Organic photovoltaic performance and structural relationship of non-fullerene small molecule acceptors based on a tetraarylphenazine core and perylene diimide†
Received
7th September 2022
, Accepted 25th October 2022
First published on 2nd November 2022
Abstract
Here we report a pair of perylene diimide (PDI) non-fullerene small molecular acceptors (SMAs) based on a phenazine core with either benzene or thiophene linkers. Our main design rationale is to introduce a relatively parallel orientation for PDI units by connecting two PDI units on the same side of the phenazine core. While we find that the phenazine core can introduce a parallel intramolecular orientation, it is also found that the choice of linker units also has a major influence on the molecular geometry. As a result, the two SMAs, TPPhen-4PDI and TTPhen-4PDI, exhibit very different performances. The SMA with benzene linkers, TPPhen-4PDI, can achieve a power conversion efficiency of up to 8.3%. The performance difference was also supported by a significant difference in charge mobility and GIWAXS analysis. Our work provides some important understanding on the linker influence of structural properties, and guidelines to the design of high performance PDI-based SMAs.
Introduction
Fullerene acceptors are limited by their poor visible light absorption, high production cost, photo-instability causing a metastable morphology and large voltage losses in organic photovoltaics (OPVs).1,2 Recently, non-fullerene small molecular acceptors (SMAs) have been proved as an alternative to fullerene derivatives and SMA based OPVs can achieve a PCE of up to 19% in a single junction device,3–12 with a stronger light absorption, tuneable absorption range and energy level, and more stable morphology. Recently, Y6-based acceptors have drawn most research interest since the excellent PCE records above are mainly achieved by them.13–15 However, the complicated mass fused-ring core of Y6 derivates also greatly increased their synthetic costs. Perylene diimide (PDI) is one of the most traditional and popular building blocks for SMAs.16–21 The high electron mobility of PDI-based molecules (in the order of 10−1 to 10−3 cm2 V−1 s−1) and the two strong electron withdrawing imide groups make it a good electron accepting material. Furthermore, PDI is a widely used pigment in industry with low production costs. Therefore, PDI-based acceptors still remain attractive to SMA developers.
However, due to the anisotropic nature, PDI tends to aggregate along the planar perylene backbone that leads to a large phase separation between donor and acceptor materials in a bulk-heterojunction (BHJ) structure. This results in excessive domain sizes (>30 nm), which in turn limits OPV performance. Various strategies were developed to reduce excessive aggregation of PDI molecules.22–29 One successful strategy is to connect PDI units to a highly congested core such that the resulting molecule has a highly twisted three-dimensional molecular geometry, which reduces its intermolecular stacking tendency.23,29 However, using an over congested core may over reduce the intermolecular PDI unit π-stacking, thus impairing the charge transportation channel through π-stacking.30,31 Recently, the reduction of intramolecular twisting to enhance intermolecular π-stacking or the improvement of intramolecular PDI π-stacking was reported to enable significant improvements in the OPV performance.26,32
In this paper, we report the synthesis, device performance and characterisation of a pair of PDI-tetramers with a phenazine (Phen) core linked by either benzene (TPPhen) or thiophene (TTPhen) linkers. We find that these PDI-tetramers exhibit very different packing, largely affecting the device PCE when blended with a donor polymer, P3TEA (Scheme 1). This result shows the importance of linker units on molecular geometry and hence the result of a significant difference of performance.
 |
| | Scheme 1 Synthetic route of 1,4,6,9-tetrabromophenazine, TPPhen-4PDI and TTPhen-4PDI, and structure of P3TEA. | |
Results and discussion
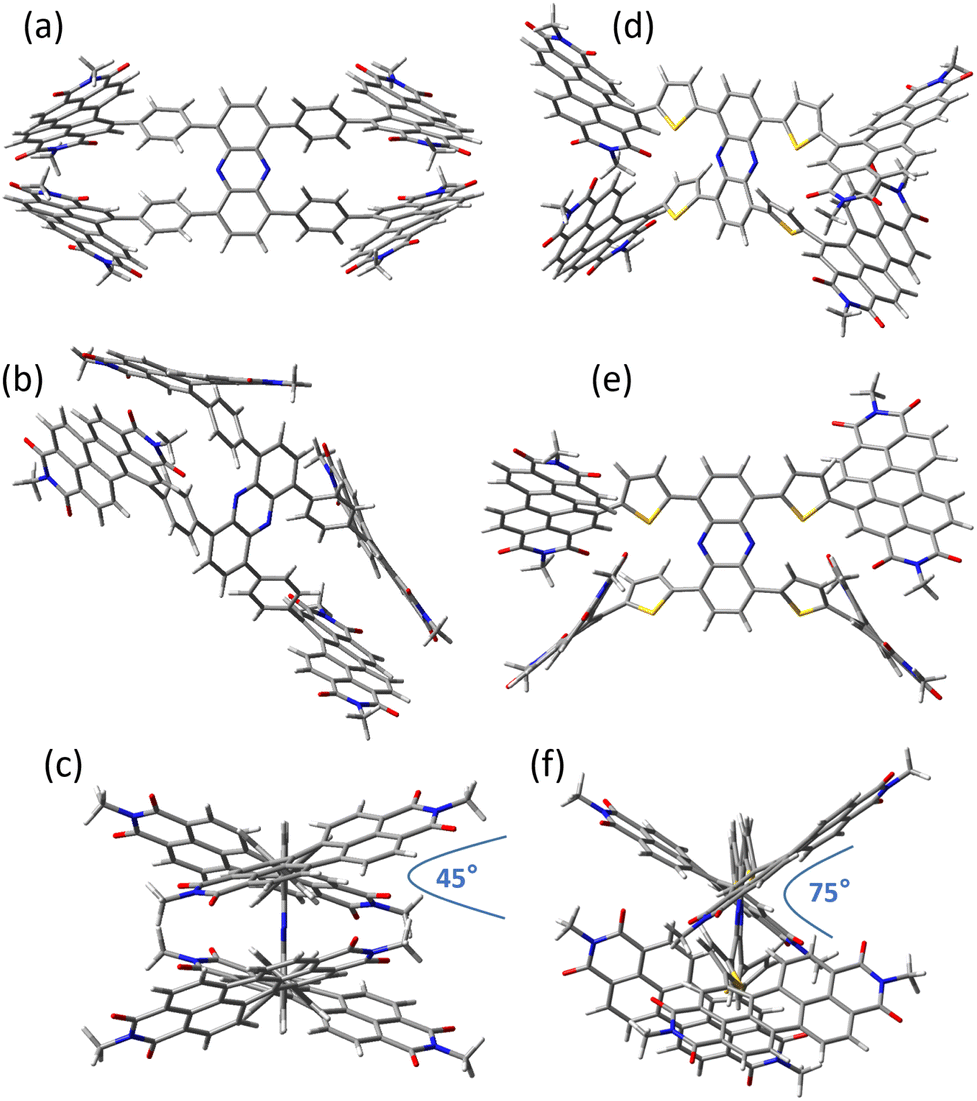
To make PDI units on the same side of the phenazine core to overlap each other to form a “double-decker” geometry,28 the linker units are attached on the long edge of the phenazine core. Furthermore, we expect that there will be a difference in the molecular geometry as phenazine and PDI units are in the para-position of the benzene linker in TPPhen-4PDI, while the bond angle of phenazine and PDI units on the thiophene linker is about 30° in TTPhen-4PDI. Density-functional theory (DFT) calculation shows that PDI units in TPPhen-4PDI exhibit a “double-decker” geometry, while PDI units in TTPhen-4PDI exhibit a more twisted geometry. The less twisted double-decker geometry of TPPhen-4PDI enables better molecular packing and leads to a better electron transport ability and higher electron mobility in blend films.
The attempt of synthesizing 1,4,6,9-tetrabromophenazine by following the reported literature33 was unsuccessful. Therefore, the bromination of phenazine was conducted in elemental bromine at room temperature. The bromine atoms on phenazine are reactive towards Suzuki–Miyaura and Stille coupling reactions. The synthetic route of TPPhen-4PDI and TTPhen-4PDI is illustrated in Scheme 1 (detailed synthetic route is in the ESI†). The structure of both target SMAs and all intermediates has been verified by NMR or mass spectroscopy (Fig. S1–S14†).
The film UV-vis absorption spectra, and the lowest unoccupied molecular orbital (LUMO) and the highest occupied molecular orbital (HOMO) energy levels of TPPhen-4PDI and TTPhen-4PDI are shown in Fig. 1, S15† and Table 1, respectively. The optical bandgap of TPPhen-4PDI is 2.10 eV while TTPhen-4PDI has a smaller optical band gap of 1.75 eV. Since the LUMO of TPPhen-4PDI is estimated to be −3.64 eV by cyclic voltammetry (CV), which is similar to the LUMO of TTPhen-4PDI (−3.68 eV), the significant difference in the optical bandgap should be due to the higher HOMO level of TTPhen-4PDI. The higher HOMO level (estimated by CV and the optical bandgap) of TTPhen-4PDI (−5.43 eV) than TPPhen-4PDI (−5.74 eV) can be explained by the HOMO distribution simulated by DFT calculation at the B3LYP/6-31G(d) level using Gaussian 09.
 |
| | Fig. 1 (a) UV-vis absorption spectra of pure and blend films (weight ratio of donor to acceptor is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5). (b) CV of TPPhen-4PDI and TTPhen-4PDI films. :1.5). (b) CV of TPPhen-4PDI and TTPhen-4PDI films. | |
Table 1 OPV performance, energy level and optical bandgap of SMAs
|
|
V
oc
(V) |
J
sc
(mA cm−2) |
FFa (%) |
PCEa (%) |
LUMOb,c (eV) |
Optical bandgapb (eV) |
HOMOb,d (eV) |
|
Average of 10 devices.
Pure SMA.
Characterized by CV.
Estimated by CV and the optical bandgap. The highest PCE values are shown in parentheses.
|
|
P3TEA:TPPhen-4PDI
|
1.057 ± 0.001 |
12.6 ± 0.2 |
60 ± 3 |
8.3 ± 0.07(8.4) |
−3.64 |
2.10 |
−5.74 |
|
P3TEA:TTPhen-4PDI
|
0.979 ± 0.002 |
10.7 ± 0.2 |
48 ± 2 |
5.1 ± 0.07(5.2) |
−3.68 |
1.75 |
−5.43 |
The optimised geometry and HOMO/LUMO distribution of both SMAs are shown in Fig. 2 and S16–S18,† respectively, with all alkyl chains replaced by a methyl group for simplicity. For both the SMAs, the LUMO is mainly localised on the PDI units and therefore both the SMAs have a similar LUMO level. However, in TTPhen-4PDI, the HOMO is mainly located on the more electron rich TTPhen core so the electron-deficit PDI units have less influence to lower the energy level, while in TPPhen-4PDI, the HOMO is delocalised on the whole molecule so the PDI units can further lower the HOMO energy level and therefore TTPhen-4PDI has a smaller bandgap than TPPhen-4PDI.
 |
| | Fig. 2 View of the DFT calculation optimised geometry of TPPhen-4PDI (a–c) and TTPhen-4PDI (d–f) in different directions. | |
For the molecular geometry, both SMAs show a two-blade propeller geometry. However, the PDI units on the long edge of phenazine in TPPhen-4PDI exhibit a face-to-face orientation (double-decker geometry), while the PDI units on the long edge of phenazine in TTPhen-4PDI exhibit an “open-glove” geometry. The dihedral angle between the two PDI units across the phenazine core influences the stacking tendency of SMA. The smaller the dihedral angle is, the stronger the tendency of SMA stacking is. The dihedral angle of two PDI planes in TPPhen-4PDI is around 45° (Fig. 2c), while that of the “open-glove” PDI plane in TTPhen-4PDI is around 75° (Fig. 2f). The smaller dihedral angle of “PDI-blade” in TPPhen-4PDI means that the PDI units in TPPhen-4PDI are more open for intermolecular π-interaction and thus TPPhen-4PDI should have a stronger tendency of molecular stacking than TTPhen-4PDI and should have better charge transportation properties.
Device performance and external quantum efficiency (EQE)
To investigate the influence of the molecular geometry on the performance of these SMAs in OPVs, the devices were fabricated with the state-of-the-art low band gap polymer P3TEA (Mw: 100.2 kDa) as the donor since it has a suitable LUMO level off-set with PDI that can facilitate charge separation at the interface, while maintaining a relatively small voltage loss. The performance and EQE of devices are summarized in Fig. 3 and Table 1, respectively. The slightly lower open-circuit voltage (Voc) of P3TEA:TTPhen-4PDI devices is expected as the LUMO energy level of TTPhen-4PDI characterised by CV and estimated by DFT calculation is slightly lower than that of TPPhen-4PDI. However, the difference in device performance originates from the short-circuit current density (Jsc) and fill factor (FF). In P3TEA:TPPhen-4PDI, the Jsc and FF can reach 12.6 mA cm−2 and 62%, while those of P3TEA:TTPhen-4PDI can only reach 11.0 mA cm−2 and 50%. The lower EQE of P3TEA:TTPhen-4PDI results in a lower Jsc. Notably, P3TEA:TTPhen-4PDI showed a significantly weakened EQE in the acceptor absorption region compared with the blend film. This phenomenon is probably due to the low electron mobility of TTPhen-4PDI and its imbalanced charge mobility with P3TEA, which will be discussed in the immediate next section.34,35
 |
| | Fig. 3 (a) J–V curve and (b) EQE spectra of P3TEA:TPPhen-4PDI and P3TEA:TTPhen-4PDI OPV devices. | |
Mobility, recombination, and light intensity
We examine the origin of the lower EQE and Jsc of TTPhen-4PDI by studying the charge mobility and recombination mechanism. The charge mobility of pure SMA and blend films was obtained by the space-charge-limited current (SCLC) method (Table 2). The similar hole mobility (μh) of both blend films (2.3 × 10−4 cm2 V−1 s−1 and 2.0 × 10−4 cm2 V−1 s−1 in P3TEA:TPPhen-4PDI and P3TEA:TTPhen-4PDI, respectively) suggests a similar polymer packing in both films, but the significant difference in the electron mobility (μe) of the two SMAs indicates a large difference of molecular packing of the SMA in the film. For TPPhen-4PDI, the pure and P3TEA-blend film electron mobility was 1.2 × 10−4 and 2.1 × 10−4 cm2 V−1 s−1 respectively. In contrast, the electron mobility of TTPhen-4PDI in pure and P3TEA-blend films was only 3.1 × 10−5 and 1.5 × 10−5 cm2 V−1 s−1, respectively, which is an order of magnitude lower than that of TPPhen-4PDI. This results in an imbalanced charge mobility in P3TEA:TTPhen-4PDI (μh/μe = 13.3) when compared with P3TEA:TPPhen-4PDI (μh/μe = 1.10). The imbalance charge mobility and low electron mobility of P3TEA:TTPhen-4PDI increase the probability of bimolecular recombination that can lead to a lower EQE, Jsc and FF.36–38 The relationship between Jsc and light intensity is log(Jsc) = logk + SlogP, where k and P are a constant and light intensity, respectively. For ideal free charge carrier collection at the electrodes without recombination, S is equal to 1. However, an S value of <1 indicates that there is some extent of bimolecular recombination prior to the charge carrier collection at the electrodes. From light intensity dependent Jsc experiments (Fig. S19†), P3TEA:TTPhen-4PDI has larger probability of bimolecular recombination (S = 0.95) when compared with P3TEA:TPPhen-4PDI (S = 0.98). This result indicates that the free charge carriers in P3TEA:TTPhen-4PDI have less chance to be collected at the electrodes, thus resulting inferior EQE and Jsc values.
Table 2 Charge mobility of pure SMAs and P3TEA-blend films
|
|
TPPhen-4PDI
|
P3TEA:TPPhen-4PDI
|
TTPhen-4PDI
|
P3TEA:TTPhen-4PDI
|
| Hole mobility (μh) (cm2 V−1 s−1) |
— |
2.3 × 10−4 |
— |
2.0 × 10−4 |
| Electron mobility (μe) (cm2 V−1 s−1) |
1.2 × 10−4 |
2.1 × 10−4 |
3.1 × 10−5 |
1.5 × 10−5 |
|
μ
h/μe |
— |
1.10 |
— |
13.3 |
RSoXS and GIWAXS
To probe the influence of molecular packing of both P3TEA and SMA to the electronic properties in films, GIWAXS39 and Resonant Soft X-ray Scattering (RSoXS)40,41 experiments were performed (Fig. 4 and Table 3). In either blend film, P3TEA has a face-on orientation relative to the substrate, which has proved that such orientation is beneficial for charge transportation.42–44 Both the d-spacing and coherence length of P3TEA are similar in either blend film (3.55 and 46.71 Å, respectively, in P3TEA:TPPhen-4PDI, and 3.61 and 38.14 Å, respectively, in P3TEA:TTPhen-4PDI), which concur with the similar values of hole mobility in both blend films. In contrast, although the d-spacing of both SMAs in the P3TEA:SMA blend film is similar (3.55 Å in TPPhen-4PDI and 3.61 Å in TTPhen-4PDI), the coherence length of TPPhen-4PDI (17.0 Å) is longer than that of TTPhen-4PDI (11.32 Å). This indicate that TPPhen-4PDI has a stronger π-stacking ability than TTPhen-4PDI. Also from RSoXS, P3TEA:TPPhen-4PDI exhibits a slightly larger domain size (13.8 nm) than P3TEA:TTPhen-4PDI (10.5 nm). Furthermore, the 32% more purer domain of P3TEA:TPPhen-4PDI than P3TEA:TTPhen-4PDI indicates that the “double-decker” geometry of TPPhen-4PDI can facilitate the phase separation of polymer:SMA solution to form a purer domain than the “open-glove” geometry of TTPhen-4PDI, while maintaining a reasonably small domain size for maximising the charge mobility and device performance.
 |
| | Fig. 4 (a) RSoXS profiles, (b) 1D GIWAXS profiles of in-plane and out-of-plane directions of P3TEA:TPPhen-4PDI and P3TEA:TTPhen-4PDI, and 2D GIWAXS pattern of (c) P3TEA:TPPhen-4PDI and (d) P3TEA:TTPhen-4PDI. | |
Table 3 GIWAXS analysis of P3TEA:TPPhen-4PDI and P3TEA:TTPhen-4PDI films
| Blend |
Assignment |
q (Å−1) |
d-Spacing (Å) |
Coherence length (Å) |
|
P3TEA:TPPhen-4PDI
|
P3TEA (010) |
1.77 |
3.55 |
46.71 |
|
TTPhen-4PDI (010) |
1.50 |
4.20 |
17.0 |
|
P3TEA:TTPhen-4PDI
|
P3TEA (010) |
1.74 |
3.61 |
38.14 |
|
TTPhen-4PDI (010) |
1.47 |
4.27 |
11.32 |
Conclusions
In conclusion, we designed and synthesized a pair of PDI tetramers with a tetraarylphenazine core. DFT calculation shows that both TPPhen-4PDI and TTPhen-4PDI have a two-blade propeller geometry, which can reduce their aggregation tendency, while the PDI units in TPPhen-4PDI are more open for intermolecular π-stacking than those in TTPhen-4PDI. RSoXS and GIWAXS analyses both showed that TPPhen-4PDI has a stronger aggregation tendency and nanophase separation in the P3TEA:SMA blend film than TTPhen-4PDI. The better π-stacking of TPPhen-4PDI facilitates a better charge transportation, while the two-blade propeller geometry limits the extent of aggregation to maintain a reasonably small domain for efficient exciton diffusion. Therefore, P3TEA:TPPhen-4PDI achieved a higher Jsc of 12.6 mA cm−2 and FF of 62%, while P3TEA:TTPhen-4PDI only achieved a Jsc of 11.0 mA cm−2 and FF of 50%. The significant difference in performance between TPPhen-4PDI and TTPhen-4PDI emphasizes the importance of linker group selection in designing high-performance PDI-based SMAs.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors acknowledge the financial support from the Guangdong Basic and Applied Basic Research Foundation (2022A1515010580), Shenzhen Science and Technology Program (GJHZ20210705142200003, JCYJ20220530141211025), Key Research and Development Program of Guangzhou City (20220602JBGS02), and Industry–Academia–Research Cooperative Project of Zhuhai City (ZH22017001210130PWC). The authors are enormously grateful to Prof. Annie Ng of Nazarbayev University for the in-depth discussions.
Notes and references
- J. T. Bloking, T. Giovenzana, A. T. Higgs, A. J. Ponec, E. T. Hoke, K. Vandewal, S. Ko, Z. Bao, A. Sellinger and M. D. McGehee, Adv. Energy Mater., 2014, 4, 1301426 CrossRef.
- J. Benduhn, K. Tvingstedt, F. Piersimoni, S. Ullbrich, Y. Fan, M. Tropiano, K. A. McGarry, O. Zeika, M. K. Riede, C. J. Douglas, S. Barlow, S. R. Marder, D. Neher, D. Spoltore and K. Vandewal, Nat. Energy., 2017, 2, 17053 CrossRef CAS.
- M. Kim, S. U. Ryu, S. A. Park, Y.-J. Pu and T. Park, Chem. Sci., 2021, 12, 14004 RSC.
- B. Schweda, M. Reinfelds, P. Hofstadler, G. Trimmel and T. Rath, ACS Appl. Energy Mater., 2021, 4, 11899–11981 CrossRef CAS PubMed.
- J. Zhao, C. Yao, M. U. Ali, J. Miao and H. Meng, Mater. Chem. Front., 2020, 4, 3487–3504 RSC.
- H. Zhang, H. Yao, J. Hou, J. Zhu, J. Zhang, W. Li, R. Yu, B. Gao, S. Zhang and J. Hou, Adv. Mater., 2018, 30, 1800613 CrossRef PubMed.
- F. Pan, X. Li, S. Bai, T. Liu, X. Wei, Y. Li, S. Chen, C. Yang, X. Chen, M. Lv and Y. Li, Chin. Chem. Lett., 2021, 32, 1257–1262 CrossRef CAS.
- W. Gao, T. Liu, R. Ming, Z. Luo, K. Wu, L. Zhang, J. Xin, D. Xie, G. Zhang, W. Ma, H. Yan and C. Yang, Adv. Funct. Mater., 2018, 28, 1803128 CrossRef.
- W. Zhao, S. Li, H. Yao, S. Zhang, Y. Zhang, B. Yang and J. Hou, J. Am. Chem. Soc., 2017, 139, 7148–7151 CrossRef CAS PubMed.
- Y. Chen, R. Cao, H. Liu, M. L. Keshtov, E. N. Koukaras, H. Dahiya, Y. Zou and G. D. Sharma, Sustainable Energy Fuels, 2020, 4, 6203–6211 RSC.
- J. Yuan, Y. Zhang, L. Zhou, G. Zhang, H.-L. Yip, T.-K. Lau, X. Lu, C. Zhu, H. Peng, P. A. Johnson, M. Leclerc, Y. Cao, J. Ulanski, Y. Li and Y. Zou, Joule, 2019, 3, 1140–1151 CrossRef CAS.
- L. Zhu, M. Zhang, J. Xu, C. Li, J. Yan, G. Zhou, W. Zhong, T. Hao, J. Song, X. Xue, Z. Zhou, R. Zeng, H. Zhu, C.-C. Chen, R. C. I. MacKenzie, Y. Zou, J. Nelson, Y. Zhang, Y. Sun and F. Liu, Nat. Mater., 2022, 21, 656–663 CrossRef CAS PubMed.
- X. Wang, Q. Sun, J. Gao, J. Wang, C. Xu, X. Ma and F. Zhang, Energies, 2021, 14, 4200 CrossRef CAS.
- S. Li, C.-Z. Li, M. Li and H. Chen, ACS Energy Lett., 2020, 5, 1554–1567 CrossRef CAS.
- K. Jiang, Q. Wei, J. Y. L. Lai, Z. Peng, H. K. Kim, J. Yuan, L. Ye, H. Ade, Y. Zou and H. Yan, Joule, 2019, 3, 3020–3033 CrossRef CAS.
- N. Zink-Lorre, E. Font-Sanchis, Á. Sastre-Santos and F. Fernández-Lázaro, Chem. Commun., 2020, 56, 3824–3838 RSC.
- V. Sharma, J. D. B. Koenig and G. C. Welch, J. Mater. Chem. A, 2021, 9, 6775–6789 RSC.
- S. Chen, D. Meng, J. Huang, N. Liang, Y. Li, F. Liu, H. Yan and Z. Wang, CCS Chem., 2021, 3, 78–84 CrossRef CAS.
- Q. Zhang, X. Xu, S. Chen, G. B. Bodedla, M. Sun, Q. Hu, Q. Peng, B. Huang, H. Ke, F. Liu, T. P. Russell and X. Zhu, Sustainable Energy Fuels, 2018, 2, 2616–2624 RSC.
- C. Yu, Y. Xu, S. Liang, X. Jiang, G. Feng, C. Li and W. Li, Chin. Chem. Lett., 2018, 29, 325–327 CrossRef CAS.
- A. D. Hendsbee, S. V. Dayneko, J. A. Pells, J. R. Cann and G. C. Welch, Sustainable Energy Fuels, 2017, 1, 1137–1147 RSC.
- H. Sun, X. Song, J. Xie, P. Sun, P. Gu, C. Liu, F. Chen, Q. Zhang, Z. K. Chen and W. Huang, ACS Appl. Mater. Interfaces, 2017, 9, 29924–29931 CrossRef CAS PubMed.
- Y. Liu, J. Y. L. Lai, S. Chen, Y. Li, K. Jiang, J. Zhao, Z. Li, H. Hu, T. Ma, H. Lin, J. Liu, J. Zhang, F. Huang, D. Yu and H. Yan, J. Mater. Chem. A, 2015, 3, 13632–13636 RSC.
- Q. Yan, Y. Zhou, Y.-Q. Zheng, J. Pei and D. Zhao, Chem. Sci., 2013, 4, 4389 RSC.
- J. Zhang, Y. Li, J. Huang, H. Hu, G. Zhang, T. Ma, P. C. Y. Chow, H. Ade, D. Pan and H. Yan, J. Am. Chem. Soc., 2017, 139, 16092–16095 CrossRef CAS PubMed.
- H. Lin, S. Chen, H. Hu, L. Zhang, T. Ma, J. Y. L. Lai, Z. Li, A. Qin, X. Huang, B. Tang and H. Yan, Adv. Mater., 2016, 28, 8546–8551 CrossRef CAS PubMed.
- W. Jiang, L. Ye, X. Li, C. Xiao, F. Tan, W. Zhao, J. Hou and Z. Wang, Chem. Commun., 2014, 50, 1024–1026 RSC.
- D. Sun, D. Meng, Y. Cai, B. Fan, Y. Li, W. Jiang, L. Huo, Y. Sun and Z. Wang, J. Am. Chem. Soc., 2015, 137, 11156–11162 CrossRef CAS PubMed.
- W. Chen, X. Yang, G. Long, X. Wan, Y. Chen and Q. Zhang, J. Mater. Chem. C, 2015, 3, 4698–4705 RSC.
- S. Ya-Rui, W. Hui-Ling, S. Ya-Ting and L. Yu-Fang, CrystEngComm, 2017, 19, 6008–6019 RSC.
- G. Grynóva, A. Nicolaï, A. Prlj, P. Ollitrault, D. Andrienko and C. Corminboeuf, J. Mater. Chem. C, 2017, 5, 350 RSC.
- Q. He, F. D. Eisner, D. Pearce, T. Hodsden, E. Rezasoltani, D. Medranda, Z. Fei, J. Nelson and M. Heeney, J. Mater. Chem. C, 2020, 8, 17237 RSC.
- H. Gilman and J. J. Dietrich, J. Am. Chem. Soc., 1957, 79, 6178–6179 CrossRef CAS.
- X. Ma, A. Zeng, J. Gao, Z. Hu, C. Xu, J. H. Son, S. Y. Jeong, C. Zhang, M. Li, K. Wang, H. Yan, Z. Ma, Y. Wang, H. Y. Woo and F. Zhang, Natl. Sci. Rev., 2021, 8, nwaa305 CrossRef PubMed.
- X. Ma, J. Wang, J. Gao, Z. Hu, C. Xu, X. Zhang and F. Zhang, Adv. Energy Mater., 2020, 10, 2001404 CrossRef CAS.
- M. M. Mandoc and L. J. A. Koster, Appl. Phys. Lett., 2007, 90, 133504 CrossRef.
- W. Xu, X. Zhu, X. Ma, H. Zhou, X. Li, S. Y. Jeong, H. Y. Woo, Z. Zhou, Q. Sun and F. Zhang, J. Mater. Chem. A, 2022, 10, 13492 RSC.
- C. Xu, K. Jin, Z. Xiao, Z. Zhao, Y. Yan, X. Zhu, X. Li, Z. Zhou, S. Y. Jeong, L. Ding, H. Y. Woo, G. Yuan and F. Zhang, Sol. RRL, 2022, 6, 2200308 CrossRef CAS.
- P. Müller-Buschbaum, Adv. Mater., 2014, 26, 7692–7709 CrossRef PubMed.
- S. Swaraj, C. Wang, H. Yan, B. Watts, J. Lüning, C. R. McNeill and H. Ade, Nano Lett., 2010, 10, 2863–2869 CrossRef CAS PubMed.
- W. Ma, J. R. Tumbleston, M. Wang, E. Gann, F. Huang and H. Ade, Adv. Energy Mater., 2013, 3, 864–872 CrossRef CAS.
- X. Guo, N. Zhou, S. J. Lou, J. Smith, D. B. Tice, J. W. Hennek, R. P. Ortiz, J. T. L. Navarrete, S. Li, J. Strzalka, L. X. Chen, R. P. H. Chang, A. Facchetti and T. J. Marks, Nat. Photonics, 2013, 7, 825–833 CrossRef CAS.
- I. Osaka, M. Saito, T. Koganezawa and K. Takimiya, Adv. Mater., 2014, 26, 331–338 CrossRef CAS PubMed.
- V. Vohra, K. Kawashima, T. Kakara, T. Koganezawa, I. Osaka, K. Takimiya and H. Murata, Nat. Photonics, 2015, 9, 403–408 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 e,
Jicheng
Yi
f,
Joshua Yuk Lin
Lai
f,
Guang
Shao
e,
Jicheng
Yi
f,
Joshua Yuk Lin
Lai
f,
Guang
Shao