
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stepwise deprotonation of truxene: structures, metal complexation, and charge-dependent optical properties†
Yumeng
Guo
 a,
Herdya S.
Torchon
b,
Yikun
Zhu
b,
Zheng
Wei
b,
Zhenyi
Zhang
c,
Haixiang
Han
*a,
Marina A.
Petrukhina
*b and
Zheng
Zhou
*a
a,
Herdya S.
Torchon
b,
Yikun
Zhu
b,
Zheng
Wei
b,
Zhenyi
Zhang
c,
Haixiang
Han
*a,
Marina A.
Petrukhina
*b and
Zheng
Zhou
*a
aSchool of Materials Science and Engineering, Tongji University, Shanghai 201804, China. E-mail: zhouzheng@tongji.edu.cn
bDepartment of Chemistry, University at Albany, State University of New York, Albany, NY 12222, USA. E-mail: mpetrukhina@albany.edu
cBruker (Beijing) Scientific Technology Co., Ltd, Shanghai 200233, China
First published on 31st October 2023
Abstract
As a planar subunit of C60-fullerene, truxene (C27H18) represents a highly symmetrical rigid hydrocarbon with strong blue emission. Herein, we used truxene as a model to investigate the chemical reactivity of a fullerene fragment with alkali metals. Monoanion, dianion, and trianion products with different alkali metal counterions were crystallized and fully characterized, revealing the core curvature dependence on charge and alkali metal coordination. Moreover, a 1proton nuclear magnetic resonance study coupled with computational analysis demonstrated that deprotonation of the aliphatic CH2 segments introduces aromaticity in the five-membered rings. Importantly, the UV-vis absorption and photoluminescence of truxenyl anions with different charges reveal intriguing charge-dependent optical properties, implying variation of the electronic structure based on the deprotonation process. An increase in aromaticity and π-conjugation yielded a red shift in the absorption and photoluminescent spectra; in particular, large Stokes shifts were observed in the truxenyl monoanion and dianion with high emission quantum yield and time of decay. Overall, stepwise deprotonation of truxene provides the first crystallographically characterized examples of truxenyl anions with three different charges and charge-dependent optical properties, pointing to their potential applications in carbon-based functional materials.
Introduction
Since the discovery of C60-fullerene in 1985,1 molecular nanocarbons have attracted enormous attention in fundamental chemistry and materials science. Over the decades, a wide variety of molecular nanocarbon architectures with different sizes and topologies have been synthesized with tunable chemical and physical properties,2–9 which greatly expands their applications in energy storage, optoelectronic devices, and superconductors.10–12 In particular, the fusion of five-membered rings into the hexagonal carbon framework can cause changes in structure and reactivity.13–18 Taking some fullerene subunits as examples (Scheme 1), the central five-membered ring in corannulene generates a Gaussian curvature with C5v symmetry, offering two distinctive π-surfaces (concave and convex) for metal binding. Corannulene (C20H10) exhibits a very rich redox chemistry and readily accepts up to four electrons upon chemical reduction, affording anionic species with interesting metal coordination or supramolecular chemistry.19–21 In contrast, sumanene (C21H12), built with three external five-membered rings, possesses a positive curvature (C3v symmetry) with site-selective reactivity,22,23 and can undergo stepwise deprotonation to form either double-concave or double-convex metal complexes.24,25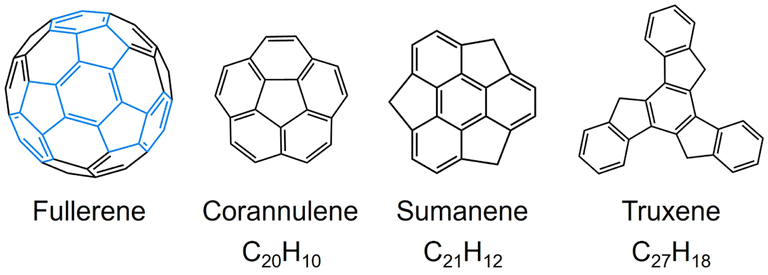 | ||
| Scheme 1 Depictions of fullerene and its subunits. | ||
Unlike bowl-shaped fragments, truxene (1, C27H18) can be considered a planar subunit of fullerene with C3h molecular symmetry.26,27 However, the external six-membered rings can be readily functionalized to generate star-shaped molecules and oligomers.28–33 Moreover, the presence of three sp3-hybridized carbon atoms on the pentagonal rings imparts additional chemical reactivity for adding alkyl chains or heteroatoms.34,35 Notably, truxene exhibits strong blue emission due to its large optical gap.36 The combination of high symmetry, rigid planar structure, site-specific reactivity, and unique optical properties make the truxene unit a highly attractive building block for the construction of diverse novel materials with broad applications, such as organic photovoltaics, liquid crystals, and nonlinear optical materials.28,29,33
Adding charges into π-conjugated systems can cause variations in chemical and electronic structures, which further allows the modulation of their optical properties. Although truxene has many promising applications, its charge-dependent optical behavior has never been explored. We have employed the chemical reduction approach to prepare charged π-conjugated carbanions whose molecular structures were successfully established by X-ray single crystal diffraction.21,37 These studies demonstrated their structural transformations, thus opening the way for investigations into the optical, electronic, and magnetic properties of the resulting solid crystalline materials.38–41 Inspired by truxene's site-specific reactivity and interesting optical properties, we report the first comprehensive study of the stepwise deprotonation of truxene with alkali metals (Na, K, and Cs). Products in different charging states have been isolated and fully characterized by single crystal X-ray diffraction, revealing the core curvature dependence on the charge and original metal complexation. Furthermore, it was revealed that the truxenyl anions demonstrate charge-dependent absorption and photoluminescence properties. Therefore, density functional theory (DFT) was performed to gain deep insights into such optical behavior. This work represents the first structural-based charge-dependent study of truxene and shows its promising applications in carbon-based functional molecular materials.
Results and discussion
Solid state structure of 1
To understand the structure–property correlation of the truxenyl framework, it is necessary to obtain the crystal structure of neutral truxene first and use it for the comparison with the reduced species. Although the synthesis of 1 was reported decades ago, its solid state structure remains unclear due to its highly disordered nature. In recent work by Zou et al.,42 truxene crystals were grown from a mixture of trichlorobenzene and methanol, but no further details were provided. Prior to a chemical reactivity study, 1 was purified by sublimation in a glass ampule in vacuo at elevated temperature (265 °C), affording nicely grown yellow needles in good yield (75%). The structural analysis shows 1 crystallizes in a P21 space group (Fig. 1a). This indicates that the molecular assembly of truxene can be chiral; however, the molecules from two orientations are overlapped and cocrystallized due to the high disorder (Fig. S23†). In the solid state structure, the molecules of 1 are stacked into a 1D column with a deck-to-deck separation of 3.477(9) Å and a slip distance of 3.142(9) Å (Fig. 1b). Unlike the fully aligned stacks of carbon bowls,22 this slipped herringbone packing is commonly seen in most planar polyarenes.43,44 It should be noted that H-atoms on the sp3 carbons weaken the π⋯π interactions in 1 (3.477(9) Å); thus, the columnar packing is dominated by C–H⋯π interactions (2.629(9)–2.674(14) Å). The 1D columns are further linked into a 2D layer through a weaker C–H⋯π contact (2.885(17) Å, Fig. 1c).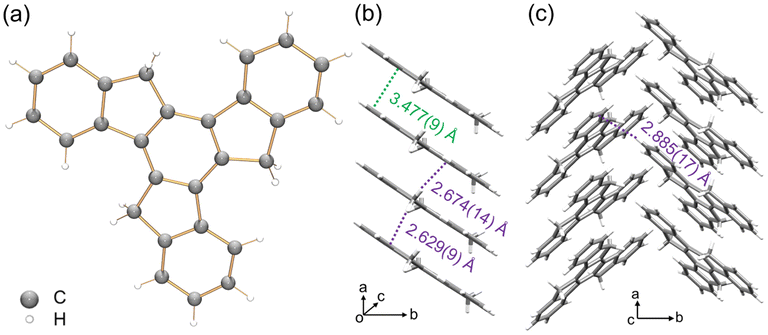 | ||
| Fig. 1 (a) Crystal structure of 1 as a ball-and-stick model, (b) 1D column and (c) solid-state structure as capped stick models with depictions of C–H⋯π (purple) and π⋯π (green) interactions. | ||
Stepwise deprotonation of 1
Truxene has a rigid carbon framework with three “exposed” five-membered rings on the edge of the molecule, so multiple deprotonations could occur to afford truxenyl anions with different charges, similar to that of π-bowl sumanene.23 Additionally, its three-fold planar structure provides more chemically-equivalent sites for metal binding, which helps stabilize the high charging states in contrast to sumanene. The chemical reactivity of truxene (C27H18, 1) was investigated in tetrahydrofuran (THF) at room temperature with Na, K, and Cs metals. The reaction first proceeds through a bright orange color, which corresponds to the monoanionic stage, followed by an orange–red color (the dianion), and finally lightens to yellow, which indicates the formation of a truxenyl trianion. By controlling the reaction time of the alkali-metal-induced deprotonation, orange, red, and yellow plate-shaped crystals corresponding to different reaction stages have been successfully isolated and characterized by single crystal X-ray diffraction (Scheme 2, see ESI† for more details). An X-ray diffraction study confirmed the formation of two solvent-separated ion pairs with Na+ counterions, [Na+(18-crown-6)(THF)2][C27H17−] (Na-1−) and [Na+(18-crown-6)(THF)2]2[C27H162−] (Na2-12−, crystallized with four interstitial THF), and three contact–ion complexes, [{Cs+(18-crown-6)}(C27H17−)] (Cs-1−), [{Cs+(18-crown-6)}2(C27H162−)] (Cs2-12−), and [K+(18-crown-6)(THF)2][{K+(18-crown-6)}2(C27H153−)] (K3-13−, crystallized with one interstitial THF).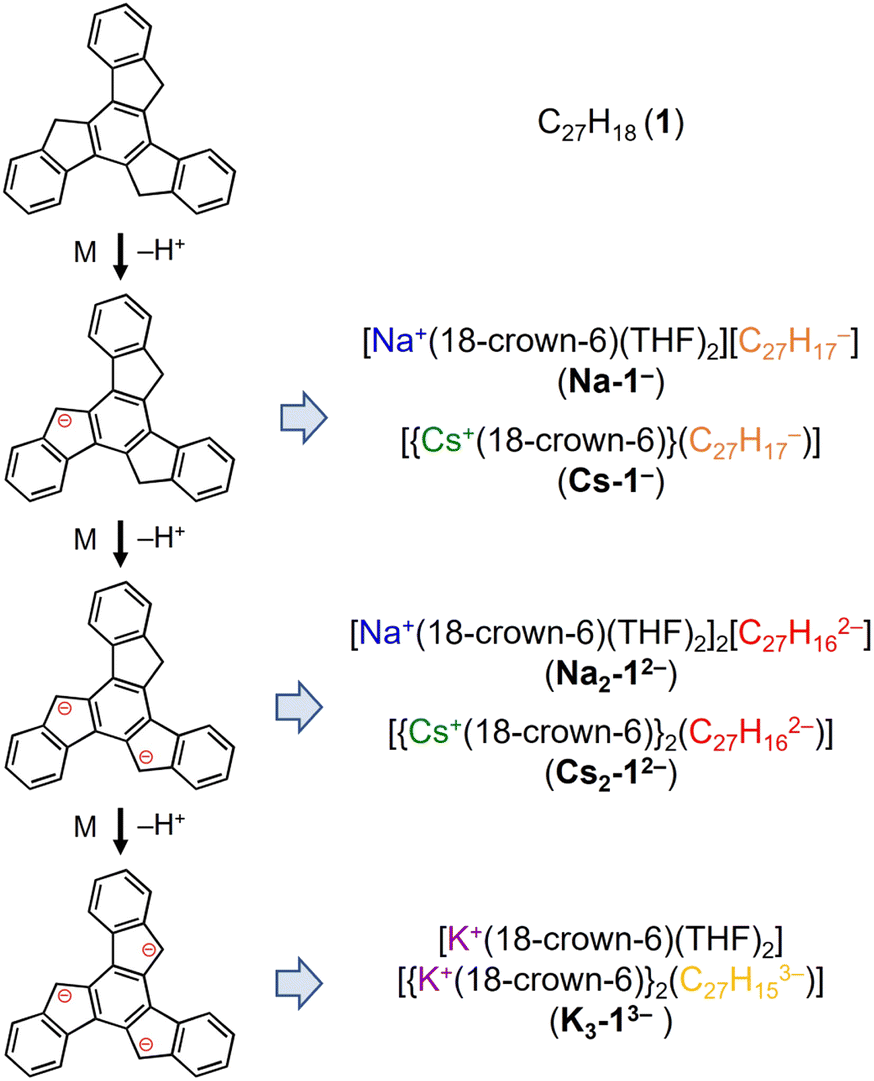 | ||
| Scheme 2 Preparation of truxenyl mono-, di-, and trianionic products with Na+, K+, and Cs+ ions. | ||
1H NMR investigation of truxenyl anions
A 1proton nuclear magnetic resonance (1H NMR) spectroscopic investigation was initially performed to understand the solution behavior of all the compounds (Fig. 2 and S1–S8†). In neutral 1, the singlet appearing at 4.34 ppm represents the H-atoms on the sp3-hybridized carbon atoms. The first deprotonation of one five-membered ring (loss of one Ha) in Na-1− affords an increase in aromaticity of the corresponding ring. As a result, the remaining Ha shifted largely downfield to the aromatic region (6.64 ppm), leaving the other two sets of aliphatic protons as singlets at 4.14 and 4.29 ppm (Hb and Hc). The same splitting was observed in the truxene monoanion complexed with a Cs+ counterion in Cs-1− (Fig. S4†). Unlike bowl-shaped sumanene, whose positive curvature enlarged the difference between the endo- and exo-protons,22 the peak Hb, which stayed close to the high electron density area, also experienced deshielding and was slightly shifted downfield (4.37 ppm vs. 4.23 ppm for Hc). Next, deprotonation occurs on the second five-membered ring to afford truxenyl dianions in Na2-12− and Cs2-12− (Fig. S5†) accompanied by the loss of one Hb and deshielding of the other. Consequently, both remaining Ha and Hb were greatly deshielded and shifted to the aromatic region, while Hc remained in the aliphatic region. Notably, the singlet for Hc confirmed the similarity of the protons on both sides and the planarity of the carbon framework. Finally, in the fully deprotonated trianion in K3-13−, the proton signals appear as a singlet observed at 6.76 ppm due to its highly symmetrical structure.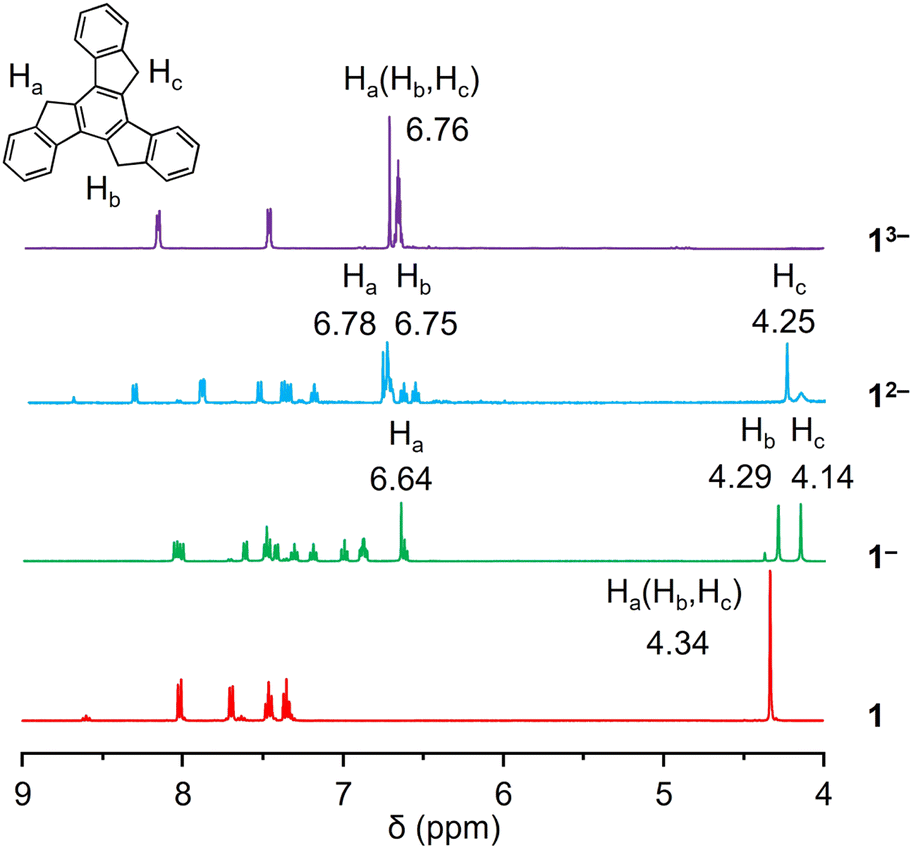 | ||
| Fig. 2 1H NMR spectra of 1, Na-1−, Na2-12−, and K3-13− in THF-d8 at 25 °C. | ||
Crystallographic analysis of truxenyl anions
In the crystal structure of Na-1− (Fig. 3a), a single deprotonation occurs at the C16 atom and the {Na+(18-crown-6)(THF)2} moiety is solvent-separated from the C27H17− core. Interestingly, in contrast to sumanene where only one-fold deprotonation was observed with Na metal,25 extending the reaction time allows the formation of a “naked” C27H162− dianion with two Na+ counterions (Fig. 3b, deprotonation at C2 and C20). In both structures, each Na+ ion is wrapped with 18-crown-6 ether and capped by two THF molecules, with the Na–O distances (Na–Ocrown: 2.575(12)–2.839(11) Å, Na–OTHF: 2.301(10)–2.350(12) Å) being comparable to the values reported in the literature.38,45–47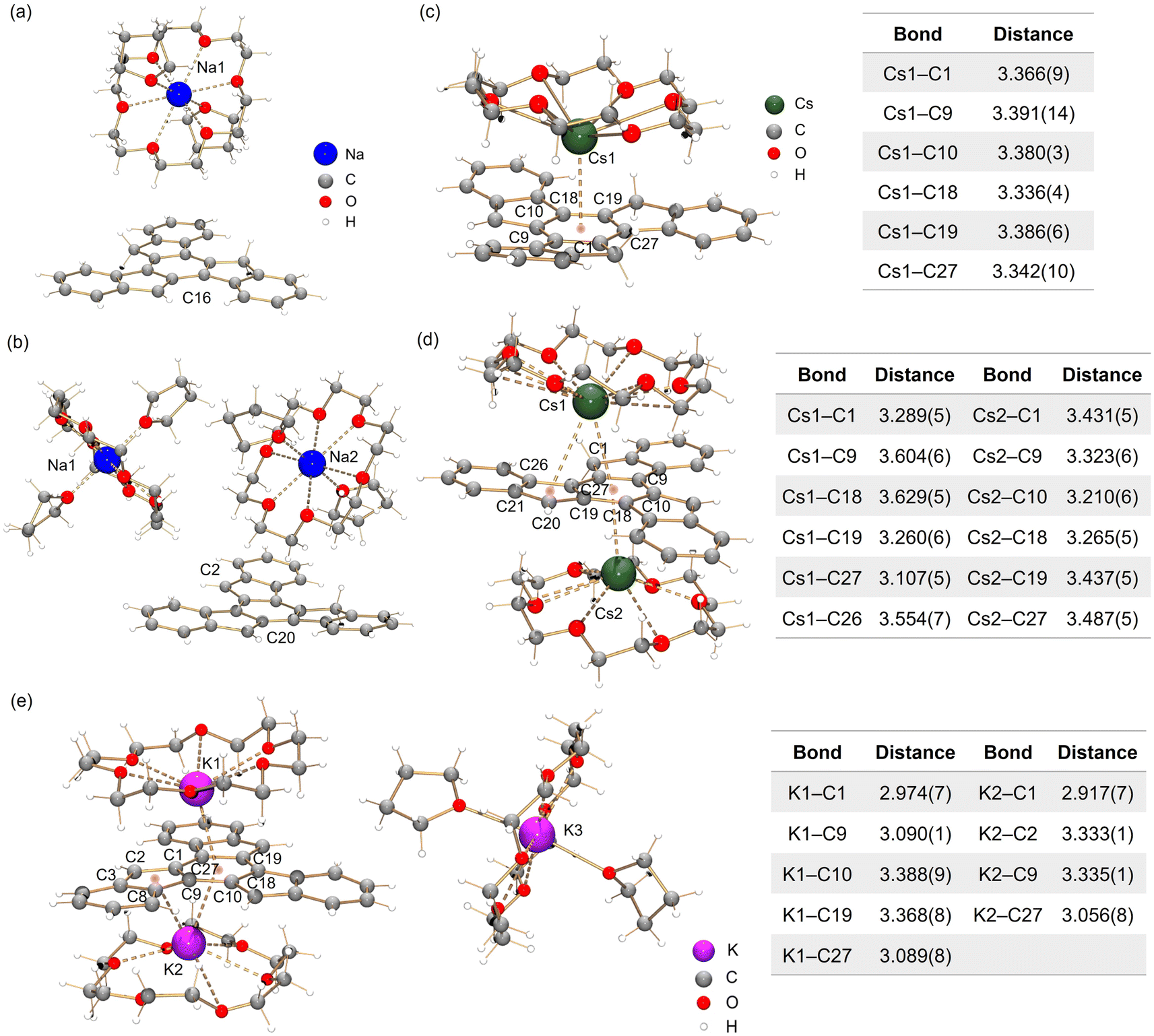 | ||
| Fig. 3 Crystal structures of (a) Na-1−, (b) Na2-12−, (c) Cs-1−, (d) Cs2-12− and (e) K3-13− as ball-and-stick models, along with tables of Cs–C and K–C bond distances (Å). | ||
By using Cs metal, the reaction undergoes the same color changes but gives rise to two contact–ion complexes. In the crystal structure of Cs-1− (Fig. 3c), the Cs+ ion is coordinated to the central six-membered ring of C27H17− in an η6-manner, with the Cs–C distances ranging over 3.336(4)–3.391(14) Å. In the crystal structure of Cs2-12− (Fig. 3d), there are two independent Cs+ ions coordinated to the truxenyl dianion, C27H162−. The Cs1 ion is bound to the central six-membered ring in an asymmetric η6-fashion, with the Cs–C distances over spanning a broad range of 3.107(8)–3.629(8) Å. Additional contact is found between Cs1 and the adjacent five-membered ring E (3.554(8) Å). The Cs2 ion is only bound to the central six-membered ring in an η6-mode (3.210(8)–3.487(8) Å). All Cs–C distances are close but shorter than the previously reported values for bowl-shaped anions.25,48 In both structures, each Cs+ ion is also asymmetrically bound to an 18-crown-6 ether molecule, with Cs–O distances of 2.983(18)–3.211(18) Å.
In contrast to the sumanenyl supramolecular aggregate with K+ counterions formed by a mixture of dianions and trianions,24 the reaction of truxene with K metal yields a pure trianionic product. In the crystal structure of K3-13− (Fig. 3e), two K+ ions are bound to the truxenyl trianion and entrapped by an 18-crown-6 ether molecule, with the K–C distances ranging from 3.107(8) Å to 3.629(8) Å. Unlike Cs ions in Cs2-12−, the K1 ion in K3-13− sits closer to the C27H153− core (2.909(8) Å), but the K2 ion is slipped over two rings (3.122(8)/3.301(8) Å). The remaining K+ ion is fully wrapped by 18-crown-6 ether and capped by two THF molecules.
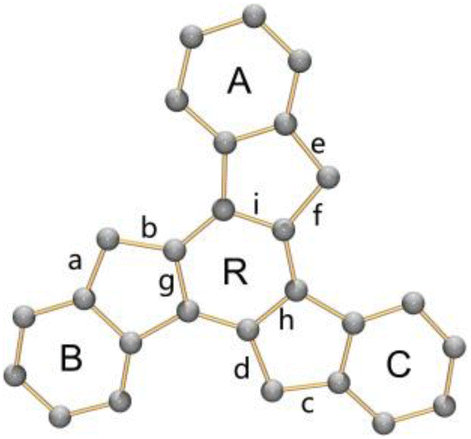
The structural perturbation of the truxenyl core upon stepwise deprotonation can be illustrated by dihedral angles and C–C bond distances compared with the neutral parent (scheme in Tables 1 and S2†). Notably, one-fold deprotonation of 1 leads to an increase in aromaticity of the deprotonated five-membered ring E in Na-1− and Cs-1−. The C–C bonds around the deprotonated carbon atom (a, b) are significantly shortened from 1.500(10) Å in C27H18 to 1.443(14) Å in Na-1− and 1.429(6) Å in Cs-1−. Additional bond length reduction is also observed in the two-fold deprotonated C27H162− (c, d in Na2-12− and Cs2-12−) and three-fold deprotonated C27H153− (c, d, e, and f in K3-13−). Moreover, C–C bonds around the central ring R (g, h, and i) in all charged species show a slight increase in length compared to 1 (e.g., 1.420(7) Å in Na2-12−vs. 1.400(7) Å in 1). In addition, the truxenyl anions experience some curvature after deprotonation, as shown by an increase in dihedral angles around the central six-membered ring (Table 1). In neutral 1, the truxene core is planar. A small curvature is observed upon one-fold deprotonation in Na-1− (2.9°). It should be noted that the direct Cs+ ion coordination in Cs-1− yields a more pronounced curvature for the truxenyl monoanion (9.9°), forming a bowl-shaped framework with a bowl depth of 0.606(6) Å. After two-fold deprotonation, the C27H162− core in Na2-12− becomes more twisted (7.9°) while that effect is less pronounced in Cs2-12−, probably due to double-sided metal binding. Finally, stemming from increased aromaticity and direct metal coordination, a curvature reduction is observed in the C27H153− core in K3-13− (4.1°).
|
|
||||||
|---|---|---|---|---|---|---|
| Dihedral angle (°) | 1 | Na-1− | Na2-12− | Cs-1− | Cs2-12− | K3-13− |
| A/R | 0 | 2.4 | 10.5 | 15.0 | 5.4 | 2.1 |
| B/R | 0 | 4.2 | 3.3 | 8.9 | 9.4 | 4.0 |
| C/R | 0 | 2.2 | 9.8 | 5.8 | 5.6 | 6.2 |
| Avg. | 0 | 2.9 | 7.9 | 9.9 | 6.8 | 4.1 |
Aromaticity evaluation of truxenyl anions
To better understand the geometric, aromatic, and electronic changes of the parent truxene (1) compared to its singly-, doubly-, and triply-deprotonated products, density functional theory (DFT) calculations were performed at the B3LYP-D3BJ/def2-TZVP level. Stanger et al. previously showed that the NICS(1.7)ZZ value is sufficient to eliminate the disturbance generated by σ-electrons when describing the aromaticity in polycyclic aromatic hydrocarbon (PAH) systems and their charged species;49 thus, it is used below for discussion and comparison. The fully optimized gas-phase neutral molecule 1 adopts a planar structure (Fig. 4a), which is similar to the geometry observed in the crystal structure. The NICS(1.7)ZZ values of four six-membered rings (A, B, C, and G) indicate that each of them has an aromatic character (avg. −19.6 ppm, or 89% of benzene; NICS(1.7)ZZ = −22 ppm for benzene at the same level of theory). In contrast, three five-membered rings D, E, and F are noticeably less aromatic. Due to the high symmetry of the molecule (D3h), no difference exists between rings A, B, and C as well as rings D, E, and F.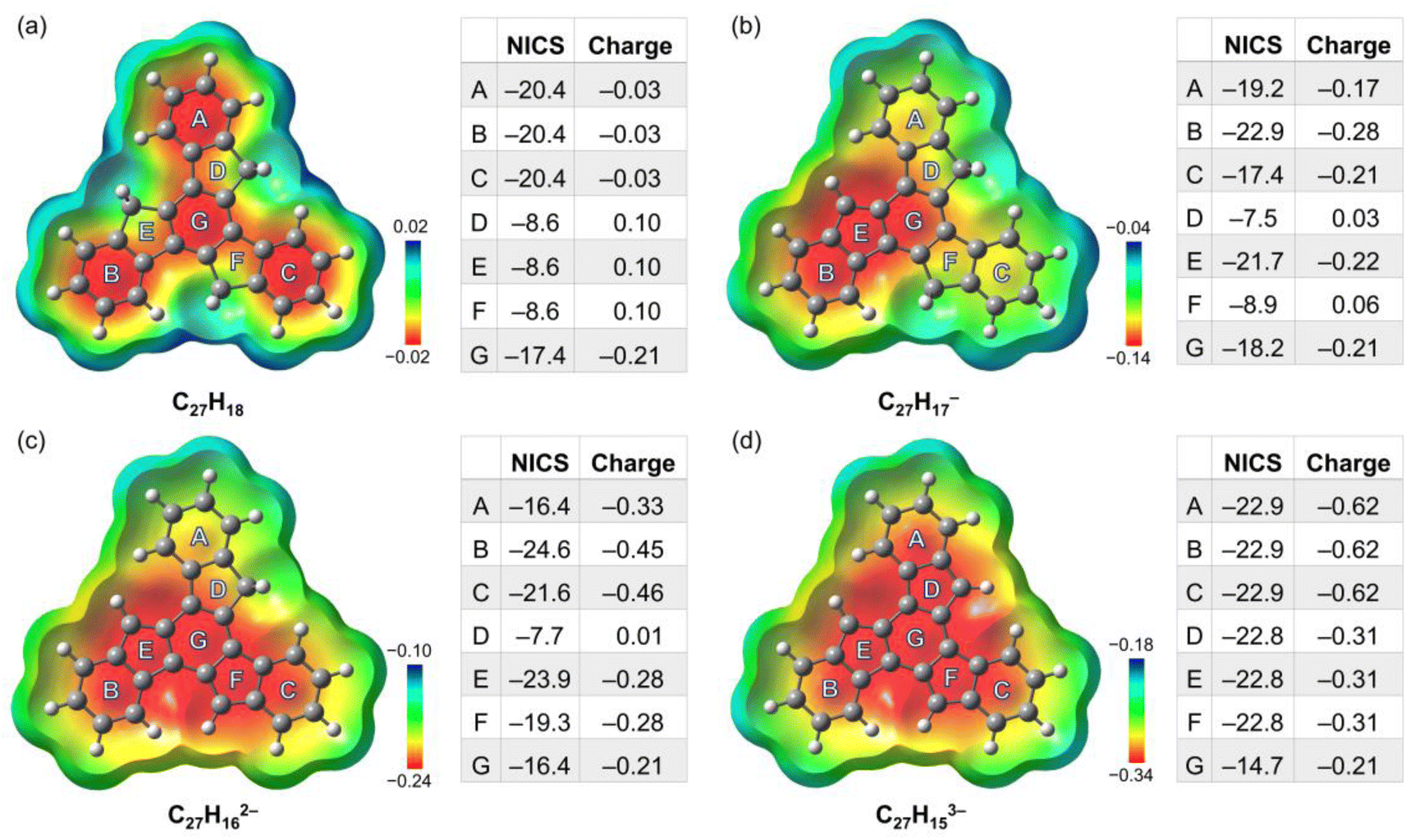 | ||
| Fig. 4 Electrostatic potential maps (mapped on the electron density using an isovalue of 0.0004 and Z-clip of 0.4) of (a) C27H18, (b) C27H17−, (c) C27H162−, and (d) C27H153−, along with tables of the corresponding NICS(1.7)ZZ values (ppm) and Lowdin partial charges for each ring. | ||
After the first deprotonation, the monoanion (C27H17−) remains planar, but the symmetry is reduced to Cs (Fig. 4b); an increase in aromaticity is observed, particularly around rings B, E, and G. Notably, the loss of a proton on ring E yields more pronounced aromaticity (−21.7 ppm), which is close to 95% that of benzene, similar to a previous investigation of sumanene, a bowl-shaped subunit of C60-fullerene.24
Following the second deprotonation to afford the “naked” dianion (Fig. 4c), the aromaticity of the system further increases. Notably, the NICS(1.7)ZZ values of rings B and E (−24.6/−23.9 ppm, 112%/108% of benzene, respectively) are more pronounced than those of rings C and F (−21.6/−19.3 ppm, 98%/88% of benzene, respectively). This can be further identified by the large difference in their partial charges (avg. −0.46 for B/C vs. −0.28 for E/F), which indicates an asymmetric charge distribution that could further affect metal coordination upon complexation. In contrast, the NICS(1.7)ZZ value of outer ring A next to ring D further reduces to −16.4 ppm.
Upon further deprotonation (Fig. 4d), the aromaticity of the trianion further increases. Apart from central ring G, all the rings have a very high NICS(1.7)ZZ of −22.8 pm (103% of benzene). The reduced NICS value for central ring G is consistent with the observation for three-fold deprotonated sumanene, where the individual contribution of the ring becomes less prominent when involved in total delocalization.50 The truxenyl trianion is reverted to D3h symmetry, and the electrons are fully delocalized.
The change in aromaticity can also be observed from the calculated anisotropy of current-induced density (ACID) plots for neutral truxene and the respective anions (Fig. 5 and S40†). In neutral 1, the presence of three protonated pentagonal rings breaks the conjugation between adjacent six-membered rings to a certain extent, enabling each of them to sustain local ring currents. Thus, four separate diatropic ring current centers are found in the ACID plot of 1 (Fig. 5a), which is more evident without σ-orbitals (Fig. S40a†). After one-fold deprotonation, one five-membered ring becomes aromatic and serves as a bridge that connects the adjacent two six-membered rings. As a result, a larger diatropic ring current is formed along one arm of the truxene monoanion (Fig. 5b and S40b†). With the increase in negative charge on the carbon framework, the second and third five-membered rings lose their proton and become aromatic, yielding a trianionic product with charge delocalization over the whole molecular core (Fig. 5c, d, S40c and d†). It is also concluded that the deprotonation of three five-membered rings changes the ring current, yielding a diatropic ring current around the periphery.
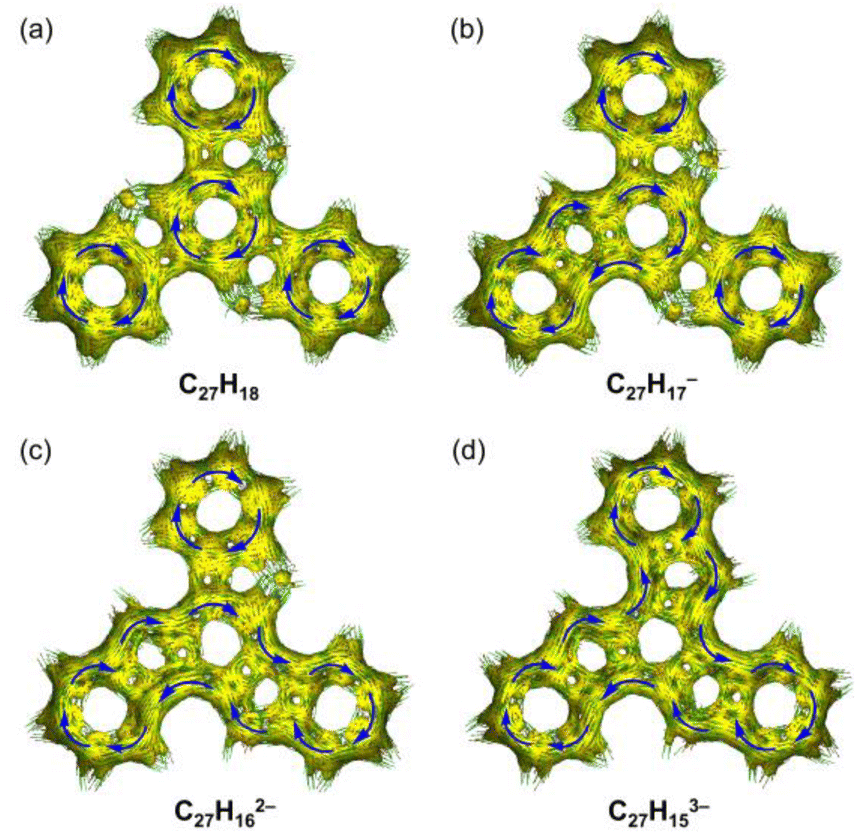 | ||
| Fig. 5 Full ACID isosurfaces for (a) C27H18, (b) C27H17−, (c) C27H162−, and (d) C27H153−. Current density vectors are plotted onto the ACID isosurface to indicate diatropic (blue) ring currents. | ||
Charge-dependent optical property of truxenyl anions
As shown above, the variation in charges achieved by controlled stepwise deprotonation provides a series of truxenyl anions with charging states ranging from 0 to −3, which possess entirely different electronic structures that further affect their optical properties. Based on the 1H NMR and UV-vis spectroscopic analyses, the Na- and Cs-complexed products behave similarly in solution, so Na-1−, Na2-12−, and K3-13− were selected as representatives for truxenyl monoanion, dianion, and trianion and used for comparison with the neutral parent.First, UV-vis spectroscopy assisted by TD-DFT calculation was used to visualize the absorption properties and natural transitions (see ESI† for more details). In general, the nearly planar truxenyl framework is dominated by the π → π* transition in neutral and in charged states. The major absorption (λabs) of 1 dissolved in THF occurs at 300 nm with a large energy gap (Fig. 6a). Upon deprotonation, Na-1− shows intense bands with an absorption maximum at 332 nm (Fig. 6b), which is correlated with the HOMO−1 → LUMO electronic transition (oscillator strength f = 0.524) according to the TD-DFT calculation, while the lowest-energy absorption band (λabs = 548 nm) comes from the HOMO → LUMO electronic transition (f = 0.205). In contrast, the largest absorption band in Na2-12− is found at 338 nm (Fig. 6c, HOMO−1 → LUMO+3, f = 1.165), while the low-energy bands in accordance with the HOMO−1 → LUMO and HOMO → LUMO electronic transitions (f = 0.160/0.048) are found at 511 and 549 nm, respectively. Finally, K3-13− shows an absorption maximum at 370 nm (Fig. 6d), originating mainly from the HOMO−2 → LUMO and HOMO−2 → LUMO+1 electronic transitions (f = 1.009/1.011). As shown in Fig. 6e, the HOMO → LUMO electron transitions in Na-1−, Na2-12−, and K3-13− occur from the deprotonated rings (bonding π-orbitals) to the protonated rings (antibonding π*-orbitals), from which the increasing π-conjugation yields a red shift in their absorption spectra.
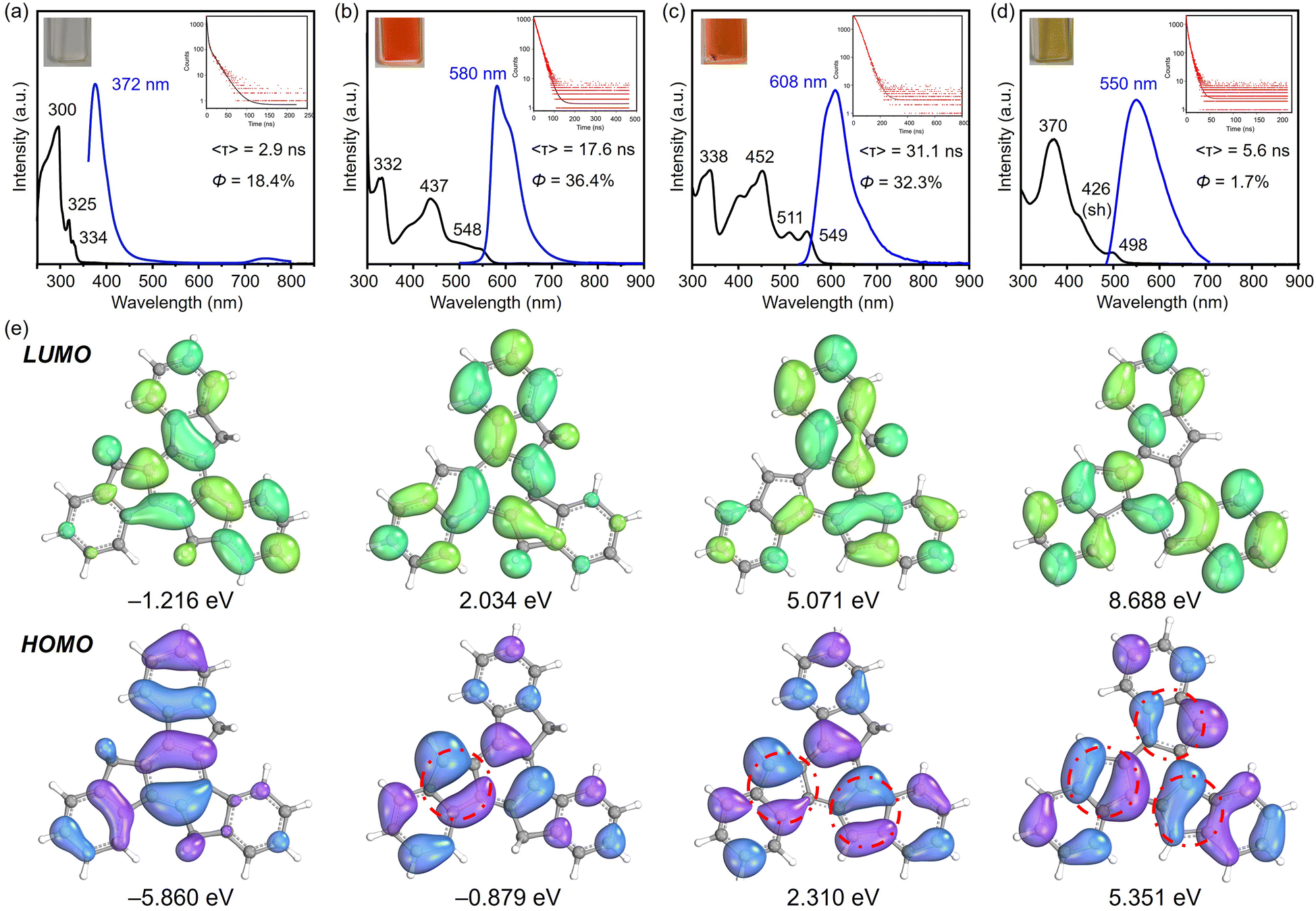 | ||
| Fig. 6 UV-vis absorption (black) and photoluminescence emission spectra (blue, excited at 350 nm) of (a) 1, (b) Na-1−, (c) Na2-12−, and (d) K3-13− in THF at 25 °C. (e) Electron density distribution of the frontier molecular orbitals (isovalue = 0.02) of C27H18, C27H17−, C27H162−, and C27H153− (B3LYP-D3BJ/def2-TZVP/CPCM(THF)). | ||
Next, to understand the light-emitting behavior of the truxenyl framework upon stepwise deprotonation, photoluminescence spectroscopy was performed. Truxene is a good monomer for building fluorescent materials with blue emission.14 In neutral truxene 1, 372 nm light was detected under excitation at 351 nm, with a fluorescence quantum yield of 18.4% (Fig. 6a). Notably, bright orange emission at 580 nm (λex = 360 nm, Fig. 6b) is observed in the monoanionic state (Na-1−), accompanied by higher emission intensity compared to 1 (Φ = 36.4%) with a decay time of 17.6 ns. In the two-fold deprotonated product Na2-12−, a bathochromic shift with an orange–red emission appears at 608 nm (λex = 351 nm, Fig. 6c) with similar emission intensity but a longer fluorescence lifetime (31.1 ns). In contrast, the fluorescence maximum of 6 is shifted back to 550 nm compared toNa2-12−, accompanied by a decrease in quantum yield to Φ = 1.7% (Fig. 6d). The large Stokes shift values in Na-1−, Na2-12−, and K3-13− are attributed to increasing π-conjugation, which increases the absorption coefficient and allows a higher chance of excitation for π-electrons.16 The decay constants kf and knr were calculated and compared for 1, Na-1−, Na2-12−, and K3-13. As shown in Table 2, Na-1− is expected to exhibit a higher Φf value among the charged species due to its faster spontaneous emission rate compared to Na2-12− and K3-13−. Compound 1 exhibits a substantially higher nonradiative decay rate (knr), affording a relatively lower quantum yield. Conversely, the low quantum yield observed in K3-13− (1.7%) can be attributed to a diminished emission rate (0.003 ns−1) and a more rapid nonradiative decay rate (0.176 ns−1).
| Compound | λ abs/nm | λ em/nm | Φ f | 〈τf〉/ns | k f/ns−1 | k nr/ns−1 |
|---|---|---|---|---|---|---|
| 1 | 300 | 372 | 0.184 | 2.9 | 0.063 | 0.281 |
| Na-1− | 332 | 580 | 0.364 | 17.6 | 0.021 | 0.036 |
| Na2-12− | 338 | 608 | 0.323 | 31.1 | 0.010 | 0.022 |
| K3-13− | 370 | 550 | 0.017 | 5.6 | 0.003 | 0.176 |
Conclusions
Using different alkali metals, we demonstrated that truxene can lose up to three protons through a controlled stepwise deprotonation process. Products of truxenyl anions with varying negative charges, namely monoanion, dianion, and trianion, have been isolated with different counterions and crystallographically characterized. A clear variation in alkali metal ion binding patterns was observed for light (Na+) vs. heavy (K+ and Cs+) counterions. The structural analysis revealed a notable core curvature of truxenyl anions affected by negative charge and metal coordination. The 1H NMR results indicated a change in aromaticity during stepwise deprotonation, as illustrated by notable upfield shifts of protons on the three five-membered rings. Moreover, computational NICS and ACID analyses were performed for truxenyl anions with different charging states with direct relevance to the experimental studies.We also observed that the increase in charge and aromaticity causes a change in optical properties, which was supported by UV-vis and photoluminescence spectroscopic investigations. The increase in aromaticity and π-conjugation yielded a red shift in the absorption and photoluminescent spectra. In particular, large Stokes shifts were observed in the truxenyl monoanion and dianion with a higher emission quantum yield and time of decay. Overall, stepwise deprotonation of truxene provides the first crystallographically characterized examples of truxenyl anions with different charges and charge-dependent optical properties. The successful generation of truxenyl anions with three different negative charges opens up their broad use in ligand exchange reactions and enables expansion of their organometallic and coordination chemistry.
Data availability
Data available in the ESI: Synthetic procedures, NMR, UV-vis, PL and TA spectra, X-ray crystallographic data and computational details.†Author contributions
Y. G. and H. S. T. synthesized the anionic products, completed their detailed structural descriptions and product characterizations; Y. Z. provided computational data analysis and wrote the corresponding manuscript sections; Z. W. and Z. Z. performed the X-ray data collection and refinement; H. H. contributed to the discussion and presentation of results; M. A. P. and Z. Z. conceived and supervised this work. Z. Z. carried out the theoretical calculations and drafted the manuscript with the support and contribution from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support for this work from the Fundamental Research Funds for the Central Universities (22120230060, Z. Z., 22120220118, H. H.), the National Natural Science Foundation of China (22301219 for Z. Z and 22101205 for H. H.), and the U.S. National Science Foundation (CHE-2003411, M. A. P.) is gratefully acknowledged. ChemMatCARS Sector 15 is supported by the National Science Foundation under grant number NSF/CHE-1834750. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC0206CH11357. We also thank Prof. Gershoni-Poranne from the Israel Institute of Technology for her scientific advice about the calculation experiment.Notes and references
- H. W. Kroto, J. R. Heath, S. C. O'Brien, R. F. Curl and R. E. Smalley, Nature, 1985, 318, 162–163 CrossRef CAS.
- S. H. Pun and Q. Miao, Acc. Chem. Res., 2018, 51, 1630–1642 CrossRef CAS PubMed.
- X.-Y. Wang, X. Yao and K. Müllen, Sci. China: Chem., 2019, 62, 1099–1144 CrossRef CAS.
- M. A. Majewski and M. Stepien, Angew. Chem., Int. Ed., 2019, 58, 86–116 CrossRef CAS PubMed.
- Q.-H. Guo, Y. Qiu, M.-X. Wang and J. F. Stoddart, Nat. Chem., 2021, 13, 402–419 CrossRef CAS PubMed.
- M. Hermann, D. Wassy and B. Esser, Angew. Chem., Int. Ed., 2021, 60, 2–26 CrossRef.
- Y. Li, H. Kono, T. Maekawa, Y. Segawa, A. Yagi and K. Itami, Acc. Mater. Res., 2021, 2, 681–691 CrossRef CAS.
- Z. Sun and K. Li, Synlett, 2021, 32, 1581–1587 CrossRef.
- L. Chao, I. A. Stepek, K. E. Yamada, H. Ito and K. Itami, Angew. Chem., Int. Ed., 2021, 60, 23508–23532 CrossRef PubMed.
- Z. Lu, J. Wang, X. Cheng, W. Xie, Z. Gao, X. Zhang, Y. Xu, D. Yi, Y. Yang, X. Wang and J. Yao, ACS Cent. Sci., 2022, 8, 905–914 CrossRef CAS PubMed.
- A. Omidvar, ACS Appl. Energy Mater., 2020, 3, 11463–11469 CrossRef CAS.
- S. Ramírez-Barroso, F. Romeo-Gella, J. M. Fernández-García, S. Feng, L. Martínez-Fernández, D. García-Fresnadillo, I. Corral, N. Martín and R. Wannemacher, Adv. Mater., 2023, 35, 2212064 CrossRef PubMed.
- X.-Q. Hou, Y.-T. Sun, L. Liu, S.-T. Wang, R.-L. Geng and X.-F. Shao, Chin. Chem. Lett., 2016, 27, 1166–1174 CrossRef CAS.
- J. M. Fernandez-Garcia, P. J. Evans, S. Medina Rivero, I. Fernandez, D. Garcia-Fresnadillo, J. Perles, J. Casado and N. Martin, J. Am. Chem. Soc., 2018, 140, 17188–17196 CrossRef CAS PubMed.
- K. Shoyama and F. Wurthner, J. Am. Chem. Soc., 2019, 141, 13008–13012 CrossRef CAS PubMed.
- M. C. Stuparu, Acc. Chem. Res., 2021, 54, 2858–2870 CrossRef CAS PubMed.
- B. Li, C. Yang, X. Wang, G. Li, W. Peng, H. Xiao, S. Luo, S. Xie, J. Wu and Z. Zeng, Angew. Chem., Int. Ed., 2021, 60, 19790–19796 CrossRef CAS PubMed.
- K. Nakamura, K. Ochiai, A. Yubuta, D. He, D. Miyajima and S. Ito, Precis. Chem., 2023, 1, 29–33 CrossRef CAS.
- M. Baumgarten, L. Gherghel, M. Wagner, A. Weitz, M. Rabinovitz, P. C. Cheng and L. T. Scott, J. Am. Chem. Soc., 1995, 117, 6254–6257 CrossRef CAS.
- A. V. Zabula, A. S. Filatov, S. N. Spisak, A. Y. Rogachev and M. A. Petrukhina, Science, 2011, 333, 1008–1011 CrossRef CAS PubMed.
- A. V. Zabula, S. N. Spisak, A. S. Filatov, A. Y. Rogachev and M. A. Petrukhina, Acc. Chem. Res., 2018, 51, 1541–1549 CrossRef CAS PubMed.
- H. Sakurai, T. Daiko and T. Hirao, Science, 2003, 301, 1878 CrossRef CAS PubMed.
- H. Sakurai, T. Daiko, H. Sakane, T. Amaya and T. Hirao, J. Am. Chem. Soc., 2005, 127, 11580–11581 CrossRef CAS PubMed.
- S. N. Spisak, Z. Wei, N. J. O'Neil, A. Y. Rogachev, T. Amaya, T. Hirao and M. A. Petrukhina, J. Am. Chem. Soc., 2015, 137, 9768–9771 CrossRef CAS PubMed.
- S. N. Spisak, Z. Wei, A. Y. Rogachev, T. Amaya, T. Hirao and M. A. Petrukhina, Angew. Chem., Int. Ed., 2017, 56, 2582–2587 CrossRef CAS PubMed.
- J. Hauausmann, Ber. Dtsch. Chem. Ges., 1889, 22, 2019 CrossRef.
- F. S. Kipping, J. Chem. Soc., 1894, 269, 269–290 RSC.
- K. Shi, J.-Y. Wang and J. Pei, Chem. Rec., 2015, 15, 52–72 CrossRef CAS PubMed.
- F. Goubard and F. Dumur, RSC Adv., 2015, 5, 3521–3551 RSC.
- W. Huang, E. Smarsly, J. Han, M. Bender, K. Seehafer, I. Wacker, R. R. Schröder and U. H. F. Bunz, ACS Appl. Mater. Interfaces, 2017, 9, 3068–3074 CrossRef CAS PubMed.
- M. Echeverri, S. Gámez-Valenzuela, R. C. González-Cano, J. Guadalupe, S. Cortijo-Campos, J. T. López Navarrete, M. Iglesias, M. C. Ruiz Delgado and B. Gómez-Lor, Chem. Mater., 2019, 31, 6971–6978 CrossRef CAS.
- Q. Zhang, Y. Sun, H. Li, K. Tang, Y.-W. Zhong, D. Wang, Y. Guo and Y. Liu, Research, 2021, 2021, 9790705 CAS.
- S. Gomez-Esteban, M. Pezella, A. Domingo, G. Hennrich and B. Gómez-Lor, Chem. – Eur. J., 2013, 19, 16080–16086 CrossRef CAS PubMed.
- G. Zhang, V. Lami, F. Rominger, Y. Vaynzof and M. Mastalerz, Angew. Chem., Int. Ed., 2016, 55, 3977–3981 CrossRef CAS PubMed.
- S. A. Wagay, I. A. Rather and R. Ali, ChemistrySelect, 2019, 4, 12272–12288 CrossRef CAS.
- H. Jiang, N. Lai, J. Tang, X. Tian, F. Wei, W. Bai and Y. Xu, J. Photochem. Photobiol., A, 2019, 369, 195–201 CrossRef CAS.
- Z. Zhou and M. A. Petrukhina, Coord. Chem. Rev., 2023, 486, 215144 CrossRef CAS.
- Y. Zhang, Y. Zhu, D. Lan, S. H. Pun, Z. Zhou, Z. Wei, Y. Wang, H. K. Lee, C. Lin, J. Wang, M. A. Petrukhina, Q. Li and Q. Miao, J. Am. Chem. Soc., 2021, 143, 5231–5238 CrossRef CAS PubMed.
- Z. Zhou, Ö. Üngör, Z. Wei, M. Shatruk, A. Tsybizova, R. Gershoni-Poranne and M. A. Petrukhina, Inorg. Chem., 2021, 60, 14844–14853 CrossRef CAS PubMed.
- Z. Zhou, Z. Wei, X. Yao, X.-Y. Wang, K. Müllen, M. Jo, M. Shatruk and M. A. Petrukhina, Cryst. Growth Des., 2023, 23, 1189–1194 CrossRef CAS.
- A. Cardenas Valdivia, Y. Dai, F. Rambaldi, J. E. Barker, J. J. Dressler, Z. Zhou, Y. Zhu, Z. Wei, M. A. Petrukhina, M. M. Haley, F. Negri and J. Casado, Chem. – Eur. J., 2023, e202300388 CrossRef PubMed.
- M. Wu, H. Liu, H. Liu, T. Lu, S. Wang, G. Niu, L. Sui, F. Bai, B. Yang, K. Wang, X. Yang and B. Zou, J. Phys. Chem. Lett., 2022, 13, 2493–2499 CrossRef CAS PubMed.
- P. R. Prasad, H. B. Singh and R. J. Butcher, J. Chem. Sci., 2014, 126, 1311–1321 CrossRef CAS.
- R. Goddard, M. W. Haenel, W. C. Herndon, C. Krueger and M. Zander, J. Am. Chem. Soc., 1995, 117, 30–41 CrossRef CAS.
- A. V. Zabula, S. N. Spisak, A. S. Filatov, V. M. Grigoryants and M. A. Petrukhina, Chem. – Eur. J., 2012, 18, 6476–6484 CrossRef CAS PubMed.
- Z. Zhou, X.-Y. Wang, Z. Wei, K. Müllen and M. A. Petrukhina, Angew. Chem., Int. Ed., 2019, 58, 14969–14973 CrossRef CAS PubMed.
- Z. Zhou, Y. Zhu, Z. Wei, J. Bergner, C. Neiß, S. Doloczki, A. Görling, M. Kivala and M. A. Petrukhina, Chem. Commun., 2022, 58, 3206–3209 RSC.
- Z. Zhou, Z. Wei, Y. Tokimaru, S. Ito, K. Nozaki and M. A. Petrukhina, Angew. Chem., Int. Ed., 2019, 58, 12107–12111 CrossRef CAS PubMed.
- R. Gershoni-Poranne and A. Stanger, in Aromaticity: Modern Computational Methods and Applications, ed. I. Fernandez, Elsevier, 2021, pp. 99–154 Search PubMed.
- Q. Xu, M. A. Petrukhina and A. Y. Rogachev, Phys. Chem. Chem. Phys., 2017, 19, 21575–21583 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, X-ray crystallographic details, NMR, UV-vis, PL and TA spectroscopic data, computational details. CCDC 2289537–2289542. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc04885c |
| This journal is © The Royal Society of Chemistry 2023 |