
Enabling a compatible Li/garnet interface via a multifunctional additive of sulfur†
Jie
Wang‡
 ab,
Saisai
Zhang‡
a,
Shaokang
Song
a,
Jintao
Liu
a,
Zhaolin
Li
ab and
Hailei
Zhao
*ab
ab,
Saisai
Zhang‡
a,
Shaokang
Song
a,
Jintao
Liu
a,
Zhaolin
Li
ab and
Hailei
Zhao
*ab
aSchool of Materials Science and Engineering, University of Science and Technology Beijing, Beijing 100083, China. E-mail: hlzhao@ustb.edu.cn
bBeijing Municipal Key Lab of Advanced Energy Materials and Technologies, Beijing 100083, China
First published on 22nd November 2022
Abstract
Ceramic-based solid-state batteries with lithium metal anode show great potential to realize a battery revolution because of their features of nonflammability and high energy density. Among the inorganic electrolytes, garnet-type Li7La3Zr2O12 attracts much attention as a promising alternative owing to its advantages of high ionic conductivity, wide electrochemical voltage window, and excellent thermal stability. However, the critical issue of the insufficient lithium–electrolyte interface contact significantly hinders its practical application. Herein, an intimate physical connection between the Li anode and garnet is achieved by improving the wettability of molten Li with multifunctional sulfur (S) addition. The generated Li2S with high ionic conductivity could also provide an extended interface area for charge transfer, enabling a reduced local current density and thus favoring the rate performance. Because of the tightly increased contact area and the homogenized interface electric-field distribution, the fabricated symmetric cells with an optimal Li–Li2S anode (LS10: 10 wt% S addition) show a significantly decreased interfacial resistance (12.4 Ω cm2) and a stable Li plating/stripping performance for over 900 h at 0.2 mA cm−2. The constructed LS10|Li6.4La3Zr1.4Ta0.6O12|LiFePO4 full cell delivers a high reversible specific capacity of 154.7 mA h g−1 at 0.1C and stable cycling with a capacity fading rate of ∼0.033% per cycle.
1 Introduction
Nowadays the development of rechargeable lithium-ion batteries (LIBs) with high energy density and good safety is of utmost importance because of the increased demand for portable electronic devices and electric vehicles (EVs). Exploring high-energy cathodes and high-capacity anodes has long been the main topic in the research field for decades.1–5 For the anode, although the use of graphite significantly contributes to the successful LIB technology, its feature of low theoretical specific capacity (372 mA h g−1) is not conducive to the construction of next-generation high energy-density LIBs. Lithium metal with the characteristics of light weight (0.53 g cm−3), low reduction potential (−3.04 V vs. standard hydrogen electrode), and high specific capacity (3861 mA h g−1) has been regarded as the ultimately optimal anode.6–8 However, the serious safety issue associated with lithium dendrite growth when using flammable organic-liquid electrolytes restricts the practical application of lithium anodes. Employing solid-state electrolytes (SSEs) shows great promise for promoting lithium metal battery development because of the intrinsic advantages of SSEs with nonflammability, good thermal stability, and mechanical strength to block lithium dendrite penetration.9–14Among the various inorganic SSEs (Li2.88PO3.86N0.14 (LiPON), Li9.54Si1.74P1.44S11.7Cl0.3, Li1.3Al0.3Ti1.7(PO4)3, Li10GeP2S12, perovskite-type (Li,La)TiO3, and garnet-type LLMO (M = Zr, Nb, Ta)),15–23 garnet-type doped Li7La3Zr2O12 (LLZO) with a cubic structure attracts much attention due to its high ionic conductivity, wide electrochemical window, and sufficient chemical stability against lithium metal.22,24–27 Although significant progress on the improved ionic conductivity of garnet SSEs was realized by lattice modulation,28–32 the challenge related to the large interfacial resistance that originates from the noncompact physical contact between the lithium anode and LLZO, which impedes the development of high-energy all-solid-state batteries (ASSBs), remains.33 The point-to-point interfacial contact with lots of voids can lead to inhomogeneous local electric field distribution and selective lithium mass deposition. The uneven Li plating/stripping directly causes lithium dendrite formation/growth that results in rapid performance degradation, especially at a high current density. To date, with the aim to improve the Li/LLZO interfacial contact, several effective approaches were devoted, including removing the LLZO surface lithiophobic impurities (e.g., Li2CO3 and LiOH),34–36 applying a lithiophilic or gel/polymer interlayer (Si, Al, Ge, Ag, Al2O3, ZnO, Cu3N, PEO, PAA, gel, etc.),33,37–45 or modifying the lithium anode composition (Li–graphite, Li–Sn, Li–g-C3N4, Li–Si3N4, etc.).46–49 For example, by wet-polishing and heat-treating garnet to eliminate surface Li2CO3 contaminants, significantly reduced interfacial resistance is obtained because of the improved wettability of molten Li against garnet.34 Luo et al. coated Li6.85La2.9Ca0.1Zr1.75Nb0.25O12 with an ultrathin amorphous Si layer, and the achieved superlithiophilicity of garnet resulting from the reaction between Li and Si ensures a decreased interfacial resistance.37 Han et al. constructed an alumina coating layer on the garnet surface, which enables a conformal interface contact of lithium metal with garnet and thus negates the lithium/garnet interfacial impedance.33 Wang et al. employed Sn as an additive to tune the surface energy of molten Li, which results in an improved Li/garnet contact with an interfacial resistance down to 7 Ω cm2.47
It is worth noting that although the above-mentioned approaches have shown great effectiveness in the improved wettability of molten Li to garnets, each strategy still presents its limitations: (1) the probably introduced surface defects or bulk phase composition change when eliminating the surface contaminated layer via polishing, acid treating, or thermal heat-treating should not be ignored. Besides, the well-treated clean garnets still suffer from the inevitable unintentional reactions that cause surface re-contamination when exposed to air;26,49,50 (2) the time-consuming and costly technologies for applying an alloyable lithiophilic interlayer significantly inhibit the manufacturing scalability.49,51 Besides, the lithiation process of the alloyable interlayer should be harmful for the physical connection stability of the Li/garnet interface,50,52 and the effects of the introduced Li-alloyed interlayer and interlayer/garnet interface on the lithium-ion diffusion kinetics need to be concerned. In addition, some reports show that the alloying interlayer does not necessarily keep stable between the Li anode and garnet during extended Li cycling;53–55 (3) when introducing lithiophilic additives into the lithium anode, the sacrificed total battery energy-density should be considered. Besides, on the premise of the realized intimate lithium/garnet interface contact, the probable negative effects of the resulting alloying phases on the lithium-ion transfer at the interface have attracted less attention. Therefore, efforts on searching for effective solutions to not only improve the lithium/garnet physical contact but also avoid the adverse effects on the anode/electrolyte interface ion transportation are still highly desired.
Herein, sulfur (S), a cheap and abundant material, was selected as the multifunctional additive into the lithium anode to address the Li/garnet physical contact problem. The spontaneity of reaction between S and Li (Gibbs free energy: ΔG < 0) could improve the wettability of molten Li on the garnet (Li6.4La3Zr1.4Ta0.6O12, LLZTO) surface and thus achieve an intimate Li/garnet interface.56 Most importantly, the generated Li2S with high ionic conductivity (∼10−5 S cm−1) can be considered as the electrolyte morphological extension,57,58 benefiting the ion diffusion kinetics at the interface over those of derived products from commonly reported additives. Moreover, Li2S with a high de-lithiation potential shows the advantage of easily realizing phase stability without any volume change during Li plating/stripping processes, which favors the interfacial structure stability. As a result, a reduced interfacial resistance (∼12.4 Ω cm2 at 25 °C) and long-term cycling over 900 h at 0.2 mA cm−2 with a small over-potential (Li–Li2S|LLZTO|Li–Li2S symmetric cell) are achieved, and the fabricated Li–Li2S|LLZTO|LiFePO4 full cell delivers a superior cycling performance from the aspects of capacity, cycling stability, and rate capability compared with the cell with a pure Li anode.
2 Experimental
2.1 Synthesis of the solid-state electrolyte and Li–Li2S composite
Li6.4La3Zr1.4Ta0.6O12 powder was synthesized via a solid-state reaction method by using the raw materials of Li2CO3, La2O3, ZrO2, and Ta2O5. To compensate for the lithium loss during calcinating/sintering processes, 10% excessive Li2CO3 was added. Briefly, the raw materials were thoroughly mechanically wet ball-milled in ethanol for 12 h. After being dried and calcined at 900 °C for 12 h in air, the obtained pure LLZTO powder was ground and ball-milled again in isopropanol. The collected powder was pressed into pellets and sintered at 1175 °C for 12 h to obtain the final dense LLZTO electrolyte.The whole experiments on the preparation of the Li–Li2S composite were carried out in an argon-filled glove box (O2 and H2O < 0.1 ppm). Specifically, S powder with different mass ratios of 5, 10, and 20 wt% was added into molten Li at 250 °C in a stainless steel crucible, which was kept under mechanical stirring for achieving a homogenous Li–Li2S composite. For simplicity, the molten Li with 5, 10, and 20 wt% S addition was denoted as LS5, LS10, and LS20, respectively.
2.2 Materials characterization
The X-ray diffraction (XRD) data were collected on a Rigaku D/max-A X-ray diffractometer (Japan) with a Cu Kα radiation (λ = 1.5406 Å) source operated at 40 kV and 40 mA. To avoid the oxidation of the Li–Li2S composite and Li metal in air, the samples were sealed with a transparent Kapton film in an argon-filled glove box before XRD tests. The surface and cross-section morphologies, as well as elemental mapping, were obtained on a SUPRA55 field emission scanning electron microscope (FESEM, Germany) coupled with an energy dispersive spectrometer (EDS). The reaction energies between the garnet and Li or Li2S were examined by density functional theory (DFT) calculations, which were obtained from the Materials Project database.592.3 Symmetric cell and full cell assembly
LLZTO electrolytes, polished with sandpaper, were smeared with the molten Li–Li2S composite on both sides, followed by sealing in a Swagelok cell. Then, the fabricated cells were heated on a hot plate at 200 °C for 30 min to further ensure a good anode/electrolyte interfacial contact before any electrochemical tests. In the control experiments, Li|LLZTO|Li cells were assembled with the same process by using molten Li as the electrode.The LiFePO4 electrode was prepared from a mixture of 75 wt% LiFePO4, 15 wt% acetylene black, and 5 wt% polyvinylidene fluoride (PVDF) using N-methyl-2-pyrrolidinone as the solvent. After spreading the uniform slurry on Al foil, the electrode was dried in a vacuum at 120 °C overnight. Then, the as-prepared dried electrode was pressed and punched into 8 mm discs for cell assembly. The mass loading of the active material in the cathode is ∼1.5 mg cm−2. The Li–Li2S|LLZTO|LiFePO4 full cell was fabricated by using the Li–Li2S composite, LLZTO, and 3 μL electrolyte (1.0 mol L−1 LiPF6 in ethylene carbonate/diethyl carbonate/dimethyl carbonate (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 vol/vol/vol)) infiltrated LiFePO4 as the anode, electrolyte, and cathode, respectively.
:1:1 vol/vol/vol)) infiltrated LiFePO4 as the anode, electrolyte, and cathode, respectively.
2.4 Electrochemical measurements
Electrochemical impedance spectroscopy (EIS) measurements were carried out on a Solartron 1260 impedance analyzer coupled with a 1287 electrochemical interface. The frequency was applied from 1 MHz to 0.01 Hz with a voltage amplitude of 10 mV. The galvanostatic plating/stripping cycling of the symmetric cells and the cycling performance of the Li–Li2S|LLZTO|LiFePO4 full cell were recorded on a Land battery tester instrument (LAND CT-2001A, China) at room temperature.3 Results and discussion
The XRD pattern of the as-prepared LLZTO electrolyte is shown in Fig. S1.† No impurity peaks are detected in the LLZTO sample. All the diffraction peaks are well indexed to garnet-type Li5La3Nb2O7 (JCPDS Card No. 80-0457), suggesting a cubic structure phase with high ionic conductivity (25 °C: 3 × 10−4 S cm−1).60 The cross-section morphology of the LLZTO pellet shows a dense microstructure with few isolated pores located at the grain boundaries (Fig. S2†), and the calculated relative density is about 96% using the Archimedes drainage method. For tuning the wettability of molten Li, commercial S powder was added into the molten Li at 250 °C in a stainless steel crucible. After the reaction between Li and S, except for the metal Li phase (JCPDS Card No. 01-1131), the XRD result of the as-cooled composite sample shows distinguished peaks at approximately 26.7°, 44.6°, and 52.9° related to the fully lithiated Li2S phase (JCPDS Card No. 77-2145), which is the only Li-rich alloy phase in the Li–S phase diagram (Fig. 1b). No observed peaks of sulfur suggest a sufficient reaction between Li and S. The surface morphologies of the as-prepared Li–Li2S composite with different S amounts were characterized by FESEM, as illustrated in Fig. 1d, e and S3.† Compared to pure Li with a clean and smooth surface (Fig. 1c), the LS10 composite displays noticeably dispersed Li2S particles in the Li matrix, which is further confirmed by the corresponding EDS analysis (Fig. 1f). The cross-section image combined with the EDS mapping result of the LS10 composite demonstrates the relatively even longitudinal distribution of Li2S, tightly embedded in the Li matrix (Fig. 1g and h). | ||
| Fig. 1 (a) XRD pattern of the Li–Li2S composite. (b) The phase diagram of Li–S.61 The surface FESEM images of (c) pure Li and (d) and (e) the LS10 composite, and (f) corresponding EDS elemental mapping images. (g) FESEM and (h) EDS elemental mapping images of the cross-section of the LS10 composite. | ||
The digital photos (Fig. 2a–d) show the effects of S addition on the wettability of molten Li on LLZTO substrates at 250 °C. It can be observed that the pure molten Li displays an insufficiently smeared nature (spherical shape) with a contact angle larger than 90°, while the Li–Li2S composite melt shows a much-reduced surface tension that results from the decreased interior Li–Li atomic interactions, probably because of the destroyed Li–Li bonding network in the molten Li, enabling well spreading on the LLZTO surface even just with 5 wt% S addition into the Li melt. When cooling down to room temperature, further interface morphology investigation results reveal an intimate physical contact without gaps between LLZTO and the Li–Li2S composite (Fig. 2g–i). EDS mapping analysis on the cross-section of the LS10 electrode in the LS10|LLZTO|LS10 symmetric cell further confirms the good dispersion of Li2S in the metallic Li (Fig. S4†). In contrast, huge gaps/voids exist at the pure Li/LLZTO interface because of the lithiophobic feature of the LLZTO surface (Fig. 2e and f). Significant improvement on the interfacial compatibility suggests the effectiveness of the S addition in modulating the physical contact between the Li anode and LLZTO electrolyte. The larger the contact area, the smaller the interfacial impedance. The homogenized current distribution is supposed to significantly enable the suppressed Li dendrite growth and thus improved cycling stability can be expected.
 | ||
| Fig. 2 The photos of melted (a) pure Li, (b) LS5, (c) LS10, and (d) LS20 on top of the garnet surface. FESEM images of (e) and (f) the garnet/Li, (g) garnet/LS5, (h) garnet/LS10, and (i) garnet/LS20 interfaces. | ||
Symmetric cells (Li–Li2S|LLZTO|Li–Li2S, Li|LLZTO|Li) were assembled to evaluate the interfacial resistance between the Li anode and LLZTO garnet, and the EIS test results are shown in Fig. 3a. The Nyquist plot results reveal a significantly decreased area-specific resistance with S addition. The calculated interfacial resistance is about 237.1 Ω cm2 for the Li|LLZTO|Li cell, while that for the LS10|LLZTO|LS10 cell is only 12.4 Ω cm2, showing a ∼95% reduction in the value. The decreased interfacial resistance value, because of the improved continuous and intimate physical contact without gaps, indicates facilitated Li-ion transfer across the interface. Besides, according to the first-principles calculation results (Fig. S5 and Table S1†), compared to the reaction energy (−6 meV per atom) between pure Li and garnet, Li2S shows a larger reaction energy of −0.38 meV per atom with garnet, suggesting a favorable thermodynamical stability without an interfacial reaction. The non-generated ion passivation phases avoid the negative effect on the charge transfer at the interface. It should be noted that when the S content increases up to 20 wt%, even though more generated Li2S at the interface can act as the morphological extension of the electrolyte for ion transfer, the reduced direct contact area between Li and garnet, resulting from the excess Li2S at the interface, is probably responsible for the increased interfacial resistance because of the inferior ionic conductivity of Li2S to LLZTO.
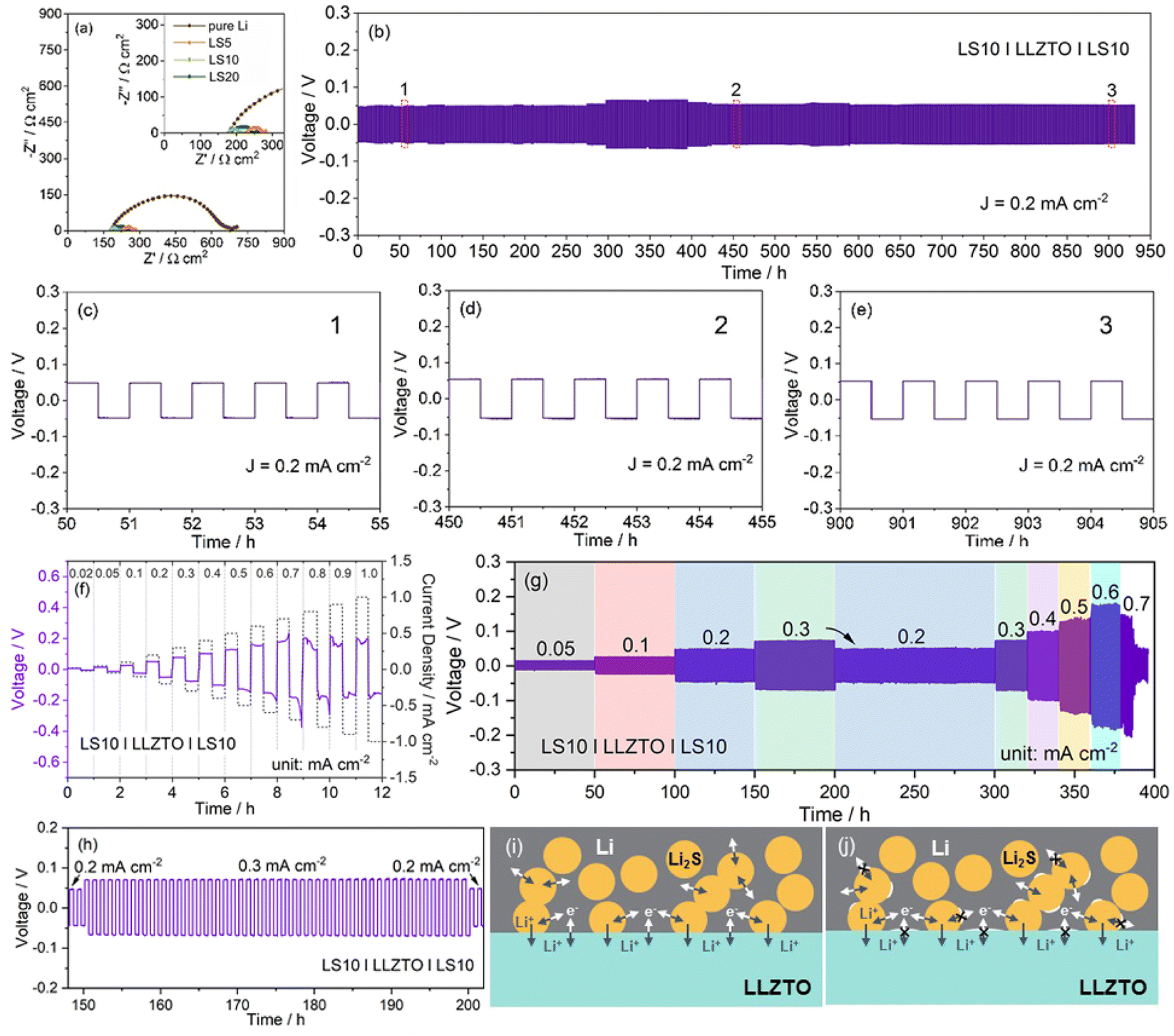 | ||
| Fig. 3 (a) Comparison of EIS plots of solid-state Li–Li2S|LLZTO|Li–Li2S and Li|LLZTO|Li symmetric cells. (b) Reversible galvanostatic Li plating/stripping curves of the symmetric LS10|LLZTO|LS10 cells under 0.2 mA cm−2 at 25 °C with a capacity of 0.1 mA h cm−2, and (c)–(e) the typically corresponded plating/stripping curves at different cycling stages marked in (b). (f) CCD for the LS10 electrode in the LS10|LLZTO|LS10 symmetric cell. (g) The stepped rate-capability of the LS10|LLZTO|LS10 symmetric cell at different current densities (0.05 mA cm−2 → 0.7 mA cm−2) with the plating/stripping capacity increasing from 0.025 mA h cm−2 to 0.35 mA h cm−2, and (h) the magnified voltage profiles of the symmetric LS10|LLZTO|LS10 cell at 0.3 mA cm−2. (i) and (j) Schematic illustration of the charge transfer at the interface for the Li–Li2S|LLZTO|Li–Li2S symmetric cell. | ||
Fig. 3b depicts the galvanostatic long-term cycling profile of the symmetric LS10|LLZTO|LS10 cell. All the tests were performed using a fixed 0.5 h for Li plating and Li stripping, respectively. The symmetric LS10|LLZTO|LS10 cell displays a stable voltage profile for at least 900 h with a small over-potential of 47.7, 48.1, 52.7, and 53 mV after 10, 50, 450, and 900 cycles at a current density of 0.2 mA cm−2, respectively. The ultra-flat voltage plateau with almost unchanged hysteresis related to the Li plating/stripping processes, shown in the zoomed-in curves at different cycling stages, demonstrates no dendrite-induced failure characteristics and superior interfacial stability between the LS10 electrode and LLZTO garnet (Fig. 3c–e). The slight variation of the interfacial impedance after different cycles (Fig. S6†) further suggests the well-maintained intimate LLZTO/LS10 contact during repeated Li plating/stripping processes, as further evidenced by the ex situ cross-sectional FESEM observation result, showing no interfacial voids/gaps after cycling (Fig. S7†). As the areal capacity increases to 1 mA h cm−2 accompanied by the same current density, a relatively stable Li plating/stripping process without an internal short circuit phenomenon over 180 h is achieved (Fig. S8†). The voltage curves of the LS10|LLZTO|LS10 symmetric cell still retain the plateau-like characteristics, and the overpotential only shows an increased value from 45.2 mV to 52.7 mV during the first deep Li stripping process. Notably, during the repeated deeper Li plating/stripping processes, the slowly increased average voltage hysteresis demonstrates that gradually accumulated interface defects could also appear. In contrast, the symmetric cell with a pure Li electrode (Fig. S9†) shows a larger over-potential in the first cycle and a rapid short circuit at 0.2 mA cm−2 (areal capacity: 0.1 mA h cm−2). Even when cycled at a lower current density of 0.1 mA cm−2, the fast voltage hysteresis increase along with the short circuit still occurs in a few hours, which should be related to the interface connection with the point-to-point contact model, leading to an inhomogeneous local electric field and lithium-ion flux distribution and thus accelerated lithium dendrite growth.
Apart from the stability, the critical current density (CCD), defined as the highest allowable current density for the solid-state electrolyte to endure lithium dendrite penetration, of the symmetric LS10|LLZTO|LS10 cell (Fig. 3f) is 0.7 mA cm−2. The increase of current density will cause an exacerbating voltage hysteresis. A smaller over-potential of 5.9, 14, 26, 77.2, 102, 128.3, 152.8, and 177.3 mV at a current density of 0.02, 0.05, 0.1, 0.3, 0.4, 0.5, 0.6, and 0.7 mA cm−2 is achieved, respectively. Stepped rate-capability tests reveal a stable cycling performance at a current density of 0.3 mA cm−2 (Fig. 3g), and the average over-potential shows a negligible increase (∼1 mV) over 50 h cycles (Fig. 3h). When the current density switches back to 0.2 mA cm−2, the displayed much stable voltage profile over 100 h with a recovered over-potential of ∼50 mV indicates the well-maintained tight connection between the Li–Li2S composite anode and LLZTO garnet in repeated Li plating/stripping processes. Subsequently returning the current density to 0.3 mA cm−2, the achieved stable cycling performance with an almost constantly flat voltage plateau across the cycles (Fig. S10a†) suggests the well-remained Li–Li2S composite morphology without pore formation.62 With further stepwise increasing the current density from 0.3 mA cm−2 to 0.6 mA cm−2, the Li plating/striping curves are still stable with cycling, demonstrating the superior rate-capability (Fig. S10b–d†). However, it should be noted that the non-constant voltage plateau at each cycle, especially under higher current density (e.g. 0.6 mA cm−2), is probably attributed to the gradually accumulated voids near the interface because of the faster adatom diffusion than lithium diffusivity in metal. When cycled with a higher current density (0.7 mA cm−2), the unstable interface leads to an increased local current density, and the promoted lithium dendrite growth rapidly leads to the short circuit of the cell with a significant voltage drop, which is consistent with the CCD test result.
A scheme in Fig. 3i and j visually illustrates the anode/garnet interface physical contact and the interfacial ion transport characteristics during Li+ stripping processes. The superior wettability of the molten Li–Li2S composite with garnet leads to an intimate interfacial connection (Fig. 3i). Thus, the lower the interface resistance, the more favorable the lithium ion transfer kinetics, and the smaller the over-potential. Besides, the more homogenized interface current distribution, the more suppressed lithium dendrites, as evidenced by the stable Li+ stripping/plating processes with a slightly increased over-potential (47.7 mV → 53 mV) (Fig. 3b–e). In contrast, the abundant voids at the pure Li/garnet interface not only impede Li+ and electron diffusivity between the pure Li anode and garnet, resulting in a higher interfacial resistance and over-potential, but also lead to an uneven electric field distribution, inducing lithium dendrite nucleation and growth and thus degrading cycling performance (Fig. S11†). For the Li–Li2S composite anode, most importantly, the generated Li2S with high ionic conductivity at the interface (I–Li2S) could also function as an ionic conductor for ion transfer. Thus, the extended interface area (Li/LLZTO + Li/I–Li2S) could reduce the local current density, benefiting the improved rate-capability and the higher CCD to some extent. Besides, at a high current density, the remained good contact Li/I–Li2S interface area enables I–Li2S to serve as a “bridge” for providing efficient ion transport paths between the garnet and Li-rich anode, ensuring a stable lower over-potential (Fig. 3j), and the over-potential increase could also be inhibited even in a deep Li stripping process. Certainly, at a higher current density, the more accumulated voids at the Li/LLZTO + Li/Li2S interface could mainly contribute to the degradation of cycling performance accompanied by the subsequently induced short circuit.
Galvanostatic cycling tests were performed on the assembled LS10|LLZTO|LiFePO4 full cells, and the results are shown in Fig. 4. Compared to the cells with a pure Li anode, the LS10|LLZTO|LiFePO4 cell shows a much-improved cycling performance. The cell delivers an initial discharge capacity of 154.7 mA h g−1 with an initial coulombic efficiency of 97% at 0.1C (Fig. 4a). After 100 cycles, a reversible specific capacity of 149.7 mA h g−1, corresponding to a capacity retention of 96.8%, with a coulombic efficiency close to 100%, is achieved, indicating excellent electrode reaction reversibility and interfacial stability. The typical discharge/charge curves with an ultra-flat LiFePO4 characteristic plateau at ∼3.4 V, related to the two-phase transfer reaction between LiFePO4 and FePO4, only show a slight polarization increase upon cycling (Fig. 4b). With stepwise increasing the current density to higher current densities, a reversible specific capacity of ∼143, ∼128, and ∼102 mA h g−1 at 0.3C, 0.5C, and 1C (Fig. 4c), corresponding to a capacity retention of 79.5%, 71.2%, and 56.5% concerning a capacity of 0.05C (Fig. 4d), is delivered, respectively. In contrast, the Li|LLZTO|LiFePO4 cell shows a rapid capacity decrease with the current density increase (Fig. S12a†). Besides, according to the discharge/charge curves (Fig. S12b and S13†), the LS10|LLZTO|LiFePO4 cell displays the well-maintained two-phase electrochemical feature of LiFePO4 even at the high current density, and the much smaller electrode polarization of the LS10|LLZTO|LiFePO4 cell than that of the Li|LLZTO|LiFePO4 cell at each current density demonstrates the facilitated reaction kinetics, resulting from the improved interfacial charge transfer. When the current density returns to 0.05C, considering the commonly known feature of insertion-type LiFePO4 cathodes with excellent phase/structure stability with cycling, the quickly recovered capacity of the LS10|LLZTO|LiFePO4 cell to the initial value further indicates its superior interfacial stability. The above-mentioned results demonstrate the great promise of the Li–Li2S anode design in the Li–metal battery application. However, since a trace amount of liquid electrolyte is applied to wet the cathode/LLZTO interface, further optimization of the cathode/LLZTO interface is still urgently important to realize the practical applications of ASSBs.
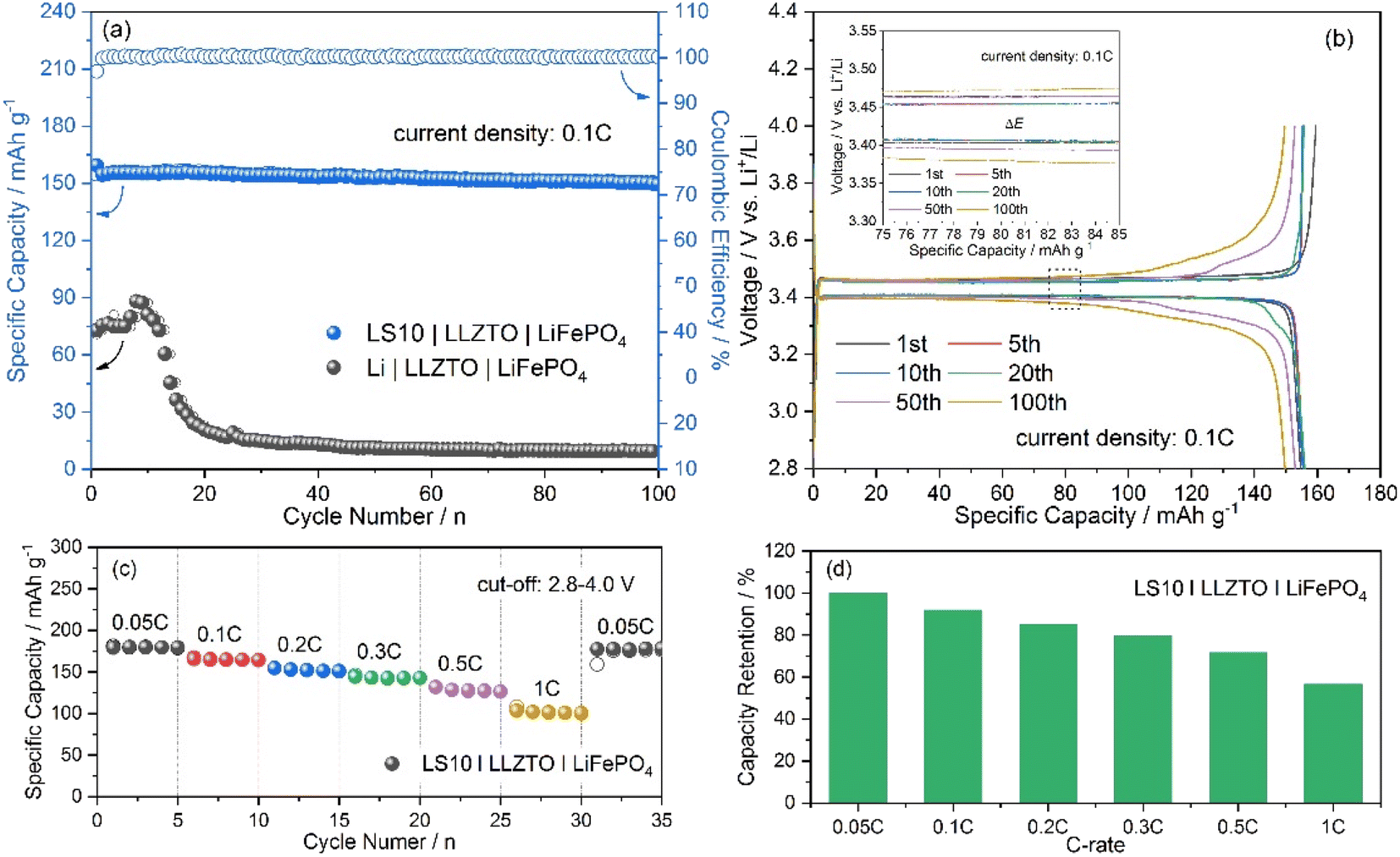 | ||
| Fig. 4 (a) Cycling performances of the fabricated LS10|LLZTO|LiFePO4 and Li|LLZTO|LiFePO4 cells at 0.1C; (b) typical discharge/charge curves for 100 galvanostatic cycles of the LS10|LLZTO|LiFePO4 cell at 0.1C and the inset shows the zoomed-in curves marked in (b); (c) rate-performance of the LS10|LLZTO|LiFePO4 cell at various current densities from 0.05C to 1C, and (d) the corresponding capacity retention at different current densities. | ||
4 Conclusions
In summary, we have developed a Li–Li2S composite by simply introducing commercialized sulfur into molten Li. The composite anode presents an intimate face-to-face physical connection with the LLZTO electrolyte because of the lowered surface tension of molten Li, ensuring the fabricated symmetric Li–Li2S|LLZTO|Li–Li2S cells with reduced interfacial resistance. The tight interface contact enables a homogenized current distribution and thus favors the suppression of lithium dendrite growth during repeated Li plating/stripping processes. Besides, Li2S with high ionic conductivity could extend the interface area for charge transfer and function as an efficient ion transport path between the Li anode and garnet, benefiting the cycling stability and rate performance. As expected, an optimized LS10|LLZTO|LS10 symmetric cell displays a low interfacial resistance of 12.4 Ω cm2 and a high lithium dendrite suppression capability, which is evidenced by the stable cycling performance over 900 h at 0.2 mA cm−2 and the achieved CCD of 0.7 mA cm−2. The fabricated LS10|LLZTO|LiFePO4 full cell can provide a superior cycling performance, including a high reversible discharge specific capacity (0.1C: 154.7 mA h g−1) and excellent cycling stability (capacity retention of 96.8% over 100 cycles) with high coulombic efficiency. This work offers an efficient strategy to enable a compatible Li/garnet interface for promoting the development of garnet-type ASSBs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (52074023 and 52102205), the Beijing Natural Science Foundation (2222062), the Interdisciplinary Research Project for Young Teachers of USTB (Fundamental Research Funds for the Central Universities) (FRF-IDRY-21-023).Notes and references
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 CrossRef CAS.
- B. Scrosati, J. Hassoun and Y. K. Sun, Energy Environ. Sci., 2011, 4, 3287–3295 RSC.
- N. S. Choi, Z. Chen, S. A. Freunberger, X. Ji, Y. K. Sun, K. Amine, G. Yushin, L. F. Nazar, J. Cho and P. G. Bruce, Angew. Chem., Int. Ed., 2012, 51, 9994–10024 CrossRef CAS.
- W. Lee, S. Muhammad, C. Sergey, H. Lee, J. Yoon, Y. M. Kang and W. S. Yoon, Angew. Chem., Int. Ed., 2020, 59, 2578–2605 CrossRef CAS PubMed.
- H. Wu and Y. Cui, Nano Today, 2012, 7, 414–429 CrossRef CAS.
- F. Han, A. S. Westover, J. Yue, X. Fan, F. Wang, M. Chi, D. N. Leonard, N. J. Dudney, H. Wang and C. Wang, Nat. Energy, 2019, 4, 187–196 CrossRef CAS.
- W. Xu, J. Wang, F. Ding, X. Chen, E. Nasybulin, Y. Zhang and J. G. Zhang, Energy Environ. Sci., 2014, 7, 513–537 RSC.
- O. Sheng, H. Hu, T. Liu, Z. Ju, G. Lu, Y. Liu, J. Nai, Y. Wang, W. Zhang and X. Tao, Adv. Funct. Mater., 2022, 32, 2111026 CrossRef CAS.
- K. Fu, Y. Gong, Z. Fu, H. Xie, Y. Yao, B. Liu, M. Carter, E. Wachsman and L. Hu, Angew. Chem., Int. Ed., 2017, 56, 14942–14947 CrossRef CAS PubMed.
- W. Zhou, S. Wang, Y. Li, S. Xin, A. Manthiram and J. B. Goodenough, J. Am. Chem. Soc., 2016, 138, 9385–9388 CrossRef CAS PubMed.
- C. Wang, Y. Gong, B. Liu, K. Fu, Y. Yao, E. Hitz, Y. Li, J. Dai, S. Xu, W. Luo, E. D. Wachsman and L. Hu, Nano Lett., 2017, 17, 565–571 CrossRef CAS PubMed.
- J. E. Ni, E. D. Case, J. S. Sakamoto, E. Rangasamy and J. B. Wolfenstine, J. Mater. Sci., 2012, 47, 7978–7985 CrossRef CAS.
- C. Monroe and J. Newman, J. Electrochem. Soc., 2005, 152, A396–A404 CrossRef CAS.
- O. Sheng, J. Zheng, Z. Ju, C. Jin, Y. Wang, M. Chen, J. Nai, T. Liu, W. Zhang, Y. Liu and X. Tao, Adv. Mater., 2020, 32, 2000223 CrossRef CAS PubMed.
- X. Yu, J. B. Bates, G. E. Jellison Jr and F. X. Hart, J. Electrochem. Soc., 1997, 144, 524–532 CrossRef CAS.
- Y. Kato, S. Hori, T. Saito, K. Suzuki, M. Hirayama, A. Mitsui, M. Yonemura, H. Iba and R. Kanno, Nat. Energy, 2016, 1, 16030 CrossRef CAS.
- N. V. Kosova, E. T. Devyatkina, A. P. Stepanov and A. L. Buzlukov, Ionics, 2008, 14, 303–311 CrossRef CAS.
- N. Kamaya, K. Homma, Y. Yamakawa, M. Hirayama, R. Kanno, M. Yonemura, T. Kamiyama, Y. Kato, S. Hama, K. Kawamoto and A. Mitsui, Nat. Mater., 2011, 10, 682–686 CrossRef CAS PubMed.
- S. Stramare, V. Thangadurai and W. Weppner, Chem. Mater., 2003, 15, 3974–3990 CrossRef CAS.
- R. Murugan, V. Thangadurai and W. Weppner, Angew. Chem., Int. Ed., 2007, 46, 7778–7781 CrossRef CAS PubMed.
- V. Thangadurai, H. Kaack and W. J. F. Weppner, J. Am. Ceram. Soc., 2003, 86, 437–440 CrossRef CAS.
- V. Thangadurai, S. Narayanana and D. Pinzaru, Chem. Soc. Rev., 2014, 43, 4714–4727 RSC.
- V. Thangadurai, D. Pinzaru, S. Narayanan and A. K. Baral, J. Phys. Chem. Lett., 2015, 6, 292–299 CrossRef CAS PubMed.
- H. Buschmann, J. Dölle, S. Berendts, A. Kuhn, P. Bottke, M. Wilkening, P. Heitjans, A. Senyshyn, H. Ehrenberg, A. Lotnyk, V. Duppel, L. Kienlee and J. Janek, Phys. Chem. Chem. Phys., 2011, 13, 19378–19392 RSC.
- F. Han, Y. Zhu, X. He, Y. Mo and C. Wang, Adv. Energy Mater., 2016, 6, 1501590 CrossRef.
- Y. Zhu, J. G. Connell, S. Tepavcevic, P. Zapol, R. Garcia-Mendez, N. J. Taylor, J. Sakamoto, B. J. Ingram, L. A. Curtiss, J. W. Freeland, D. D. Fong and N. M. Markovic, Adv. Energy Mater., 2019, 9, 1803440 CrossRef.
- J. Wolfenstine, J. L. Allen, J. Read and J. Sakamoto, J. Mater. Sci., 2013, 48, 5846–5851 CrossRef CAS.
- S. Ohta, T. Kobayashi and T. Asaoka, J. Power Sources, 2011, 196, 3342–3345 CrossRef CAS.
- Y. Wang and W. Lai, Electrochem. Solid-State Lett., 2012, 15, A68–A71 CrossRef CAS.
- L. J. Miara, S. P. Ong, Y. Mo, W. D. Richards, Y. Park, J. M. Lee, H. S. Lee and G. Ceder, Chem. Mater., 2013, 25, 3048–3055 CrossRef CAS.
- E. Rangasamy, J. Wolfenstine, J. Allen and J. Sakamoto, J. Power Sources, 2013, 230, 261–266 CrossRef CAS.
- S. Qin, X. Zhua, Y. Jiang, M. Ling, Z. Hu and J. Zhu, Appl. Phys. Lett., 2018, 112, 113901 CrossRef.
- X. Han, Y. Gong, K. Fu, X. He, G. T. Hitz, J. Dai, A. Pearse, B. Liu, H. Wang, G. Rubloff, Y. Mo, V. Thangadurai, E. D. Wachsman and L. Hu, Nat. Mater., 2017, 16, 572–579 CrossRef CAS PubMed.
- A. Sharafi, E. Kazyak, A. L. Davis, S. Yu, T. Thompson, D. J. Siegel, N. P. Dasgupta and J. Sakamoto, Chem. Mater., 2017, 29, 7961–7968 CrossRef CAS.
- H. Huo, Y. Chen, N. Zhao, X. Lin, J. Luo, X. Yang, Y. Liu, X. Guo and X. Sun, Nano Energy, 2019, 61, 119–125 CrossRef CAS.
- Y. Li, X. Chen, A. Dolocan, Z. Cui, S. Xin, L. Xue, H. Xu, K. Park and J. B. Goodenough, J. Am. Chem. Soc., 2018, 140, 6448–6455 CrossRef CAS PubMed.
- W. Luo, Y. Gong, Y. Zhu, K. K. Fu, J. Dai, S. D. Lacey, C. Wang, B. Liu, X. Han, Y. Mo, E. D. Wachsman and L. Hu, J. Am. Chem. Soc., 2016, 138, 12258–12262 CrossRef CAS PubMed.
- K. Fu, Y. Gong, B. Liu, Y. Zhu, S. Xu, Y. Yao, W. Luo, C. Wang, S. D. Lacey, J. Dai, Y. Chen, Y. Mo, E. Wachsman and L. Hu, Sci. Adv., 2017, 3, e160165 Search PubMed.
- W. Luo, Y. Gong, Y. Zhu, Y. Li, Y. Yao, Y. Zhang, K. Fu, G. Pastel, C. F. Lin, Y. Mo, E. D. Wachsman and L. Hu, Adv. Mater., 2017, 29, 1606042 CrossRef PubMed.
- W. Feng, X. Dong, P. Li, Y. Wang and Y. Xia, J. Power Sources, 2019, 419, 91–98 CrossRef CAS.
- C. Wang, Y. Gong, B. Liu, K. Fu, Y. Yao, E. Hitz, Y. Li, J. Dai, S. Xu, W. Luo, E. D. Wachsman and L. Hu, Nano Lett., 2017, 17, 565–571 CrossRef CAS PubMed.
- H. Huo, Y. Chen, R. Li, N. Zhao, J. Luo, J. G. Pereira da Silva, R. Mücke, P. Kaghazchi, X. Guo and X. Sun, Energy Environ. Sci., 2020, 13, 127–134 RSC.
- W. Zhou, S. Wang, Y. Li, S. Xin, A. Manthiram and J. B. Goodenough, J. Am. Chem. Soc., 2016, 138, 9385–9388 CrossRef CAS PubMed.
- H. Huo, J. Gao, N. Zhao, D. Zhang, N. G. Holmes, X. Li, Y. Sun, J. Fu, R. Li, X. Guo and X. Sun, Nat. Commun., 2021, 12, 176 CrossRef CAS PubMed.
- B. Liu, Y. Gong, K. Fu, X. Han, Y. Yao, G. Pastel, C. Yang, H. Xie, E. D. Wachsman and L. Hu, ACS Appl. Mater. Interfaces, 2017, 9, 18809–18815 CrossRef CAS PubMed.
- J. Duan, W. Wu, A. M. Nolan, T. Wang, J. Wen, C. Hu, Y. Mo, W. Luo and Y. Huang, Adv. Mater., 2019, 31, 1807243 CrossRef PubMed.
- C. Wang, H. Xie, L. Zhang, Y. Gong, G. Pastel, J. Dai, B. Liu, E. D. Wachsman and L. Hu, Adv. Energy Mater., 2018, 8, 1701963 CrossRef.
- Y. Huang, B. Chen, J. Duan, F. Yang, T. Wang, Z. Wang, W. Yang, C. Hu, W. Luo and Y. Huang, Angew. Chem., Int. Ed., 2020, 59, 3699–3704 CrossRef CAS PubMed.
- M. Du, Y. Sun, B. Liu, B. Chen, K. Liao, R. Ran, R. Cai, W. Zhou and Z. Shao, Adv. Funct. Mater., 2021, 31, 2101556 CrossRef CAS.
- W. Feng, X. Dong, X. Zhang, Z. Lai, P. Li, C. Wang, Y. Wang and Y. Xia, Angew. Chem., Int. Ed., 2020, 59, 5346–5349 CrossRef CAS PubMed.
- S. H. Wang, J. Yue, W. Dong, T. T. Zuo, J. Y. Li, X. Liu, X. D. Zhang, L. Liu, J. L. Shi, Y. X. Yin and Y. G. Guo, Nat. Commun., 2019, 10, 4930 CrossRef PubMed.
- J. Fu, P. Yu, N. Zhang, G. Ren, S. Zheng, W. Huang, X. Long, L. Hong and X. Liu, Energy Environ. Sci., 2019, 12, 1404–1412 RSC.
- T. Krauskopf, R. Dippel, H. Hartmann, K. Peppler, B. Mogwitz, F. H. Richter, W. G. Zeier and J. Janek, Joule, 2019, 3, 2030–2049 CrossRef CAS.
- T. Krauskopf, B. Mogwitz, C. Rosenbach, W. G. Zeier and J. Janek, Adv. Energy Mater., 2019, 9, 1902568 CrossRef CAS.
- M. Hiratani, K. Miyauchi and T. Kudo, Solid State Ionics, 1988, 28–30, 1406–1410 CrossRef.
- Z. J. Zheng, H. Ye and Z. P. Guo, Adv. Sci., 2020, 7, 2002212 CrossRef CAS PubMed.
- R. A. Huggins, Electrochim. Acta, 1977, 22, 773–781 CrossRef CAS.
- H. Chen, A. Pei, D. Lin, J. Xie, A. Yang, J. Xu, K. Lin, J. Wang, H. Wang, F. Shi, D. Boyle and Y. Cui, Adv. Energy Mater., 2019, 9, 1900858 CrossRef.
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder and K. A. Persson, APL Mater., 2013, 1, 011002 CrossRef.
- K. Zhang, T. Xu, H. Zhao, S. Zhang, Z. Zhang, Y. Zhang, Z. Du and Z. Li, Int. J. Energy Res., 2020, 44, 9177–9184 CrossRef CAS.
- PAULING FILE in: Inorganic Solid Phases, SpringerMaterials (Online Database), ed. P. Villars, Springer, Heidelberg, Springer Materials, Li-S binary phase diagram 0-100 at.% S, 2012, https://materials.springer.com/isp/phase-diagram/docs/c_0901521 Search PubMed.
- G. V. Alexander, O. V. Sreejith, M. S. Indu and R. Murugan, ACS Appl. Energy Mater., 2020, 3, 9010–9017 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ta05912f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |