
Advanced trifunctional electrodes for 1.5 V-based self-powered aqueous electrochemical energy devices†
Ran
Xiao
a,
Peng
Huang
a,
Tuzhi
Xiong
a,
Jingjing
Wei
a,
Feng
Wang
b,
Jianqiu
Deng
 b,
Zhongmin
Wang
*c and
M.-Sadeeq
Balogun
*ab
b,
Zhongmin
Wang
*c and
M.-Sadeeq
Balogun
*ab
aCollege of Materials Science and Engineering, Hunan Joint International Laboratory of Advanced Materials and Technology for Clean Energy, Hunan University, Changsha, 410082, People's Republic of China. E-mail: balogun@hnu.edu.cn
bGuangxi Key Laboratory of Information Materials, Guilin University of Electronic Technology, Guilin 541004, People's Republic of China
cGuangxi Academy of Sciences, Nanning, Guangxi 530007, China. E-mail: zmwang@mail.gxas.cn
First published on 28th November 2022
Abstract
Energy devices with multifunctional applications, such as integrating two different energy devices, are emerging as promising strategies to elevate energy technology. This is, however, an alarming challenge that appeals to a single electrode material with multifunctional applications to minimize cost. Herein, a novel and cost-effective dual-functional self-powered aqueous electrochemical energy device (DAEED) was successfully assembled by integrating asymmetric supercapacitor (ASC) and overall water splitting (OWS) devices using a unique P-doped NiMoO4/MoO2 (denoted NMO-MO-P) nanorods as the trifunctional electrode. NMO-MO-P exhibits excellent supercapacitor (SC) storage and oxygen evolution reaction (OER) catalytic ability owing to the tuning of the electronic structure synergistic interfacial and doping engineering. The as-prepared NMO-MO-P trifunctional electrode offers a 1.50 V operating NMO-based ASC device and a 1.50 V bifunctional NMO-based overall water splitting (OWS) in aqueous alkaline KOH solution. The similar operating potentials of both ASC and OWS devices allow the electrical energy stored by the NMO-based ASC device to simultaneously and in a self-powered way generate H2 and O2 from the as-assembled DAEED. This present work creates more opportunities towards achieving cost-effective EEDs technology.
Introduction
Due to the exasperating energy crisis as well as environmental issues, one of the major alternatives to fossil fuels is the development of renewable energy sources.1,2 The irregular nature has inspired rapid research of clean and inexpensive electrical energy storage and energy conversion devices.3–6 Recently, water electrolysis, especially in aqueous alkaline electrolytes, is one of the key advanced energy conversion systems that usually require electrical energy to split water into clean hydrogen energy.7,8 Compared to the instability and direct application of the solar and wind energy as power grid,9–11 lithium-ion batteries (LIBs) and supercapacitors (SCs) are of particular interest and remain as the promising energy storage systems.12,13 Despite the advantages of LIBs in terms of high energy density,14 they have safety problems that originate from the organic electrolytes and high voltages15 that has granted SCs to also become promising energy storage devices as they possess favorable characteristics, such as the use of aqueous electrolytes, high power density, fast charging, and discharging capability, and long cycle life.16–18 Thus, SCs should be able to store the electricity generated by unstable renewable energy sources, and then safely use it in the alkaline water electrolyzers to produce hydrogen (H2).19–21 Thus, there is a need to develop sustainable and cost-effective dual-functional aqueous electrochemical energy device (DAEED) technologies.The soul of EEDs is the electrode material. Over the last few years, great efforts have been focussed on searching for high-performance electrodes that can efficiently and simultaneously function in both SCs and OWS.22–26 Transition metal oxides (TMOs) have become promising electrode materials for SCs due to their multiple valence states and distinct morphological features.27,28 Moreover, the high electrochemical activities of the complex chemical compositions and synergistic effects between two metal elements create more opportunity for ternary transition metal oxides (TTMOs).29 Compared to the spinel-type TTMOs (such as AxB3−xO4; A, B = Co, Ni, Zn, Mn, Fe),30,31 molybdate-based (XMoO4; X = Co, Ni) TTMOs have been extensively studied owing to several chemical oxidation states of Mo between +3 and +6 (ref. 29 and 32) and the better conductivity of Mo over Co and Ni.33 Computational and experimental studies have proved that NiMoO4 (NMO) could improve the capacitance and also possesses stronger electrostatic repulsion between Ni cations because of its higher redox potential.34,35 However, the generally known inferior electronic conductivity of NMO still inhibits the practical applications either in SCs or OWS,36,37 and has been significantly improved by various strategies, especially by doping and interfacial engineering.38–40 Nevertheless, there is still much opportunity for further enhancing the energy properties of NMO and its application in SCs and OWS towards achieving efficient dual-functional aqueous electrochemical energy devices (DAEEDs).
Herein, we systematically utilized NMO nanorods as the trifunctional electrode to design a novel and cost-effective DAEED for the first time. NMO nanorods were first modulated to achieve excellent SC storage ability and OER catalytic performances via interfacial and heteroatom doping approaches. The as-prepared P-doped NiMoO4/MoO2 (denoted NMO-MO-P) not only exhibits excellent performance as an SC-positive electrode but when coupled with a commercially available activated carbon (AC) anode (NMO-MO-P//AC ASC device) also operates at voltage window of up to 1.50 V. The ASC delivers a maximum energy density of 55.36 W h kg−1, a maximum power density of 2556.3 W kg−1, and remarkable cyclic performance up to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles with ∼100% capacity retention. In addition, the optimized NMO-MO-P also delivers an impressive OER catalytic activity with an overpotential of 240 mV at 10 mA cm−2 in 1.0 M KOH alkaline solution. When used as bifunctional catalysts for OWS (i.e., NMO-MO-P//NMO-MO-P OWS), the device achieved an overall potential of 1.50 V also at 10 mA cm−2 in the same electrolyte. Thus, by coupling both NMO-MO-P//AC and NMO-MO-P//NMO-MO-P OWS, a cost-effective NMO-MO-P-based 1.50 V-DAEED was assembled. The electrical energy from the ASC could generate both H2 and O2 from the bifunctional NMO-MO-P//NMO-MO-P OWS for up to 2.5 min in a KOH electrolyte. This work creates more opportunities for assembling cost-effective aqueous EED technology using a single high-performance electrode.
000 cycles with ∼100% capacity retention. In addition, the optimized NMO-MO-P also delivers an impressive OER catalytic activity with an overpotential of 240 mV at 10 mA cm−2 in 1.0 M KOH alkaline solution. When used as bifunctional catalysts for OWS (i.e., NMO-MO-P//NMO-MO-P OWS), the device achieved an overall potential of 1.50 V also at 10 mA cm−2 in the same electrolyte. Thus, by coupling both NMO-MO-P//AC and NMO-MO-P//NMO-MO-P OWS, a cost-effective NMO-MO-P-based 1.50 V-DAEED was assembled. The electrical energy from the ASC could generate both H2 and O2 from the bifunctional NMO-MO-P//NMO-MO-P OWS for up to 2.5 min in a KOH electrolyte. This work creates more opportunities for assembling cost-effective aqueous EED technology using a single high-performance electrode.
Results and discussion
Morphological and phase characterization
Our attempt to develop the DAEED involves designing a highly efficient and cost-effective multifunctional electrode (such as NMO) with self-supportive and tunable electronic structure characteristics to improve its electrical conductivity. Herein, both interfacial and doping engineering were simultaneously employed to achieve this goal. The above-mentioned properties were achieved in NMO by two-step approaches that include hydrothermal reaction followed by partial phosphorization. The three-dimensional nickel foam (NF) (ESI, Fig. S1†) was used as the starting material for the self-supportive growth of the samples. First, yellow films of NiMo precursor nanorods (Fig. 1a and S2†) were formed on the NF through a hydrothermal reaction. The nanorod morphology was retained upon the annealing of the precursor in the air (Fig. S3†) to form the pristine NMO sample (X-ray diffraction, XRD in Fig. 1b-i). However, the nanorod morphology became rough and scattered. XRD studies confirmed the formation of both NiMoO4 and MoO2 in NMO-MO and NMO-MO-P samples (Fig. 1b-ii and iii).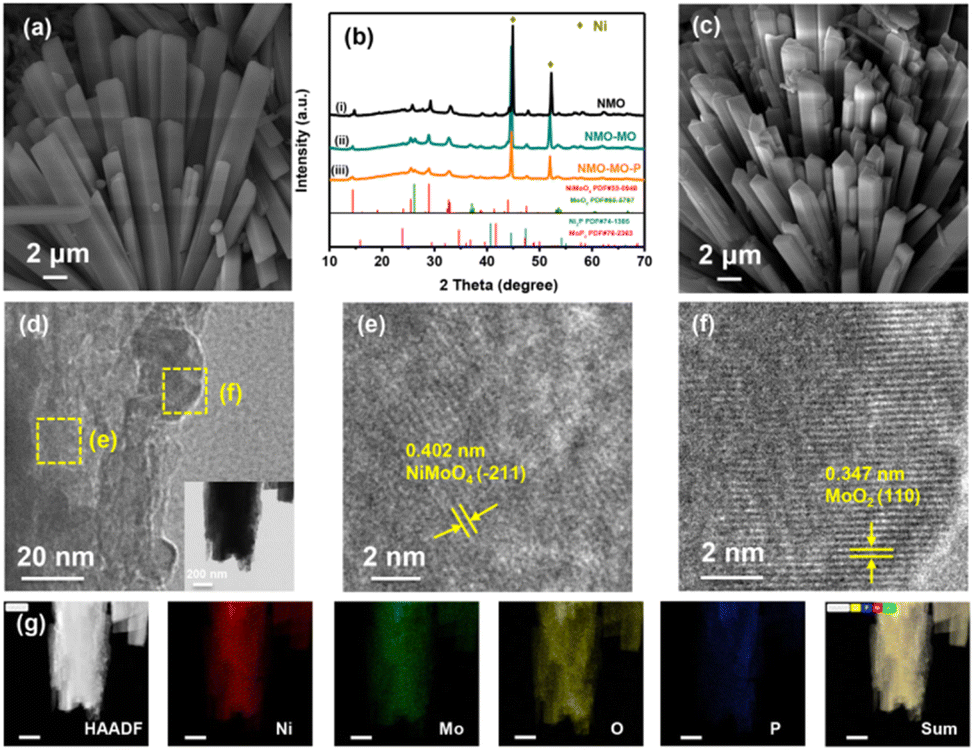 | ||
| Fig. 1 (a) SEM image of the NMO precursor. (b) The XRD patterns of NMO, NMO-MO, and NMO-MO-P. (c) SEM image of NMO-MO-P. (d–f) TEM images of NMO-MO-P. Inset in “d” is the TEM image of NMO-MO-P showing its nanorod morphology. (g) Elemental mapping images of NMO-MO-P. | ||
As shown in Fig. 1b, the two strong peaks of the NF substrate (PDF#04-0850) at 44.4° and 51.8°, the characteristic diffraction peaks at 14.3°, 25.3°, 28.8°, and 32.6° were indexed to (110), (−112), (220) and (022) of NiMoO4 (PDF#33-0948), another three diffraction peaks at 26.0°, 37.0°, and 53.5° are assigned to (011), (−211) and (−311) of MoO2 (PDF#65-5787), respectively. Furthermore, no peaks of Ni2P (PDF#74-1385) or MoP2 (PDF#76-2363) were observed in the XRD pattern, indicating that NMO-MO-P was successfully prepared. By annealing the precursor in an argon (Ar) atmosphere without and in the presence of a phosphorus source NMO-MO and NMO-MO-P nanorods were also retained, as shown in Fig. S4a, b† and 1c, respectively. The thickness of each nanorod was ≈2 μm. Based on the elemental mapping, the percentages of every atom (Ni:Mo:O:P) were 19.4%:43.7%:31.9%:5.0% with a Ni:Mo ratio of 1:2.25 (Fig. S5†), while that of the inductively coupled plasma-optical emission spectrometry (ICP-OES) results gave 19.8%:43.1%:30.2%:6.9%, respectively, with the Ni:Mo ratio of 1:2.18. In addition, ICP-OES results gave 23.0%:42.6%:34.4% of Ni:Mo:O, respectively, with the Ni:Mo ratio of 1:1.85 for NMO, and 22.3%:39.1%:38.6% of Ni:Mo:O, respectively, with the Ni:Mo ratio of 1:1.75 for NMO-MO (Table S1†). The elemental mapping and ICP-OES results were very similar, justifying the uniformity of our electrode design. The transmission electron microscopy (TEM) result of the NMO-MO-P nanorods further confirmed the nanorod morphology (Fig. 1d-inset). The high-resolution transmission electron microscopy (HRTEM) image of the NMO-MO-P nanorods (Fig. 1d) identified both NiMoO4 (Fig. 1e) and MoO2 (Fig. 1f) phases, which is indicative of the interfacial engineering in NMO-MO-P sample. Similar results were observed for the NMO-MO sample (Fig. S4c and d†). The doping of P was confirmed from the elemental mapping data shown in Fig. 1g (scale = 200 nm). The SEM, TEM, and XRD characterizations confirmed the formation of the interfacial and doping engineering in NMO-MO-P.
Electronic structure and interaction
X-ray photoelectron spectroscopy (XPS) measurements were performed to investigate the composition and electronic structure of the NMO, NMO-MO, and NMO-MO-P nanorods (Fig. S6†). The XPS analysis provides important information on the modulation of the electronic structure of the catalysts upon in situ hybridization and phosphorization. As shown in Fig. 2a, Ni 2p XPS spectra of NMO and NMO-MO display the peaks at 855.8 and 873.6 eV fitting with Ni2+ 2p3/2 and Ni2+ 2p1/2, respectively, of NiMoO4.41 At first, Ni 2p XPS peaks of NMO-MO-P display a slight peak shift to a higher binding energy compared with that of the NMO and NMO-MO. Fig. 2b shows the Mo 3d XPS spectra of the samples, in which the NMO-MO-P sample exhibits two doublet peaks at 232.5 eV (3d5/2) and 235.5 eV (3d3/2), and 229.1 eV (3d5/2) and 231.5 eV (3d3/2), assigned to Mo6+ and Mo4+, respectively. Apart from the Mo4+ and Mo6+ peaks, the intermediate oxidation states of Mo5+ at binding energies of 230.2 eV and 232.5 eV are observed, which proves the formation of Mo5+ species after the thermal treatment in a reducing atmosphere.42 More importantly, for the Mo 3d XPS spectra, both the peaks of NMO-MO and NMO-MO-P samples also shifted to the higher binding energy compared to the pristine NMO. Such a shift possesses the possibility of electron transfer between the cations in the hybrid as well as a change in the electronic configuration.43,44 In the O 1s XPS spectra in Fig. 2c, the peaks located at 529.5, 530.3, and 532.2 eV suggest the presence of the lattice O of M–O species (M = Ni, Mo) for NMO, oxygen vacancy and OH− species, respectively.45 The peaks at 529.6, 530.8, and 532.3 eV could be ascribed to the lattice O of M–O species for NMO-MO-P, oxygen vacancy, and OH− species, respectively, and the introduction of a new peak of P–Ox at 533.6 eV in NMO-MO-P,46,47 which could promote charge transfer and facilitate H adsorption.37,44 Moreover, compared to the single O 1s XPS peaks of the NMO and NMO-MO samples, the O 1s XPS spectrum of NMO-MO-P was featured with a positive shift (Fig. 2c), further justifying the stronger electronic interaction in the NMO-MO-P sample.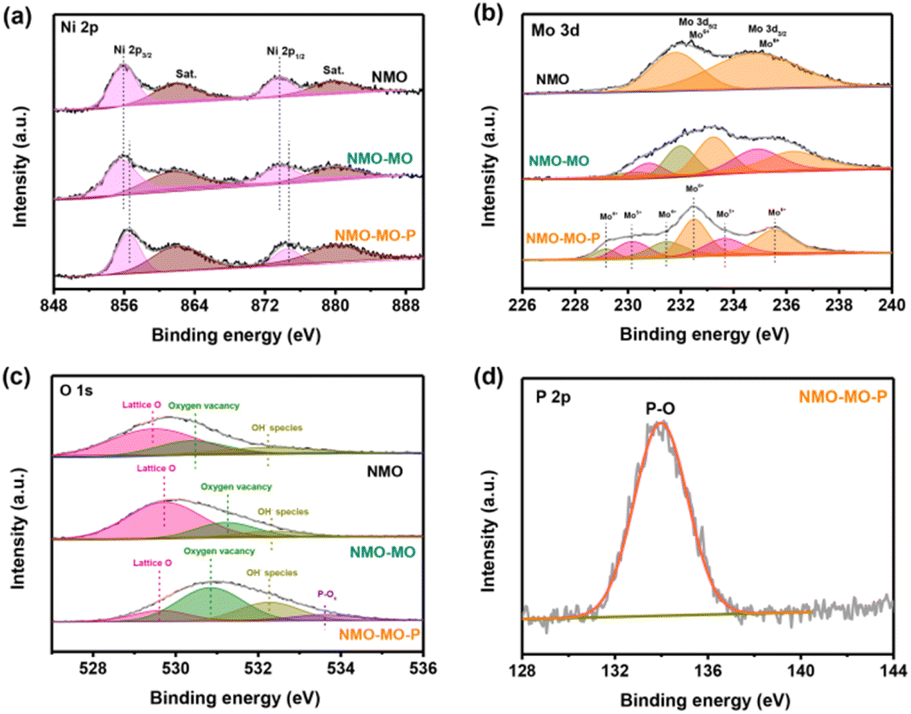 | ||
| Fig. 2 XPS analysis. (a) Ni 2p, (b) Mo 3d, (c) O 1s, and (d) P 2p XPS spectra of NMO, NMO-MO, and NMO-MO-P samples. | ||
Thus, there exists a strong electronic interaction between Ni and Mo, and the strong electronic interaction was confirmed by the changes in the electronic state of NMO-MO-P compared to NMO and NMO-MO. Strong electronic interaction plays a crucial role in tuning the electronic configuration of the metal orbital centers, resulting in the optimized storage performance and adsorption energy of the reaction intermediates. The P 2p XPS peaks at 134.1 eV, which corresponded to the P–O species (Fig. 2d).46,48 Moreover, P usually remains due to elemental doping as a single P–O peak at ≈134.1 eV was seen in the XPS spectra,49,50 whereas, multiple peaks are observed if there are other bonds, as reported in other studies.51,52 In addition, there was no low valence state of the P element, and no peaks of Ni2P or MoP2 were observed from the XRD pattern (Fig. 1b), demonstrating that the successful doping of P, which could capture more protons in the OER electrocatalysis process.46
SC storage properties of the electrodes
The SC storage properties of NMO, NMO-MO, and NMO-MO-P electrodes were first compared in a three-electrode configuration using the as-prepared electrodes as the working electrode, carbon rod as the counter electrode, and Ag/AgCl as a reference electrode in 3.0 M KOH electrolyte. The electrochemically active surface area (ECSA) was first evaluated to study the effect of P doping and interfacial engineering. By plotting the anodic current densities (I) derived from the CV curves in Fig. S7a–c† against their corresponding scan rates at a potential of 0.3 V versus SCE, the slope of the NMO-MO-P sample showed a much higher capacitance of 137.8 mF cm−2, predicting larger ECSA and enhanced surface properties of the NMO-MO-P electrode than both NMO-MO (62.0 mF cm−2) and pristine NMO (33.8 mF cm−2) (Fig. 3a). The representative CV curves of the three electrodes at scan rates of 100 mV s−1 showed that NMO-MO-P was characterized by both larger current density and curve area (Fig. S7d†), suggesting enhanced electrochemical reactivity.53,54 Moreover, the IR-drop plot (IR-drop measurement obtained from the galvanostatic charge/discharge profiles shown in Fig. S7a–c†) of both NMO and NMO-MO are the same and characterized with bigger slopes (Fig. 3b). However, the smaller slope obtained in NMO-MO-P reveals its better kinetics owing to the heteroatom P modulation. The boosted kinetics in an electrode material leads to enhanced rate performance. | ||
| Fig. 3 (a) Scan rate dependence of the current densities of the different electrodes at 0.3 V and their corresponding linear fittings. (b) IR drop of the different electrodes measured at different current densities. (c) Cycling performance of the different electrodes. The areal capacitances of the different electrodes as a function of (d) scan rates and (e) current densities. (f) The areal capacitance of the NMO-MO-P electrode as a function of current densities in different electrolyte concentrations. | ||
Indeed, the rate performance of the NMO-MO-P electrode was superior to both NMO-MO and NMO electrodes irrespective of the areal capacitances derived from the CV curves at different scan rates or galvanostatic charge/discharge profiles at different current densities. For example, based on the scan rates, the NMO-MO-P electrode yielded an outstanding areal capacitance of 9.20 C cm−2 (11.51 F cm−2) at 5 mV s−1, which is more than 2 folds those of NMO-MO (4.54 C cm−2; 5.67 F cm−2) and NMO (4.29 C cm−2; 5.36 F cm−2) electrodes (Fig. 3d and S7e†).
Moreover, based on the current densities, the NMO-MO-P electrode delivered a remarkable areal capacitance of 7.32 C cm−2 (18.29 F cm−2) and a specific capacitance of 239.1 C g−1 (597.7 F g−1) at 6 mA cm−2, which are superior to 2.46 C cm−2 (6.14 F cm−2) and 100.7 C g−1 (251.7 F g−1) of NMO-MO and 82.24 C g−1 (205.6 F g−1) of NMO electrodes (Fig. 3e and S8a–f†). The corresponding rate capability values of the NMO-MO-P electrode were significantly higher than those of the recently reported NMO-based electrodes, even at a higher current density of 30 mA cm−2 (2.56 C cm−2 and 6.39 F cm−2) (Table S2†). After the rate test, the NMO-MO-P electrode could retain an areal capacitance of 2.51 C cm−2 (6.27 F cm−2) at 30.0 mA cm−2, which is the same as the initial capacitance at the same current density, indicating its good reversibility and cyclic stability (Fig. S9a†).
Therefore, the cyclic stability of the NMO-MO-P electrode was not only outstanding by displaying stability between that of the pristine NMO and NMO-MO but also exhibited capacitance retention of 100% after 2000 cycles (Fig. 3c). Its morphology was perfectly retained after the stability test (Fig. S9b†). The outstanding rate performance and cyclic stability can not only be attributed to the synergistic effect between NiMoO4 and MoO2 but also the electronic modulation caused by the P-doping.37,45,55 Considering the fact that NMO-based positive electrodes can operate at different molar concentrations of KOH, we also studied the SC storage performance of NMO-MO-P in 1.0 and 2.0 M KOH electrolytes. Although NMO-MO-P displayed its best performance in 3.0 M KOH compared to other KOH concentrations (Fig. 3f and S10†), NMO-MO-P in 1.0 M KOH still revealed superior storage capability than some other reported NMO-based electrodes at the same and higher KOH concentrations (Table S2†).45,56,57 In order to explore the influence of the different doping amount of phosphorus and annealing times on the percentage of P doping on SC performance. Two additional phosphorization conditions of 100 mg and 240 mg NaH2PO2·H2O were selected, and the samples were named NMO-MO-P(100) and NMO-MO-P(240), respectively. Furthermore, in addition, 0.5 h and 2 h annealing times were set to explore the effect of annealing time on SC performance, and the samples were named NMO-MO-P-0.5 and NMO-MO-P-2, respectively. The results of CV and GCD curves (Fig. S11 and S12†) showed that the areal capacitance of NMO-MO-P (optimized sample with 170 mg NaH2PO2·H2O and 1 h annealing time) was the highest compared to NMO-MO-P(100), NMO-MO-P(240), NMO-MO-P-0.5 and NMO-MO-P-2. A series of controlled experiments were also performed to investigate the effect of the heterojunction and P-doping by synthesizing Mo and Ni-free and their corresponding P-doped samples, such as pristine MoO2 and P-doped MoO2 (Fig. S13†), and NiO and P-doped NiO (Fig. S14†). The results show that the Mo-based samples show better electrochemical properties than the Ni-based samples, while the P-doped samples revealed superior SC performance than the un-doped electrodes (Fig. S15†). These results further justify the contribution of interfacial and P-doping engineering.
Storage properties of the NMO-MO-P asymmetric device
To justify the practicability of the NMO-MO-P as a positive electrode in the ASC device, commercially available activated carbon (AC) was employed as the negative electrode and our as-prepared NMO-MO-P was the positive electrode to fabricate the NMO-MO-P//AC ASC device (Fig. 4a). The performances of the AC as the negative electrode in 3.0 M KOH are displayed in Fig. S16 and Table S3.† Then, both the AC negative and NMO-MO-P positive electrodes were balanced, as shown in Fig. S17a,† and evaluated to be 1:0.994. At a sweep rate of 100 mV s−1, the CV curves of the NMO-MO-P//AC ASC device can work at different operating voltages between 0 and 1.8 V (Fig. 4b). The CV shapes remain intact when the potential windows are below 1.6 V. However, when the potential window is extended to more than 1.8 V, the shape becomes slightly polarized, showing a typical characteristic of either a positive or negative electrode, and confirming that the optimal potential is no other than 1.6 V. Moreover, the device exhibited typical SC behaviour at varying sweep rates of 5.0–100 mV s−1 (Fig. S17b†). Based on the galvanostatic charge/discharge profiles of the as-assembled ASC device (Fig. S17c†), the areal capacitance of the NMO-MO-P//AC ASC device was estimated, as shown in Fig. 4c. The device delivered maximum and minimum areal capacitances of 4.20 F cm−2 @ 6 mA cm−2 (155.71 F g−1) and 2.14 F cm−2 @ 40 mA cm−2 (79.14 F g−1), respectively. Interestingly, according to the areal capacitance of the device, our as-assembled ASC device achieved a maximum energy density of 55.36 W h kg−1 at a power density of 359.4 W kg−1 according to the Ragone plot in Fig. 4d and outperforms most of the recently reported NMO-based ASC devices (Table S4†). At different electrolyte concentrations, the NMO-MO-P//AC ASC device still achieved excellent storage performances (Fig. S18†). More importantly, the power density of the device attains 2556.3 W kg−1 @ 30 mA cm−2, which is higher than that previously reported for most of the NMO-based devices and also comparable with that of the NiCo2O4-UNSA@NiMoO4 device (≈1318.6 W kg−1 @ 40 mA cm−2).58
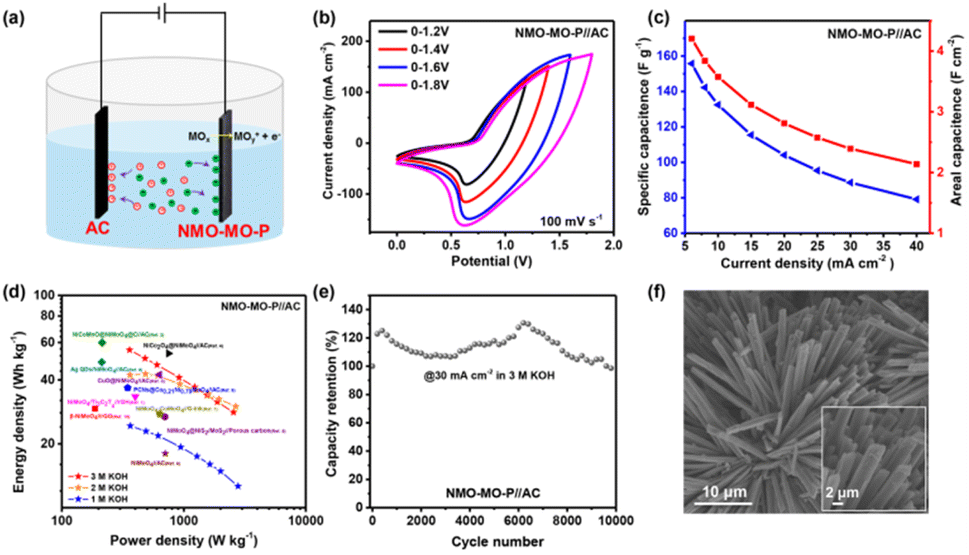 | ||
| Fig. 4 (a) Schematic representation of the NMO-MO-P//AC ASC. (b) CV curves for the NMO-MO-P//AC ASC at different potential windows. (c) Specific capacitance of NMO-MO-P//AC device as a function of current density in 3.0 M KOH. (d) Ragone plots of the NMO-MO-P//AC ASC and other recently reported supercapacitor devices. (e) Cycling stability showing the coulombic efficiency and capacity retention of the NMO-MO-P//AC ASC at a current density of 30 mA cm−2. (f and inset) SEM images of NMO-MO-P after 10000 cycles. | ||
Moreover, the NMO-MO-P//AC ASC device also achieved a long-term shelf-life of 100% capacitance retention after 10000 cycles (Fig. 4e) and the nanorod morphology of the NMO-MO-P (Fig. 4f), as well as the phase structure, were retained (Fig. S17d†), indicating excellent structural stability of the as-designed NMO-MO-P positive electrode.
OER and overall water-splitting properties
Generally, OER is a half-reaction process of the OWS. The crucial challenge is to explore non-noble transition metal-based catalysts for promoting the sluggish transfer of the four excitons during OER.59 Therefore, we further compared the OER performance of our as-prepared NMO-MO-P with those of pristine NMO and NMO-MO. NiFe-LDH coated on NF (denoted NiFe-LDH/NF) (Fig. S19†) was introduced for comparison. Fig. 5a shows the linear sweep voltammetry (LSV) profiles of these electrocatalysts. To deliver a current density of 10 mA cm−2, NMO-MO-P only requires an overpotential of 241 mV, which is significantly lower than that of NMO-MO (309 mV), pristine NMO (357 mV), NiFe-LDH/NF (255 mV) and most of the recently reported NMO-based OER electrocatalysts (Table S5†). | ||
| Fig. 5 OER and OWS properties. (a) OER LSV curves and (b) Tafel slopes of the NMO, NMO-MO, NMO-MO-P, and NiFe-LDH catalysts. (c) OER stability curves of NMO-MO-P catalyst at a fixed overpotential of 240 mV. Inset is the corresponding SEM image after the stability test. (d) The OER free-energy diagram on the structural models. Inset is the optimized NiMoO4–MoO2–P with the intermediates adsorbed molecules. (e) Schematic representation and (f) LSV curve of the NMO-MO-P//NMO-MO-P OWS. Inset is its corresponding stability test. (g) Comparison of the NMO-MO-P//NMO-MO-P device with NMO-based OWS devices. | ||
The OER kinetics and mechanisms were studied using Tafel plots, ECSA and TOF. The Tafel slope can be used to reveal the rate-determining steps that are critical to understanding the electrocatalytic reaction mechanisms. Generally, a smaller Tafel slope implies faster electrochemical kinetics. According to Fig. 5b, the Tafel slope of the NMO-MO-P (130 mV dec−1) is much lower than those of NMO-MO (159 mV dec−1) and NMO (190 mV dec−1) but slightly inferior to that of NiFe-LDH/NF (100 mV dec−1), indicating the outstanding OER kinetics of the OER process of the NMO-MO-P catalyst. In addition, the synergistic effect between NiMoO4 and MoO2 in the NMO-MO-P sample is also a crucial reason to improve its OER kinetics compared with NMO. ECSA is a quantitative activity parameter to evaluate the catalytic activity. Meanwhile, the double-layer capacitances (Cdl) of the different samples obtained by fitting the cyclic voltammetry curves (Fig. S20a–c†) are shown in Fig. S20e,† which is proportional to the ECSA value. The ECSA obtained from the CV curves (at a potential of 0.10 V versus SCE) shows the largest ECSA and AECSA values for NMO-MO-P (7.96 mF cm−2; 199 cmECSA2) in comparison with NMO-MO (2.29 mF cm−2; 57.25 cmECSA2) and NMO (1.80 mF cm−2; 45 cmECSA2). This ECSA result indicates that the specific surface area of the sample can be increased to a certain extent by simple phosphating treatment, which is conducive to exposing a large number of active sites. As shown in Fig. S20f,† the normalized ECSA of the NMO-MO-P still showed smaller overpotential than those of NMO-MO and NMO, indicating that the higher catalytic performance of the NMO-MO-P electrode is mainly attributed to its larger ECSA compared to the than electrodes. The turnover frequency (TOF) values calculated for each catalyst revealed that the NMO-MO-P electrocatalyst significantly exhibited a much higher TOF value (1.65 s−1) than NMO-MO (1.10 s−1) and pristine NMO (0.69 s−1) at a fixed potential of 1.60 V, corresponding to that of NMO (Fig. S21a-experimental section and S21b†).
These results explain part of the OER mechanisms and solidify the superior catalytic kinetics and better electrical conductivity of the NMO-MO-P catalyst towards efficient HER catalytic capability.
Details on the OER mechanism were studied using density functional theory (DFT) calculations. To quantitatively illustrate the contribution of the electronic interaction within NiMoO4–MoO2–P during the OER process, the classical single-site mechanism was applied to calculate the adsorption-free energy. The structural models of the catalysts, including the adsorption of their intermediates, are shown in Fig. S22.† As shown in Fig. 5d, the rate-determining step (RDS) is the formation of *OOH owing to its maximum free energy. The free energies of the OER intermediates on the NiMoO4–MoO2–P catalyst site decrease compared with those of MoO2, NiMoO4, and NiMoO4–MoO2, which can be attributed to the doping of P. The doping of P could enhance the conductivity, optimize the adsorption of oxygen-containing intermediates, and reduce the kinetic barrier. Therefore, the accepted OER mechanism in alkaline media that includes the transformation and formation of OH, O, and OOH intermediates with Mo catalytic active site can be shown as follows;
| Mo + OH− → Mo – OH + e− | (1) |
| Mo – OH + OH− → Mo – O + H2O + e− | (2) |
| Mo – O + OH− → Mo – OOH + e− | (3) |
| Mo – OOH + OH− → Mo + O2 + H2O + e− | (4) |
Meanwhile, NMO-MO-P could also achieve impressive cyclic stability of up to 10 h (Fig. 5c). The OER LSV curves, Tafel plots, and stability test results affirmed the better catalytic performance of NMO-MO-P as a result of the simultaneous presence of rich epitaxial in-growth phase boundaries and P integration. After the OER stability test, the introduction of new peaks corresponded to the Ni3+ species in the Ni 2p XPS spectra of NMO-MO-P after OER (Fig. S23a†) corresponds to the formation of NiOOH. In addition, TEM images (Fig. S23d–f†) also confirmed the thin layer observed on the edge of the nanorods with the fringe spacing of 0.208 nm corresponds to the (105) phase of the NiOOH layer associated with the redox reaction of Ni surface species, generating in situ efficient OER active sites.60 For P 2p XPS spectra (Fig. S23b†), the intensity of the P–O peak was reduced after OER indicating that the surface P was oxidized under high anodic oxidation and the phosphate group on the catalyst surface was partially dissolved in the alkaline electrolyte.61,62 However, the P–O peak disappeared (Fig. S23c†), presumably due to the dissolution of the surface P in the alkaline electrolyte and the formation of a NiOOH thin layer. These results are consistent with the reduction of P–O signal intensity in Fig. S23b.†
To explore if different doping amounts of phosphorus show an effect on the structure and electrocatalytic activity, other phosphorization experimental conditions, such as 100 and 240 mg NaH2PO2·H2O at a constant annealing time of 1 h were introduced. In addition, annealing times of 0.5 h and 2 h at a constant NaH2PO2·H2O content of 170 mg were also introduced. The samples were named NMO-MO-P(100), NMO-MO-P(240), NMO-MO-P-0.5 and NMO-MO-P-2. We performed a comprehensive characterization of the different electrodes, including SEM and XRD (Fig. S24, S25 and Table S6†), and XPS (Fig. S26†). The results confirmed that the phase structure, bulk composition, and morphology of the nanorod arrays were almost the same. The electrochemical results showed that the OER electrocatalytic performance of NMO-MO-P (optimized sample with 170 mg NaH2PO2·H2O at constant annealing time of 1 h) was the best (Fig. S27a–c, S28, and Table S7†). To understand the reason for the better OER performance of the NMO-MO-P catalyst better than that of NMO-MO and the effect of P species on the enhanced OER activity, further experimental analyses were carried out. For example, the effect of the NF substrate on the OER catalytic activity confirmed that the P introduction improved the OER catalytic property (Fig. S27d and e†). In addition powder NMO-MO-P was prepared (Fig. S29†), the nanorod morphology was retained and its catalytic performance was inferior to that of the real NMO-MO-P sample, further affirming the significance of the heteroatom integration and self-support electrode approaches (Fig. S30†).
OWS in alkaline solution, also known as alkaline electrolysis has captured significant interest using bifunctional electrocatalysts.63 Thus, motivated by the impressive OER catalytic performance of the NMO-MO-P electrocatalyst, an OWS was assembled by employing NMO-MO-P, as both HER (cathode) and OER (anode), in 1.0 M KOH (Fig. 5e). Our previous report has shown that NMO-MO-P exhibited excellent HER performance with an overpotential of 23 mV at 10 mA cm−2.44 Therefore, to attain an electrolytic process up to the current density of 10 mA cm−2, an overall voltage of 1.50 V was required (Fig. 5f), which is also superior to the recent NMO-based alkaline electrolyzers and comparable to some others (Fig. 5g). After OWS stability, the bifunctional NMO-MO-P catalysts revealed excellent performance, as shown in Fig. S31.† All these results further affirmed the excellent stability of the as-designed NMO-MO-P catalyst.
Dual-functional aqueous electrochemical energy device (DAEED)
As mentioned above, we aimed to develop a DAEED using a highly efficient and cost-effective single electrode with multifunctional applications, which we achieved by engineering NMO with heterojunctions and heteroatom doping. The DAEED involves the energy storage device and an energy conversion system operating simultaneously to generate H2. To achieve this, we have designed an SC, which is our as-assembled NMO-MO-P//AC ASC device to store the electrical energy and a water splitter that is our NMO-MO-P//NMO-MO-P OWS device demands electrical energy to produce H2, as schematically represented in Fig. 6a. The digital image of the DAEED device is shown in Fig. 6b. After charging the NMO-MO-P//AC ASC device for 300 s (enlarged image in Fig. 6c) and connecting to the NMO-MO-P//NMO-MO-P OWS device, the electrical energy stored drives the OWS device to produce H2 and O2 for about 148 s. The video demonstration of the DAEED is shown in Fig. SV1,† where the bubbles in Fig. 6d represent H2 and O2 gases. | ||
| Fig. 6 Dual-functional self-powered aqueous electrochemical energy device (DAEED). (a) Schematic illustration and (b) digital image of the DAEED. (c and d) Actual conditions before and after connecting to the OWS device when ASC is charged to 1.5 V. | ||
Conclusions
A kind of multifunctional energy device, namely a dual-functional self-powered aqueous electrochemical energy device (DAEED) that involves integrating energy conversion and energy storage devices was developed in this work. The as-prepared NMO-MO-P electrode served as the trifunctional electrode out of the four electrodes required to operate the cost-effective DAEED. The DAEED simultaneously generates H2 and O2 from the OWS device from the electrical energy stored by the ASC device. As the cathode material in SC, the NMO-MO-P//AC ASC device could successfully operate at a working potential of 1.50 V, delivering a maximum energy and power densities of 55.36 W h kg−1 and 2556.3 W kg−1, respectively, and excellent stability of up to 10000 cycles. In addition, an OWS device assembled using NMO-MO-P as a bifunctional electrode also required a very low potential of 1.50 V to reach a current density of 10 mA cm−2. The excellent performance of the electrode can be attributed to the interfacial and doping engineering synergistic effect. The similar operating voltages of both devices allow the simultaneous generation of H2 and O2 from the NMO-MO-based OWS using the electrical energy generated by the NMO-MO-based ASC. This work creates more opportunities towards the development of cost-effective aqueous EEDs technology and beyond.
Experimental
Materials
Nickel nitrate hexahydrate [Ni(NO3)2·6H2O, 99%], ammonium molybdate tetrahydrate [(NH4)6Mo7O24·4H2O, 99%] and sodium hypophosphite [NaH2PO2·H2O, 99%] were purchased from Sinopharm Group Chemical Co. Ltd, Shanghai Aladdin Biochemical Technology Co., Ltd and Shanghai Maclin Biochemical Technology Co., Ltd.All reagents were used as received without any further purification. Nickel foam (NF) was purchased from Changsha Liyuan New Material Co., Ltd. An ultra-pure purification system (Master-S15Q, Hitech Instruments Co. Ltd., Shanghai, China, and The Lab) was used to produce 18.2 MΩ cm−1 water in all experiments.
Synthesis of NiMo precursor
0.75 mmol Ni(NO3)2·6H2O and 0.75 mmol (NH4)6Mo7O24·4H2O were mixed and dissolved in 25 mL deionized (DI) water. Before the hydrothermal reaction, nickel foam (2 × 3 cm2) was first cleaned with 3 M HCl solution in an ultrasound bath for 3 min to remove the surface oxide layer. Then, the NF was cleaned by sonication sequentially with ethanol and deionized (DI) water for 4 min each. The mixture and a piece of NF (2 × 3 cm2) were transferred to a 25 mL Teflon-lined stainless-steel autoclave and warmed up to 160 °C and maintained for 4 h. The NiMo precursor was washed with DI water and ethanol several times and dried in a vacuum oven at 60 °C.Synthesis of NMO-MO-P, NMO-MO, and NMO
To prepare the NMO-MO-P sample, the NiMo precursor and 170.1 mg NaH2PO2·H2O were placed in quartz boats, and NaH2PO2·H2O was placed at the upstream side of the NiMo precursor. Subsequently, the samples were heated at 500 °C for 1 h at a ramping rate of 10 °C min−1 in a 200 sccm Ar atmosphere and then cooled naturally. The obtained product was NMO-MO-P. For comparison, NMO-MO was obtained by annealing the NiMo precursor in Ar without the presence of a phosphorus source. The NMO sample was retained upon annealing the NiMo precursor in air. NMO-MO-P powder was also obtained by collecting the powder NiMo precursor left in the autoclave and subjected to the same annealing conditions as that of NMO-MO-P. The detailed characterization of the powder-NMO-MO-P is shown in Fig. S29.†Characterization
X-ray diffraction (XRD) patterns were collected using an X-ray diffractometer (XRD, SMARTLAB, 9 kW). The structures and morphologies of the materials were characterized using a field emission scanning electron microscope (FESEM, TESCAN MALA3). Transmission electron microscopy (TEM) and energy dispersive X-ray spectroscopy (EDX) of NMO-MO-P were performed using an FEI Tecnai G2 F30. The XPS spectra were collected on a Thermo Scientific K-Alpha instrument with an Al Kα X-ray source (1486.6 eV photons). The X-ray source was operated at 12 kV and 6 mA. The sample was mounted on the sample tray with double-sided tape, and pumped into the preparation chamber less than 2 × 10−7 mbar before being introduced into the XPS chamber. The full spectrum scan (narrow spectrum scan) had a pass energy of 150 eV (30 eV), a step size of 1 eV (0.1 eV), and a spot size of about 500 μm. Raw XPS bands could be fitted using a non-linear least-square fitting procedure using XPSPEAK41 software with Shirley-type background and Gaussian–Lorentzian peak shapes for all the peaks. The binding energy (BE) of the core-level C 1s peak was set at 284.8 eV to compensate for surface-charging effects. The final uncertainty of BE was not greater than 0.1 eV. ICP-OES was performed on an Agilent 720ES instrument.Supercapacitor measurement and calculations
The electrochemical performances of the electrodes were tested on a CHI760E electrochemical workstation in a three-electrode configuration using 3 M KOH aqueous solution as the electrolyte, where the as-prepared samples were directly used as the working electrode, carbon rod as the counter electrode, and Ag/AgCl (saturated KCl) electrode as the reference electrode. Moreover, the areal-specific capacitance (Cs, C cm−2) and mass-specific capacitance (Cm, C g−1) were calculated according to the following equations,64 respectively. | (5) |
 | (6) |
The asymmetric supercapacitors were assembled using NMO-MO-P as the positive electrode, AC (by mixing with carbon black and PVDF at a weight ratio of 8:1:1) coated on the nickel foam substrate as the negative electrode, and 3 M KOH aqueous solution as the electrolyte. To obtain the optimal capacitance performance the charge between the positive and negative electrodes should be balanced according to the following equation:
| Q = Cm × m | (7) |
 | (8) |
The specific capacitance of the two-electrode asymmetrical supercapacitor (C, F g−1) was computed from the GCD curves according to the equations:
| C = It/(mV) | (9) |
The energy density (E, W h kg−1) and power density (P, W kg−1) were calculated using the following equations:
| E = 1/2C(V)2 | (10) |
| P = E/t | (11) |
Electrocatalytic performance measurements
All electrochemical measurements were carried out on a CHI760E electrochemical analyzer, using a three-electrode system in 1.0 M KOH with a carbon rod as the counter electrode, Ag/AgCl (saturated KCl) electrode as the reference electrode, and the synthesized samples as the working electrodes directly. The equation used to calibrate the measured overpotential (V vs. RHE) is shown below:| E(RHE) = E(Ag/AgCl) + 0.197 V + 0.059 V × pH | (12) |
The Tafel slope was calculated following this equation:
| η = blogj + a | (13) |
Chronopotentiometry was used to test the long-term stability of the materials. Electrochemical impedance spectroscopy was performed between the frequency ranges of 10 mHz to 1.0 MHz with an AC excitation amplitude of 5 mV. The LSV curves were not IR-compensated.
For the electrochemical surface area (ECSA), CV curves were collected in the non-faradaic region to estimate the electrochemical double-layer-specific capacitance (Cdl). The Cdl was calculated according to Cdl = |ja − jc|/2ν, where ja and jc are charging and discharging current densities, respectively, and ν is the scan rate. The potential range of the measurements was from 0.05 to 0.15 V vs. RHE in a non-faradaic region. The scan rates were from 10 to 200 mV s−1 (10, 20, 50, 100, 200 mV s−1). The difference between the charging and discharging current densities at 0.1 V vs. RHE was used for the calculation.
ECSA can then be calculated using the following equation:
| ECSA = Cdl/Cs | (14) |
Turnover frequency (TOF) values were calculated using the following equation:
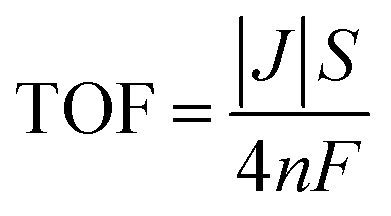 | (15) |
 | (16) |
485 C mol−1), i is the current density (A), V is the voltage (V) and u is the scanning rate (V s−1).
Author contributions
Ran Xiao: conceptualization, investigation, formal analysis, data curation, writing-original draft. Peng Huang: formal analysis, resources, software. Tuzhi Xiong: investigation, data curation. Jingjing Wei: data curation. Feng Wang: methodology, validation, funding acquisition. Jianqiu Deng: methodology, validation, funding acquisition. Zhongmin Wang: validation, funding acquisition, supervision, writing-review & editing. M.-Sadeeq (Jie Tang) Balogun: conceptualization, project administration, funding acquisition, supervision, writing-review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Science Foundation of Hunan Province (2021JJ30087), the Hunan Joint International Laboratory of Advanced Materials and Technology for Clean Energy (2020CB1007), and the Fundamental Research Funds for the Central Universities and Guangxi Key Laboratory of Information Materials and Guilin University of Electronic Technology, China (211011K).References
- K. N. Dinh, Q. Liang, C.-F. Du, J. Zhao, A. I. Y. Tok, H. Mao and Q. Yan, Nano Today, 2019, 25, 99–121 Search PubMed.
- N. Kittner, F. Lill and D. M. Kammen, Nat. Energy, 2017, 2, 17125 Search PubMed.
- X. F. Lu, Y. Fang, D. Luan and X. W. D. Lou, Nano Lett., 2021, 21, 1555–1565 CrossRef CAS.
- K. R. G. Lim, A. D. Handoko, S. K. Nemani, B. Wyatt, H.-Y. Jiang, J. Tang, B. Anasori and Z. W. Seh, ACS Nano, 2020, 14, 10834–10864 Search PubMed.
- M.-S. Balogun, Y. Huang, W. Qiu, H. Yang, H. Ji and Y. Tong, Mater. Today, 2017, 20, 425–451 CrossRef CAS.
- J. Li, Z. Zhu, Y. Huang, F. Wang and M.-S. Balogun, Mater. Today Energy, 2022, 26, 101001 CrossRef CAS.
- X. Y. Yu and X. W. Lou, Adv. Energy Mater., 2018, 8, 1701592 CrossRef.
- Y. Wang, F. Chu, J. Zeng, Q. Wang, T. Naren, Y. Li, Y. Cheng, Y. Lei and F. Wu, ACS Nano, 2021, 15, 210–239 CrossRef CAS PubMed.
- M. Yousif, Q. Ai, W. A. Wattoo, Z. Jiang, R. Hao and Y. Gao, J. Power Sources, 2019, 412, 710–716 Search PubMed.
- A. Al-Dousari, W. Al-Nassar, A. Al-Hemoud, A. Alsaleh, A. Ramadan, N. Al-Dousari and M. Ahmed, Energy, 2019, 176, 184–194 Search PubMed.
- M. Mehrasa, E. Pouresmaeil, B. Pournazarian, A. Sepehr, M. Marzband and J. P. S. Catalão, Energies, 2018, 11, 2469 Search PubMed.
- M. K. Aslam, Y. Niu and M. Xu, Adv. Energy Mater., 2021, 11, 2000681 CrossRef CAS.
- S. Yu, V. M. Hong Ng, F. Wang, Z. Xiao, C. Li, L. B. Kong, W. Que and K. Zhou, J. Mater. Chem. A, 2018, 6, 9332–9367 RSC.
- Y. Huang, H. Yang, T. Xiong, D. Adekoya, W. Qiu, Z. Wang, S. Zhang and M.-S. Balogun, Energy Storage Mater., 2020, 25, 41–51 CrossRef.
- Z. Liu, Y. Huang, Y. Huang, Q. Yang, X. Li, Z. Huang and C. Zhi, Chem. Soc. Rev., 2020, 49, 180–232 Search PubMed.
- D. Chao, W. Zhou, F. Xie, C. Ye, H. Li, M. Jaroniec and S.-Z. Qiao, Sci. Adv., 2020, 6, eaba4098 CrossRef CAS.
- M. Yu, Y. Lu, H. Zheng and X. Lu, Chem. - Eur. J., 2018, 24, 3639–3649 CrossRef CAS PubMed.
- Y. Zhang, H.-x. Mei, Y. Cao, X.-h. Yan, J. Yan, H.-l. Gao, H.-w. Luo, S.-w. Wang, X.-d. Jia, L. Kachalova, J. Yang, S.-c. Xue, C.-g. Zhou, L.-x. Wang and Y.-h. Gui, Coord. Chem. Rev., 2021, 438, 213910 CrossRef CAS.
- L. Hu, Y. Hu, R. Liu, Y. Mao, M. S. Balogun and Y. Tong, Int. J. Hydrogen Energy, 2019, 44, 11402–11410 CrossRef CAS.
- Y. Huang, M. Zhu, Y. Huang, Z. Pei, H. Li, Z. Wang, Q. Xue and C. Zhi, Adv. Mater., 2016, 28, 8344–8364 Search PubMed.
- Y. Huang, L. Hu, R. Liu, Y. Hu, T. Xiong, W. Qiu, M. S. Balogun, A. Pan and Y. Tong, Appl. Catal., B, 2019, 251, 181–194 CrossRef CAS.
- S. A. Hesse, K. E. Fritz, P. A. Beaucage, R. P. Thedford, F. Yu, F. J. DiSalvo, J. Suntivich and U. Wiesner, ACS Nano, 2020, 14, 16897–16906 CrossRef CAS PubMed.
- T. Xiong, X. Yao, Z. Zhu, R. Xiao, Y.-w. Hu, Y. Huang, S. Zhang and M. S. Balogun, Small, 2022, 18, 2105331 CrossRef CAS PubMed.
- T.-F. Hung, Z.-W. Yin, S. B. Betzler, W. Zheng, J. Yang and H. Zheng, Chem. Eng. J., 2019, 367, 115–122 CrossRef CAS.
- Y. Zhang, C.-r. Chang, X.-d. Jia, Q.-y. Huo, H.-l. Gao, J. Yan, A.-q. Zhang, Y. Ru, H.-x. Mei, K.-z. Gao and L.-z. Wang, Inorg. Chem. Commun., 2020, 111, 107631 CrossRef CAS.
- P. Liu, W. Pan, R. Yao, L. Zhang, Q. Wu, F. Kang, H. J. Fan and C. Yang, J. Mater. Chem. A, 2022, 10, 3760–3770 RSC.
- Y. Li, J. Zhang, Q. Chen, X. Xia and M. Chen, Adv. Mater., 2021, 33, 2100855 CrossRef CAS PubMed.
- M. Zheng, H. Tang, L. Li, Q. Hu, L. Zhang, H. Xue and H. Pang, Adv. Sci., 2018, 5, 1700592 CrossRef PubMed.
- Y. Zhao, X. Li, B. Yan, D. Xiong, D. Li, S. Lawes and X. Sun, Adv. Energy Mater., 2016, 6, 1502175 Search PubMed.
- M.-S. Park, J. Kim, K. J. Kim, J.-W. Lee, J. H. Kim and Y. Yamauchi, Phys. Chem. Chem. Phys., 2015, 17, 30963–30977 Search PubMed.
- Y. Zhang, C.-r. Chang, X.-d. Jia, Y. Cao, J. Yan, H.-w. Luo, H.-l. Gao, Y. Ru, H.-x. Mei, A.-q. Zhang, K.-z. Gao and L.-z. Wang, Inorg. Chem. Commun., 2020, 112, 107697 CrossRef CAS.
- Y. Zhang, H.-l. Gao, X.-d. Jia, S.-w. Wang, J. Yan, H.-w. Luo, K.-z. Gao, H. Fang, A.-q. Zhang and L.-z. Wang, J. Renewable Sustainable Energy, 2018, 10, 054101 CrossRef.
- X. Xu, T. Wei, X. Zhang, X. Xing, L. Liang, H. Wang and Y. Zhao, Int. J. Energy Res., 2020, 44, 2196–2207 CrossRef CAS.
- T. Watcharatharapong, M. Minakshi Sundaram, S. Chakraborty, D. Li, G. M. Shafiullah, R. D. Aughterson and R. Ahuja, ACS Appl. Mater. Interfaces, 2017, 9, 17977–17991 Search PubMed.
- C. V. V. M. Gopi, S. Sambasivam, K. V. G. Raghavendra, R. Vinodh, I. M. Obaidat and H.-J. Kim, J. Energy Storage, 2020, 30, 101550 Search PubMed.
- C. Qing, C. Yang, M. Chen, W. Li, S. Wang and Y. Tang, Chem. Eng. J., 2018, 354, 182–190 Search PubMed.
- F. Wang, K. Ma, W. Tian, J. Dong, H. Han, H. Wang, K. Deng, H. Yue, Y. X. Zhang and W. Jiang, J. Mater. Chem. A, 2019, 7, 19589–19596 Search PubMed.
- G. Solomon, A. Landström, R. Mazzaro, M. Jugovac, P. Moras, E. Cattaruzza, V. Morandi, I. Concina and A. Vomiero, Adv. Energy Mater., 2021, 11, 2101324 CrossRef CAS.
- J. Yuan, D. Yao, L. Jiang, Y. Tao, J. Che, G. He and H. Chen, ACS Appl. Energy Mater., 2020, 3, 1794–1803 CrossRef CAS.
- L. Ma, Z. Liu, T. Chen, Y. Liu and G. Fang, Electrochim. Acta, 2020, 355, 136777 CrossRef CAS.
- X. Zhang, Z. Li, Z. Yu, L. Wei and X. Guo, Appl. Surf. Sci., 2020, 505, 144513 CrossRef CAS.
- Z. Zhang, X. Ma and J. Tang, J. Mater. Chem. A, 2018, 6, 12361–12369 RSC.
- M.-I. Jamesh and M. Harb, J. Energy Chem., 2021, 56, 299–342 CrossRef CAS.
- T. Xiong, B. Huang, J. Wei, X. Yao, R. Xiao, Z. Zhu, F. Yang, Y. Huang, H. Yang and M. S. Balogun, J. Energy Chem., 2022, 67, 805–813 Search PubMed.
- Z. Zhu, W. Tian, X. Lv, F. Wang, Z. Hu, K. Ma, C. Wang, T. Yang and J. Ji, J. Colloid Interface Sci., 2021, 587, 855–863 CrossRef CAS PubMed.
- Y. Song, W. Sha, M. Song, P. Liu, J. Tian, H. Wei, X. Hao, B. Xu, J. Guo and J. Liang, Ceram. Int., 2021, 47, 19098–19105 CrossRef CAS.
- S. Zhuang, S. Tong, H. Wang, H. Xiong, Y. Gong, Y. Tang, J. Liu, Y. Chen and P. Wan, Int. J. Hydrogen Energy, 2019, 44, 24546–24558 CrossRef CAS.
- R. Jiang, D. Zhao, H. Fan, Y. Xie, M. Li, H. Lin and Z. S. Wu, J. Colloid Interface Sci., 2022, 606, 384–392 CrossRef CAS PubMed.
- Z. Guo, Y. Pang, H. Xie, G. He, I. P. Parkin and G. L. Chai, ChemElectroChem, 2020, 8, 135–141 CrossRef.
- R.-L. Zhang, J.-J. Feng, Y.-Q. Yao, K.-M. Fang, L. Zhang, Z.-Z. Yin and A.-J. Wang, Appl. Surf. Sci., 2021, 548, 149280 CrossRef CAS.
- K. Xu, Y. Sun, X. Li, Z. Zhao, Y. Zhang, C. Li and H. J. Fan, ACS Mater. Lett., 2020, 2, 736–743 CrossRef CAS.
- W. Xu, G. Fan, S. Zhu, Y. Liang, Z. Cui, Z. Li, H. Jiang, S. Wu and F. Cheng, Adv. Funct. Mater., 2021, 31, 2107333 CrossRef CAS.
- P. M. Shafi, N. Joseph, A. Thirumurugan and A. C. Bose, Chem. Eng. J., 2018, 338, 147–156 CrossRef CAS.
- T.-F. Yi, L.-Y. Qiu, J. Mei, S.-Y. Qi, P. Cui, S. Luo, Y.-R. Zhu, Y. Xie and Y.-B. He, Sci. Bull., 2020, 65, 546–556 CrossRef CAS.
- W. Chu, Z. Shi, Y. Hou, D. Ma, X. Bai, Y. Gao and N. Yang, ACS Appl. Mater. Interfaces, 2020, 12, 2763–2772 CrossRef CAS PubMed.
- D. Zhu, X. Sun, J. Yu, Q. Liu, J. Liu, R. Chen, H. Zhang, D. Song, R. Li and J. Wang, J. Colloid Interface Sci., 2020, 574, 355–363 Search PubMed.
- S. Chen, S. Chandrasekaran, S. Cui, Z. Li, G. Deng and L. Deng, J. Electroanal. Chem., 2019, 846, 113153 CrossRef CAS.
- P. Zhang, J. Zhou, W. Chen, Y. Zhao, X. Mu, Z. Zhang, X. Pan and E. Xie, Chem. Eng. J., 2017, 307, 687–695 CrossRef CAS.
- J. M. Gonçalves, M. Ireno da Silva, L. Angnes and K. Araki, J. Mater. Chem. A, 2020, 8, 2171–2206 RSC.
- J. Choi, D. Kim, W. Zheng, B. Yan, Y. Li, L. Y. S. Lee and Y. Piao, Appl. Catal., B, 2021, 286, 119857 CrossRef CAS.
- M. Yang, Y. Jiang, M. Qu, Y. Qin, Y. Wang, W. Shen, R. He, W. Su and M. Li, Appl. Catal., B, 2020, 269, 118803 Search PubMed.
- Q. Wang, H. Zhao, F. Li, W. She, X. Wang, L. Xu and H. Jiao, J. Mater. Chem. A, 2019, 7, 7636–7643 Search PubMed.
- A. Ali and P. K. Shen, Electrochem. Energy Rev., 2020, 3, 370–394 Search PubMed.
- K. Zhou, W. Zhou, L. Yang, J. Lu, S. Cheng, W. Mai, Z. Tang, L. Li and S. Chen, Adv. Funct. Mater., 2015, 25, 7530–7538 CrossRef.
- W. Zhu, W. Chen, H. Yu, Y. Zeng, F. Ming, H. Liang and Z. Wang, Appl. Catal., B, 2020, 278, 119326 Search PubMed.
- D. Merki, S. Fierro, H. Vrubel and X. Hu, Chem. Sci., 2011, 2, 1262–1267 RSC.
- J. Tian, Q. Liu, A. M. Asiri and X. Sun, J. Am. Chem. Soc., 2014, 136, 7587–7590 CrossRef CAS PubMed.
- L. Zeng, K. Sun, X. Wang, Y. Liu, Y. Pan, Z. Liu, D. Cao, Y. Song, S. Liu and C. Liu, Nano Energy, 2018, 51, 26–36 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ta05872c |
| This journal is © The Royal Society of Chemistry 2023 |