.
- C. Feng, F. Wang, Z. Liu, M. Nakabayashi, Y. Xiao, Q. Zeng, J. Fu, Q. Wu, C. Cui and Y. Han, Nat. Commun., 2021, 12, 1–10 CrossRef.
- Z. Lu, W. Zhu, X. Yu, H. Zhang, Y. Li, X. Sun, X. Wang, H. Wang, J. Wang and J. Luo, Adv. Mater., 2014, 26, 2683–2687 CrossRef CAS.
- D. Zhou, P. Li, X. Lin, A. McKinley, Y. Kuang, W. Liu, W.-F. Lin, X. Sun and X. Duan, Chem. Soc. Rev., 2021, 50, 8790–8817 RSC.
- C. Spöri, J. T. H. Kwan, A. Bonakdarpour, D. P. Wilkinson and P. Strasser, Angew. Chem., Int. Ed., 2017, 56, 5994–6021 CrossRef.
- F. D. Speck, K. E. Dettelbach, R. S. Sherbo, D. A. Salvatore, A. Huang and C. P. Berlinguette, Chem, 2017, 2, 590–597 CAS.
- L. Peng, N. Yang, Y. Yang, Q. Wang, X. Xie, D. Sun-Waterhouse, L. Shang, T. Zhang and G. I. Waterhouse, Angew. Chem., 2021, 133, 24817–24824 CrossRef.
- C. Andronescu, S. Seisel, P. Wilde, S. Barwe, J. Masa, Y. T. Chen, E. Ventosa and W. Schuhmann, Chem.–Eur. J., 2018, 24, 13773–13777 CrossRef CAS PubMed.
- S. Jin, ACS Energy Lett., 2017, 2, 1937–1938 CrossRef CAS.
- Y. Zhang, L. Gao, E. J. M. Hensen and J. P. Hofmann, ACS Energy Lett., 2018, 3, 1360–1365 CrossRef CAS.
- Y. Zhu, H.-C. Chen, C.-S. Hsu, T.-S. Lin, C.-J. Chang, S.-C. Chang, L.-D. Tsai and H. M. Chen, ACS Energy Lett., 2019, 4, 987–994 CrossRef CAS.
- H. Xiao, H. Shin and W. A. Goddard, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 5872–5877 CrossRef CAS.
- F. Dionigi, Z. Zeng, I. Sinev, T. Merzdorf, S. Deshpande, M. B. Lopez, S. Kunze, I. Zegkinoglou, H. Sarodnik and D. Fan, Nat. Commun., 2020, 11, 1–10 CrossRef.
- W. Xu, Z. Lu, X. Sun, L. Jiang and X. Duan, Acc. Chem. Res., 2018, 51, 1590–1598 CrossRef CAS.
- F. Zhao, H. Liu, H. Zhu, X. Jiang, L. Zhu, W. Li and H. Chen, J. Mater. Chem. A, 2021, 9, 10169–10179 RSC.
- V. Wang, N. Xu, J. Liu, G. Tang and W. Geng, Comput. Phys. Commun., 2021, 267, 108033 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ta06514b |
| This journal is © The Royal Society of Chemistry 2023 |