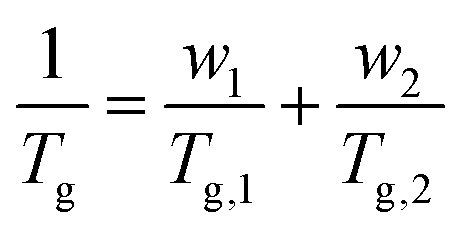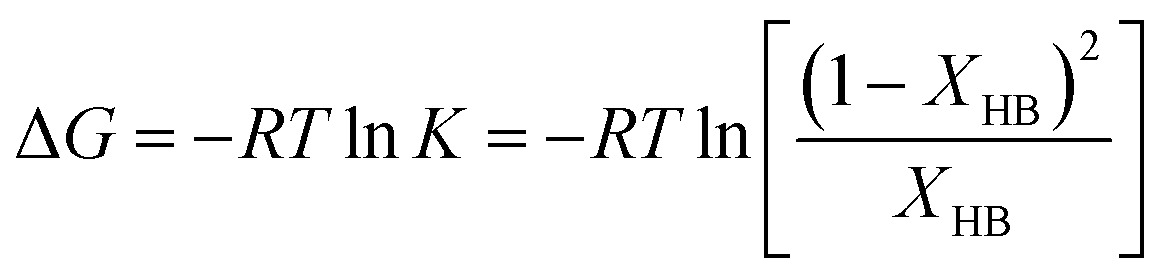DOI:
10.1039/D2SM01229D
(Paper)
Soft Matter, 2023,
19, 128-136
Effect of the polar group content on the glass transition temperature of ROMP copolymers†
Received
10th September 2022
, Accepted 18th November 2022
First published on 21st November 2022
Abstract
Polar groups have long been recognized to greatly influence the glass transition temperature (Tg) of polymers, but understanding the underlying physical mechanism remains a challenge. Here, we study the glass formation of ring-opening metathesis polymerization (ROMP) copolymers containing polar groups by employing all-atom molecular dynamics simulations. We show that although the number of hydrogen bonds (NHB) and the cohesive energy density increase linearly as the content of polar groups (fpol) increases, the Tg of ROMP copolymers increases with the increase of fpol in a nonlinear fashion, and tends to plateau for sufficiently high fpol. Importantly, we find that the increase rate of Gibbs free energy for HB breaking gradually slows down with the increase of fpol, indicating that the HB is gradually stabilized. Therefore, Tg is jointly determined by NHB and the strength of HBs in the system, while the latter dominates. Although NHB increases linearly with increasing fpol, the HB strength increases slowly with increasing fpol, which leads to a decreasing rate of increase in Tg.
Introduction
Glass transition temperature (Tg) of polymers is an important intrinsic quantity that is closely related to their applications.1 This quantity can be strongly influenced by many factors such as chain length,2,3 chain flexibility,4–9 and intermolecular interactions.10–13 Among them, modifying the monomer with polar groups can dramatically change the intermolecular interactions and thus the Tg of the polymer.14,15 In particular, Tg can be adjusted by changing the type and content of polar groups (fpol). However, how non-covalent interactions, such as hydrogen bonds (HBs), between polar groups affect the Tg of polymers is not well understood.
Pan et al.16 found that Raman spectroscopy can be used to monitor in situ ring-opening metathesis polymerization (ROMP) of cyclic olefins, enabling real-time monitoring of the incorporation rate of individual monomers into polymers to obtain the desired chain compositions. Thus, the number of polar groups available for HB in the polymer can be precisely controlled. Norbornene (NB) with a bicyclic structure and modified with polar functional groups are important monomers for ROMP17,18 to obtain cyclic olefin polymers (COPs).19 The COPs possess the properties of excellent transparency and low birefringence, while at the same time these materials can be processed into thin films for display fabrication due to their high Tg.20,21 Experimentally, Dennis et al.22 synthesized homopolymers and copolymers of 5-ethylidene-2-norbornene (ENB) and 5-methanol-2-norbornene (NBCH2OH) using ROMP. They systematically varied the NBCH2OH content and found that the increasing trend of Tg with the NBCH2OH content was in good agreement with the prediction of the Kwei23–25 model. Their results indicate that there are strong HBs in the system. However, this work does not reveal how the HB interaction strength affects the variation trend of Tg, and why Tg gradually increases with the increase of the NBCH2OH content, but the increase rate gradually slows down. In order to explain this phenomenon at the molecular scale, we use molecular dynamics (MD) simulations to study the influence of the type and fpol of polar groups on Tg from the perspective of HBs.
Inspired by experimental calorimetry and dilatometry, linear temperature quenching is traditionally used in MD simulations to estimate Tg based on the temperature at which the temperature dependence of the system's energy or volume undergoes a change in the slope from liquid to glassy states.26–28 Although this method is simple and easy to implement, the linear cooling method keeps the cooling rate constant throughout the process, resulting in the system being simulated for times that exceed that required to reach equilibrium and therefore low computational efficiency at high temperatures. In contrast, the structure is highly unrelaxed at low temperatures since the simulation time is much less than the relaxation time of the system. Simmons and coworkers29 developed a so-called Predictive Stepwise Quenching (PreSQ) algorithm, which provides both easy access to supercooled states and high-throughput simulations of supercooled liquid dynamics. The method employs a designed quench process to obtain configurations within a range of temperatures, performs an isothermal annealing step on each of these configurations to obtain a well-defined equilibrium state, and then calculates relaxation times at different temperatures. This method can predictively find the appropriate equilibrium time at each temperature, so that each conformation is sufficiently relaxed, and avoids a simulation time exceeding the required equilibrium time. Because the annealing at different temperatures is completely parallel, this strategy also makes better use of modern computer hardware, greatly improving the simulation efficiency. For these reasons, this is the most advanced method for modern simulations to study supercooled state dynamics and the glass transition process.29
In this study, we first predict Tg of cyclic olefin copolymers (COC) composed of ethylene, NB and its derivatives by all-atom molecular dynamics simulation to verify the effectiveness of the PreSQ approach and the chosen force field. The simulation results are in good agreement with the reported experimental results. Subsequently, we investigate the effect of polar groups on the Tg of COPs from the ROMP reaction by focusing on systems with two different polar groups, namely NBCH2OH (labelled as COP1) and NBCOOH (COP2). We find that the Tg of COP2 is higher than that of COP1 at the same fpol. After systematically varying the content of the two polar groups in COP, it is found that the Tg increases with the increase of fpol, and tends to plateau for high fpol, which is consistent with the previous experimental results.22 The observed variation trend in Tg can be described reasonably by the Kwei23–25 model, but the classic Fox23 equation significantly underestimates Tg. The deviation between the Fox equation and predicted Tg can be attributed to the existence of strong hydrogen bonding (HB) interactions in the system. Therefore, we analyze the cohesive energy density (ΠCED) and HB interactions in the system. We find that ΠCED increases linearly with fpol, and the increase rate of the COP2 system is larger than that of the COP1 system, resulting in a higher Tg in the COP2 system. The increase rate of Tg gradually slows down with the increase of ΠCED, which is similar to the variation trend with fpol. In addition, the number of HBs (NHB) also increases linearly with fpol, as does ΠCED, and has a linear relationship with ΠCED. Consequently, the variation trend of Tg with NHB is similar to that with ΠCED. To further understand the intrinsic factors from HB interactions, we calculate the Gibbs free energy (ΔG) for HB breaking. Importantly, we find that the variation trend of ΔG with fpol also gradually slows down, indicating that the HB strength is gradually stable. Therefore, even if NHB increases linearly, their strength gradually stabilizes, resulting in a nonlinear increase of Tg with the increase of the polar groups.
Simulation methods
All-atomic force field
The all-atom optimized potentials for the liquid simulations (OPLS-AA)30,31 force field is considered to be the best comprehensive force field for describing liquid organic molecules due to its ability to reproduce the thermodynamic properties of the liquid phase. However, simulations for n-alkanes with the original OPLS-AA force-field show an overestimation of the intermolecular attractive force and therefore result in a higher heat of vaporization than the experiment.32 To solve the above problem, Siu et al.33 developed an optimized version called L-OPLS, where partial charges and the coefficients defining the potential function of several dihedral angles are modified. In ref. 34, Zangi found that simulations with the original OPLS-AA force-field will lead to unphysical spontaneous crystallization of 1-octanol, 1-nonanol, and 1-decanol at temperatures which are about 35–55 K higher than the experimental melting temperatures, while using a mixed version (adopts L-OPLS parameters for the hydrocarbon tail of the alcohols, while using the original OPLS-AA force-field parameter for the hydroxyl head group) can avoid the crystallization of long-chain alcohols above the experimental melting temperature. Inspired by their work, we also use a combination of the original OPLS-AA and L-OPLS force fields for norbornene with polar groups. We use the L-OPLS force field for the alkane part, and the original OPLS-AA for the polar groups, and this combinational usage of the original OPLS and L-OPLS is labeled as mix-OPLS in the following text.
Simulated systems and preparation of the initial configuration
As mentioned above, benchmark simulations are performed for COC polymers based on ethylene (Eth) and NB (labelled as COC1), Eth and NBCH2OH (labelled as COC2), and the results of Tg are compared with the available experimental values. Thereafter the effect of polar groups is studied in COP polymers of NB and NBCH2OH (labelled as COP1), and that of NB and NBCOOH (COP2). Note that COC1 with different chain lengths are studied. Details of these systems are listed in Table 1. For the initial configuration, firstly a self-avoiding walk chain configuration is constructed, which is subsequently duplicated several times according to the system size, and thereafter these chains are packed into the simulation box using the PACKMOL program35 at a low density of 0.02 g cm−3 and subjected to an energy minimization process. Subsequently, we equilibrate the system at 1000 K for a long time under the NPT ensemble to eliminate local stresses and to attain an equilibrated density, and the equilibration times teq of different systems are shown in the Table 1. When the polymer chains are appropriately equilibrated, bond vectors that are far apart along the chain are uncorrelated. This feature can be characterized by the Flory's characteristic ratio (Cn).36| |  | (1) |
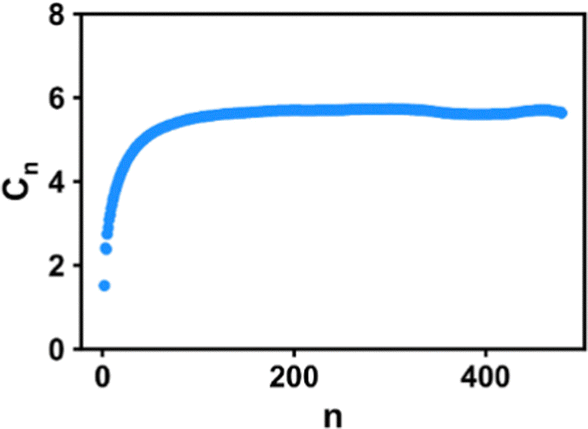
where n is the number of backbone bonds, and θij is the angle between the bond vector ri and rj. As an example, the results for the COC1 polymer with a chain length of 240 are shown in Fig. 1, where a fast convergence is shown, demonstrating the appropriate equilibration of the chain conformation.
Table 1 Chain length, number of chains, and the initial equilibration times at 1000 K for the simulated COC and COP polymers containing ethylene (Eth), NB and its derivatives
| Systems |
Chain length |
Number of chains |
t
eq (ns) |
| COC1 (Eth/NB) |
60 |
20 |
300 |
| 240 |
20 |
770 |
| 500 |
8 |
1000 |
| 1000 |
4 |
1690 |
| COC2 (Eth/NB/NBCH2OH) |
130 |
20 |
360 |
| COP1 (NB/NBCH2OH) |
40 |
20 |
100 |
| COP2 (NB/NBCOOH) |
40 |
20 |
100 |
 |
| | Fig. 1 Flory's characteristic ratio Cn saturates at C∞ for the Eth/NB system with a chain length of 240. The x-axis is the number of backbone bonds. | |
Relaxation time calculations
We calculate the incoherent intermediate scattering function Fs(q,t)37–45 to quantify the segmental relaxation time from the simulation. This function is computed as| |  | (2) |
where q is a wavevector, rj(t) is the position of atom j at time t, the summation is over all N atoms in the system, and the brackets denote an ensemble average. We chose the wavenumber q* corresponding to the first peak in the static structure factor. We define the relaxation time as the time at which Fs(q,t) relaxes to 0.2.40,46–48 Note that we use a consistent value of q* throughout our calculation for all the systems, unless otherwise noted.
Upon cooling, segmental dynamics of polymers often exhibit a super-Arrhenius behavior, especially at low temperatures approaching Tg, where relaxation times become highly sensitive to temperature. As discussed in ref. 49, the temperature dependence of segmental dynamics of polymer systems can be well captured by the Vogel–Fulcher–Tammann (VFT) equation,50–52
| | 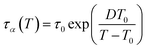 | (3) |
where
τα(
T) is the segmental relaxation time at temperature
T,
τ0 is an extrapolated high-temperature relaxation time,
T0 is the characteristic temperature at which
τα is extrapolated to diverge, and
D is a thermal breadth parameter. Due to this highly sensitive super-Arrhenius temperature dependence of segmental dynamics at low temperatures, supercooled liquid dynamics approaching
Tg are therefore not feasible in conventional linear quench simulations, since the method is simulated for times that exceed those needed to reach equilibrium at high temperatures while the simulation at low temperatures is far from enough for the equilibrium.
Simulation strategy and details of the PreSQ algorithm
We used the PreSQ algorithm29 to simulate the Tg of polymers, the basic idea of this approach is to progressively increase the simulation time at lower temperatures during a cooling process. In practice, this algorithm divides relaxation time τα(T) space into different windows (τ1 = 100–101 ps, τ2 = 101–101.5 ps, τ3 = 101.5–102 ps,…) located sequentially at successive temperature ranges. A priori choice of appropriate equilibration simulation times in these temperature ranges is the major challenge, which is solved by an iterative process in the PreSQ algorithm. Here are the detailed steps we adopt in our simulation:
(i) The first step is to take the equilibrated configuration from the NPT simulation at 1000 K as the initial configuration for the first relaxation time window. We preset the temperature range for the 1st window from 980 K to 900 K. The equilibration time (τe) is set to 10 times the segmental relaxation time at 1000 K, we calculate the segmental relaxation time (τα) at each temperature point in this range and compare it with the equilibration time, if τe < 10τα, we prolong the simulation at this point until τe > 10τα. The segmental relaxation time at the last temperature point is recorded as 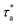 .
.
(ii) After the simulations at these temperature points are finished, the segmental relaxation time (τα) data in this window are fitted with the VFT equation. Thereafter we extend this VFT relationship to estimate the next temperature range where the last temperature point in this range satisfies the condition 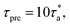 where τpre is the predicted segmental relaxation time at this last temperature point from the VFT equation.
where τpre is the predicted segmental relaxation time at this last temperature point from the VFT equation.
(iii) The above two steps are iteratively performed along the temperature axes until the segmental relaxation time reaches the order of 104 ps. We find that the predicted Tg value can have a reasonable convergence on this simulation timescale. A detailed discussion can be found in Fig. S1 (ESI†).
(iv) We fit all the obtained structural relaxation time data at different temperatures from the above procedure with the VFT equation and thereafter extrapolate the relaxation time data to a timescale of 100 s for the experimental glass transition, from which the Tg of the system is obtained.
Note that during the above process, the temperature is cooled down step-wise with a step size of ΔT = −20 K. At each temperature, a 3 ns NPT MD simulation is performed. When the system reaches an equilibrium density, the last frame of the conformation is annealed under NVT conditions for sampling. The Berendsen thermostat with a coupling time of 0.5 ps and Berendsen barostat with a coupling time of 5.0 ps are used to control the temperature and pressure, respectively. A cutoff of 1.0 nm is used for the calculation of the van der Waals interactions and the particle-mesh Ewald method is used to treat the electrostatic interactions. The energy and pressure calculations are corrected by the long-range dispersion. The leap-frog algorithm is used to integrate the Newton's equations of motion with a time step of 1 fs for all the simulations. All all-atom MD simulations are performed using the GROMACS 2018 molecular dynamics package.53
Results
Verification of the validity of the chosen force-field and the PreSQ algorithm
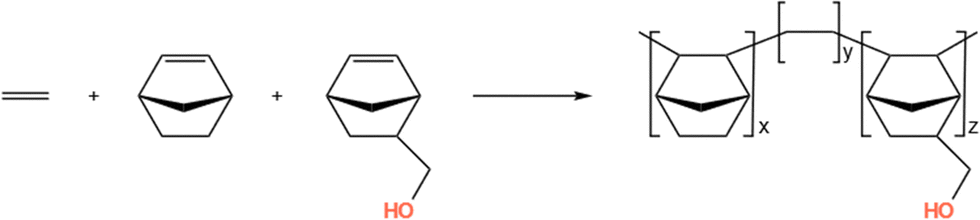
In this section, we simulate a copolymer system of ethylene and norbornene, namely COC1 in Table 1 with different chain lengths, and compare the simulated Tg value with the available experimental data.54 In particular, we simulate alternating copolymers with various molecular weights (Mn = 3660 g mol−1, 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 640 g mol−1, 30500 g mol−1, 61000 g mol−1), where the ratio of ethylene and norbornene in the block copolymer chain is 0.5/0.5 (Scheme 1). Considering the computational efficiency, the number of chains in the simulated systems with different chain lengths is different, and is set to 20, 20, 8, and 4, respectively.
640 g mol−1, 30500 g mol−1, 61000 g mol−1), where the ratio of ethylene and norbornene in the block copolymer chain is 0.5/0.5 (Scheme 1). Considering the computational efficiency, the number of chains in the simulated systems with different chain lengths is different, and is set to 20, 20, 8, and 4, respectively.
 |
| | Scheme 1 Ethylene and norbornene copolymerization. | |
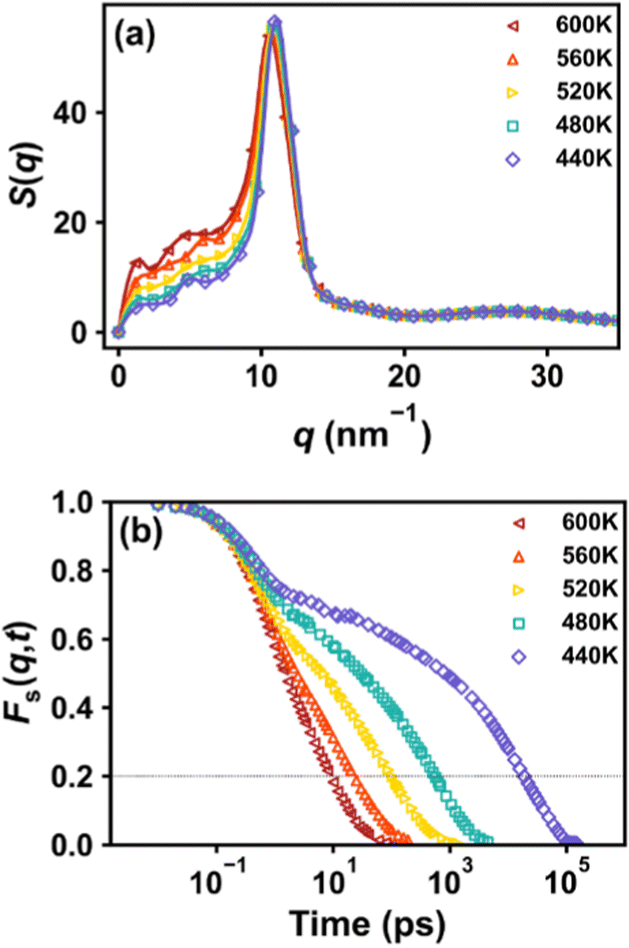
As described above, an incoherent intermediate scattering function Fs(q,t) is calculated to analyze the alpha relaxation behavior. ‘q’ is taken from the first peak of the static structure factor, S(q), as shown in Fig. 2a for an example with a molecular weight of Mn = 3660 g mol−1 at 480 K. Fig. 2b shows the results of Fs(q,t) at different temperatures. These scattering functions show characteristic temperature dependence and a two-step relaxation behavior at lower temperatures typical for glass formers. From these curves, the time at which Fs(q,t) relaxes to 0.2 is recorded as the segmental relaxation time, τα(T).
 |
| | Fig. 2 (a) Static structure factor S(q) at 480 K, and (b) incoherent intermediate scattering function Fs(q,t) at different temperatures, for the system with a molecular weight of Mn = 3660 g mol−1. The dotted line in (b) indicates the definition of the structural relaxation time. | |
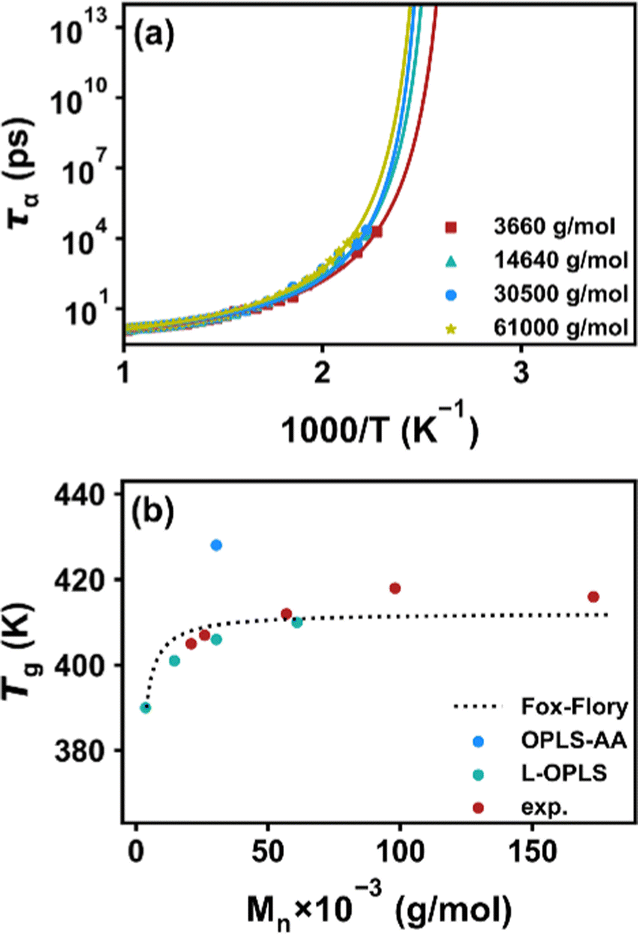
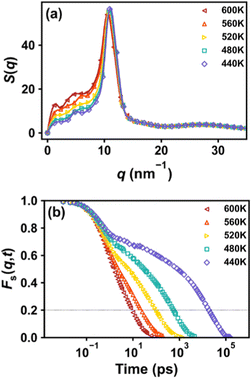
The results of τα(T) are plotted in Fig. 3a, and Tg is obtained by extrapolating the functional form of the VFT equation to the experimental 100-second time scale. The predicted Tg values for systems with different chain lengths are listed in Table 2. For comparison, we additionally use the original OPLS-AA force field to simulate the system with a molecular weight of 30500 g mol−1. All the data points and experimental values from ref. 54 are plotted in Fig. 3b. We see that the Tg of different chain lengths obtained by the L-OPLS force field are quite consistent with the experimental value, and follow the Fox–Flory relationship,55Tg = T∞g − K/Mn. However, the Tg value predicted from the original OPLS-AA force field is much higher due to the excessive intermolecular attraction. Therefore, the use of the L-OPLS force field and PreSQ algorithm is suitable for the purpose of this study.
Table 2 Glass transition temperature for alternating COC1 polymers of ethylene and NB with different molecular weights using original OPLSAA and L-OPLSAA force fields
|
M
n × 10−3 (g mol−1) |
3.66 |
14.64 |
30.50 |
61.00 |
| Sim. Tg (K) (L-OPLS) |
390 |
401 |
406 |
410 |
| Sim. Tg (K) (OPLS-AA) |
|
|
428 |
|
 |
| | Fig. 3 (a) Temperature dependence of segmental relaxation time for COC1 polymers with different molecular weights, lines represent the least-squares fitting with the VFT equation. (b) Comparison of the simulated data using L-OPLS and the original OPLS force-field, and that of the experimental data from ref. 54. The dotted line represents the Fox–Flory relationship between molecular weight and Tg of the copolymers. | |
Glass transition temperature of polar group functionalized COC
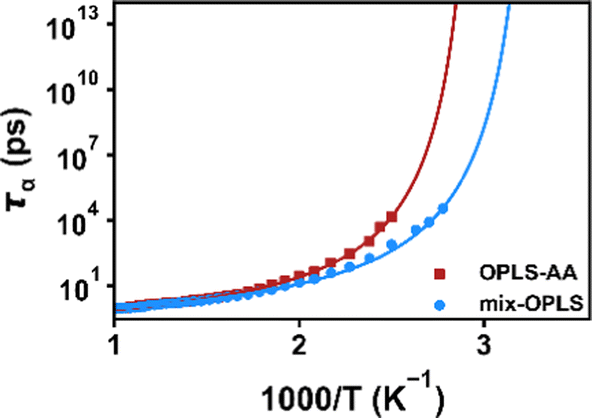
Before studying the effect of polar groups on the Tg for the COP system, a comparison with available experimental data is performed to verify the validity of the method and the force field for predicting the Tg of systems with polar groups. We simulate a block copolymer (COC2 listed in Table 1) composed of three monomers: Eth/NB/NBCH2OH (Scheme 2), where Eth is the ethylene unit. We setup the simulation according to the experimental system details in ref. 56, i.e., the ratio of Eth:NB:NBCH2OH is 80%:10%:10%, the ratio of NBCH2OH(endo) and NBCH2OH(exo) is 5:8, and the molecular weight is 5758 g mol−1. To be consistent with the experiment, the copolymer chain in our simulation has a degree of polymerization of N = 130, and each chain contains 13 NB, 13 NBCH2OH (5 endo conformation, 8 exo conformation), and 104 Eth units. To control the stereoisomerism of the NBCH2OH group, we set the improper dihedral angle in the topology file to control its stereoisomerism. For simplicity, Eth and NB (or NBCH2OH) units are evenly arranged in one chain as much as possible in our simulation. Twenty block copolymer chains are placed in a simulation box. For comparison, the original OPLS-AA force field and the mix-OPLS force field are used for comparison. Fig. 4 shows the segmental relaxation time data for simulations with both force fields and corresponding fitting with the VFT equation. The Tg obtained by using the original OPLS-AA force field is 351 K, and the Tg obtained from the mix-OPLS force field is 321 K, which are both higher than Tg = 316 K measured experimentally by DSC. Since our method for predicting the Tg is based on the kinetic properties of the system, it is comparable to the experimental DMTA results. The Tg measured by DMTA is usually higher than that of DSC. Therefore, the results obtained using mix-OPLS are in reasonable agreement with the experiment.
 |
| | Scheme 2 Ethylene, norbornene and 5-methanol-2-norbornene copolymer. | |
 |
| | Fig. 4 Temperature dependence of relaxation time for the Eth/NB/NBCH2OH copolymer using both the original OPLSAA and mix-OPLS force fields. Lines represent the least-squares fitting with the VFT equation. | |
Influence of the polar group content on the glass transition temperature of COP polymers
As discussed in the introduction, the Tg of the polymer will increase with the increase of fpol, but the increasing rate is gradually slowed down. The HBs caused by the introduction of polar groups must play an important role, but the underlying mechanism remains unknown. The influence of polar groups on the Tg is significant, and there is no doubt that both type and fpol are important influencing factors determining the value of Tg. Therefore, in this section, we will use the above-validated method and force field to study the effect of polar groups on the Tg, and to determine the variation trend of Tg with fpol from the perspective of HB interactions. The systems we simulated are ROMP22,57–59 COP of norbornene partially functionalized with polar groups of CH2OH or COOH, as demonstrated in Scheme 3. The content of 5-methanol-2-norbornene (NBCH2OH) or 5-carboxylic-2-norbornene (NBCOOH) is varied systematically. Each system contains 20 monomers, and there are 20 chains in total.
 |
| | Scheme 3 Ring Opening Metathesis Polymerization of norbornene and its derivative functionalized with CH2OH or COOH. | |
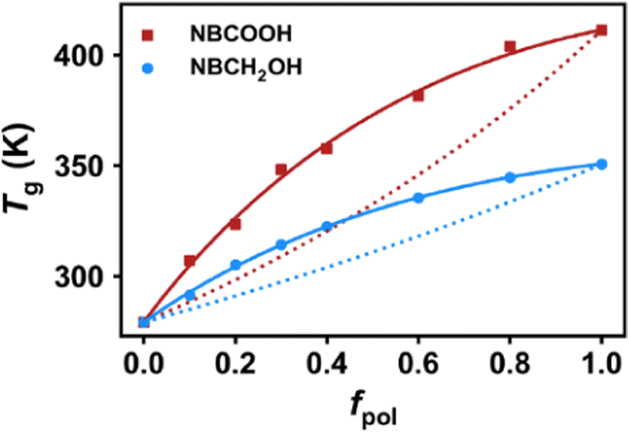
Fig. 5 shows the glass transition temperature as a function of fpol. From the simulated results, the Tg obtained for the COP2 system containing the NBCOOH group is higher than that of the COP1 containing NBCH2OH group, indicating that the COOH polar group has a greater influence on Tg than the CH2OH polar group. And the two prediction models, Fox equation eqn (4)23 and Kwei eqn (5)23–25 models are used to fit with the variation trend of Tg,
| |  | (4) |
| |  | (5) |
The classic Fox equation eqn (4), which assumes a simple mixing rule for the two components where
wi and
Tg,I are the weight fraction and
Tg of the constituent components. We find that fitting with the Fox equation fails for both systems and there are obvious underestimations of
Tg, suggesting that the system must involve strong specific interactions that are not taken into account by the Fox equation.
60 In contrast, the Kwei eqn (5) model can accurately predict the variation trend of the
Tg with
fpol over the whole range. The parameter
k is the unequal contribution to
Tg from the individual components. The parameter
p counts the contribution from HBs in the system.
23–25,61,62 The
p value obtained from the fitting of the Kwei equation is positive, indicating strong intermolecular HB interactions in the system. The
p value in the COP1 and COP2 systems are 16 and 28, respectively, indicating that the HB strength between COOH groups in the COP2 system is greater than that between CH
2OH groups in the COP1 system.
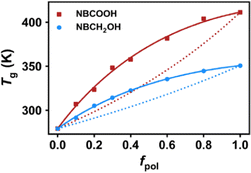 |
| | Fig. 5 Simulated glass transition temperature for COP1 and COP2 systems with different content of NBCH2OH or NBCOOH groups, respectively. The x-axis is the polar group content. Solid lines represent least-squares fitting with the Kwei model, while dotted lines are for the Fox equation. | |
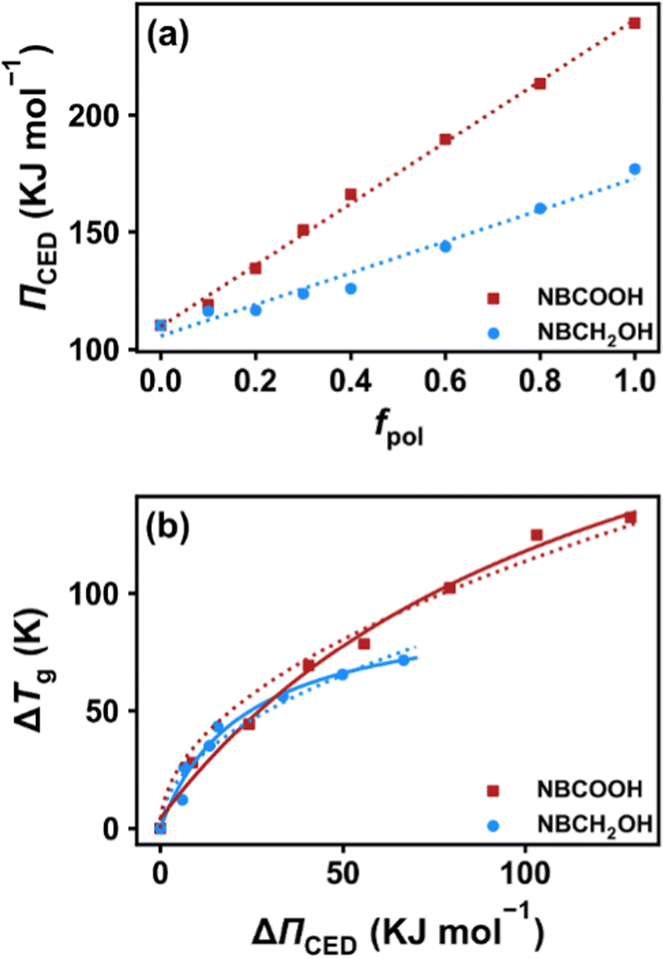
A direct consequence of raising the content of HB interaction agent, polar groups, in the system will be an increase in cohesive energy density (ΠCED), which is an important parameter determining the Tg of polymers.13 We estimate ΠCED in our simulation as,63,64
| |  | (6) |
where
Ψinter is the total intermolecular potential energy and
V is the volume of the simulation box. The results for both COP1 and COP2 systems with different
fpol at a fixed temperature of 480 K are plotted in
Fig. 6a. This shows that
ΠCED increases with increasing
fpol in a linear fashion for both COP1 and COP2 systems. As expected, the COP2 system has a much higher slope than the COP1 system due to stronger HB interactions in the former. In
Fig. 6b, we plot the increase of the glass transition temperature (Δ
Tg) as a function of that of the cohesive energy density (Δ
ΠCED) for both systems. We find that Δ
Tg increases in a sub-linear manner with Δ
ΠCED. Two empirical models are used to fit the data. The first model we used was proposed early by Tsutsui and Tanaka,
65 Δ
Tg =
K (Δ
ΠCED)
1/2, the variation trend is nicely captured by this model, as demonstrated by the dotted lines in the figure. The fitting parameter
K = 9 and 11 for the COP1 and COP2 systems, respectively. A larger value for the COP2 system indicates a stronger dependence. Another equation we used is the one proposed by Xu
et al.7,8 based on the generalized entropy theory, Δ
Tg = (
b +
cΔ
ΠCED)/(1 +
dΔ
ΠCED), we see that this model works much better, as indicated by solid lines in the figure. In their original model, an interaction strength parameter is used. Here we directly used Δ
ΠCED in the equation since both parameters have a linear relationship with each other according to their calculations.
7,8 Note that this relationship was originally used by Broadhurst
66,67 to describe the dependence of the melting temperature
Tm of the alkane polymers on their molecular weight. We speculate that the direct consequence of the variation in molecular weight will be the variation in
ΠCED, therefore our usage of this equation on the correlation between Δ
Tg ∼ Δ
ΠCED is reasonable.
 |
| | Fig. 6 (a) The cohesive energy density plotted as a function of polar group content in both COP1 and COP2 systems at 480 K. (b) The increase of glass transition temperature (ΔTg) plotted as a function of the increase of cohesive energy density (ΔΠCED) at 480 K. Dotted lines represent least-squares fitting with the model of Tsutsui and Tanaka,65 while solid lines represent the model of Xu et al.7,8 | |
Hydrogen bonding interaction in both COP systems
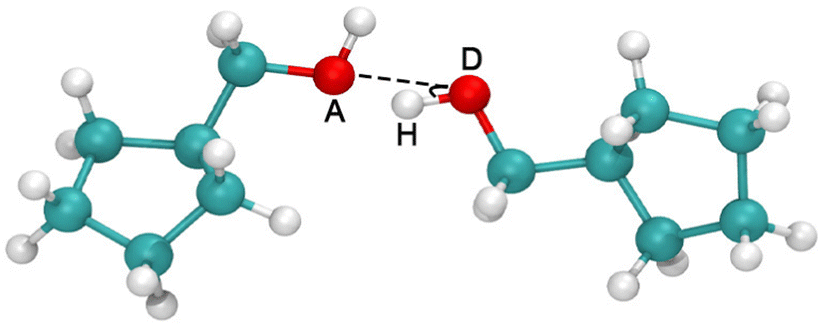
The results in Fig. 6b demonstrate that the variations in Tg are directly determined by the change in cohesive energy density, ΠCED. However, the ΠCED of polymers with polar groups is the sum of the contributions from various interactions, such as van der Waals interactions, Coulombic interactions, and HB interactions etc. For the current systems, HB interactions have a major contribution. In order to analyze the HBs interactions quantitatively, we define two criteria for the HB formation in our calculation, (i) the distance between the acceptor A and the donor D, r ≤ 0.35 nm, and (ii) the angle between the hydrogen, the donor D, and the acceptor A, α ≤ 30° (Fig. 7). Once both conditions are met, it is counted as a formed HB pair.
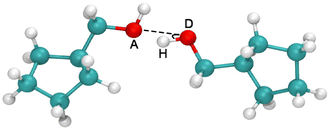 |
| | Fig. 7 Schematic illustration of the geometrical criterion for the HB formation. | |
By the above criteria, we calculate the number of HBs (NHB) formed between NBCH2OH groups in COP1 and between NBCOOH groups in the COP2 system respectively. The results are denoted as NHB as shown in Fig. 8a, where these data are calculated at a fixed temperature of 480 K. The NHB increases linearly with polar groups. At a fixed fpol, the NHB of NBCOOH is higher than that of NBCH2OH, resulting in higher ΠCED and hence higher Tg in the COP2 system than in the COP1 system. Since COOH contains two different kinds of oxygen atoms, HBs formed by these two different oxygen atoms are plotted in Fig. 8b. It can be seen that the HBs are dominated by those formed by carbonyl oxygen, while the HBs formed by hydroxyl oxygen are negligible. In Fig. 8c, we plot the correlation between ΠCED and NHB, where a linear relationship is found as expected. Not surprisingly, Tg increases with the NHB in a sub-linear manner (Fig. 8d) and this trend is consistent with the variation trend of Tg with ΠCED as shown in Fig. 6b. Again, we show that Xu's equation7,8 performs much better fitting with the data than that of Tsutsui and Tanaka.65 In addition, it shows that NHB increases with fpol, resulting in an increase in ΠCED and therefore an increase in Tg. The above results indicate that the hydrogen bonding interaction plays a dominant role in determining ΠCED and consequently the Tg of the system. By analyzing the variation trend of Tg with ΠCED and NHB, it shows that there must be a factor in the system that slows down the rate of Tg rise.
 |
| | Fig. 8 (a) The number of HBs formed in COP1 and COP2 systems with different polar group contents at 480 K. (b) HBs formed by carbonyl and hydroxyl oxygen atoms in the COP2 system. (c) The cohesive energy density as a function of the number of HBs for both systems at 480 K. (d) The increase of glass transition temperature (ΔTg) plotted as a function of the increase of the number of HBs (ΔNHB) at 480 K. Dotted lines represent least-squares fitting with the model of Tsutsui and Tanaka,65 while solid lines represent the model of Xu et al.7,8 | |
In order to further understand the underlying mechanism for the influence of HBs on Tg, we adopt the model proposed by Schroeder and Cooper,68,69 where the fraction of HBs (XHB) can be related to the Gibbs free energy (ΔG) for HB breaking. The equilibrium for the hydrogen bonding can be expressed as
| | (OH⋯OH)bonded ![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) 2(OH)free 2(OH)free | (7) |
The associated equilibrium constant
K is given by,
| |  | (8) |
The Gibbs free energy can be written as,
| |  | (9) |
where
R is the gas constant. According to the above equations, a higher value of Δ
G indicates a higher stability of HB pair.
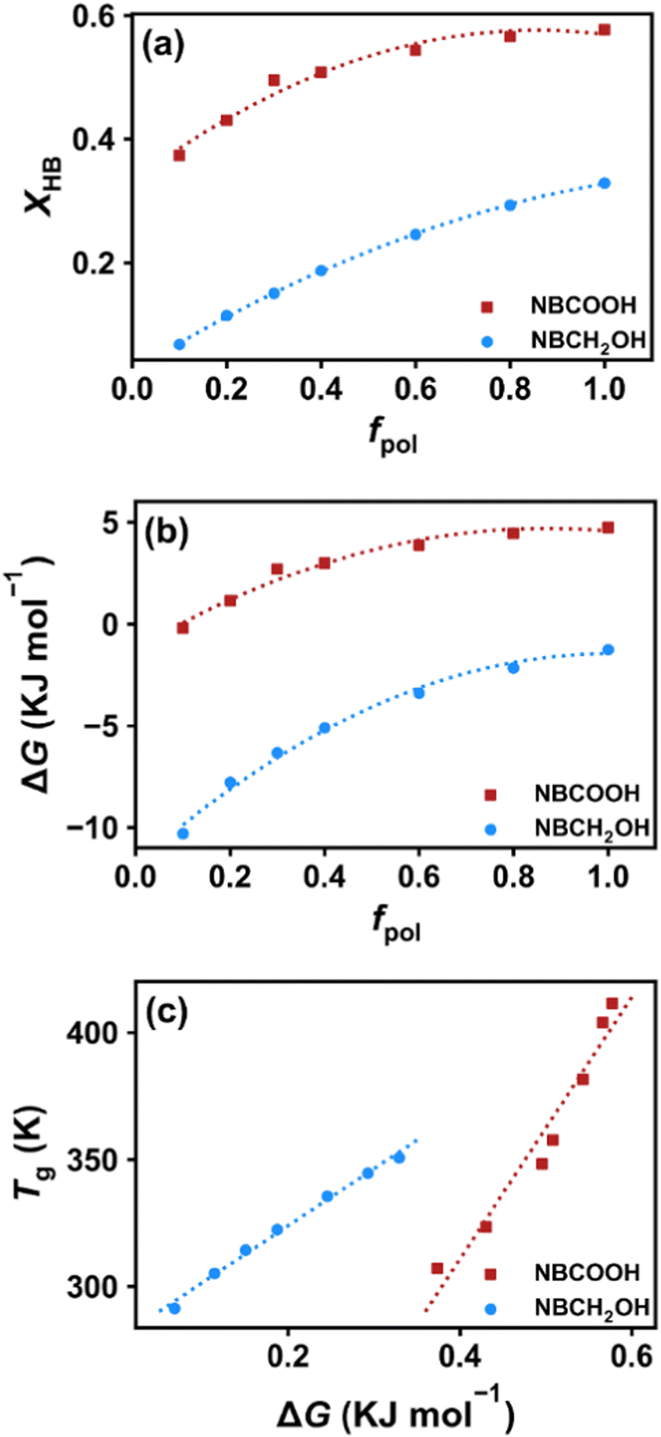
XHB is the hydrogen bond fraction, which is the number of hydrogen bonds divided by the number of polar groups, as shown in
Fig. 9a. By calculating
XHB, Δ
G at each temperature can be obtained using eqn (9). For the COP2 system containing NBCOOH, the HBs formed by the carbonyl oxygen dominates, therefore we calculate the Δ
G for HB breaking formed by the carbonyl oxygen. We also plot the Δ
G for systems with different
fpol at temperatures of 480 K (
Fig. 9b). The Δ
G of the COP2 system is higher than that of the COP1 system containing the NBCH
2OH group, which is direct evidence that the hydrogen bond strength in the COP2 system is stronger than that of the COP1 system. Importantly, Δ
G in both systems increase in a sub-linear manner with
fpol with the increase rate gradually slowing down, indicating that hydrogen bond stability increases in a sub-linear manner. Therefore, even though the
NHB and the
ΠCED both increase linearly with
fpol, the
Tg does increase nonlinearly, with a gradually decreasing rate of increase. Not surprisingly, we find that
Tg increases with Δ
G in a linear fashion for both COP1 and COP2 systems (
Fig. 9c). From the aspect of the slope of
Tg with Δ
G, the COP2 system is larger than the COP1 system. This is because
Tg is determined collectively by the number and strength of HBs. The number of HBs in the COP2 system is more than that in the COP1 system. Therefore, when the change in Δ
G is the same, the change of
Tg of the COP2 system is greater than that of the COP1 system.
 |
| | Fig. 9 (a) The fraction of HBs formed in COP1 and COP2 systems with different polar group contents at 480 K. (b) The Gibbs free energy of HB breaking plotted at different polar group contents for both COP1 and COP2 systems at temperatures of 480. (c) Glass transition temperature plotted as a function of the Gibbs free energy of HB breaking. | |
Conclusions
Atomistic molecular dynamics simulations are carried out for ROMP COP containing NB, a non-polar aliphatic monomer, NBCH2OH or NBCOOH, polar monomers capable of forming HBs. Our aim is to elucidate the effects of the types and contents of polar functional groups on the Tg of ROMP polymers. First, we successfully establish a Tg prediction model by using the PreSQ algorithm. Tg values of cyclic olefin copolymers composed of ethylene, NB and its derivatives are successfully predicted, and the obtained results are highly consistent with the experiments. Based on the above results, we investigate the effect of two kinds of polar groups (CH2OH and COOH) on the Tg of COP polymers. It is found that enhancement in Tg introduced by including the COOH group is much higher than that of the CH2OH group. However, the rate of increase in Tg decreases gradually with the content of both groups. In order to explain this phenomenon, we analyze the cohesive energy density ΠCED and hydrogen bond interactions in both systems. We find that both ΠCED and the number of NHB increase linearly with fpol. Not surprisingly, they have a linear relationship, since the increase in ΠCED is mainly caused by NHB. As expected, Tg does not increase linearly with ΠCED and NHB, and the growth rate gradually slows down. In order to further figure out the factor that slows down the growth rate of Tg, we calculate the free energy ΔG for hydrogen bond breaking at 480K. We find that the ΔG in the COP2 system containing the COOH group is larger than that in the COP1 system containing CH2OH, indicating that the COP2 system has stronger HBs and hence a higher Tg at a given fraction of polar groups. Importantly, we find that the increase rate of ΔG gradually slows down with the increase of fpol, and Tg increases with ΔG in a linear fashion. Therefore, even though NHB increases linearly with fpol, the increasing rate in its strength gradually slows down, which in turn leads to Tg increasing sub-linearly. Our results demonstrate that the HB interaction has a strong effect on Tg, while the HB strength plays a dominant role. Overall, our work successfully predicts the Tg of ROMP COP polymers and provides a new understanding of the effect of polar groups on polymer Tg.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work is supported by the National Natural Science Foundation of China (21873040, 22133002 and 21833008). H.-J. Q. and Z.-Y. L. also acknowledge the support from the Program for JLU Science and Technology Innovative Research Team. We are also grateful for the financial support from ExxonMobil Asia Pacific Research & Development Co., Ltd. The authors acknowledge the high performance computing center of Jilin University.
Notes and references
- S.-W. Kuo, W.-P. Liu and F.-C. Chang, Macromolecules, 2003, 36, 5165–5173 CrossRef CAS.
- A. R. Baljon, G. Mendoza, N. Balabaev and A. Lyulin, Polym. Sci., Ser. A, 2021, 63, 356–362 CrossRef CAS.
- S.-J. Xie, H.-J. Qian and Z.-Y. Lu, J. Chem. Phys., 2014, 140, 044901 CrossRef PubMed.
- H. A. Schneider, J. Appl. Polym. Sci., 2003, 88, 1590–1599 CrossRef CAS.
- S. Della Sciucca, G. Spagnoli, M. Penco, S. Battiato, F. Samperi and R. Mendichi, J. Polym. Sci., Part B: Polym. Phys., 2009, 47, 596–607 CrossRef CAS.
- P. Carbone, A. Rapallo, M. Ragazzi, I. Tritto and D. R. Ferro, Macromol. Theory Simul., 2006, 15, 457–468 CrossRef CAS.
- X. Xu, J. F. Douglas and W.-S. Xu, Macromolecules, 2021, 54, 6327–6341 CrossRef CAS.
- W.-S. Xu, J. F. Douglas and X. Xu, Macromolecules, 2020, 53, 9678–9697 CrossRef CAS.
- W.-S. Xu, J. F. Douglas and X. Xu, Macromolecules, 2020, 53, 4796–4809 CrossRef CAS.
- A. Askadskii, T. Matseevich and V. Markov, Polym. Sci., Ser. A, 2016, 58, 506–516 CrossRef CAS.
- J. Konieczkowska, H. Janeczek, J. G. Malecki and E. Schab-Balcerzak, Eur. Polym. J., 2018, 109, 489–498 CrossRef CAS.
- S. Pawlus, A. Grzybowski, S. Kołodziej, M. Wikarek, M. Dzida, P. Góralski, S. Bair and M. Paluch, Sci. Rep., 2020, 10, 1–8 CrossRef.
- Z. Yang, X. Xu and W.-S. Xu, Macromolecules, 2021, 54, 9587–9601 CrossRef CAS.
- F. Krohn, C. Neuber, E. A. Rössler and H.-W. Schmidt, J. Phys. Chem. B, 2019, 123, 10286–10293 CrossRef CAS.
- K. Li, L. Zhou, S. Wu, Q. Yu and L. Yang, High Perform. Polym., 2020, 32, 316–323 CrossRef CAS.
- Y. Pan, T. Li, Y. Zhou and L. Li, Polymer, 2022, 242, 124613 CrossRef CAS.
- M. Abubekerov, S. M. Shepard and P. L. Diaconescu, Eur. J. Inorg. Chem., 2016, 2016, 2634–2640 CrossRef CAS.
- B. Liu, Y. Li, B. G. Shin, D. Y. Yoon, I. Kim, L. Zhang and W. Yan, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 3391–3399 CrossRef CAS.
- J. Mol, J. Mol. Catal. A: Chem., 2004, 213, 39–45 CrossRef CAS.
- S. H. Moon, K. L. Park, J. W. Baek, H. J. Lee, J. H. Choi, Y. Lee and B. Y. Lee, J. Polym. Sci., 2020, 58, 1253–1261 CrossRef CAS.
- M. Rosenberger, S. Kefer, M. Girschikofsky, G.-L. Roth, S. Hessler, S. Belle, B. Schmauss and R. Hellmann, Opt. Lett., 2018, 43, 3321–3324 CrossRef CAS PubMed.
- J. M. Dennis, T. R. Long, A. Krishnamurthy, N. T. Tran, B. A. Patterson, C. E. Busch, K. A. Masser, J. L. Lenhart and D. B. Knorr Jr, ACS Appl. Polym. Mater., 2020, 2, 2414–2425 CrossRef CAS.
- L. Weng, R. Vijayaraghavan, D. R. MacFarlane and G. D. Elliott, Cryobiology, 2014, 68, 155–158 CrossRef CAS PubMed.
- A. A. Lin, T. Kwei and A. Reiser, Macromolecules, 1989, 22, 4112–4119 CrossRef CAS.
- X. Lu and R. Weiss, Macromolecules, 1992, 25, 3242–3246 CrossRef CAS.
- A. Cavagna, Phys. Rep.-Rev. Sec. Phys. Lett., 2009, 476, 51–124 CAS.
- P. G. Debenedetti and F. H. Stillinger, Nature, 2001, 410, 259–267 CrossRef CAS.
- M. Ediger and P. Harrowell, J. Chem. Phys., 2012, 137, 080901 CrossRef CAS PubMed.
- J.-H. Hung, T. K. Patra, V. Meenakshisundaram, J. H. Mangalara and D. S. Simmons, Soft Matter, 2019, 15, 1223–1242 RSC.
- M. L. Price, D. Ostrovsky and W. L. Jorgensen, J. Comput. Chem., 2001, 22, 1340–1352 CrossRef CAS.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, J. Am. Ceram. Soc., 1996, 118, 11225–11236 CAS.
- L. L. Thomas, T. J. Christakis and W. L. Jorgensen, J. Phys. Chem. B, 2006, 110, 21198–21204 CrossRef CAS PubMed.
- S. W. Siu, K. Pluhackova and R. A. Böckmann, J. Chem. Theory Comput., 2012, 8, 1459–1470 CrossRef CAS.
- R. Zangi, ACS Omega, 2018, 3, 18089–18099 CrossRef CAS PubMed.
- L. Martínez, R. Andrade, E. G. Birgin and J. M. Martínez, J. Comput. Chem., 2009, 30, 2157–2164 CrossRef.
- M. Vacatello and P. Flory, Macromolecules, 1986, 19, 405–415 CrossRef CAS.
- F. W. Starr, F. Sciortino and H. E. Stanley, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 1999, 60, 6757 CrossRef CAS PubMed.
- B. A. P. Betancourt, P. Z. Hanakata, F. W. Starr and J. F. Douglas, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 2966–2971 CrossRef.
- Y. Cheng, J. Yang, J.-H. Hung, T. K. Patra and D. S. Simmons, Macromolecules, 2018, 51, 6630–6644 CrossRef CAS.
- R. J. Lang and D. S. Simmons, Macromolecules, 2013, 46, 9818–9825 CrossRef CAS.
- J. Baschnagel and F. Varnik, J. Phys.: Condes. Matter, 2005, 17, R851 CrossRef CAS.
- F. W. Starr, J. F. Douglas and S. Sastry, J. Chem. Phys., 2013, 138, 12A541 CrossRef PubMed.
- D. S. Simmons and J. F. Douglas, Soft Matter, 2011, 7, 11010–11020 RSC.
- D. D. Hsu, W. Xia, J. Song and S. Keten, ACS Macro Lett., 2016, 5, 481–486 CrossRef CAS PubMed.
- A. Shavit and R. A. Riggleman, Macromolecules, 2013, 46, 5044–5052 CrossRef CAS.
- P. Z. Hanakata, J. F. Douglas and F. W. Starr, J. Chem. Phys., 2012, 137, 244901 CrossRef PubMed.
- J.-H. Hung, J. H. Mangalara and D. S. Simmons, Macromolecules, 2018, 51, 2887–2898 CrossRef CAS.
- J. H. Mangalara and D. S. Simmons, ACS Macro Lett., 2015, 4, 1134–1138 CrossRef CAS.
- B. Schmidtke, M. Hofmann, A. Lichtinger and E. Rössler, Macromolecules, 2015, 48, 3005–3013 CrossRef CAS.
- G. S. Fulcher, J. Am. Ceram. Soc., 1925, 8, 339–355 CrossRef CAS.
- G. Tammann and W. Hesse, Z. Anorg. Allg. Chem., 1926, 156, 245–257 CrossRef CAS.
- C. Rodríguez-Tinoco, J. Ràfols-Ribé, M. González-Silveira and J. Rodríguez-Viejo, Sci. Rep., 2016, 6, 1–8 CrossRef.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1, 19–25 CrossRef.
- N. Ekizoglou, K. Thorshaug, M. L. Cerrada, R. Benavente, E. Pérez and J. M. Pereña, J. Appl. Polym. Sci., 2003, 89, 3358–3363 CrossRef CAS.
- T. G. Fox Jr and P. J. Flory, J. Appl. Phys., 1950, 21, 581–591 CrossRef.
- R. A. Wendt and G. Fink, Macromol. Chem. Phys., 2000, 201, 1365–1373 CrossRef CAS.
- K. Szwaczko, I. Czeluśniak and K. Grela, J. Organomet. Chem., 2017, 847, 146–153 CrossRef CAS.
- K. Nomura, S. Takahashi and Y. Imanishi, Polymer, 2000, 41, 4345–4350 CrossRef CAS.
- S. I. Subnaik and C. E. Hobbs, Polym. Chem., 2019, 10, 4524–4528 RSC.
- J. Prinos and C. Panayiotou, Polymer, 1995, 36, 1223–1227 CrossRef CAS.
- S. W. Kuo, H. Xu, C. F. Huang and F. C. Chang, J. Polym. Sci., Part. B: Polym. Phys., 2002, 40, 2313–2323 CrossRef CAS.
- S. W. Kuo and F. C. Chang, Macromolecules, 2001, 34, 5224–5228 CrossRef CAS.
- W.-S. Xu, J. F. Douglas and K. F. Freed, Macromolecules, 2016, 49, 8341–8354 CrossRef CAS.
- J. K. Maranas, M. Mondello, G. S. Grest, S. K. Kumar, P. G. Debenedetti and W. W. Graessley, Macromolecules, 1998, 31, 6991–6997 CrossRef CAS.
- T. Tsutsui and T. Tanaka, Polymer, 1977, 18, 817–821 CrossRef CAS.
- M. G. Broadhurst, J. Chem. Phys., 1962, 36, 2578–2582 CrossRef CAS.
- M. G. Broadhurst, J. Res. Natl. Bur. Stand., Sect. A, 1966, 70, 481 CrossRef CAS.
- L. Schroeder and S. L. Cooper, J. Appl. Phys., 1976, 47, 4310–4317 CrossRef CAS.
- H. A. Karimi-Varzaneh, P. Carbone and F. Müller-Plathe, Macromolecules, 2008, 41, 7211–7218 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sm01229d |
| ‡ Current address: South China Advanced Institute for Soft Matter Science and Technology, School of Emergent Soft Matter, South China University of Technology, Guangzhou 510640, China. |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 and
Hu-Jun
Qian
and
Hu-Jun
Qian
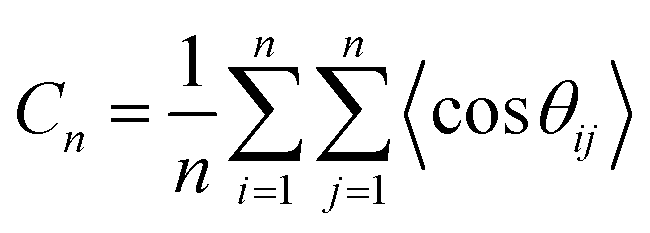
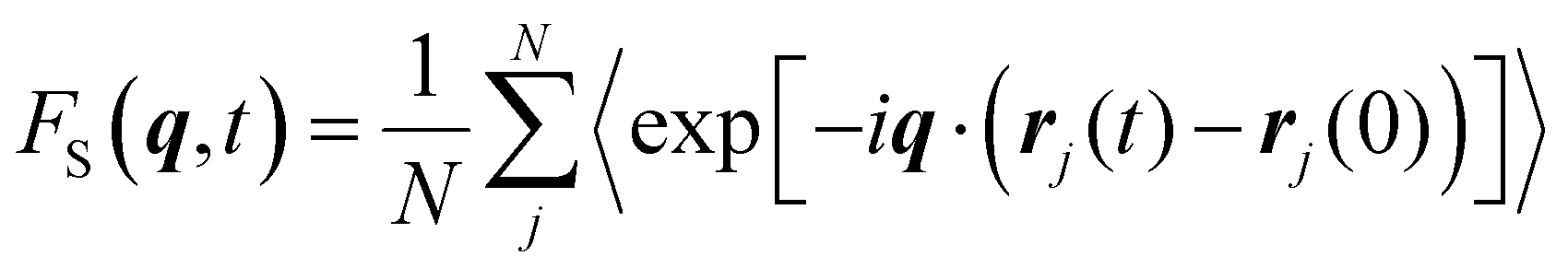
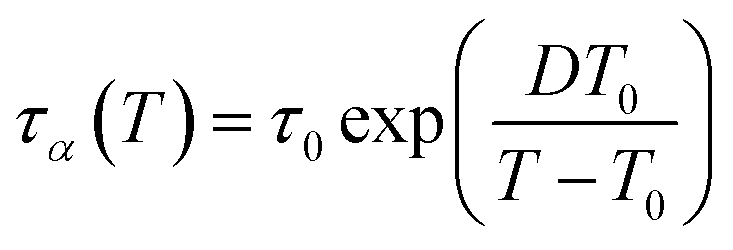
 .
.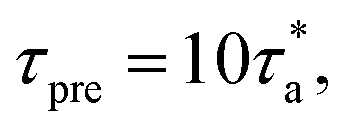 where τpre is the predicted segmental relaxation time at this last temperature point from the VFT equation.
where τpre is the predicted segmental relaxation time at this last temperature point from the VFT equation.