
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePoly-pnictogen bonding: trapping halide ions by a tetradentate antimony(III) Lewis acid†
J. Louis
Beckmann
 ,
Jonas
Krieft
,
Yury V.
Vishnevskiy
,
Beate
Neumann
,
Hans-Georg
Stammler
and
Norbert W.
Mitzel
*
,
Jonas
Krieft
,
Yury V.
Vishnevskiy
,
Beate
Neumann
,
Hans-Georg
Stammler
and
Norbert W.
Mitzel
*
Chair of Inorganic and Structural Chemistry, Center for Molecular Materials CM2 Faculty of Chemistry, Bielefeld University Universitätsstrasse 25, Bielefeld 33615, Germany. E-mail: mitzel@uni-bielefeld.de
First published on 10th November 2023
Abstract
A highly halide affine, tetradentate pnictogen-bonding host-system based on the syn-photodimer of 1,8-diethynylanthracene was synthesized by a selective tin-antimony exchange reaction. The host carries four C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–Sb(C2F5)2 units and has been investigated regarding its ability to act as a Lewis acidic host component for the cooperative trapping of halide ions (F−, Cl−, Br−, I−). The chelating effect makes this host-system superior to its bidentate derivative in competition experiments. It represents a charge-reversed crown-4 and has the ability to dissolve otherwise poorly soluble salts like tetra-methyl-ammonium chloride. Its NMR-spectroscopic properties make it a potential probe for halide ions in solution. Insights into the structural properties of the halide adducts by X-ray diffraction and computational methods (DFT, QTAIM, IQA) reveal a complex interplay of attractive pnictogen bonding interactions and Coulomb repulsion.
C–Sb(C2F5)2 units and has been investigated regarding its ability to act as a Lewis acidic host component for the cooperative trapping of halide ions (F−, Cl−, Br−, I−). The chelating effect makes this host-system superior to its bidentate derivative in competition experiments. It represents a charge-reversed crown-4 and has the ability to dissolve otherwise poorly soluble salts like tetra-methyl-ammonium chloride. Its NMR-spectroscopic properties make it a potential probe for halide ions in solution. Insights into the structural properties of the halide adducts by X-ray diffraction and computational methods (DFT, QTAIM, IQA) reveal a complex interplay of attractive pnictogen bonding interactions and Coulomb repulsion.
Introduction
The application of cooperatively binding σ-hole systems for the complexation of nucleophiles is of increasing interest as it paves the way for compounds used in anion-abstraction catalysis, substrate recognition in host–guest chemistry, or anion transport.1,2 The application of σ-hole donors for complexation of Lewis bases offers the possibility of non-covalent interactions, which are mainly characterized by a high directionality of the Lewis acid–base interaction.3,4 Halogen bonds are the most prominent representatives of σ-hole interactions. They are usually represented as R–X⋯Y and describe the attractive, non-covalent interaction of a halogen X with a Lewis base Y, while R should preferably be an electronegative substituent.5 Using suitable organic backbones, the directionality of σ-hole donor sites can be prearranged to achieve cooperativity as well as selectivity. On this basis, various multidentate host systems and catalysts based on halogen bonding have been designed.6–21 Cooperative chalcogen bonding or pnictogen bonding systems have only been explored to a lesser extent.22–34Recent results suggest that the application of chalcogen or pnictogen bonding may open up exciting new opportunities for catalysis and anion recognition.22–48 While the lighter group 15 elements are unsuitable candidates due to steric overloading, antimony turns out to be an exceptionally good pnictogen bond donor due to its size and easy polarizability.48–54 Almost all group 15 based host systems known in the literature are bidentate and based on Sb(V), such as the distiborane A of Gabbaï et al. (Scheme 1).55 One of the few examples of a bidentate Sb(III) host is an oxo-bridged distibane of Cozzolino et al.56,57
 | ||
| Scheme 1 Fluoride complex of the bidentate Sb(V) host system A55 and halide complexes of our Sb(III) host systems investigated previously and herein. | ||
Building on this, we have previously explored the enormous potential of antimony(III) in the form of the bidentate pnictogen-bonding host system 1.58 By using pentafluoroethyl substituents as extremely electron-withdrawing groups, we could demonstrate that the bis(pentafluoroethyl)stibanyl-ethynyl function exhibits two strongly pronounced σ-holes at the antimony(III) atom, approximately perpendicular to the alkyne. We found host 1 to be a promising tool for the chelation of various group-15-, -16-, and -17-based Lewis bases. It shows the highest affinity towards halide ions. Following this idea, in this work we introduce the tetradentate photodimer 2, the ‘big brother’ of 1. The arrangement of four antimony atoms in a rectangular pocket facilitates the cooperativity of multiple pnictogen bonding interactions. The chelating effect is used to construct a particularly affine receptor for halide ions. In the following, we describe the synthesis and complexation behavior of this partly fluorinated charge-reversed crown ether, also with respect to a competition with 1.
Results and discussion
Synthesis and study of the host system
To chelate a halide ion by four Lewis acidic antimony atoms, our approach uses a roughly square planar coordination environment. The four σ-holes should be positioned in a rectangle and have the ability to align to its center. In previous work, we established the synthesis and application of the syn-photodimer of diethynylanthracene as an organic backbone for poly-Lewis acids.59 This tetraalkyne brings four functions into close proximity and thus provides a suitable arrangement necessary for quadruple interactions. The diagonal spacing of the alkyne carbon atoms of about 6.2 Å in the tetraalkyne59 should provide a suitable binding pocket for halide ions. The electrophilic binding sites opposite the pentafluoroethyl substituents and the alkyne form approximately a right angle.58 This is essential for the functionality of our fourfold host system. The free rotatability around the alkyne-axes allows one of the positively polarized regions of each of the four Sb(III) atoms to align into the central cavity of the host.The synthesis of host system 2 was achieved by following a protocol established for the bidentate system 1: reaction of the corresponding tin compound 3 in a metal exchange reaction with four equivalents of chlorobis(pentafluoroethynyl)stibane (4) with liberation of chlorotrimethylstannane (Scheme 2).58,60 After recrystallization, the tetrastibane 2 was obtained in 74% yield. Like the bidentate system 1, system 2 decomposes on exposure to air by oxidation of Sb(III) and hydrolyses on contact with moisture forming pentafluoroethane. The determination of the solid-state structure of 2 by single-crystal X-ray diffraction experiments confirmed the desired arrangement of four antimony atoms approximately in one plane (Fig. 1), for all calculations, only the main positions of the atoms were considered in the case of disorder.
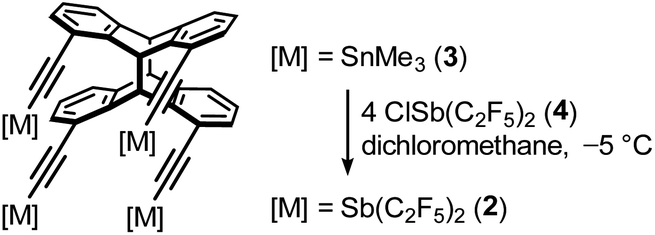 | ||
| Scheme 2 Synthesis of the tetradentate host system 2. | ||
 | ||
| Fig. 1 Molecular structure of 2 in the solid state from two perspectives. Hydrogen atoms, a disorder of one pentafluoroethyl group, and fluorine atoms (bottom) are omitted for clarity. Red dotted lines (top) indicate Sb⋯F contacts below the sum of van der Waals radii.61,62 Ellipsoids are set to 50% probability. Selected distances [Å] and angles [°]: Sb(1)⋯Sb(2) 5.731(1), Sb(2)⋯Sb(4) 5.615(1), Sb(3)⋯Sb(4) 5.722(1), Sb(1)⋯Sb(3) 4.217(1), Sb(1)⋯F(21) 3.223(3), Sb(2)⋯F(7) 3.255(4), Sb(2)⋯F(37) 3.044(1), Sb(3)⋯F(31) 3.220(4), Sb(3)⋯F(36) 3.311(1), Sb(3)–C(43) 2.230(5), Sb(3)–C(45) 2.244(6); C(42)–Sb(3)–C(43) 87.9(2), C(42)–Sb(3)–C(45) 94.5(2), C(41)–C(42)–Sb(3) 169.7(5), C(27)–C(41)–C(42) 174.5(5), C(40)–C(27)–C(41) 118.1(4), C(28)–C(27)–C(41) 122.6(4). Root mean square deviation of the four Sb atoms from their best plane: 0.001 Å. | ||
The Calkyne–Sb–CF angles average 92.0° over all eight angles, suggesting the desired perpendicular orientation of the σ-holes to the alkyne. Since the diagonal Sb⋯Sb distances are 7.28(1) Å and 7.74(1) Å, the cavity is wider than in the unfunctionalized tetraalkyne (d(Sb⋯Sb) ≈ 6.2 Å).13 This indicates a certain, albeit limited, flexibility of the alkyne groups and thus ensures some adaptability of the binding pocket towards different halide ions. The Sb–CF bond lengths, which will later serve as a reference for a donation into the σ*(Sb–CF)-orbital, are 2.229 Å on average (range 2.198(5)–2.244(6) Å). Some Sb⋯F distances in host 2 are shorter than the sum of their van der Waals radii (ΣdvdW(Sb,F) = 3.53 Å),61,62 including 16 Sb⋯F distances between each antimony atom and the four geminal fluorine atoms of their adjacent C2F5 groups.
Of greater interest are the additional five Sb⋯F contacts between two Sb(C2F5)2 functions, which indicate weak intramolecular pnictogen bonding interactions between the fluorine atoms of CF2 groups and the antimony atoms of another group. Four out of five of these interactions show a directionality by adopting F–Sb–CF angles between 162.8(2)° and 170.6(2)°, which is typical for σ-hole interactions of pnictogens. On a larger scale, the molecules arrange back-to-back, forming alternating layers of perfluorinated Sb(C2F5)2 groups and organic backbone, exhibiting different aryl–aryl interactions (see ESI†).
In a first attempt at complexation experiments with halide ions, we used tetra-butyl-ammonium halides (TBAX, X = Cl, Br, I). Since attempts to obtain single crystals of the chloride adduct with TBACl failed, but were successful with the tetramethylammonium cation, we used tetramethylammonium chloride (TMACl) instead. Attempts to synthesize the corresponding fluoride adduct by reaction with anhydrous TMAF or TASF resulted in decomposition.
For each reaction (Scheme 3), the selective formation of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1-adduct of the composition [2·X]−[R4N]+ could be verified by NMR titration, X-ray diffraction of single crystals, elemental analyses, and IR spectroscopy.
:1-adduct of the composition [2·X]−[R4N]+ could be verified by NMR titration, X-ray diffraction of single crystals, elemental analyses, and IR spectroscopy.
 | ||
| Scheme 3 Adduct formation of [2·X]− in benzene, dichloromethane or THF and competitive reaction of host 2 with up to two equivalents of monomer 1. | ||
NMR studies
The host system forms stable adducts with the tetra-alkyl-ammonium halides (X = Cl, Br, I), resulting in salts containing the complex anions [2·X]− in quantitative yield. In solution, the bromide and iodide adducts are stable for up to a week, whereas the chloride adduct begins to decompose after a few days.Although the three tetraalkylammonium halides as well as the host are poorly soluble in benzene, a mixture of the suspensions immediately forms the 1:1-adduct, which is first dissolved as a (probably supersaturated) solution, from which the adduct precipitates after seconds. In THF, however, all three adducts are soluble and can be characterized by NMR spectroscopy.
Due to the formation of the anionic complex, the cavity-side bridgehead protons receive a characteristic shift in the 1H NMR spectrum. In the 19F NMR spectrum, the resonances of the pentafluoroethyl groups split in a diagnostic way (Fig. 2). Due to the limited rotatability of the Sb(C2F5)2 groups, both pentafluoroethyl groups are chemically inequivalent to each other, resulting in two sets of signals for these groups. In addition, signals with roof effect for a geminal 2JF,F-coupling are present for both sets of AB-systems (2JF,F = 301 Hz and 305 Hz) due to the prochirality of antimony and thus the diastereotopic of the CF2 fluorine atoms (Fig. 2, top). While for the free host, the chemical inequivalence of the two fluorine atoms is less pronounced, leading to a partial signal overlap (Fig. 2, bottom), this effect is much more noticeable in the three adducts due to the coordination of the halide ion.
 | ||
| Fig. 2 Sections of the 19F NMR spectra (471 MHz) of compound 2 and its titration with TBAI in THF-d8. Depicted are the signals of the CF3-groups (left) and the CF2-groups (right, intensities not in the corresponding ratio). Titration experiments of 2 with TMACl and TBABr show a similar pattern (see ESI†). | ||
The resulting, very characteristic signal pattern serves here as an NMR sensor for a complete 1:1-adduct formation. Titration of the host system with halide salts leads to a strong broadening due to exchange reactions in the case of a substoichiometric amount of halide ions; the fully resolved signal pattern appears only with equimolar or higher concentrations of halide ions. In contrast, an excess of halide salt does not lead to further changes, indicating that the system is saturated with one equivalent. The signal indicates an equal participation of each Sb(C2F5)2 group and therefore a dynamic exchange can be inferred. These features can be observed for each halide adduct. The adducts differ from each other only by slightly different shifts in the 19F NMR spectrum.
It should also be mentioned that TMACl (unlike the tetrabutylammonium halides) is insoluble in THF. When host system 2 is added, exactly one equivalent of TMACl is complexed and dissolved, while excess TMACl remains undissolved. Thus, the host acts as a complexing agent and the anionic complex must have a significantly better solubility product than TMACl.
To demonstrate the effect of more efficient chelation, the binding properties of host 2 were investigated by NMR titration experiments. In titration experiments with TBAX (X = Cl, Br, I), the cavity-side bridgehead protons of the backbone showed a significant shift (up to 0.5 ppm) in the 1H NMR spectra during titration. Binding isotherms were obtained confirming a 1:1 binding mode of complexation and the binding constants Ka were calculated using the WinEQNMR 2 program63 (see ESI† for further details and data). The Ka values of 2 (Ka(Cl−): 2140(296) M−1, Ka(Br−): 2007(560) M−1, Ka(I−): 1786(519) M−1) are significantly higher than those of the bidentate system 1 (Ka(Cl−): 76(6) M−1, Ka(Br−): 28(1) M−1, Ka(I−): 16(1) M−1).58
Note, however, that the binding isotherm shows a sharp bend at 1.0 equivalents of halide salt and almost no further change of the chemical shift above 1.0 equivalents. Because of this rather atypical feature, which we suspect is due to the unique binding situation of the four stibanyl functions as well as the special position of the bridgehead protons, the fit function does not fit perfectly around 1.0 equivalents, resulting in a comparatively large error for Ka.
In addition, competitive reactions of the tetradentate host 2 with two equivalents of the bidentate host 1 were performed.
Assuming that complexes of the form [12·X]− are present in solution, two equivalents of monomer 1 were mixed with the corresponding halide salt (Scheme 3). In this way, the only differences would be the pre-organization of either two or four Sb(C2F5)2 groups as well as an entropic contribution that further disadvantages the trimolecular process of [12·X]− formation. According to NMR spectroscopic data, ligand 1 is almost completely displaced and the adducts [2·X]− are formed. Since the 19F NMR spectra clearly show when host 2 is saturated with halide ions (see above), it can be assumed that the equilibrium is far on the side of [2·X]−. Regarding TMACl, another interesting observation is made: in contrast to 2, which dissolves exactly one equivalent of TMACl, host 1 dissolves only traces of up to 0.15 equivalents.
Consequently, the chelating effect of four pre-organized antimony functions in 2, as opposed to only two in 1, has a pronounced effect on the complexation abilities of 2, which can be seen in the significantly higher binding constants Ka, in the competition experiments with 1 as well as in the ability of the tetradentate system to dissolve quantitative amounts of TMACl.
Solid state studies
Recrystallization from benzene yielded single crystals of all three adducts. The solid-state structures of each adduct show the expected structural motif of a halogen atom surrounded by four antimony atoms in a roughly square-planar fashion (Fig. 3). This coordination geometry of halide ions is rare, but not unknown in the literature. For example, the solid-state structure of the complex [K([18]crown-6)][(PhSbI2)4I] by Huttner et al. can be described as an iodide ion surrounded by four PhSbI2 units in a square-planar coordination.64 The spontaneous assembly of this structural motif in the solid state suggests that this unusual coordination geometry of iodide may sometimes be favorable. In the halide adducts of host 2, this square planar coordination geometry of Sb4X is partially distorted. | ||
| Fig. 3 Molecular structures of [2·Cl]−[Me4N]+, [2·Br]−[nBu4N]+ and [2·I]−[nBu4N]+ in the solid state. Hydrogen atoms, solvent molecules (benzene) and disordered fragments are omitted for clarity. Ellipsoids are set at 50% probability. In the top row, the ammonium ion is omitted. In the bottom row, only the Sb4X-plane and the carbon atoms of the pentafluoroethyl groups are depicted. Distances [Å] and angles [°] of [2·Cl]−[Me4N]+: Sb(1)–C(17) 2.302(13), Sb(1)–C(19) 2.234(10), Sb(2)–C(23) 2.252(10), Sb(2)–C(25) 2.248(11), Sb(3)–C(43) 2.299(6), Sb(3)–C(45) 2.242(5), Sb(4)–C(49) 2.362(10), Sb(4)–C(51) 2.223(8); Sb(1)–Sb(2)–Sb(4) 62.7(1), Sb(2)–Sb(4)–Sb(3) 103.9(1), Sb(4)–Sb(3)–Sb(1) 90.6(1), Sb(3)–Sb(1)–Sb(2) 102.8(1); [2·Br]−[nBu4N]+: Sb(1)–C(37) 2.304(5), Sb(1)–C(40) 2.237(5), Sb(2)–C(41) 2.255(5), Sb(2)–C(43) 2.251(5), Sb(3)–C(45) 2.248(5), Sb(3)–C(47) 2.271(6), Sb(4)–C(49) 2.223(6), Sb(4)–C(51) 2.267(5); Sb(1)–Sb(2)–Sb(4) 82.5(1), Sb(2)–Sb(4)–Sb(3) 97.5(1), Sb(4)–Sb(3)–Sb(1) 73.7(1), Sb(3)–Sb(1)–Sb(2) 106.2(1); [2·I]−[nBu4N]+: Sb(1)–C(37) 2.276(3), Sb(1)–C(39) 2.253(3), Sb(2)–C(41) 2.274(3), Sb(2)–C(43) 2.248(3), Sb(3)–C(45) 2.331(7), Sb(3)–C(47) 2.311(19), Sb(4)–C(49) 2.257(3), Sb(4)–C(51) 2.242(3); Sb(1)–Sb(2)–Sb(4) 87.8(1), Sb(2)–Sb(4)–Sb(3) 90.5(1), Sb(4)–Sb(3)–Sb(1) 99.1(1), Sb(3)–Sb(1)–Sb(2) 82.5(1). | ||
The eight pentafluoroethyl groups are oriented outwards. The CC–Sb units show different degrees of curvature, which is why on the one hand the CC–Sb angles vary between 166.7(9)° and 176.8(8)°, while on the other hand the Calkyne–Sb⋯X angles vary between 71.1(3)° and 87.7(1)°. Each of the halogen atoms in the three structures are close to the calculated geometrical center of the four antimony atoms (d(X⋯Sb4-centroid) Cl: 0.387(3), Br: 0.116(1), I: 0.390(2) Å). A closer comparison shows an increasing distortion in the arrangement of the antimony atoms within the Sb4X-plane along the series iodide (rectangle) and bromide to chloride (rhombus), which is indicated by an increasing deviation of the corners' angles from 90°. At the same time, the halogen moves deeper into the molecule, i.e. in the direction of the aromatic backbone. While the iodide is still 0.101(3) Å above the plane defined by the four antimony atoms, the bromide is only 0.025(1) Å and the chloride 0.316(3) Å below this plane. With a smaller anion radius, the antimony atoms move closer together, causing the pentafluoroethyl groups in turn to increasingly sterically repel another. A possible interpretation is a competition between an attractive Sb⋯X interaction and a repulsion of the pentafluoroethyl substituents, with an increasingly repulsive contribution with decreasing anion size.
A comparison with the complexes of monomer 1 shows, that the two Sb⋯X distances in the adducts [1·X]−[nBu4N]+ (X = Br, I) are slightly shorter than the four Sb⋯X distances in [2·X]−[nBu4N]+ (X = Br, I). It appears, that the ‘ideal’ distance cannot be realized in the adducts of 2, since the four antimony atoms cannot sufficiently approach the central halide ion due to repulsion of their substituents. In this context, decisive differences between the iodide, bromide and chloride adducts also become apparent when one looks more closely at the formation of pnictogen bonding contacts in the solid state. A common criterion for a σ-hole interaction is a distance between donor and acceptor atoms smaller than the sum of their van der Waals radii.5 In the case of [2·I]−, all four distances are below this sum, while for [2·Cl]− as well as [2·Br]−, only three of their Sb⋯X distances are smaller than this sum (Fig. 4). Another characteristic feature of σ-hole interactions is the collinearity of the donor–acceptor bond with an electronegative ligand at the electron acceptor. In classical halogen bonding, the electron-withdrawing substituent, the halogen atom and the electron pair donor span an angle of 180°. In pnictogen bonding, this linearity is usually distorted by the presence of the lone pair and falls within a range of 165° to 175°.48,58,66 When the three adducts are examined in terms of these angles, the iodide adduct has one angle that is strongly out of linearity, while the bromide and chloride adducts each have two strongly deviating angles. The X⋯Sb–CF motifs with these deviating angles correlate with the longest Sb⋯X distances mentioned above. A strongly developed interaction comprises a substantial attractive electrostatic component, but also a charge-transfer component of a lone pair → σ* donation type or, in other words, an incomplete 3c-4e-type bonding.3,4,67 Such a donation of the electron pair of the nucleophile populates the σ*(Sb–CF)-orbital and results in an elongation of that bond. Consistent with theory, the longest Sb–CF bonds of each adduct are found to be associated with the strongest Sb⋯X interactions, i.e. the shortest Sb⋯X distances, as well as the straightest CF–Sb⋯X angles (Fig. 4). To investigate the affinity for halide ions in more detail, we also studied host 2 and its adducts by means of quantum-chemical calculations.
 | ||
| Fig. 4 Diagram of the four Sb⋯X distances of [2·Cl]−[Me4N]+, [2·Br]−[nBu4N]+ and [2·I]−[nBu4N]+ in the solid state. The red area marks an Sb⋯X distance below the sum of the covalence radii,65 the blue area a distance above the sum of the van der Waals radii.61,62 In addition to these distances, the corresponding angle of each X⋯Sb–CF motif is given. Sb⋯X distances, for which the corresponding Sb–CF bond is not elongated (compared to the longest Sb–CF distance in free host 2; 2.244(6) Å) are marked with a circle. The numerical values of this figure are provided in the ESI.† | ||
Computational studies
All adduct formations (including the fluoride adduct, which was not realized experimentally) were investigated by DFT calculations at the PBE0-D3BJ/def2-TZVPD level of theory. The enthalpies of formation (Table 1) for the complexes were obtained using the formal reaction scheme with a reference reaction shown in Scheme 4. The calculation for this reference was performed in our previous work, in which we already calculated the enthalpies of formation for reactions of 1 with F−, Cl−, Br−, I−.58| ΔH [kcal mol−1] | ||
|---|---|---|
| Adduct | [2·X]− | [1·X]− |
| F | −91.1 | −87.0 |
| Cl | −93.2 | −51.8 |
| Br | −95.0 | −45.6 |
| I | −94.4 | −39.3 |
 | ||
| Scheme 4 Isodesmic reactions of host 2 with halides X− (X = F, Cl, Br, I). Enthalpies of formation of the adducts are provided in Table 1, those of the calibration complexes [Sb(CF3)3·X]− are provided in the ESI† of our previous work.58 | ||
The interatomic Sb⋯X interactions found in the adducts [2·X]− are not the only stabilizing factors in the calculated adducts, but they are clearly the strongest interactions in each adduct. Table 1 lists the enthalpies of formation (ΔH) of the halide adducts [2·X]− compared to those of the bidentate host 1. While the enthalpy of formation of [1·F]− is identical to that of [2·F]− (within the assumed error), the enthalpies of formation of [2·X]− (X = Cl, Br, I) are higher than those for the corresponding adducts of 1. This result is consistent with our observations in the NMR competition experiments (see above): adducts with 2 are more stable in solution and able to abstract halide ions from complexes with 1. Unexpectedly, the enthalpies of formation of [2·X]− (X = F, Cl, Br, I) are indistinguishable within the assumed error. This is particularly surprising when comparing these values with the enthalpies of formation of host 1 with the halides. For the adducts [1·X]−, we clearly demonstrated that the adducts with lighter halides (i.e., the anions with a higher charge density) are the more stable ones.58 This is because the Sb⋯X interaction strength increases according to the trend I < Br < Cl < F. Since the enthalpies of formation of the complexes [2·X]− (X = F, Cl, Br, I) are equal, this stabilizing tendency must somehow be compensated for in host 2. The reason for this is the different number of strongly attractive Sb⋯X interactions in [2·X]−, which increases as the halide becomes heavier.
To demonstrate this, we applied QTAIM68 and IQA69 analyses to classify the bonding situation in the adducts of 2. Fig. 5 shows the molecular graphs and electron density contours of the four calculated structures of [2·X]− (X = F, Cl, Br, I) along with the four Sb⋯X distances as well as the electron density and Laplacian values at the bond critical points. In general, the well-defined pnictogen bonding interactions (for example, Sb1⋯Cl1 or Sb1⋯Br1) are very similar to the Sb⋯X interactions that we found in halide adducts of 1 (in terms of the electron density and Laplacian values). Also, as mentioned above, Sb⋯X interactions with lighter halides are generally stronger, but only if a suitable distance can be achieved (e.g. observed in the series Sb4⋯I < Sb4⋯Br < Sb4⋯Cl < Sb4⋯F). However, such a matching Sb⋯X distance cannot be achieved in every case. The optimized structures show a similar effect as already observed in the solid state structures: the lighter, i.e. the smaller the halide, the closer the four stibanyl groups must be to each other to ensure an effective Sb⋯X interaction. However, this is countered by the steric repulsion of the pentafluoroethyl substituents, which makes it increasingly difficult for the Sb(C2F5)2 functions to approach spatial proximity to the anion in case of the lighter halides. As a result, for the lighter halide ions, fewer favorable Sb⋯X distances, Sb–X–C angles, and thus less (comparatively) strong Sb⋯X interactions can be realized. This counteracting effect appears to offset the generally higher interactions with the lighter halides, so that effectively all enthalpies of formation of host 2 are similar.
 | ||
| Fig. 5 QTAIM molecular graphs of [2·X]− (X = F, Cl, Br, I) showing the bond paths and bond critical points. Depicted are just bond paths with ρBCP > 0.06 e Å−3. Additionally, the electron density contours of the plane defined by Sb2, X and Sb4 are depicted. Below each structure, the corresponding Sb⋯X distances (in Å), electron densities at the bond critical point (ρBCP in e Å−3) and Laplacian values at the bond critical point (∇2ρBCP in e Å−5) are provided. Full molecular graphs (with additional weaker bond paths) and full contour plots are provided in the ESI† (note, that the labels may differ). | ||
Therefore, in [2·I]− there are about four relatively strong interactions, which are similar to each other. In contrast, for [2·Br]− and [2·Cl]−, there are two relatively strong Sb⋯X interactions and two relatively weak Sb⋯X interactions, which are opposite to each other. For the two stronger interactions, the Sb⋯X distance is shorter, the electron densities at the bond critical points are higher and the X–Sb–C angles are closer to 180°. This trend is consistent with our observations for the solid state structures (Fig. 3 and 4). The fluoride adduct [2·F]− is clearly different and most extreme: the most stable calculated structure does not show a ‘centrally’ chelated fluoride ion, but an only bidentate chelated F− anion on one side of the host molecule. The fluoride ion shows very strong Sb⋯F interactions with two antimony atoms (comparable to the electron density and Laplacian values of [1·F]−) but no stabilizing interactions at all with the other two antimony atoms. Because the Sb⋯F⋯Sb bonding situation is very similar in [1·F]− and [2·F]−, the enthalpies of formation of these two are equal within the error.
For fluoride, we additionally calculated the conventionally used (gas-phase) fluoride ion affinity (FIA) via the isodesmic reference reaction with Me3SiF instead of Sb(CF3)3 (compare with Scheme 4, see ESI†). The FIA of host 2 is 92.8 kcal mol−1, which is slightly higher than that of host 1 (88.6 kcal mol−1)58 and remarkably higher than the FIA of Sb(C2F5)3 (75.3 kcal mol−1).70
Conclusions
Solid state structures and quantum-chemical calculations suggest that with decreasing anion radius, the spatial alignment required for an optimal interaction with the tetradentate host system 2 becomes increasingly difficult. For the lighter halides, it is energetically more favorable to avoid steric hindrance with one or two stibanyl functions and thus waive the energetically favorable formation of additional Sb⋯X interactions. This can be seen in the distortion of the Sb4-quadrangle towards a rhombus shape, the different Sb⋯X distances, the elongated Sb–CF bonds, as well as the X⋯Sb–C angles. This counteracting effect compensates for the stronger Sb⋯X interaction of the lighter halides, making system 2 an equally strong acceptor for F, Cl, Br, and I.The tetradentate host 2 is a significantly stronger acceptor than the bidentate host 1, as demonstrated both experimentally in solution by determination of the binding constants Ka and by competition reactions with the monomer 1 as well as by the calculated enthalpies of formation. Thus, 2 represents an impressive example of the chelating effect for poly-Lewis acids, that is well known for Lewis-basic ligands. Therefore, host 2 can be considered as a charge-reversed case of [12]crown-4 and also represents (to the best of our knowledge) the first tetradentate poly-Lewis acid based on pnictogen bonding interactions. Its high affinity for halides is also reflected in the ability of 2 to dissolve equimolar amounts of Me4NCl, which is itself insoluble in THF. In addition, the unique 19F NMR spectroscopic properties make host 2 a potential probe for halide ions, although the hydrolysis sensitivity of the system may prevent its useful application in aqueous media. The effects and correlations observed for our system provide a detailed perspective for the application of multiple, cooperating pnictogen bonding sites.
Data availability
The data that supports the findings of this study are available in the ESI† of this article. See the file cartesianXYZ.txt for the coordinates of the optimized structures. CCDC 2284089–2284092† contain the supplementary crystallographic data for this paper.Author contributions
J. L. Beckmann: investigation, methodology, validation, visualization, writing (original draft), J. Krieft: investigation (supporting synthesis), Y. V. Vishnevskiy: investigation (DFT, QTAIM, IQA), B. Neumann: investigation (SCXRD), H.-G. Stammler: investigation (SCXRD), N. W. Mitzel: funding acquisition, project administration, supervision, reviewing and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Marco Wißbrock and Dr Andreas Mix for recording NMR spectra, Barbara Teichner for performing elemental analyses, as well as Dr Jan-Hendrik Lamm for fruitful discussions. This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – grant Mi477/25-3, project no. 248859450 and Mi477/39-1, project no. 424957011 and grant Vi713/1-3, project no. 243500032. We furthermore thank the Regional Computing Centre of the University of Cologne (RRZK) for providing computing time and support on the DFG-funded (Funding number: INST 216/512/1FUGG) HPC system CHEOPS.Notes and references
- M. Breugst and J. J. Koenig, Eur. J. Org Chem., 2020, 2020, 5473–5487 CrossRef CAS.
- J. Y. Lim and P. D. Beer, Chem, 2018, 4, 731–783 CAS.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2013, 15, 11178–11189 RSC.
- G. R. Desiraju, P. S. Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati and K. Rissanen, Pure Appl. Chem., 2013, 85, 1711–1713 CrossRef CAS.
- A. Borissov, J. Y. C. Lim, A. Brown, K. E. Christensen, A. L. Thompson, M. D. Smith and P. D. Beer, Chem. Commun., 2017, 53, 2483–2486 RSC.
- A. Brown and P. D. Beer, Chem. Commun., 2016, 52, 8645–8658 RSC.
- D. Bulfield and S. M. Huber, Chem.–Eur. J., 2016, 22, 14434–14450 CrossRef CAS PubMed.
- G. Cavallo, P. Metrangolo, T. Pilati, G. Resnati, M. Sansotera and G. Terraneo, Chem. Soc. Rev., 2010, 39, 3772–3783 RSC.
- S. P. Cornes, M. R. Sambrook and P. D. Beer, Chem. Commun., 2017, 53, 3866–3869 RSC.
- J.-P. Gliese, S. H. Jungbauer and S. M. Huber, Chem. Commun., 2017, 53, 12052–12055 RSC.
- F. Heinen, D. L. Reinhard, E. Engelage and S. M. Huber, Angew. Chem., Int. Ed., 2021, 60, 5069–5073 CrossRef CAS PubMed.
- S. H. Jungbauer, D. Bulfield, F. Kniep, C. W. Lehmann, E. Herdtweck and S. M. Huber, J. Am. Chem. Soc., 2014, 136, 16740–16743 CrossRef CAS PubMed.
- S. H. Jungbauer and S. M. Huber, J. Am. Chem. Soc., 2015, 137, 12110–12120 CrossRef CAS PubMed.
- F. Kniep, S. H. Jungbauer, Q. Zhang, S. M. Walter, S. Schindler, I. Schnapperelle, E. Herdtweck and S. M. Huber, Angew. Chem., Int. Ed., 2013, 52, 7028–7032 CrossRef CAS PubMed.
- C. J. Massena, N. B. Wageling, D. A. Decato, E. Martin Rodriguez, A. M. Rose and O. B. Berryman, Angew. Chem., Int. Ed., 2016, 55, 12398–12402 CrossRef CAS PubMed.
- P. Metrangolo, H. Neukirch, T. Pilati and G. Resnati, Acc. Chem. Res., 2005, 38, 386–395 CrossRef CAS PubMed.
- J. Pancholi and P. D. Beer, Coord. Chem. Rev., 2020, 416, 213281 CrossRef CAS.
- M. G. Sarwar, B. Dragisic, S. Sagoo and M. S. Taylor, Angew. Chem., Int. Ed., 2010, 49, 1674–1677 CrossRef CAS PubMed.
- R. L. Sutar and S. M. Huber, ACS Catal., 2019, 9, 9622–9639 CrossRef CAS.
- J. Wolf, F. Huber, N. Erochok, F. Heinen, V. Guérin, C. Y. Legault, S. F. Kirsch and S. M. Huber, Angew. Chem., Int. Ed., 2020, 59, 16496–16500 CrossRef CAS PubMed.
- S. Benz, J. Mareda, C. Besnard, N. Sakai and S. Matile, Chem. Sci., 2017, 8, 8164–8169 RSC.
- R. Hein, A. Docker, J. J. Davis and P. D. Beer, J. Am. Chem. Soc., 2022, 144, 8827–8836 CrossRef CAS PubMed.
- K. T. Mahmudov, M. N. Kopylovich, M. F. C. Da Guedes Silva and A. J. L. Pombeiro, Dalton Trans., 2017, 46, 10121–10138 RSC.
- L. Vogel, P. Wonner and S. M. Huber, Angew. Chem., Int. Ed., 2019, 58, 1880–1891 CrossRef CAS PubMed.
- W. Wang, H. Zhu, L. Feng, Q. Yu, J. Hao, R. Zhu and Y. Wang, J. Am. Chem. Soc., 2020, 142, 3117–3124 CrossRef CAS PubMed.
- H. Zhao and F. P. Gabbaï, Nat. Chem., 2010, 2, 984–990 CrossRef CAS PubMed.
- B. Zhou and F. P. Gabbaï, Organometallics, 2021, 40, 2371–2374 CrossRef CAS.
- M. Hirai, J. Cho and F. P. Gabbaï, Chem.–Eur. J., 2016, 22, 6537–6541 CrossRef CAS PubMed.
- M. Hirai and F. P. Gabbaï, Angew. Chem., Int. Ed., 2015, 54, 1205–1209 CrossRef CAS PubMed.
- S. Moaven, M. C. Andrews, T. J. Polaske, B. M. Karl, D. K. Unruh, E. Bosch, N. P. Bowling and A. F. Cozzolino, Inorg. Chem., 2019, 58, 16227–16235 CrossRef CAS PubMed.
- H. J. Trubenstein, S. Moaven, M. Vega, D. K. Unruh and A. F. Cozzolino, New J. Chem., 2019, 43, 14305–14312 RSC.
- M. Yang, M. Hirai and F. P. Gabbaï, Dalton Trans., 2019, 48, 6685–6689 RSC.
- D. You, B. Zhou, M. Hirai and F. P. Gabbaï, Org. Biomol. Chem., 2021, 19, 4949–4957 RSC.
- H. Chen, A. Frontera, M. Ángeles Gutiérrez López, N. Sakai and S. Matile, Helv. Chim. Acta, 2022, 105, e202200119 CrossRef CAS.
- A. Gini, M. Paraja, B. Galmés, C. Besnard, A. I. Poblador-Bahamonde, N. Sakai, A. Frontera and S. Matile, Chem. Sci., 2020, 11, 7086–7091 RSC.
- V. M. Gonzalez, G. Park, M. Yang and F. P. Gabbaï, Dalton Trans., 2021, 50, 17897–17900 RSC.
- M. Hirai and F. P. Gabbaï, Chem. Sci., 2014, 5, 1886–1893 RSC.
- H. V. Humeniuk, A. Gini, X. Hao, F. Coelho, N. Sakai and S. Matile, JACS Au, 2021, 1, 1588–1593 CrossRef CAS PubMed.
- L. M. Lee, M. Tsemperouli, A. I. Poblador-Bahamonde, S. Benz, N. Sakai, K. Sugihara and S. Matile, J. Am. Chem. Soc., 2019, 141, 810–814 CrossRef CAS PubMed.
- Y. Li, L. Meng, C. Sun and Y. Zeng, J. Phys. Chem. A, 2020, 124, 3815–3824 CrossRef CAS PubMed.
- G. Park and F. P. Gabbaï, Chem. Sci., 2020, 11, 10107–10112 RSC.
- D. Tofan and F. P. Gabbaï, Chem. Sci., 2016, 7, 6768–6778 RSC.
- P. Wonner, A. Dreger, L. Vogel, E. Engelage and S. M. Huber, Angew. Chem., Int. Ed., 2019, 58, 16923–16927 CrossRef CAS PubMed.
- P. Wonner, L. Vogel, M. Düser, L. Gomes, F. Kniep, B. Mallick, D. B. Werz and S. M. Huber, Angew. Chem., Int. Ed., 2017, 56, 12009–12012 CrossRef CAS PubMed.
- M. Yang, D. Tofan, C.-H. Chen, K. M. Jack and F. P. Gabbaï, Angew. Chem., Int. Ed., 2018, 57, 13868–13872 CrossRef CAS PubMed.
- J. Zhang, J. Wei, W.-Y. Ding, S. Li, S.-H. Xiang and B. Tan, J. Am. Chem. Soc., 2021, 143, 6382–6387 CrossRef CAS PubMed.
- S. Benz, A. I. Poblador-Bahamonde, N. Low-Ders and S. Matile, Angew. Chem., Int. Ed., 2018, 57, 5408–5412 CrossRef CAS PubMed.
- L. de Azevedo Santos, T. A. Hamlin, T. C. Ramalho and F. M. Bickelhaupt, Phys. Chem. Chem. Phys., 2021, 23, 13842–13852 RSC.
- A. Frontera and A. Bauzá, Crystals, 2021, 11, 1205 CrossRef CAS.
- L. Lu, Y. Lu, Z. Zhu and H. Liu, J. Mol. Model., 2019, 26, 16 CrossRef PubMed.
- J. S. Murray, P. Lane and P. Politzer, Int. J. Quantum Chem., 2007, 107, 2286–2292 CrossRef CAS.
- D. Sharma, S. Balasubramaniam, S. Kumar, E. D. Jemmis and A. Venugopal, Chem. Commun., 2021, 57, 8889–8892 RSC.
- D. Sharma, A. Benny, R. Gupta, E. D. Jemmis and A. Venugopal, Chem. Commun., 2022, 58, 11009–11012 RSC.
- C.-H. Chen and F. P. Gabbaï, Angew. Chem., Int. Ed., 2017, 56, 1799–1804 CrossRef CAS PubMed.
- J. Qiu, B. Song, X. Li and A. F. Cozzolino, Phys. Chem. Chem. Phys., 2017, 20, 46–50 RSC.
- J. Qiu, D. K. Unruh and A. F. Cozzolino, J. Phys. Chem. A, 2016, 120, 9257–9269 CrossRef CAS PubMed.
- J. L. Beckmann, J. Krieft, Y. V. Vishnevskiy, B. Neumann, H.-G. Stammler and N. W. Mitzel, Angew. Chem., Int. Ed., 2023, 62, e202310439 CrossRef CAS PubMed.
- P. Niermeier, K. A. M. Maibom, J.-H. Lamm, B. Neumann, H.-G. Stammler and N. W. Mitzel, Chem. Sci., 2021, 12, 7943–7952 RSC.
- S. Legoupy, L. Lassalle, J.-C. Guillemin, V. Metail, A. Senio and G. Pfister-Guillouzo, Inorg. Chem., 1995, 34, 1466–1471 CrossRef CAS.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- M. Mantina, A. C. Chamberlin, R. Valero, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. A, 2009, 113, 5806–5812 CrossRef CAS PubMed.
- M. J. Hynes, J. Chem. Soc., Dalton Trans., 1993, 311–312 RSC.
- J. von Seyerl, O. Scheidsteger, H. Berke and G. Huttner, J. Organomet. Chem., 1986, 311, 85–89 CrossRef CAS.
- P. Pyykkö and M. Atsumi, Chem.–Eur. J., 2009, 15, 186–197 CrossRef PubMed.
- S. Scheiner, J. Phys. Chem., 2011, 115, 11202–11209 CrossRef CAS PubMed.
- H. A. Bent, Chem. Rev., 1968, 68, 587–648 CrossRef CAS.
- R. F. W. Bader, Atoms in Molecules – A Quantum Theory, Oxford University Press, Oxford, 1990 Search PubMed.
- M. A. Blanco, A. Martín Pendás and E. Francisco, J. Chem. Theory Comput., 2005, 1, 1096–1109 CrossRef CAS PubMed.
- P. Erdmann, J. Leitner, J. Schwarz and L. Greb, ChemPhysChem, 2020, 21, 987–994 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic protocols, spectra, crystallographic data, details on calculations. CCDC 2284089–2284092. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc04594c |
| This journal is © The Royal Society of Chemistry 2023 |