
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMulticonfigurational photodynamics simulations reveal the mechanism of photodecarbonylations of cyclopropenones in explicit aqueous environments†
Daniel M.
Adrion
 ,
Waruni V.
Karunaratne
and
Steven A.
Lopez
*
,
Waruni V.
Karunaratne
and
Steven A.
Lopez
*
Department of Chemistry and Chemical Biology, Northeastern University, Boston, Massachusetts 02115, USA. E-mail: s.lopez@northeastern.edu
First published on 10th November 2023
Abstract
Gas-evolving photochemical reactions use light and mild conditions to access strained organic compounds irreversibly. Cyclopropenones are a class of light-responsive molecules used in bioorthogonal photoclick reactions; their excited-state decarbonylation reaction mechanisms are misunderstood due to their ultrafast (<100 femtosecond) lifetimes. We have combined multiconfigurational quantum mechanical (QM) calculations and non-adiabatic molecular dynamics (NAMD) simulations to uncover the excited-state mechanism of cyclopropenone and a photoprotected cyclooctyne-(COT)-precursor in gaseous and explicit aqueous environments. We explore the role of H-bonding with fully quantum mechanical explicitly solvated NAMD simulations for the decarbonylation reaction. The cyclopropenones pass through asynchronous conical intersections and have dynamically concerted photodecarbonylation mechanisms. The COT-precursor has a higher quantum yield of 55% than cyclopropenone (28%) because these trajectories prefer to break a σCC bond to avoid the strained trans-cyclooctene geometries. Our solvated simulations show an increased quantum yield (58%) for the systems studied here.
Introduction
Photochemistry is an effective means by which strained, energy-dense compounds can be accessed while facilitating green chemistry through mild reaction conditions. Gas-evolving reactions produce gases such as oxygen or carbon dioxide (CO2) and are utilized for processes such as the oxygen reduction reaction (ORR) and CO2 reduction.1–4 Photochemical decarbonylation has been utilized to promote cross-coupling and form biaryl compounds,5 promote synthetic pathways towards strained compounds such as tetrahedrane,6,7 and facilitate transition-metal-catalyzed C–C bond cleavage.1,2,8 Small concentrations of CO have been shown to have therapeutic effects, including it being used as an anti-inflammatory agent,9 and used to treat cancer and bacterial infections.9–12 CO delivery can be activated through irradiation of a photoactive CO-releasing molecule (CORM).13,14 Light enables spatiotemporal activation of different chemical processes, including energy storage in materials chemistry15–26 and pro-drugs in drug delivery.27–29 Reactive cycloaddends (e.g., cyclooctynes and 1,3-dipoles) can be accessed using light through a class of biorthogonal reactions known as ‘photoclick chemistry.’ Photoclick reactions require light-promoted activation to access the reactive species and a thermal strain-promoted cycloaddition reaction in the biochemical milieu. Strain-promoted and distortion-accelerated30 cycloadditions can occur without Cu(I) catalysts and benefit from high reaction rates.31 The first photoclick reaction was reported by Lin and co-workers in 2008 and involved the tetrazole-ene cycloaddition reaction.32–34 Upon irradiation, N2 is photochemically extruded from tetrazole and the remaining 1,3-dipole, nitrile imine can undergo reactions with 2π-cycloaddends. Other photoclick reactants include photoprotected cyclooctynes,35–42 syndones,43 azirines,34,43–46 Diels Alder reactions,47–53 and thiol click reactions.54–59Scheme 1 illustrates two examples of photoclick reactions reported by Song, Poloukhtine, and Wang.34,37,38,44,45,60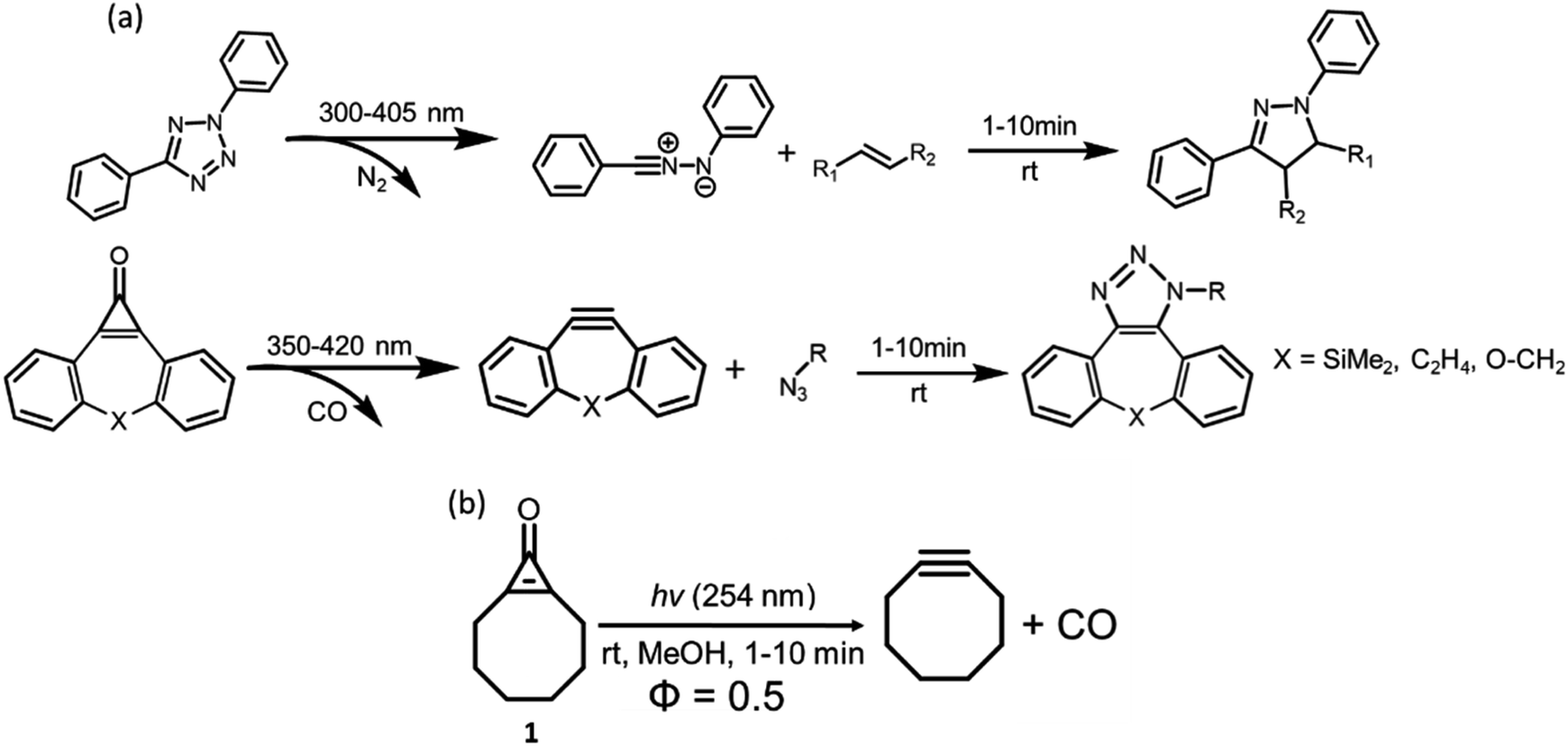 | ||
| Scheme 1 Examples of different photoclick reactions including tetrazole denitrogenation and cyclopropenone photodecarbonylation toward a strained alkyne (a). Photochemical decarbonylation reaction towards carbon monoxide and cyclooctyne from a cyclooctyne pre-cursor (1), (b). | ||
Functionalizing the vinyl positions of 1 with aryl groups redshifts the absorption wavelength to activate decarbonylation at 350 nm. The quantum yield (QY) of 4-dibenzocyclooctynol after irradiation with 350 nm light was measured to be 0.33 by Popik and co-workers.60 The substituent effects on both cyclopropenone (2) and cyclooctynol were also reported by Popik and co-workers, who used multi-photon absorption with a near-IR laser to generate reactive intermediates to undergo a strain-promoted alkyne–azide cycloaddition (SPAAC) reaction (i.e., photoclick reaction).61–63 Recently, Kunishima and co-workers have used a visible-light responsive photocatalyst to redshift the absorption wavelength required for the decarbonylation of substituted cyclopropenones.64,65
While the thermal cycloadditions involved in bio-orthogonal chemistry have been intensely researched by experimentalists36,66–68 and theorists,69–72 the photochemical steps of the reactions have received less attention. We hypothesize this is due to the challenge of experimentally studying molecular excited states and reactive intermediates in the typically short timescale of photochemical reactions (10−13 to 10−8 seconds). This timescale is too short for the atomistic resolution of excited-state geometries. Fortunately, multiconfigurational NAMD simulations offer high-fidelity structural information about the rapidly interconverting excited-state structures and mechanisms of organic photochemical reactions.
Computational approaches provide valuable structural information of 2 on the 1–10 picosecond timescale. Fang and co-workers investigated the photochemical decarbonylation of 2 in 2014 using gas phase multiconfigurational calculations (CASSCF and multistate-CASPT2) to mechanistic critical points on the S0 and S1 potential energy surfaces (PESs).73Ab initio multiple spawning (AIMS)74 non-adiabatic molecular dynamics (NAMD) simulations were performed to investigate the photochemical reaction dynamics along the S1 surface. The authors located one S1/S0 conical intersection, leading to the dissociation mechanism in 72% of trajectories. The authors also located a second S1/S0 conical intersection that reverts to the reactant (cyclopropenone) in 93% of trajectories. Fang reports an S1 lifetime of 125 fs for 2. This past study only simulated cyclopropenone in the gas phase. To best represent biological environments, the simulation conditions should be as close to the experimental conditions as possible (i.e., replicating solvent and temperature). We now compute the vertical excitation energies, absorption spectra, photochemical mechanism, and quantum yields for 1 and cyclopropenone (2). The NAMD simulations are performed with multiconfigurational quantum chemical calculations in the gas phase and explicitly solvated aqueous environments. We compare our results with those presented in the 2003 work by Poloukhtine and Popik.75
Results and discussion
Cyclopropenone is the prototypical pseudo-aromatic molecule on the ground state. We first computed the ground state geometries of 1, 2, and cyclopropenone (3) with density functional theory (DFT) calculations (Fig. 1).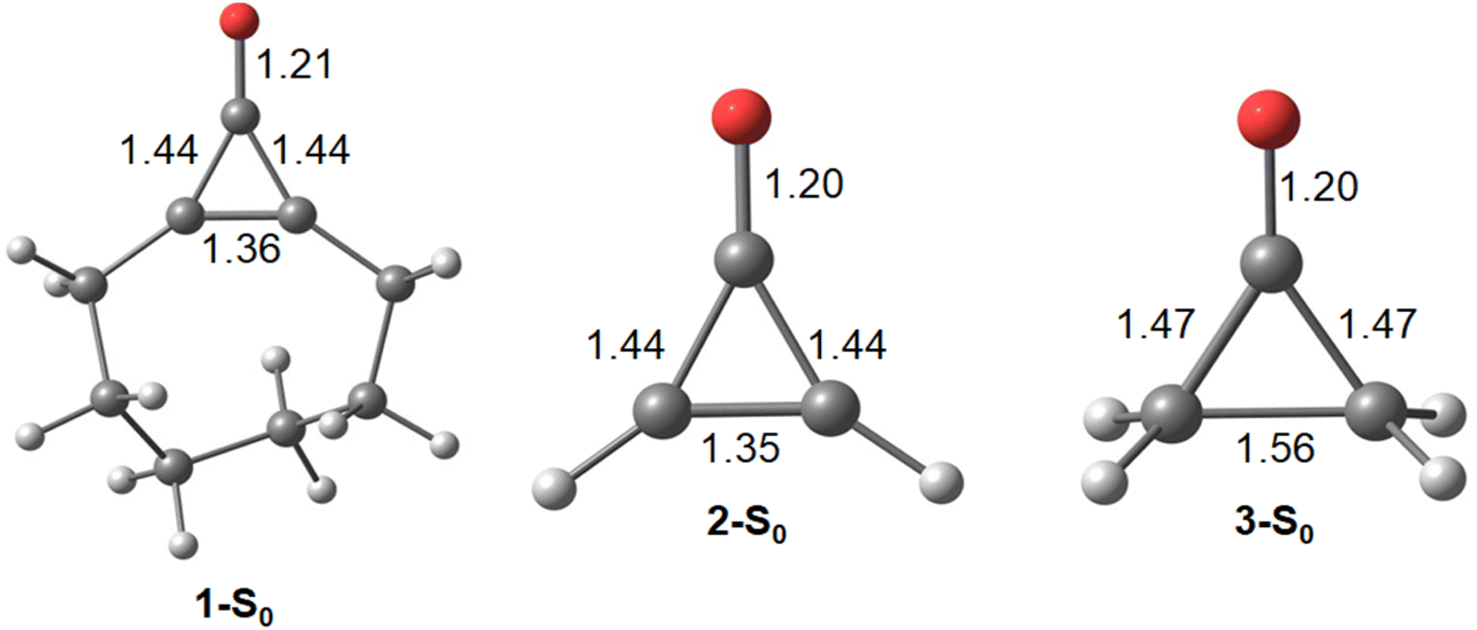 | ||
| Fig. 1 Optimized S0 global minimum geometries for 1-S0, 2-S0, and 3-S0. Bond lengths are presented in Angstroms (Å). All structures were optimized using PBE0-D3BJ/cc-pVDZ. | ||
We first compare the πCO bondlengths of the pseudo-aromatic cyclopropenones (1-S0, 2-S0); they are nearly identical (1.20 and 1.21 Å, respectively). The 1-S0 and 2-S0σCC bonds are 1.44 Å in 1-S0 and 2-S0. The relatively short σCC bonds in 1-S0 and 2-S0 is consistent with conjugated πCC and πCO. 3-S0 is non-aromatic and has notably longer σCC bonds (1.47 and 1.56 Å); the πCO bondlength is 1.20 Å. Next, we computed the excitation energies and electronic transitions of 1 and 2 with time-dependent density functional theory (TD-DFT) and multiconfigurational complete active space self-consistent field (CASSCF). We used extended multi-state complete active space second-order perturbation theory (XMS-CASPT2) to account for dynamic correlation of the electronic states.76 Our TD-DFT calculations included the lowest 10 singlet excited states and used the range-separated density functional CAM-B3LYP77 with the triple-ζ aug-cc-pVTZ78,79 basis set. We included 10 electrons and 9 orbitals in the CASSCF active space for 1 and 2; Fig. 2 shows the orbitals and average orbital occupancies for 2. The same active space was used for 1.
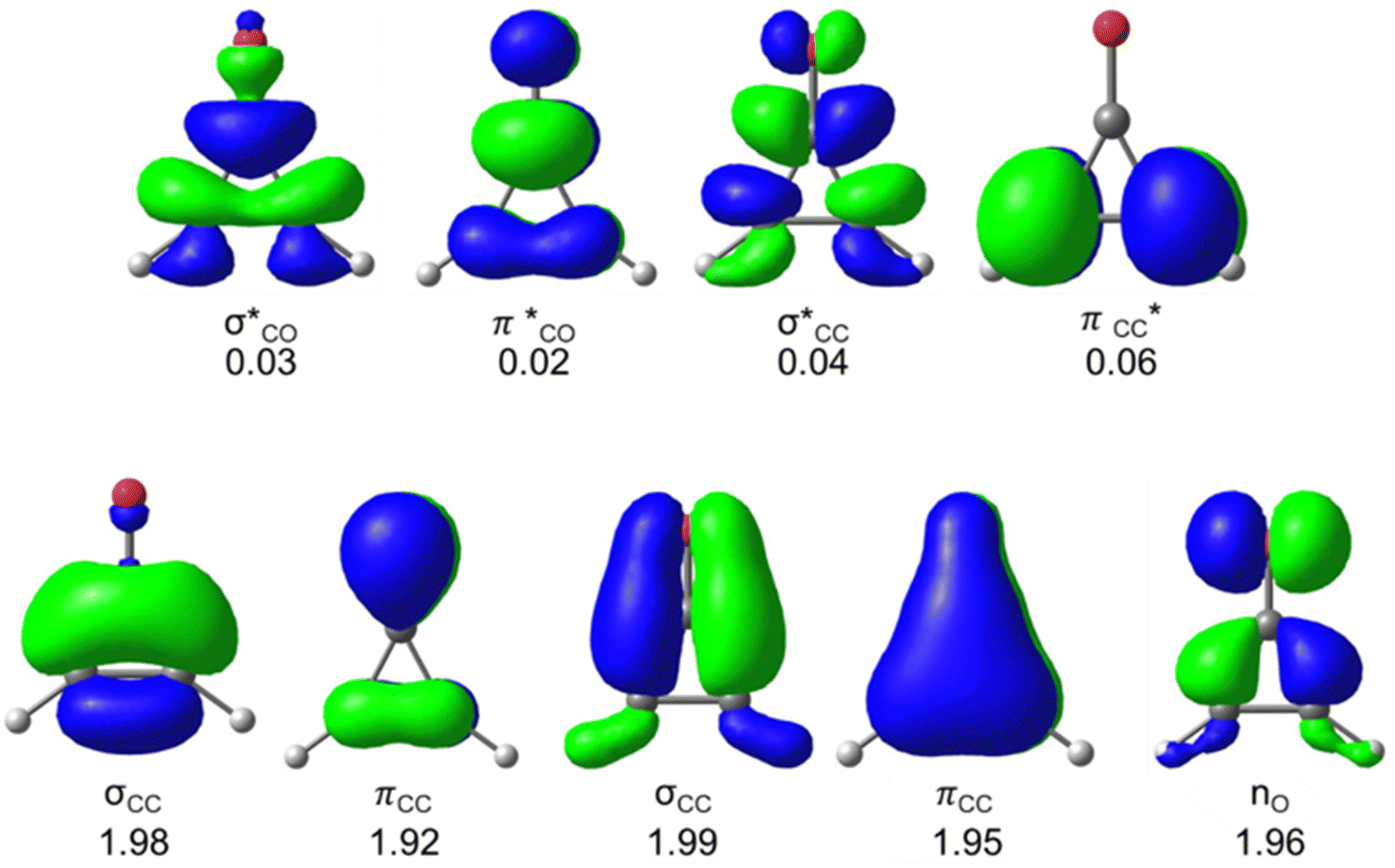 | ||
| Fig. 2 CASSCF(10,9) active space of 2 with average electron occupancies. Orbitals are shown with an isosurface value of 0.06 and computed with SA(4)-CASSCF(10,9)/ANO-S-VDZP. The excitations, oscillator strengths, and electronic transition characters for 1 and 2 were computed with SA(4)-CASSCF(10,9)/ANO-VDZP, SA(4)-XMS-CASPT2(10,9)/ANO-S-VDZP, and TD-DFT (Tables 1 and 2). | ||
| Method | State | Energy (eV) | Wavelength (nm) | Oscillator strength | Nature |
|---|---|---|---|---|---|
| CAM-B3LYP/aug-cc-pVTZ | S1 | 4.80 | 258 | 0.0008 | nO → π* (S1) |
| S2 | 5.42 | 229 | 0.0001 | nO → π* (S2) | |
| S3 | 5.58 | 222 | 0.0002 | π → Ryd (S3) | |
| XMS-CASPT2(10,9)/ANO-S-VDZP | S1 | 4.93 | 281 | nO → π* (S1) | |
| S2 | 5.66 | 248 | nO → π* (S2) | ||
| S3 | 7.21 | 167 | π → π* (S3) | ||
| SA(4)-CASSCF(10,9)/ANO-S-VDZP | S1 | 5.13 | 242 | 0.0086 | nO → π* (S1) |
| S2 | 5.75 | 216 | 0.0000 | nO → π* (S2) | |
| S3 | 7.38 | 168 | 0.0690 | π → π* (S3) |
| Method | State | Energy (eV) | Wavelength (nm) | Oscillator strength | Nature |
|---|---|---|---|---|---|
| CAM-B3LYP/aug-cc-pVTZ | S1 | 4.36 | 284 | 0.0004 | nO → π* (S1) |
| S2 | 5.46 | 227 | 0.0000 | nO → π* (S2) | |
| S3 | 6.20 | 200 | 0.0062 | π → Ryd (S3) | |
| XMS-CASPT2(10,9)/ANO-S-VDZP | S1 | 4.52 | 274 | nO → π* (S1) | |
| S2 | 5.68 | 218 | nO → π* (S2) | ||
| S3 | 7.14 | 174 | π → π* (S3) | ||
| SA(4)-CASSCF(10,9)/ANO-S-VDZP | S1 | 4.61 | 269 | 0.0069 | nO → π* (S1) |
| S2 | 5.70 | 218 | 0.0000 | nO → π* (S2) | |
| S3 | 6.95 | 178 | 0.0484 | π → π* (S3) |
The S0 → S1 excitation energies computed with XMS-CASPT2(10,9)/ANO-S-VDZP for 1 and 2 are 4.42 and 4.52 eV, respectively. Both transitions are n → π* and have oscillator strengths of 0.0094 (1) and 0.0069 (2). The S0 → S2 excitations have energies of 5.00 eV (1) and 5.68 eV (2); both are π → π* transitions. The oscillator strengths of these transitions are 0 for 1 and 2. The S0 → S3 electronic excitations of 1 and 2 are π → π* transitions, with excitation energies and oscillator strengths of 7.42 eV and 7.14 eV and 0.07 and 0.05, respectively. We generated an ensemble of 500 Wigner-sampled non-equilibrium geometries, computed the S0 →Sn (n = 1,2,3) vertical excitation energies, and oscillator strengths to generate an absorption spectrum for 1 and 2. Fig. 3 shows an overlay of select non-equilibrium geometries for 1 and 2 and the absorption spectrum; the intensities are normalized for the S0 → S3 transition.
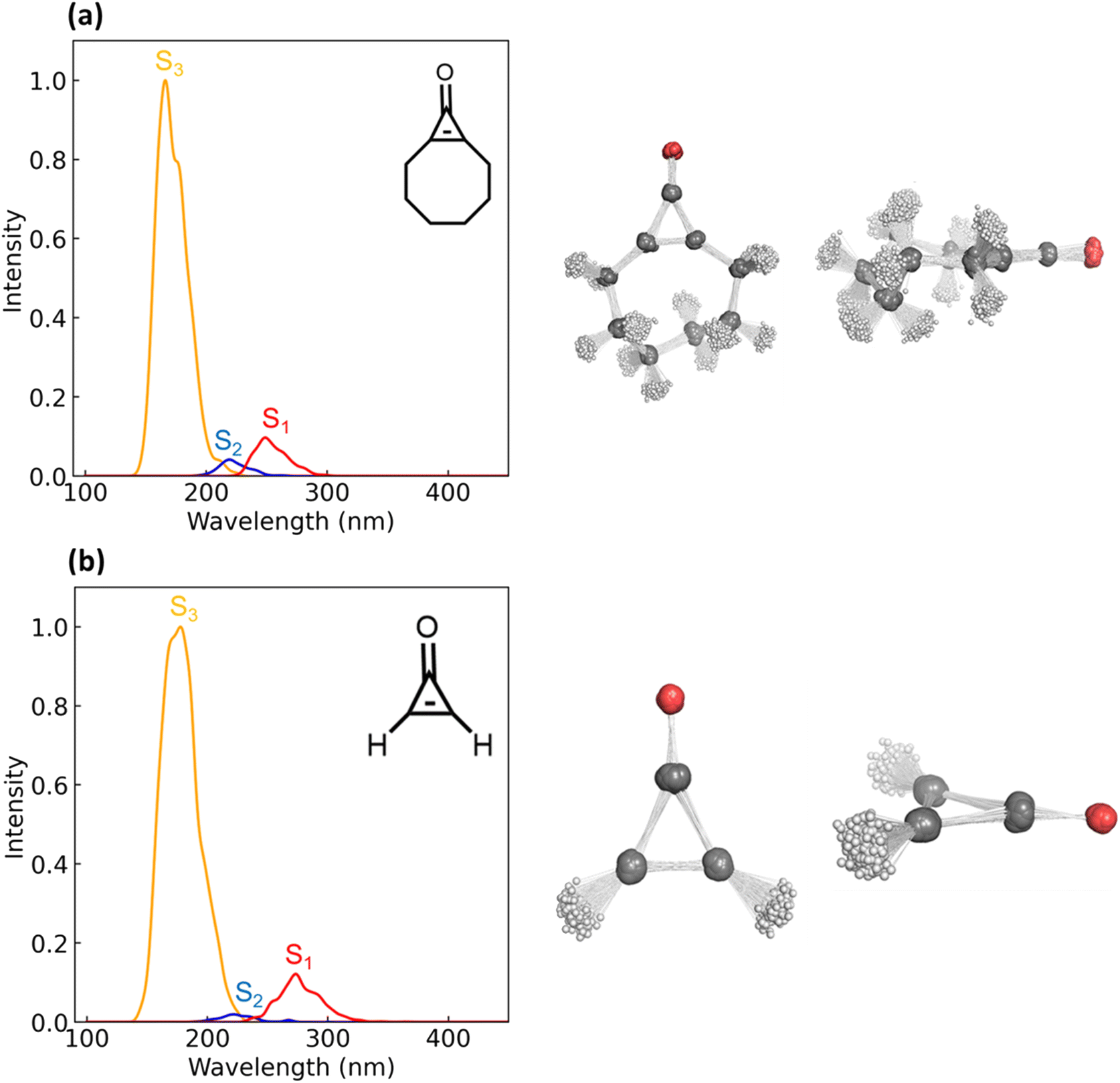 | ||
| Fig. 3 Absorption spectra and geometric overlays corresponding to Wigner sampled geometries of 1 (a) and 2 (b). Both spectra were computed in the gas-phase using SA(4)-CASSCF(10,9)/ANO-S-VDZP, and the Wigner sampled geometries were generated from using the vibrational frequencies of a ground-state geometry optimized with SA(4)-CASSCF(10,9)/ANO-S-VDZP. | ||
The absorption spectra for 1 and 2 show three λmax peaks centered at 190, 220, and 260 nm. The peak centered at 190 nm corresponds to a π → π* transition; the λmax peaks centered at 220 and 260 nm correspond to n → π* transitions. The λmax peaks near 190 nm correspond to S0 → S3 excitations and have the highest intensity because they are ‘allowed’ transitions. The n → π* transition at 220 nm corresponds to an S0 → S2 excitation and has a relatively low intensity. The S0 → S1 excitation for 1 and 2 (n → π*) has a peak centered near 260 nm. While the intensity of this peak (shown in red) is substantially lower than that of the π → π* transition, it is sufficiently low in energy to undergo photoexcitation with the experimental light source (254 nm). The peaks corresponding to the second-and third-lowest excited states (shown in blue and yellow, respectively) correspond to excitation energies higher than the experimental light source. As such, we decided to focus on the S1-state to explore the static and dynamic components of the photo-decarbonylation reaction mechanism of 1 and 2.
Cyclopropenone photodissociation mechanism
We first performed a minimum energy path (MEP) calculation along the S1-state to determine the dominant mechanistic pathway from the FC-point for 1 and 2. Fig. 4a and b show the MEP, S0- and S1-minimum geometries, and the final structure of the MEP for 1 and 2.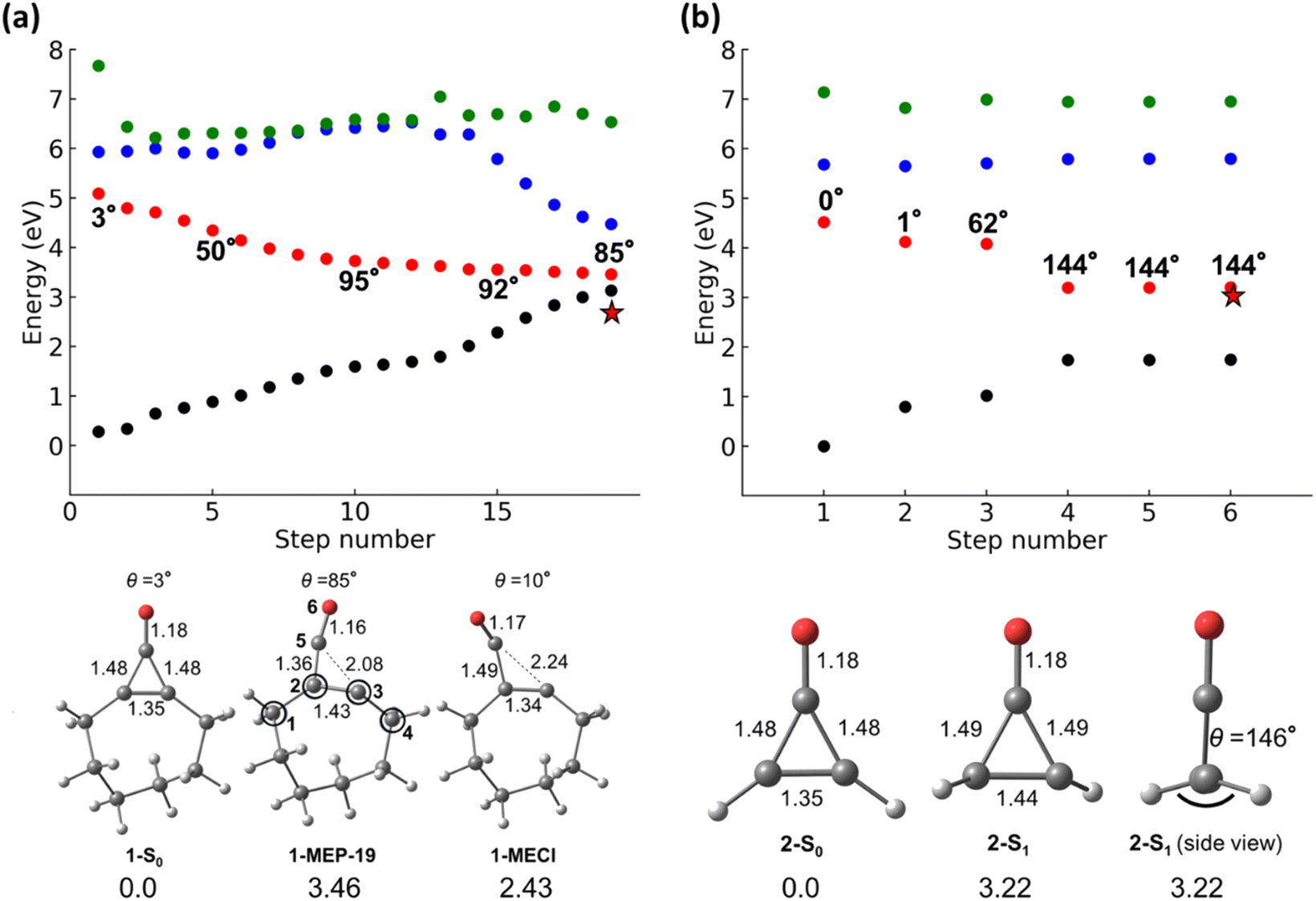 | ||
| Fig. 4 Minimum energy path calculations for 1 (a) and 2 (b). The dots on the graphs correspond to the S0 (black), S1 (red), S2 (blue), and S3 (green) states. Relevant structures (S0 and S1 minima, MECI structures, and endpoints of the MEP calculations) are shown below the plots. 1-MECI (left) and 2-S1 (right) are shown on the plots with red stars. For both optimized points, the final MEP geometry from the calculation was used as the initial guess structure. The MEP calculations were run with SA(4)-CASSCF(10,9)/ANO-S-VDZP, and SA(4)-XMSCASPT2(10,9) single-point energy corrections were performed on each MEP step. All optimized structures were performed with SA(4)-CASSCF(10,9)/ANO-S-VDZP with SA(4)-XMS-CASPT2(10,9) energy corrections. | ||
Fig. 4a shows the MEP along S1-surface and the energies for the corresponding S0–3 states; it contains 19 geometries. This MEP demonstrates that the S0 and S1 states approach a degeneracy by the 19th step (S1–S0 energy gap of 0.33 eV) for 1. The steps after the FC-point reflect the geometrical changes resulting from the n → π* electronic transition. We defined a torsional angle parameter, θ, across C1–C2–C3–C4 for 1 and 2 (atoms labels are given in Fig. 4). 1-S0 and 2-S0 are nearly planar (θ = 3 and 0°, respectively). θ increases from θ0 = 3° to θ25 = 80°. The πC2–C3 bond length increases from 1.35 Å to 1.43 Å, and C5 is visibly pyramidalized in 1-MEP-19 (O–C5–C2 angle of 169°). The pyramidalization of C5 is accompanied by a shortening of the C5![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond (1.18 to 1.16 Å), which resembles a C5
O bond (1.18 to 1.16 Å), which resembles a C5![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O triple bond. The C5–C3 bond is nearly broken in 1-MEP-19 (2.08 Å), suggesting different mechanisms for 1 and 2.
O triple bond. The C5–C3 bond is nearly broken in 1-MEP-19 (2.08 Å), suggesting different mechanisms for 1 and 2.
The S1–S0 energy gap at the end of MEP calculations can suggest the dominant reaction mechanism in photochemical reactions. Large energy gaps suggest an S1-minimum and radiative decay to S0; small energy gaps suggest a degeneracy region, indicating a possible productive pathway to photoproducts. Unlike the large S1–S0 energy gap observed in 2-MEP-6, 1-MEP-19 has a relatively small S1–S0 energy gap (0.33 eV). Prompted us to locate an S1/S0 minimum energy conical intersection (1-MECI). 1-MECI is 2.43 eV above 2-S0 and has C3–C5 and C2–C5 bond lengths of 2.24 and 1.49 Å, respectively. Although there is a broken C–C bond in the cyclopropenone ring, the alkene carbons (C2 and C3) are slightly pyramidalized (θ = 10°).
Fig. 4b shows the MEP along the S1–3 surfaces for 2. The MEP includes 6 steps and leads to a final structure (2-MEP-6) with an S1–S0 energy gap of 1.46 eV, substantially larger than that of 1-MEP-19 (S1–S0 energy gap of 0.33 eV). The large S1–S0 energy gap of 2-MEP-6 suggests that the pseudo-dominant mechanistic pathway for 2 is towards an S1-minimum. We optimized an S1 minimum using the final MEP geometry as an input (shown in Fig. 4b). 2-S1 is 3.22 eV above 2-S0 and nearly degenerate with 2-MEP-6. The πCC of 2-S1 is pyramidalized, and the vinyl-hydrogens adopt a trans-relationship.
The S1-MEPs of 1 and 2 lead to a conical intersection and an S1-minimum, respectively. While both molecules are theoretically capable of undergoing cis–trans isomerization, this reaction is highly disfavored in 1 because of the resulting trans-cyclooctene, which is highly strained. Trans-cyclooctene is higher in energy than cis-cyclooctene by 16 kcal mol−1.802, however, avoids the additional strain energy and undergoes the cis–trans isomerization to 2-S1. Although MEP calculations provide mechanistic information, they omit dynamical effects, which control photochemical reaction mechanisms and quantum yields. As such, we performed NAMD simulations starting from the S1 FC region for 1 and 2 to enumerate all possible mechanistic pathways.
Non-adiabatic molecular dynamics simulations
We performed 1 picosecond (ps) NAMD simulations using the FSSH algorithm81,82 on 1 and 2, using Wigner-sampled initial conditions. For 1, 497 of the trajectories crossed to the S0 after 1 ps (99%), while 3 (1%) remained on the S1. Of the 497 trajectories that passed through S1/S0 hopping points, 274 underwent decarbonylation (Φcomp = 55%). The remaining 223 trajectories (45%) reverted to 1. For 2, 464 trajectories crossed to the ground state after 1 ps. 130 of the 464 trajectories corresponded to the decarbonylation reaction (Φcomp = 28%), while 334 trajectories (72%) reverted to cyclopropenone. We plotted the two σCC bond lengths in 1 and 2 to determine whether the reaction was concerted or stepwise. Fig. 5 shows these bond lengths throughout the 1 ps trajectories; the black dots correspond to the S1/S0 hopping points.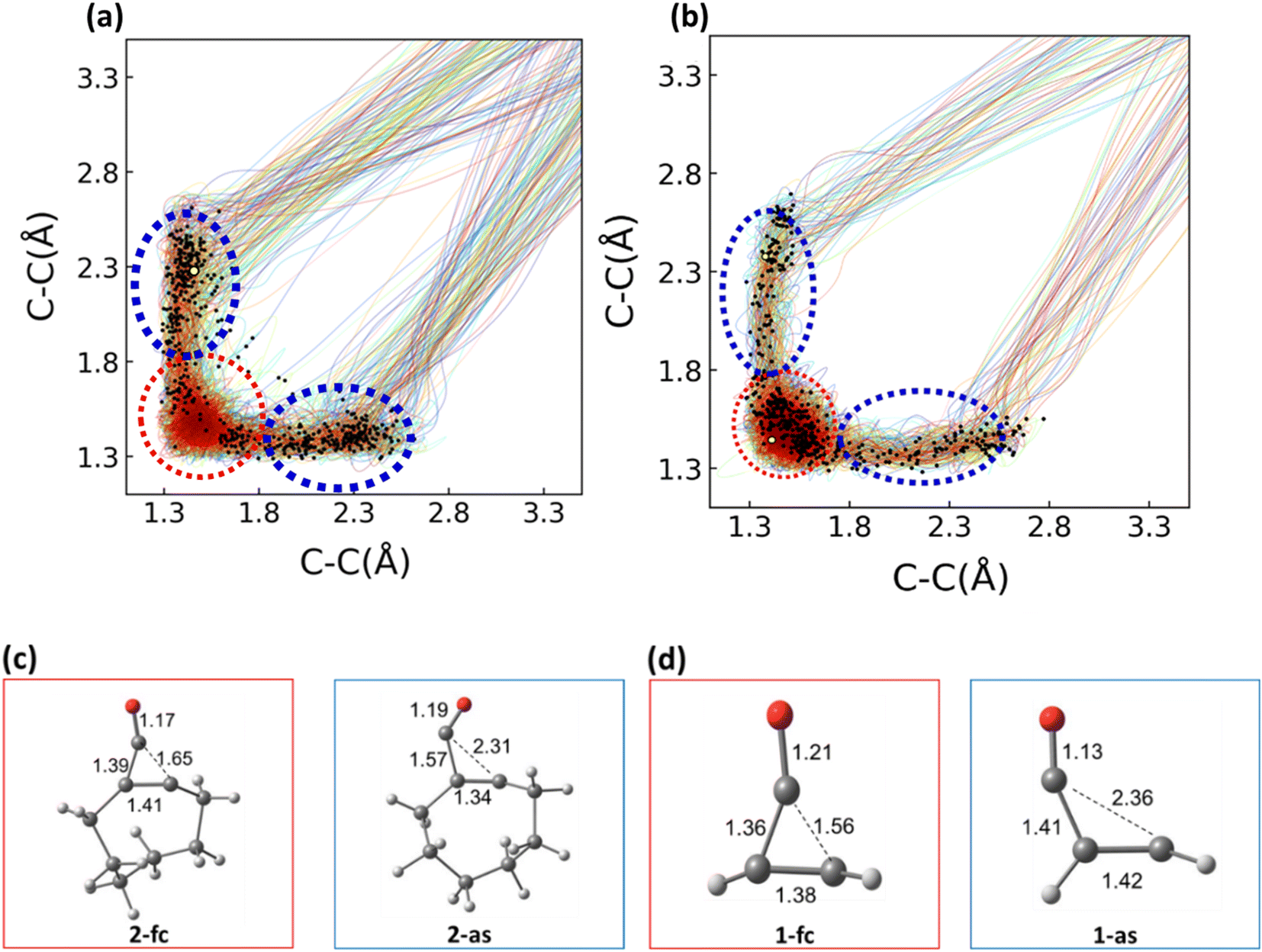 | ||
| Fig. 5 C–C bonds plotted against each other for 1 (a) and 2 (b). The bond lengths we calculated are depicted by red dotted lines. The solid multicolor lines on each plot show the bond lengths over time, and the black dots represent the S1/S0 surface hopping points for each trajectory. The minimum energy conical intersections (MECIs) are marked on the plots with a yellow point. Representative hopping points are given for the reactant (red) and asynchronous (blue) regions for 1 (c) and 2 (d). Hopping points from the FC region have the suffix ‘fc’ and hopping points from the asynchronous region have the suffix ‘as’. NAMD simulations were run with SA(4)-CASSCF(10,9)/ANO-S-VDZP. 500 initial conditions were sampled by an S0 Wigner distribution. Trajectory simulations were run for 1 ps with a 0.5 fs timestep. The hopping points in (c) and (d) are snapshots obtained directly from the NAMD simulations. | ||
Fig. 5 shows the σCC distances throughout each of the trajectories. Examples of hopping point structures from both seam regions are shown in Fig. 5c and d. Both the asynchronous hopping point geometries (1-as and 2-as) contain one σCC bond in the cyclopropenone ring that is fully broken (2.31 Å and 2.36 Å, respectively). In contrast, the other cyclopropenone σCC bond is lengthened from 1-S0 (1.57 and 1.41 Å, respectively). All trajectories with σCC bonds exceeding 3.3 Å were considered dissociated. The QYs for the dissociation reaction of 1 and 2 are 55% and 28%, respectively. Optimized MECI structures for 1 and 2 are shown as yellow points on both plots, and the black dots represent S1/S0 hopping points. The S1/S0 hopping points for 1 and 2 have a large range of σCC bond distances (1.36–2.77 Å). We grouped the hopping points structures based on their structural similarities. The blue region corresponds to hopping points with at least one σCC > 1.80 Å and are labeled asynchronous because the breaking σCC distances are different by 0.5 Å. The hopping points in the red region correspond to structures with both σCC bonds <1.80 Å (labeled FC-region).
We computed the branching ratios for asynchronous and FC-region hopping points to determine if the hopping points' structures influenced the QYs of each reaction. Only 87 (18%) of the 497 NAMD trajectories of 1 passed through a FC-region hopping point. All 87 trajectories that passed through a hopping point in the FC-region reformed 1. The remaining 410 (82%) NAMD trajectories passed through asynchronous hopping points. The strain energy associated witxh trans-cyclooctene geometries redirects many of the trajectories (82%) away from the FC-region conical intersection seam and towards through more productive asynchronous conical intersection seam. These hopping points in the asynchronous region 274 (67%) dissociate, while 136 (33%) reformed the reactant. 269 (58%) of the 464 trajectories of 2 passed through a FC-region hopping point, and 195 (42%) passed through asynchronous hopping points. Of those 269 FC-region hopping points, 262 hopping points revert to the reactant, and only 7 trajectories dissociate to the product. 123 of the trajectories (63%) from the asynchronous region undergo photo-decarbonylation dissociate, and 72 trajectories (37%) reform the reactant. For 1 and 2, there is an overwhelming preference to reform the reactant after surface hopping occurs in the FC-region, and a ∼2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 preference to dissociate after surface hopping occurs in the asynchronous region. We hypothesize that the preference for reactant reformation is due to the energy required to break the cyclopropenone ring structure on the ground state. We also present the branching space for an MECI optimized along the FC-region of the S1/S0 hopping seam in the ESI.† For trajectories that hop in the asynchronous region, there is a slight preference for breaking the second C–C bond on the S0 state after hopping, as these bonds are already extended in the excited state.
:1 preference to dissociate after surface hopping occurs in the asynchronous region. We hypothesize that the preference for reactant reformation is due to the energy required to break the cyclopropenone ring structure on the ground state. We also present the branching space for an MECI optimized along the FC-region of the S1/S0 hopping seam in the ESI.† For trajectories that hop in the asynchronous region, there is a slight preference for breaking the second C–C bond on the S0 state after hopping, as these bonds are already extended in the excited state.
The θ values of 1-fc and 1-as are 95° and 74°, respectively. This hopping point preference for 1 is nearly double that of 2 (42%). This agrees with the MEP results in Fig. 4, which suggest a direct path to an asynchronous S1/S0 hopping point (one broken σCC bond in the cyclopropenone ring). We find an overwhelming preference for a non-productive reaction outcome from hopping points in the FC-region (100% and 97% for 1 and 2, respectively). Our interest in understanding 2 as a bioorthogonal photoclick reagent drove us to understand the role of solvation on the QYs and mechanisms. To that end, we generated solvated initial conditions for 1 and 2 and ran 250 fs NAMD simulations from their respective S1 FC-regions.
Explicitly solvated NAMD simulations
We used an explicitly solvated fully quantum mechanical NAMD simulations to determine the photodecarbonylation mechanism of 1 and 2 because multiscale QM/MM simulations cannot capture the electronic effect of intermolecular hydrogen bonding between the water molecules and the cyclopropenones. Historically, dynamics simulations involving solvent utilize a multiscale approach where the chromophore is treated with QM method, and the surrounding environment is treated using MM. QM/MM approaches are particularly useful for calculating large systems, which generally include proteins and solvent molecules. To compare to the explicitly solvated all-QM method, we used a QM/MM approach for simulating the photochemical pathways along the S1 for 1 and 2; the presentation and discussion of these results are given in the ESI.† Initial conditions were sampled in line with the report by Gonzalez and co-workers last year.83 We observed an increased quantum yield for the decarbonylation reaction for both systems compared to the gas-phase simulations (58% for 1 and 2).We performed 250 fs NAMD simulations on solvated spheres (20 water molecules) of 1 and 2, starting with initial conditions sampled using MD simulations in CP2K. The NAMD simulations utilized the FSSH surface hopping algorithm and were propagated from the S1 state. The σCC bond lengths in the cyclopropenone ring were measured throughout the trajectories and are represented in Fig. 6.
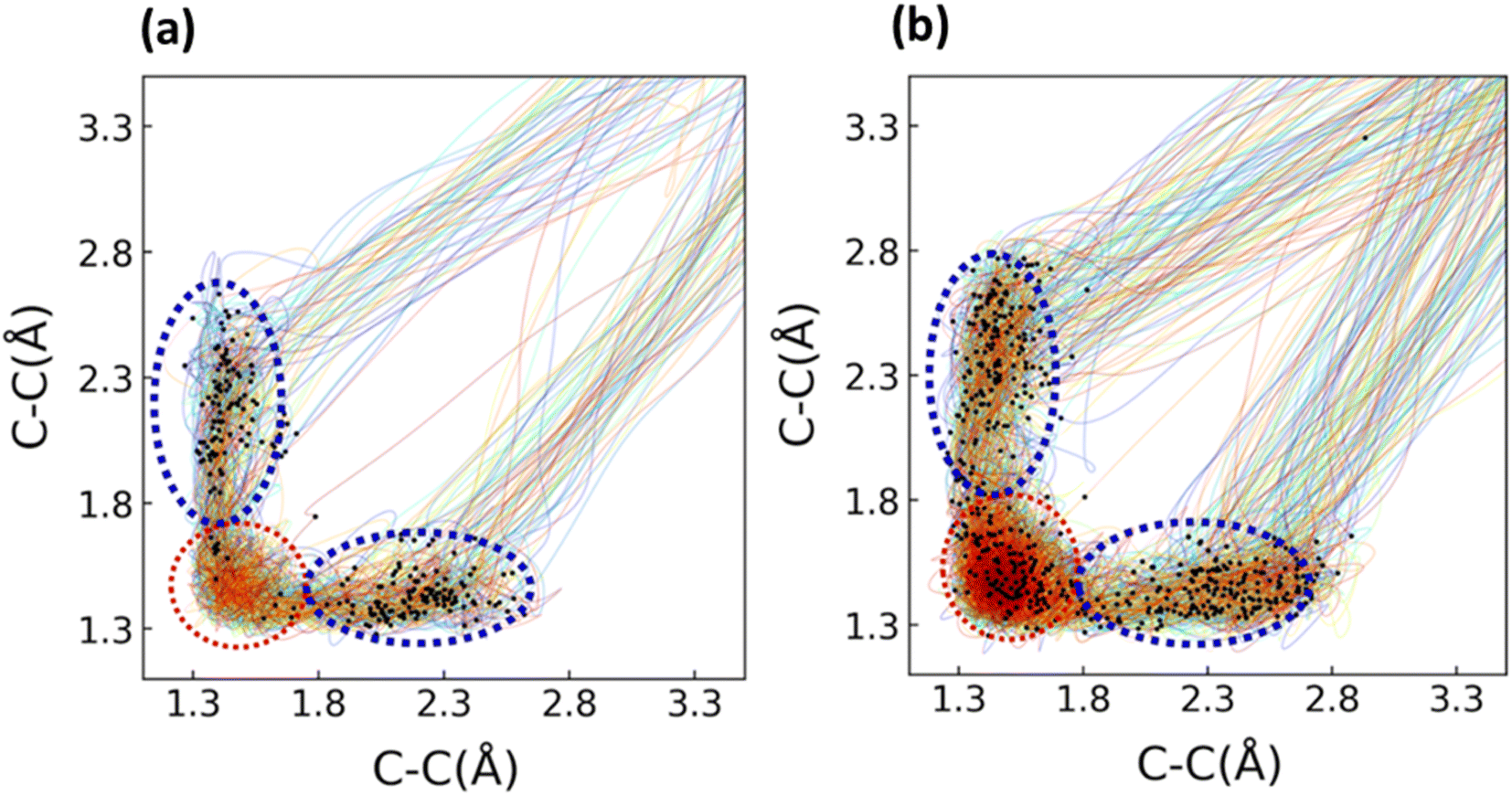 | ||
| Fig. 6 Trajectory plots of 1 (a) and 2 (b) were solvated with 20 water molecules. The bonds being measured over are the breaking C–C σ bonds in the cyclopropenone ring. The NAMD simulations were run using SA(4)-CASSCF(10,9). The ANO-S-VDZP basis set was used for the chromophores 1 and 2, and the ANO-S-MB was used for the water molecules. The S1/S0 crossing seams for the solvated systems of 1 and 2 are indicated with black dots. The distribution of S1/S0 hopping points for 1 and 2 have a large range of σCC bond distances (1.42–3.25 Å). For 1, there were 276 initial conditions propagated from the S1. Of these 276, 253 crossed to the S0 by the end of the simulation. Of these 253, there were 19 trajectories where the endpoint was an intermediate structure on the ground state (one σCC bond in the cyclopropenone ring >1.80 Å and the other σCC bond in the cyclopropenone ring <1.60 Å). These intermediate structures were not counted in the final quantum yield. An example intermediate structure is provided in the ESI.† Of the remaining 234 trajectories, we observed 135 trajectories that underwent decarbonylation (Φcomp = 58%) and 99 that reformed the reactant (42%). For the solvated system of 2, we ran simulations on 481 initial conditions. Of these 481, 20 trajectories (4%) ended at an intermediate structure on the ground state (one σCC bond in the cyclopropenone ring >1.80 Å and the other σCC bond in the cyclopropenone ring <1.60 Å). 277 trajectories underwent decarbonylation (Φcomp = 58%), and 173 trajectories (36%) reformed the reactant. We also calculated the ratio of trajectories for 1 and 2 that crossed through the asynchronous (one σCC bond of 1.80 Å or longer) and FC-regions (both σCC bonds less than 1.80 Å) of the S1/S0 crossing seam. For the solvated system of 1, 237 trajectories (94%) went through a hopping point in the asynchronous region, while 16 (6%) crossed at a hopping point in the FC-region. There were 114 trajectories from the asynchronous hopping region that dissociated (48%) and 88 trajectories that reformed the reactant (37%). From the FC-region of the solvated system of 1, 9 trajectories formed the decarbonylation product (56%), and 4 reformed the reactant (25%). For the solvated system of 2, there were 337 trajectories (70%) that crossed through hopping points in the asynchronous region (one σCC bond of 1.80 Å or longer), while 144 trajectories (30%) crossed through hopping points in the FC-region. From the asynchronous region, 198 trajectories (59%) went on to form the products, whereas 116 (34%) reformed the reactant structure. The 20 trajectories with intermediate structures (6%) exclusively went through hopping points in the asynchronous region. We located 78 (54%) trajectories from the FC region that showed decarbonylation and 57 (40%) which reformed the reactant. The hopping point structures from each region of the hopping seam resemble those from the gas phase simulations; the asynchronous hopping points feature a broken σCC bond in the cyclopropenone ring, whereas the FC region contains points where both bonds are still intact. | ||
The inclusion of an explicit solvent in our NAMD simulations resulted in the Φcomp for the dissociation products increasing from 53% to 58% for 1, and from 28% to 58% for 2. This increased yield can be explained by the ratio of trajectories going through the asynchronous and FC regions of the S1/S0 seam. For the solvated NAMD simulations of 1, nearly all trajectories crossed to the S0 in the asynchronous region (94%), corresponding to a 12% increase compared to the gas-phase simulations. While the decarbonylation yield from the asynchronous region is smaller than for the gas-phase simulations (48% and 64%, respectively), the overwhelming preference for the asynchronous region ultimately leads to a higher quantum yield. From the FC-region of 1, there was a larger number of trajectories (9) that dissociated than those that reformed the reactant (4), which was significantly different from the gas phase. For 2, the number of trajectories hopping in the asynchronous region increased for the solvated and gas-phase systems (70% and 52%, respectively); the quantum yield from the asynchronous region was slightly smaller for the solvated system compared to the gas-phase (59% and 63%, respectively), the higher ratio of trajectories crossing through this region leads to a higher Φcomp. To understand the solvent effect on the reactivities of 1 and 2, we hypothesized that hydrogen bonding intermolecular interactions between the solvent and the chromophores were important in the S0, S1-surface, and conical intersection seams.
Hydrogen bonding analysis of solvated trajectory simulations
We analyzed the hydrogen bonding throughout all the explicitly solvated NAMD trajectories 1 and 2 with the VMD software.84 We noted a hydrogen bond when the donor–acceptor distance (denoted as the Owater − Ocarbonyl distance) was 3.1 Å, and the donor–acceptor angle (Ocarbonyl − Hwater − Owater) was near linearity (25°). These criteria were chosen based on previous work examining and measuring intermolecular H-bonding.85–87Fig. 7 shows a snapshot for the solvated systems of 1 and 2 and the H bonding interaction between the carbonyl oxygen (the hydrogen bond acceptor) and a water molecule (the hydrogen bond donor) for 2.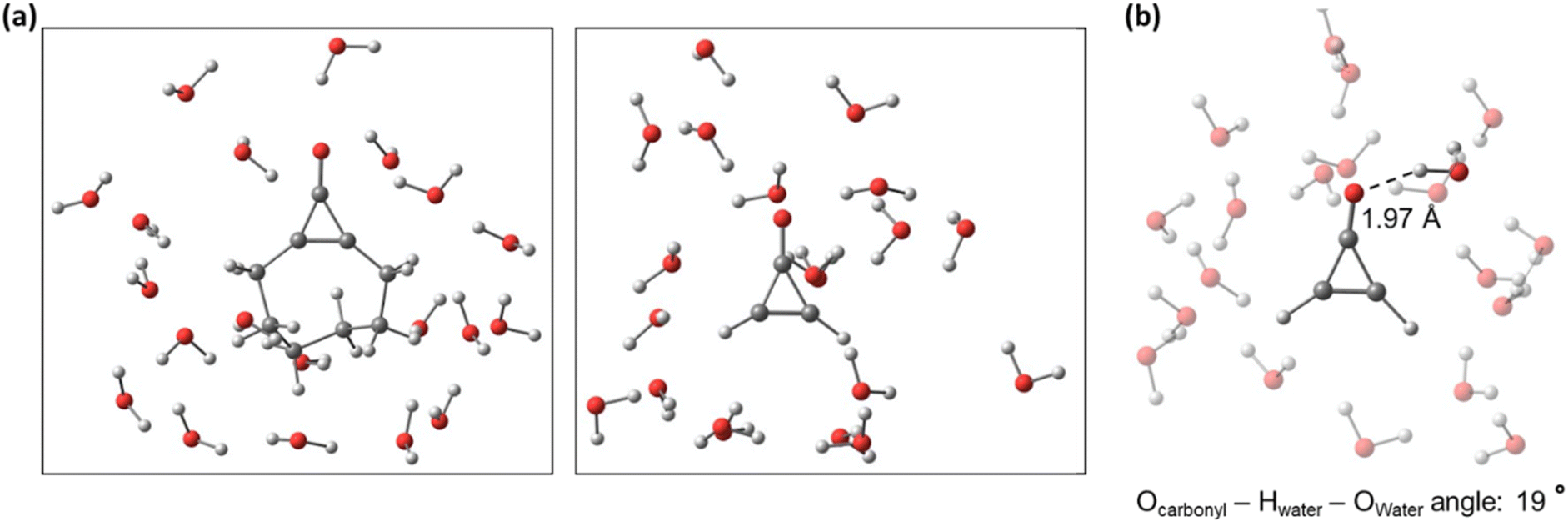 | ||
| Fig. 7 Snapshots of solvated trajectories of 1 and 2 (a) and an example of a solvated structure with intermolecular hydrogen bonding present between the Ocarbonyl and a water molecule (b). | ||
Our analysis found that the initial condition geometries 64% of 1 and 54% of 2 had H-bonding interactions. To evaluate if H-bonding in the initial condition geometry affects the mechanistic outcome, we divided the initial condition geometries into productive and non-productive trajectories. For 1, 64% of the non-reactive initial conditions and 72% of the reactive initial conditions had H-bonding, respectively. For 2, 54% of the non-reactive initial conditions and 57% of the reactive initial conditions featured H-bonding, respectively. While the reactive initial conditions have slightly more H-bonding for both 1 and 2, there is no clear correlation between H-bonding in the ground state and photochemical decarbonylation.
While H-bonds are a strongly stabilizing intermolecular force on the ground state between the oxygen and water molecule(s), this interaction should become repulsive in the S1. To test this hypothesis, we quantified the degree of H-bonding in the productive and non-productive trajectory pathways to determine how this electronic structure affected the intermolecular H-bonding interactions. After analyzing the full trajectory pathways of 1, we computed that 22% of productive trajectories have hydrogen bonding, while 34% of non-productive trajectories have hydrogen bonding. For the trajectory pathways of 2, we computed that 17% of productive trajectories have hydrogen bonding, while 43% of non-productive trajectories have hydrogen bonding. The productive trajectories overwhelmingly access the asynchronous hopping point geometries, which feature a broken σCC bond in the cyclopropenone ring. The lack of hydrogen bonding throughout the productive trajectories suggests that it is not stabilizing for the Ocarbonyl to be interacting with the surrounding water molecules. The non-productive trajectories have much higher levels of H-bonding, we hypothesize this is because the Ocarbonyl possesses two lone pairs to participate in intermolecular H-bonding.
Conclusion
We have used multiconfigurational quantum mechanical calculations and NAMD simulations to predict the photochemical decarbonylation of cyclopropenones with direct applications in photomedicine, bioorthogonal, and photoclick chemistry. We identified that the mechanistically relevant electronic transitions are S0 → S1 (n → π*) transitions and have excitation energies of 5.13 eV (1) and 4.61 eV (2). Our MEP calculations suggest that 1 will prefers to proceed to an S1/S0 crossing region while 2 will approach a minimum on the S1 surface. Our gas-phase NAMD simulations provide dynamical mechanistic information and predict a quantum yield for the dissociation products of 55% and 28% for 1 and 2, respectively. The time constants of the S1 were calculated to be 77 and 89 fs for 1 and 2 in the gas phase. We conclude from these simulations that nearly all productive trajectories involve an ultrafast dynamically asynchronous reaction mechanism with one σCC bond breaking in the S1-state and the second σCC bond breaking in the S0-state. The cyclopropenones immediately undergo cis–trans isomerization after excitation to S1, leading to the cleavage of one σCC bond. The fused cyclopropenone of 1 has higher QYs because these trajectories avoid the FC-region that lead to highly strained trans-cyclooctene, thus preferring to proceed through the productive asynchronous region of the S1/S0 seam. We also report the first explicitly solvated NAMD simulations needed to emulate the aqueous environment of the in vivo reaction. In solvated environments, we observed increased quantum yields of 58% for both 1 and 2 and S1 time constants of 56 fs (1) and 68 fs (2). We found that H-bonding between the OCarbonyl and explicit water molecules was energetically stabilizing hopping points in the FC-region. There were significantly higher rates of H-bonding in trajectories that revert and overwhelmingly undergo surface hopping in the FC-region (34% and 43% for 1 and 2, respectively) compared to those that dissociate and prefer to hop in the asynchronous region (22% and 17% for 1 and 2, respectively).Computational methods
Single reference methods
We used density functional theory (DFT) to optimize the ground-state global minima of 1, 2, and 3. Geometries were optimized using the PBE0-D3BJ/cc-pvdz.88–90 Time-dependent density functional theory (TDDFT)91 was used to calculate the excitation energies and wavelengths for the first 10 excited states of 1, 2, and 3. The TD-DFT calculations were run using the CAM-B3LYP77 functional with the aug-cc-pVTZ basis set.78,79 DFT includes dynamical correlation between electronic states, which provides accurate vertical excitations and transition natures for small organic molecules. This is an improvement over the multiconfigurational method (CASSCF) which omits dynamical correlation and tends to overestimate excitation energies. While it can accurately predict excitation energies into the Franck–Condon region where there is very little coupling between states, DFT cannot accurately predict the electronic structure on regions of a PES where there is significant coupling between two electronic states (e.g., avoided crossings and conical intersections. We used multiconfigurational methods to predict molecular electronic structure more accurately near these energetic degeneracies. All DFT calculations were run using the Gaussian 16 software.92Multiconfigurational methods
The multiconfigurational calculations were performed with a state-averaged complete active space self-consistent field (SA-CASSCF) using OpenMolcas 21.02.93 The methods are described with the format SA(N)-CASSCF(m,n), where N is the number of singlet states the calculation is averaged over. The variables m and n denote the number of electrons and orbitals used in the active space, respectively. For all included molecules, we used a (10,9) active space consisting of two σ and σ* orbitals, two π and π* orbitals, and one lone-pair orbital, including a lone pair on the oxygen atom in the cyclopropenone moiety. The active space is shown in Fig. 2. We used SA(4)-CASSCF(10,9)/ANO-S-VDZP multiconfigurational calculations to optimize the geometries for the ground and excited states and to run minimum energy path (MEP) calculations to track the steepest path along the excited state, starting from the Franck–Condon (FC) point. We used the extended multi-state complete active space perturbation theory (XMS-CASPT2)76 single-point energy correction to account for dynamic correlation for all multiconfigurational calculations. All optimized structures had their vibrational frequencies computed to confirm that they were stationary points (zero imaginary frequencies).Non-adiabatic molecular dynamics
We used NAMD to track the photochemical pathways of 1 and 2 along the PESs of the ground and excited states. We sampled the initial conditions (nuclear positions and velocities) from the vibrational modes using the Wigner distribution at the zero-point energy (ZPE). The electronic structure calculations used SA(4)-CASSCF (10,9)/ANO-S-VDZP. The trajectories started from the S1 (FC)-point, and the simulation time was 1 ps with a 0.5 fs time step. We computed the surface hopping probability at each step using Tully's fewest switches surface hopping method82,94 in OpenMolcas.93Molecular dynamics simulations
All molecular dynamics (MD) simulations of 1 and 2 in the presence of explicit water molecules were performed in CP2K version 9.1 (ref. 95 and 96) using Generalized Amber Force Field (GAFF)97 for the chromophore and SPC/Fw98 flexible water forcefield. First, a single geometry of the chromophore was solvated by 200 water molecules in a cubic box. Next, the systems were energy minimized and equilibrated in an isobaric-isothermal (NPT) ensemble for 0.5 ns and a canonical (NVT) ensemble for 1 ns. The last snapshot of this pre-equilibration process was taken, and respective Wigner geometries replaced the chromophore to initialize 500 parallel MD simulations for each molecule (1 and 2). During these MD simulations, the chromophores were kept frozen at their Wigner positions. The systems were minimized and equilibrated with a 0.1 ns NPT simulation followed by a 2 ns NVT simulation. All MD simulations used the velocity Verlet algorithm to integrate the equations of motion with a time step of 0.5 fs except during the energy minimization steps in which the conjugate gradient algorithm was used. All short-range non-bonded interactions were evaluated with a cutoff of 8 Å. To compute the coulombic interactions, Smooth Particle-Mesh Ewald (SPME)99 summation was used. To maintain a temperature of 300 K and 1.0 bar pressure during the MD simulations, Nose–Hoover thermostat100,101 and barostat were used as needed with time constants of 0.2 ps and 1.0 ps, respectively. The final snapshots of the NVT simulations were used to generate droplets of 20 water molecules surrounding the chromophore for subsequent ground and excited state CASSCF dynamics simulations.Explicitly solvated NAMD simulations
After running the gas-phase NAMD simulations, we next prepared the solvated NAMD simulations to be run. The production-run solvated trajectories contained our chromophores of interest (1 and 2) centered within a droplet of 20 water molecules. For 1 and 2, the initial geometries for the chromophore were obtained from Wigner sampling using the ground state vibrational frequencies. The initial conditions of the water molecules were taken from the final snapshots of the NVT simulations described in the MD section. The non-adiabatic coupling terms (NACTs) were computed between neighboring states at a given timestep using Tully's FSSH algorithm in OpenMolcas at the SA(4)-CASSCF(10,9) level of theory. To avoid overcoherence between states, we implemented a decoherence correction of 0.1 Hartree, as reported by Persico and Granucci.102 The velocity rescaling was handled at a constant temperature of 300 K using the Nosé–Hoover method. To leverage computational resources and accuracy, the ANO-S-VDZP basis set was used only for the chromophores (1 and 2), and ANO-S-MB was used for all 20 water molecules throughout the NAMD simulations. 276 initial conditions were simulated for the solvated system of 1, and 500 initial conditions were computed for the solvated system of 2. All simulations were 250 fs in length using a timestep of 0.5 fs.Data availability
The ESI† is available on Figshare and includes all static and dynamics calculations (https://doi.org/10.6084/m9.figshare.21667808.v1).Author contributions
D. Adrion – calculations, data analysis, manuscript writing, figure preparation. W. V. Karunaratne – calculations, manuscript writing. S. A. Lopez – project conceptualization, research supervision, and manuscript editing.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
W. V. K acknowledges support from Harold Alfond Foundation and the Roux Institute at Northeastern University. D. M. A. and S. A. L. acknowledge the National Science Foundation CAREER award (NSF-CHE-2144556). All authors appreciate the assistance from the Northeastern Research Computing Team and the computing resources provided by the Massachusetts Life Science Center grant (G00006360).References
- B. Rowden and N. Garcia-Araez, A review of gas evolution in lithium ion batteries, Energy Rep., 2020, 6, 10–18 CrossRef.
- D. Zhou, P. Li, X. Lin, A. McKinley, Y. Kuang, W. Liu, W. F. Lin, X. Sun and X. Duan, Layered double hydroxide-based electrocatalysts for the oxygen evolution reaction: identification and tailoring of active sites, and superaerophobic nanoarray electrode assembly, Chem. Soc. Rev., 2021, 50(15), 8790–8817 RSC.
- J. Li, X. Gao, L. Zhu, M. N. Ghazzal, J. Zhang, C.-H. Tung and L.-Z. Wu, Graphdiyne for crucial gas involved catalytic reactions in energy conversion applications, Energy Environ. Sci., 2020, 13(5), 1326–1346 RSC.
- J. He, Y. Zou and S. Wang, Defect engineering on electrocatalysts for gas-evolving reactions, Dalton Trans., 2018, 48(1), 15–20 RSC.
- C. Liu, C. L. Ji, Z. X. Qin, X. Hong and M. Szostak, Synthesis of Biaryls via Decarbonylative Palladium-Catalyzed Suzuki-Miyaura Cross-Coupling of Carboxylic Acids, iScience, 2019, 19, 749–759 CrossRef CAS PubMed.
- G. Li, Q. Li, Y. Xie and R. King, B; Schaefer III, H., F, (Acetylene)dicobalt Carbonyl Derivatives: Decarbonylation of the H2C2Co2(CO)6 Tetrahedrane, Organomettalics, 2009, 28, 3390–3394 CrossRef CAS.
- T. Krachko, A. W. Ehlers, M. Nieger, M. Lutz and J. C. Slootweg, Synthesis and Reactivity of the Phosphorus Analogues of Cyclopentadienone, Tricyclopentanone, and Housene, Angew Chem. Int. Ed. Engl., 2018, 57(6), 1683–1687 CrossRef CAS PubMed.
- H. Lu, T. Y. Yu, P. F. Xu and H. Wei, Selective Decarbonylation via Transition-Metal-Catalyzed Carbon-Carbon Bond Cleavage, Chem. Rev., 2021, 121(1), 365–411 CrossRef CAS PubMed.
- X. Yang, W. Lu, C. P. Hopper, B. Ke and B. Wang, Nature's marvels endowed in gaseous molecules I: Carbon monoxide and its physiological and therapeutic roles, Acta Pharm. Sin. B, 2021, 11(6), 1434–1445 CrossRef CAS PubMed.
- S. W. Ryter and A. M. Choi, Carbon monoxide: present and future indications for a medical gas, Korean J. Intern. Med., 2013, 28(2), 123–140 CrossRef CAS PubMed.
- X. Yang, W. Lu, M. Wang, C. Tan and B. Wang, "CO in a pill": Towards oral delivery of carbon monoxide for therapeutic applications, J. Controlled Release, 2021, 338, 593–609 CrossRef CAS PubMed.
- J. Cheng, G. Gan, Z. Shen, L. Gao, G. Zhang and J. Hu, Red Light-Triggered Intracellular Carbon Monoxide Release Enables Selective Eradication of MRSA Infection, Angew Chem. Int. Ed. Engl., 2021, 60(24), 13513–13520 CrossRef CAS PubMed.
- P. Peng, C. Wang, Z. Shi, V. K. Johns, L. Ma, J. Oyer, A. Copik, R. Igarashi and Y. Liao, Visible-light activatable organic CO-releasing molecules (PhotoCORMs) that simultaneously generate fluorophores, Org. Biomol. Chem., 2013, 11(39), 6671–6674 RSC.
- E. M. Sánchez-Carnerero, M. Russo, A. Jakob, L. Muchová, L. Vítek and P. Klán, Effects of Substituents on Photophysical and CO-Photoreleasing Properties of 2,6-Substituted meso-Carboxy BODIPY Derivatives, Chemistry, 2021, 3(1), 238–255 CrossRef.
- M. A. Gerkman and G. G. D. Han, Toward Controlled Thermal Energy Storage and Release in Organic Phase Change Materials, Joule, 2020, 4(8), 1621–1625 CrossRef.
- Q. Qiu, S. Yang, M. A. Gerkman, H. Fu, I. Aprahamian and G. G. D. Han, Photon Energy Storage in Strained Cyclic Hydrazones: Emerging Molecular Solar Thermal Energy Storage Compounds, J. Am. Chem. Soc., 2022, 144(28), 12627–12631 CrossRef CAS PubMed.
- J. L. Greenfield, M. A. Gerkman, R. S. L. Gibson, G. G. D. Han and M. J. Fuchter, Efficient Electrocatalytic Switching of Azoheteroarenes in the Condensed Phases, J. Am. Chem. Soc., 2021, 143(37), 15250–15257 CrossRef CAS PubMed.
- R. S. L. Gibson, J. Calbo and M. J. Fuchter, Chemical Z – E Isomer Switching of Arylazopyrazoles Using Acid, ChemPhotoChem, 2019, 3(6), 372–377 CrossRef CAS.
- M. A. Gerkman, R. S. L. Gibson, J. Calbo, Y. Shi, M. J. Fuchter and G. G. D. Han, Arylazopyrazoles for Long-Term Thermal Energy Storage and Optically Triggered Heat Release below 0 degrees C, J. Am. Chem. Soc., 2020, 142(19), 8688–8695 CrossRef PubMed.
- G. G. D. Han, J. H. Deru, E. N. Cho and J. C. Grossman, Optically-regulated thermal energy storage in diverse organic phase-change materials, Chem. Commun., 2018, 54(76), 10722–10725 RSC.
- A. E. Hillers-Bendtsen, P. G. Iuel Lunoe Dunweber, L. H. Olsen and K. V. Mikkelsen, Prospects of Improving Molecular Solar Energy Storage of the Norbornadiene/Quadricyclane System through Bridgehead Modifications, J. Phys. Chem. A, 2022, 126(17), 2670–2676 CrossRef CAS PubMed.
- M. Le and G. G. D. Han, Stimuli-Responsive Organic Phase Change Materials: Molecular Designs and Applications in Energy Storage, Acc. Mater. Res., 2022, 3(6), 634–643 CrossRef CAS.
- E. Durgun and J. C. Grossman, Photoswitchable Molecular Rings for Solar-Thermal Energy Storage, J. Phys. Chem. Lett., 2013, 4(6), 854–860 CrossRef CAS PubMed.
- A. Lennartson, A. Roffey and K. Moth-Poulsen, Designing photoswitches for molecular solar thermal energy storage, Tetrahedron Lett., 2015, 56(12), 1457–1465 CrossRef CAS.
- A. Pradeep, R. Varadharajan and V. Ramamurthy, Reversible Photoisomerization of Norbornadiene-Quadricyclane within a Confined Capsule(dagger), Photochem. Photobiol., 2022, 99, 624–636 CrossRef PubMed.
- Q. Li, H. Qian, B. Shao, R. P. Hughes and I. Aprahamian, Building Strain with Large Macrocycles and Using It To Tune the Thermal Half-Lives of Hydrazone Photochromes, J. Am. Chem. Soc., 2018, 140(37), 11829–11835 CrossRef CAS PubMed.
- P. Pan, D. Svirskis, S. W. P. Rees, D. Barker, G. I. N. Waterhouse and Z. Wu, Photosensitive drug delivery systems for cancer therapy: Mechanisms and applications, J. Controlled Release, 2021, 338, 446–461 CrossRef CAS PubMed.
- Z. Guo, Y. Ma, Y. Liu, C. Yan, P. Shi, H. Tian and W.-H. Zhu, Photocaged prodrug under NIR light-triggering with dual-channel fluorescence: in vivo real-time tracking for precise drug delivery, Sci. China: Chem., 2018, 61(10), 1293–1300 CrossRef CAS.
- Y. Venkatesh, Y. Rajesh, S. Karthik, A. C. Chetan, M. Mandal, A. Jana and N. D. Singh, Photocaging of Single and Dual (Similar or Different) Carboxylic and Amino Acids by Acetyl Carbazole and its Application as Dual Drug Delivery in Cancer Therapy, J. Org. Chem., 2016, 81(22), 11168–11175 CrossRef CAS PubMed.
- F. J. Hernandez, J. M. Cox, J. Li, R. Crespo-Otero and S. A. Lopez, Multiconfigurational Calculations and Photodynamics Describe Norbornadiene Photochemistry, J. Org. Chem., 2023, 88(9), 5311–5320 CrossRef CAS PubMed.
- J. C. Jewett and C. R. Bertozzi, Cu-free click cycloaddition reactions in chemical biology, Chem. Soc. Rev., 2010, 39(4), 1272–1279 RSC.
- L. Yu, C. Xu and C. Zhu, Probing the pi?>pi* photoisomerization mechanism of cis-azobenzene by multi-state ab initio on-the-fly trajectory dynamics simulation, Phys. Chem. Chem. Phys., 2015, 17(27), 17646–17660 RSC.
- W. G. Dauben and M. S. Kellogg, Photochemistry of cis-fused bicyclo[4.n.0]-2,4-dienes. Ground state conformational control, J. Am. Chem. Soc., 2002, 102(13), 4456–4463 CrossRef.
- W. Song, Y. Wang, J. Qu, M. M. Madden and Q. Lin, A photoinducible 1,3-dipolar cycloaddition reaction for rapid, selective modification of tetrazole-containing proteins, Angew Chem. Int. Ed. Engl., 2008, 47(15), 2832–2835 CrossRef CAS PubMed.
- Z. G. Wu, X. J. Liao, L. Yuan, Y. Wang, Y. X. Zheng, J. L. Zuo and Y. Pan, Visible-Light-Mediated Click Chemistry for Highly Regioselective Azide-Alkyne Cycloaddition by a Photoredox Electron-Transfer Strategy, Chemistry, 2020, 26(25), 5694–5700 CrossRef CAS PubMed.
- B. D. Fairbanks, L. J. Macdougall, S. Mavila, J. Sinha, B. E. Kirkpatrick, K. S. Anseth and C. N. Bowman, Photoclick Chemistry: A Bright Idea, Chem. Rev., 2021, 6915–6990 CrossRef CAS PubMed.
- A. Poloukhtine, N. E. Mbua, M. A. Wolfert, G. Boons and V. V. Popik, Selective Labeling of Living Cells by a Photo-Triggered Click Reaction, J. Am. Chem. Soc., 2009, 131, 15769–15776 CrossRef CAS PubMed.
- M. Martinek, L. Filipova, J. Galeta, L. Ludvikova and P. Klan, Photochemical Formation of Dibenzosilacyclohept-4-yne for Cu-Free Click Chemistry with Azides and 1,2,4,5-Tetrazines, Org. Lett., 2016, 18(19), 4892–4895 CrossRef CAS PubMed.
- M. A. Tasdelen and Y. Yagci, Light-induced copper(I)-catalyzed click chemistry, Tetrahedron Lett., 2010, 51(52), 6945–6947 CrossRef CAS.
- B. J. Adzima, Y. Tao, C. J. Kloxin, C. A. DeForest, K. S. Anseth and C. N. Bowman, Spatial and temporal control of the alkyne-azide cycloaddition by photoinitiated Cu(II) reduction, Nat. Chem., 2011, 3(3), 256–259 CrossRef CAS PubMed.
- S. Jiang, X. Wu, H. Liu, J. Deng, X. Zhang, Z. Yao, Y. Zheng, B. Li and Z. Yu, Ring-Strain-Promoted Ultrafast Diaryltetrazole–Alkyne Photoclick Reactions Triggered by Visible Light, ChemPhotoChem, 2020, 4(5), 327–331 CrossRef CAS.
- J. Li, H. Kong, L. Huang, B. Cheng, K. Qin, M. Zheng, Z. Yan and Y. Zhang, Visible Light-Initiated Bioorthogonal Photoclick Cycloaddition, J. Am. Chem. Soc., 2018, 140(44), 14542–14546 CrossRef CAS PubMed.
- L. Zhang, X. Zhang, Z. Yao, S. Jiang, J. Deng, B. Li and Z. Yu, Discovery of Fluorogenic Diarylsydnone-Alkene Photoligation: Conversion of ortho-Dual-Twisted Diarylsydnones into Planar Pyrazolines, J. Am. Chem. Soc., 2018, 140(24), 7390–7394 CrossRef CAS PubMed.
- Y. Wang, W. J. Hu, W. Song, R. K. Lim and Q. Lin, Discovery of Long-Wavelength Photoactivatable Diaryltetrazoles for Bioorthogonal 1,3-Dipolar Cycloaddition Reactions, Org. Lett., 2008, 10(17), 3725–3728 CrossRef CAS PubMed.
- W. Song, Y. Wang, J. Qu and Q. Lin, Selective Functionalization of a Genetically Encoded Alkene-Containing Protein via "Photoclick Chemistry" in Bacterial Cells, J. Am. Chem. Soc., 2008, 130, 9654–9655 CrossRef CAS PubMed.
- R. K. Lim and Q. Lin, Azirine ligation: fast and selective protein conjugation via photoinduced azirine-alkene cycloaddition, Chem. Commun., 2010, 46(42), 7993–7995 RSC.
- F. Feist, L. L. Rodrigues, S. L. Walden, T. W. Krappitz, T. R. Dargaville, T. Weil, A. S. Goldmann, J. P. Blinco and C. Barner-Kowollik, Light-induced Ligation of o-Quinodimethanes with Gated Fluorescence Self-reporting, J. Am. Chem. Soc., 2020, 142(17), 7744–7748 CrossRef CAS PubMed.
- K. K. Oehlenschlaeger, J. O. Mueller, N. B. Heine, M. Glassner, N. K. Guimard, G. Delaittre, F. G. Schmidt and C. Barner-Kowollik, Light-induced modular ligation of conventional RAFT polymers, Angew Chem. Int. Ed. Engl., 2013, 52(2), 762–766 CrossRef CAS PubMed.
- K. Hiltebrandt and K. Elies, D'Hooge D, R.; Blinco, J. P.; Barner-Kowollik, C., A Light-Activated Reaction Manifold, J. Am. Chem. Soc., 2016, 138(22), 7048–7054 CrossRef CAS PubMed.
- S. V. Mayer, A. Murnauer, M. K. von Wrisberg, M. L. Jokisch and K. Lang, Photo-induced and Rapid Labeling of Tetrazine-Bearing Proteins via Cyclopropenone-Caged Bicyclononynes, Angew Chem. Int. Ed. Engl., 2019, 58(44), 15876–15882 CrossRef CAS PubMed.
- H. Zhang, W. S. Trout, S. Liu, G. A. Andrade, D. A. Hudson, S. L. Scinto, K. T. Dicker, Y. Li, N. Lazouski, J. Rosenthal, C. Thorpe, X. Jia and J. M. Fox, Rapid Bioorthogonal Chemistry Turn-on through Enzymatic or Long Wavelength Photocatalytic Activation of Tetrazine Ligation, J. Am. Chem. Soc., 2016, 138(18), 5978–5983 CrossRef CAS PubMed.
- V. X. Truong, K. M. Tsang, F. Ercole and J. S. Forsythe, Red Light Activation of Tetrazine–Norbornene Conjugation for Bioorthogonal Polymer Cross-Linking across Tissue, Chem. Mater., 2017, 29(8), 3678–3685 CrossRef CAS.
- S. Arumugam and V. V. Popik, Light-induced hetero-Diels-Alder cycloaddition: a facile and selective photoclick reaction, J. Am. Chem. Soc., 2011, 133(14), 5573–5579 CrossRef CAS PubMed.
- R. A. Prasath, M. T. Gokmen, P. Espeel and F. E. Du Prez, Thiol-ene and thiol-yne chemistry in microfluidics: a straightforward method towards macroporous and nonporous functional polymer beads, Polym. Chem., 2010, 1(5), 685–692 RSC.
- B. D. Fairbanks, E. A. Sims, K. S. Anseth and C. N. Bowman, Reaction Rates and Mechanisms for Radical, Photoinitated Addition of Thiols to Alkynes, and Implications for Thiol–Yne Photopolymerizations and Click Reactions, Macromolecules, 2010, 43(9), 4113–4119 CrossRef CAS.
- C. E. Hoyle and C. N. Bowman, Thiol-ene click chemistry, Angew Chem. Int. Ed. Engl., 2010, 49(9), 1540–1573 CrossRef CAS PubMed.
- C. E. Hoyle, T. Y. Lee and T. Roper, Thiol-enes: Chemistry of the past with promise for the future, J. Polym. Sci., Part A: Polym. Chem., 2004, 42(21), 5301–5338 CrossRef CAS.
- H. Choi, M. Kim, J. Jang and S. Hong, Visible-Light-Induced Cysteine-Specific Bioconjugation: Biocompatible Thiol-Ene Click Chemistry, Angew. Chem., Int. Ed. Engl., 2020, 59(50), 22514–22522 CrossRef CAS PubMed.
- H. He, X. Meng, Q. Yue, W. Yin, Y. Gao, P. Fang and L. Shen, Thiol-ene click chemistry synthesis of a novel magnetic mesoporous silica/chitosan composite for selective Hg(II) capture and high catalytic activity of spent Hg(II) adsorbent, Chem. Eng. J., 2021, 405, 126743 CrossRef CAS.
- N. K. Urdabayev, A. Poloukhtine and V. V. Popik, Two-photon induced photodecarbonylation reaction of cyclopropenones, Chem. Commun., 2006, 4, 454–456 RSC.
- J. Calbo, C. E. Weston, A. J. White, H. S. Rzepa, J. Contreras-Garcia and M. J. Fuchter, Tuning Azoheteroarene Photoswitch Performance through Heteroaryl Design, J. Am. Chem. Soc., 2017, 139(3), 1261–1274 CrossRef CAS PubMed.
- D. A. Sutton and V. V. Popik, Sequential Photochemistry of Dibenzo[a,e]dicyclopropa[c,g][8]annulene-1,6-dione: Selective Formation of Didehydrodibenzo[a,e][8]annulenes with Ultrafast SPAAC Reactivity, J. Org. Chem., 2016, 81(19), 8850–8857 CrossRef CAS PubMed.
- C. D. McNitt, H. Cheng, S. Ullrich, V. V. Popik and M. Bjerknes, Multiphoton Activation of Photo-Strain-Promoted Azide Alkyne Cycloaddition "Click" Reagents Enables in Situ Labeling with Submicrometer Resolution, J. Am. Chem. Soc., 2017, 139(40), 14029–14032 CrossRef CAS PubMed.
- K. Mishiro, T. Kimura, T. Furuyama and M. Kunishima, Phototriggered Active Alkyne Generation from Cyclopropenones with Visible Light-Responsive Photocatalysts, Org. Lett., 2019, 21(11), 4101–4105 CrossRef CAS PubMed.
- K. Mishiro, M. Nomura, T. Furuyama and M. Kunishima, Efficiency Enhancement of a Photocatalytic Decarbonylation of an Aminocyclopropenone by Benzothiophene Substitution, J. Org. Chem., 2021, 86(4), 3625–3636 CrossRef CAS PubMed.
- S. L. Scinto, D. A. Bilodeau, R. Hincapie, W. Lee, S. S. Nguyen, M. Xu, C. W. Am Ende, M. G. Finn, K. Lang, Q. Lin, J. P. Pezacki, J. A. Prescher, M. S. Robillard and J. M. Fox, Bioorthogonal chemistry, Nat. Rev. Methods Primers, 2021, 1, 30 CrossRef CAS PubMed.
- Kenry and B. Liu, Bio-orthogonal Click Chemistry for In Vivo Bioimaging, Trends Chem., 2019, 1(8), 763–778 CrossRef CAS.
- M. Smeenk, J. Agramunt and K. M. Bonger, Recent developments in bioorthogonal chemistry and the orthogonality within, Curr. Opin. Chem. Biol., 2021, 60, 79–88 CrossRef CAS PubMed.
- T. Deb, J. Tu and R. M. Franzini, Mechanisms and Substituent Effects of Metal-Free Bioorthogonal Reactions, Chem. Rev., 2021, 121(12), 6850–6914 CrossRef CAS PubMed.
- J. Ravasco and J. A. S. Coelho, Predictive Multivariate Models for Bioorthogonal Inverse-Electron Demand Diels-Alder Reactions, J. Am. Chem. Soc., 2020, 142(9), 4235–4241 CrossRef CAS PubMed.
- H. Wu and N. K. Devaraj, Advances in Tetrazine Bioorthogonal Chemistry Driven by the Synthesis of Novel Tetrazines and Dienophiles, Acc. Chem. Res., 2018, 51(5), 1249–1259 CrossRef CAS PubMed.
- F. Liu, Y. Liang and K. N. Houk, Bioorthogonal Cycloadditions: Computational Analysis with the Distortion/Interaction Model and Predictions of Reactivities, Acc. Chem. Res., 2017, 50(9), 2297–2308 CrossRef CAS PubMed.
- L. Liu, S. Xia and W. H. Fang, Photodecarbonylation mechanism of cyclopropenone in the gas phase: electronic structure calculation and AIMS dynamics simulation, J. Phys. Chem. A, 2014, 118(39), 8977–8985 CrossRef CAS PubMed.
- M. Ben-Nun, J. Quenneville and T. J. Martinez, Ab Inition Multiple Spawning: Photochemistry from First Principles Quantum Molecular Dynamics, J. Phys. Chem. A, 2000, 104(22), 5161–5175 CrossRef CAS.
- A. Poloukhtine and V. V. Popik, Highly Efficient Photochemical Generation of a Triple Bond: Synthesis, Properties, and Photodecarbonylation of Cyclopropenones, J. Org. Chem., 2003, 68, 7833–7840 CrossRef CAS PubMed.
- T. Shiozaki, W. Gyorffy, P. Celani and H. J. Werner, Communication: extended multi-state complete active space second-order perturbation theory: energy and nuclear gradients, J. Chem. Phys., 2011, 135(8), 081106 CrossRef PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP), Chem. Phys. Lett., 2004, 393(1–3), 51–57 CrossRef CAS.
- R. A. Kendall, T. H. Dunning and R. J. Harrison, Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions, J. Chem. Phys., 1992, 96, 224108 CrossRef.
- T. H. Dunning, Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen, J. Chem. Phys., 1989, 90(2), 1007–1023 CrossRef CAS.
- R. Walker, R. M. Conrad and R. H. Grubbs, The Living ROMP of trans-Cyclooctene, Macromolecules, 2009, 42(3), 599–605 CrossRef CAS PubMed.
- J. C. Tully and R. K. Preston, Trajectory Surface Hopping Approach to Nonadiabatic Molecular Collisions: The Reaction of H+ with D2, J. Chem. Phys., 1971, 55(2), 562–572 CrossRef CAS.
- J. C. Tully, Molecular dynamics with electronic transitions, J. Chem. Phys., 1990, 93(2), 1061–1071 CrossRef CAS.
- D. Avagliano, E. Lorini and L. Gonzalez, Sampling effects in quantum mechanical/molecular mechanics trajectory surface hopping non-adiabatic dynamics, Philos. Trans. R. Soc., A, 2022, 380(2223), 20200381 CrossRef PubMed.
- W. Humphrey, A. Dalke and K. Schulten, VMD – Visual Molecular Dynamics, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- C. N. Pace, H. Fu, K. Lee Fryar, J. Landua, S. R. Trevino, D. Schell, R. L. Thurlkill, S. Imura, J. M. Scholtz, K. Gajiwala, J. Sevcik, L. Urbanikova, J. K. Myers, K. Takano, E. J. Hebert, B. A. Shirley and G. R. Grimsley, Contribution of hydrogen bonds to protein stability, Protein Sci., 2014, 23(5), 652–661 CrossRef CAS PubMed.
- C. N. Pace, Energetics of protein hydrogen bonds, Nat. Struct. Mol. Biol., 2009, 16(7), 681–682 CrossRef CAS PubMed.
- K. P. Tan, K. Singh, A. Hazra and M. S. Madhusudhan, Peptide bond planarity constrains hydrogen bond geometry and influences secondary structure conformations, Curr. Res. Struct. Biol., 2021, 3, 1–8 CrossRef CAS PubMed.
- C. Adamo and V. Barone, Toward reliable density functional methods without adjustable parameters: The PBE0 model, J. Chem. Phys., 1999, 110(13), 6158–6170 CrossRef CAS.
- J. C. Slater and J. C. Phillips, Quantum Theory of Molecules and Solids Vol. 4: The Self-Consistent Field for Molecules and Solids, Phys. Today, 1974, 27(12), 49–50 CrossRef.
- D. E. Woon and T. H. Dunning, Gaussian basis sets for use in correlated molecular calculations. V. Core-valence basis sets for boron through neon, J. Chem. Phys., 1995, 103(11), 4572–4585 CrossRef CAS.
- F. Furche and R. Ahlrichs, Adiabatic time-dependent density functional methods for excited state properties, J. Chem. Phys., 2002, 117(16), 7433–7447 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz; A. F. Izmaylov; J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian 16 Rev. C.01, Wallingford, CT, 2016 Search PubMed.
- I. Fdez Galvan, M. Vacher, A. Alavi, C. Angeli, F. Aquilante, J. Autschbach, J. J. Bao, S. I. Bokarev, N. A. Bogdanov, R. K. Carlson, L. F. Chibotaru, J. Creutzberg, N. Dattani, M. G. Delcey, S. S. Dong, A. Dreuw, L. Freitag, L. M. Frutos, L. Gagliardi, F. Gendron, A. Giussani, L. Gonzalez, G. Grell, M. Guo, C. E. Hoyer, M. Johansson, S. Keller, S. Knecht, G. Kovacevic, E. Kallman, G. Li Manni, M. Lundberg, Y. Ma, S. Mai, J. P. Malhado, P. A. Malmqvist, P. Marquetand, S. A. Mewes, J. Norell, M. Olivucci, M. Oppel, Q. M. Phung, K. Pierloot, F. Plasser, M. Reiher, A. M. Sand, I. Schapiro, P. Sharma, C. J. Stein, L. K. Sorensen, D. G. Truhlar, M. Ugandi, L. Ungur, A. Valentini, S. Vancoillie, V. Veryazov, O. Weser, T. A. Wesolowski, P. O. Widmark, S. Wouters, A. Zech, J. P. Zobel and R. Lindh, OpenMolcas: From Source Code to Insight, J. Chem. Theory Comput., 2019, 15(11), 5925–5964 CrossRef CAS PubMed.
- J. C. Tully, Perspective: Nonadiabatic dynamics theory, J. Chem. Phys., 2012, 137(22), 22A301 CrossRef PubMed.
- J. Hutter, M. Iannuzzi, F. Schiffmann and J. VandeVondele, cp2k: atomistic simulations of condensed matter systems, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4(1), 15–25 CAS.
- T. D. Kuhne, M. Iannuzzi, M. Del Ben, V. V. Rybkin, P. Seewald, F. Stein, T. Laino, R. Z. Khaliullin, O. Schutt, F. Schiffmann, D. Golze, J. Wilhelm, S. Chulkov, M. H. Bani-Hashemian, V. Weber, U. Borstnik, M. Taillefumier, A. S. Jakobovits, A. Lazzaro, H. Pabst, T. Muller, R. Schade, M. Guidon, S. Andermatt, N. Holmberg, G. K. Schenter, A. Hehn, A. Bussy, F. Belleflamme, G. Tabacchi, A. Gloss, M. Lass, I. Bethune, C. J. Mundy, C. Plessl, M. Watkins, J. VandeVondele, M. Krack and J. Hutter, CP2K: An electronic structure and molecular dynamics software package – Quickstep: Efficient and accurate electronic structure calculations, J. Chem. Phys., 2020, 152(19), 194103 CrossRef PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, Development and testing of a general amber force field, J. Comput. Chem., 2004, 25(9), 1157–1174 CrossRef CAS PubMed.
- Y. Wu, H. L. Tepper and G. A. Voth, Flexible simple point-charge water model with improved liquid-state properties, J. Chem. Phys., 2006, 124(2), 024503 CrossRef PubMed.
- J. C. Phillips, D. J. Hardy, J. D. C. Maia, J. E. Stone, J. V. Ribeiro, R. C. Bernardi, R. Buch, G. Fiorin, J. Henin, W. Jiang, R. McGreevy, M. C. R. Melo, B. K. Radak, R. D. Skeel, A. Singharoy, Y. Wang, B. Roux, A. Aksimentiev, Z. Luthey-Schulten, L. V. Kale, K. Schulten, C. Chipot and E. Tajkhorshid, Scalable molecular dynamics on CPU and GPU architectures with NAMD, J. Chem. Phys., 2020, 153(4), 044130 CrossRef CAS PubMed.
- S. Nosé, A molecular dynamics method for simulations in the canonical ensemble, Mol. Phys., 2006, 52(2), 255–268 CrossRef.
- S. Nosé, A unified formulation of the constant temperature molecular dynamics methods, J. Chem. Phys., 1984, 81(1), 511–519 CrossRef.
- G. Granucci and M. Persico, Critical appraisal of the fewest switches algorithm for surface hopping, J. Chem. Phys., 2007, 126(13), 134114 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The ESI contains the (10,9) active space for 1 and an analysis and visualization of the branching plane for an MECI structure of 2 in the FC-region of the S1/S0 hopping region. We also present two concerted hopping point structures for 1, a bond length analysis of the hopping points for the NAMD simulations of 1 and 2 in the gas-phase, and an analysis of the S1 lifetimes for 1 and 2 for the gas-phase and solvated dynamics trajectories. We also tested two different active spaces, presented a conformational analysis of 1, and compared SA(2) NAMD simulations of 2 to SA(4) NAMD simulations. Finally, we present QM/MM NAMD simulations for solvated systems of 1 and 2. All optimized structures and NAMD simulations are available online (DOI: https://doi.org/10.6084/m9.figshare.21667808). See DOI: https://doi.org/10.1039/d3sc03805j |
| This journal is © The Royal Society of Chemistry 2023 |