
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA highly diastereoselective strain-release Doyle–Kirmse reaction: access to functionalized difluoro(methylene)cyclopropanes†
Suparnak
Midya
and
Durga Prasad
Hari
 *
*
Department of Organic Chemistry, Indian Institute of Science, Bangalore 560012, India. E-mail: dphari@iisc.ac.in
First published on 2nd November 2023
Abstract
Difluoro(methylene)cyclopropanes (F2MCPs) show better anti-cancer properties and chemical reactivities compared to their nonfluorinated analogues. However, catalytic stereoselective methods to access these privileged motifs still remain a challenging goal. The Doyle–Kirmse reaction is a powerful strategy for the concomitant formation of carbon–carbon and carbon–sulfur bonds. Although the enantioselective variants of this reaction have been achieved with high levels of selectivity, the methods that control the diastereoselectivity have been only moderately successful. Herein, we report a catalytic, highly diastereoselective strain-release Doyle–Kirmse reaction for synthesizing functionalized F2MCPs using an inexpensive copper catalyst. The transformation proceeds under mild conditions and displays excellent functional group compatibility on both diazo compounds and difluorocyclopropenyl methyl sulfane/selane derivatives. Furthermore, the obtained products were efficiently transformed into valuable building blocks, such as functionalized spiroheterocycles, difluorocyclopropanes, and skipped dienes.
Introduction
Methylenecyclopropanes are not only present in several biologically active natural products but are also useful reactive intermediates in organic synthesis.1 Owing to their high ring strain and unique reactivity, these privileged motifs participate in a plethora of transformations that build molecular complexity rapidly and selectively through ring-opening, ring-expansion, or cycloaddition reactions.2 Introducing a fluorine atom onto methylenecyclopropanes can significantly alter their biological properties and chemical reactivities because of the strongly electron-withdrawing nature of fluorine. Indeed, compounds with an embedded difluoro(methylene)cyclopropane (F2MCP) motif exhibit a wide spectrum of activities, including the Towne strain of human cytomegalovirus propagated in human foreskin fibroblasts and inhibitory activity against nematode-induced root damage and enoyl-CoA hydratase (Fig. 1A).3 Furthermore, F2MCPs could undergo a variety of transformations either on the distal or proximal bonds of the difluorocyclopropane ring and the exo-methylene moiety and showed superior reactivity to the nonfluorinated analogues.4 Nevertheless, the synthesis of F2MCPs is arduous due to their chemical instability under basic conditions, leading to more thermodynamically stable difluorocyclopropenes that undergo hydrolysis into cyclopropenones with an aromatic character.5 Taguchi and co-workers first reported the synthesis of F2MCPs through the elimination reaction of the selenoxide derived from difluorocyclopropylmethanol (Fig. 1B).4b,6 These motifs could also be accessed from allenes via difluorocyclopropanation using difluorocarbene precursors at very high temperatures.7 In 2017, Cossy and co-workers reported an efficient one-pot difluorocyclopropenation/Ireland–Claisen rearrangement strategy for the synthesis of F2MCPs.8 However, several drawbacks remain with the reported methods, such as the need for multiple reaction steps, elevated temperatures, strong base, and limited substrate scope, which hampered the application of these methodologies in organic synthesis. Furthermore, to the best of our knowledge, there are no examples of catalytic stereoselective methods for accessing F2MCPs. Consequently, the development of modular, catalytic stereoselective methods that rely on the use of readily available feedstocks under ambient conditions is highly desirable and would be valuable for drug design and discovery. | ||
| Fig. 1 (A) Bioactive molecules containing the F2MCP moiety; (B) reported methods for the synthesis of the F2MCP moiety; (C) Doyle–Kirmse reaction involving allylic sulfonium ylides; and (D) this work: catalytic highly diastereoselective strain-release Doyle–Kirmse reaction. | ||
The [2,3]-sigmatropic rearrangement of allyl sulfonium ylides is a powerful strategy for the concomitant formation of carbon–carbon and carbon–sulfur bonds. Among the methods used for the generation of sulfonium ylides, in situ formation of ylides via transition-metal catalyzed reaction of diazo compounds with allylic sulfides – the Doyle–Kirmse reaction – has found numerous applications in organic chemistry to rapidly access functionalized building blocks with the applications in the synthesis of biologically active molecules and natural products (Fig. 1C).9 The enantioselective variants of this reaction have been achieved with excellent levels of selectivity, but the strategies that control the diastereoselectivity have been only moderately successful.10 This is attributed to the fact that the stereo-determining step is the [2,3]-sigmatropic rearrangement of the sulfonium ylide without the metal catalyst attached.10d As a result, the selectivity of the rearrangement does not depend on the metal catalyst. In this context, strategies that provide good diastereocontrol over the rearrangement step would be highly desirable since they would give access to highly substituted homoallylic sulfides containing two contiguous stereocenters. In the last few years, ring strain in organic molecules has emerged as a powerful tool to promote reactivity through strain-release, allowing the facile construction of a myriad of useful scaffolds.11 We considered the possibility of a strain-release strategy to promote the Doyle–Kirmse reaction. Wiberg and co-workers reported that introducing each sp2 carbon center into a cyclopropane ring creates an additional ring strain of around 12–15 kcal mol−1.12 For example, the strain energy of cyclopropene is estimated to be 14.6 kcal mol−1 higher than that of methylenecyclopropane. Consequently, the exocyclic double bond in the three-membered ring is more stable than the endocyclic double bond. Furthermore, owing to cyclopropene's conformational rigidity, better diastereocontrol over the [2,3]-sigmatropic rearrangement step could be anticipated. Taking these factors into account, we envisaged that the reaction of difluorocyclopropene derivative 2 with a metal carbene would generate an ylide I, which then undergoes strain-release [2,3]-sigmatropic rearrangement to give stereoselective access to highly functionalized F2MCPs 3 (Fig. 1D).
This proposal offers a mechanistically different method to access challenging substituted F2MCPs and opens up significant chemical space due to the readily diversifiable positions. Herein, we report our success in developing a catalytic highly diastereoselective strain-release Doyle–Kirmse reaction under mild conditions using an inexpensive copper catalyst. The reaction scope was broad with respect to both diazo compounds and difluorocyclopropene derivatives and tolerated a wide range of functional groups. Furthermore, the obtained F2MCPs are easily converted into other important building blocks through the transformations of the reactive exo-methylene moiety and difluorocyclopropane ring.
Results and discussion
Our investigations began by studying the strain-release Doyle–Kirmse reaction using tert-butyl diazoacetate (1a) and ((3,3-difluorocycloprop-1-en-1-yl)methyl)(phenyl)sulfane (2a), which is readily obtained by the reaction of phenyl propargyl sulfide with TMSCF3.13 First, we evaluated metal catalysts used for both metallocarbene formation and Lewis acid activation. Pleasingly, in the presence of Rh2(OAc)4 catalyst, the desired F2MCP 3a was obtained in 22% yield with excellent diastereoselectivity along with a large amount of C–H insertion product 4 (Table 1, entry 1). With a more electrophilic rhodium catalyst, Rh2(TFA)4, only a trace amount of the product was detected (Table 1, entry 2). The yield could be improved to 32% using Rh2(esp)2 (Table 1, entry 3). No reaction was observed when using AgOTf, Fe(TPP)Cl, or Cu(OTf)2 (Table 1, entries 4–6). CuCl and Cu(acac)2 gave comparable yields to Rh2(esp)2, but dimerization product 5 was the major product (Table 1, entries 7 and 8). In copper-catalyzed reactions, chelating ligands often display enhancement in the reactivity.14 Next, the effect of various ligands on this transformation was examined. The results showed that the employed ligands reduced the catalytic activity, resulting in slightly lower yields (Table 1, entries 9–11). Among the solvents tested (Table 1, entries 12–14), dichloromethane was found to be the best solvent. By performing the reaction at 40 °C, we observed a significantly improved yield of 59% without affecting the diastereoselectivity (Table 1, entry 15). Finally, by using 2 equiv. of diazo, the desired product could be obtained in 80% yield (Table 1, entry 16). Without Cu(acac)2, no product formation was observed, demonstrating that the copper catalyst is necessary for the reaction (Table 1, entry 17).|
|
|||||
|---|---|---|---|---|---|
| S. no. | Catalyst | Solvent | Temp. (°C) | 3a yieldb | d.r.c |
| a Reaction conditions: 0.15 mmol of 1a, 0.15 mmol of 2a, catalyst (5 mol%), ligand (10 mol%), and solvent (0.1 M). b NMR yields using dibromomethane as the internal standard. c d.r. was determined by 19F-NMR of the crude reaction mixture. d With 0.30 mmol of 1a. NR = no reaction. | |||||
| 1 | Rh2(OAc)4 | DCM | 25 | 22 | >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
| 2 | Rh2(TFA)4 | DCM | 25 | Trace | >20:1 |
| 3 | Rh2(esp)2 | DCM | 25 | 32 | >20:1 |
| 4 | AgOTf | DCM | 25 | NR | — |
| 5 | Fe(TPP)Cl | DCM | 25 | NR | — |
| 6 | Cu(OTf)2 | DCM | 25 | NR | — |
| 7 | CuCl | DCM | 25 | 30 | >20:1 |
| 8 | Cu(acac)2 | DCM | 25 | 32 | >20:1 |
| 9 | CuCl, L1 | DCM | 25 | 13 | >20:1 |
| 10 | CuCl, L2 | DCM | 25 | 19 | >20:1 |
| 11 | CuCl, L3 | DCM | 25 | 10 | >20:1 |
| 12 | Cu(acac)2 | DCE | 25 | 19 | >20:1 |
| 13 | Cu(acac)2 | PhCH3 | 25 | 26 | >20:1 |
| 14 | Cu(acac)2 | THF | 25 | 20 | >20:1 |
| 15 | Cu(acac)2 | DCM | 40 | 59 | >20:1 |
| 16d | Cu(acac)2 | DCM | 40 | 80 | >20:1 |
| 17 | No catalyst | DCM | 40 | NR | — |

|
|||||
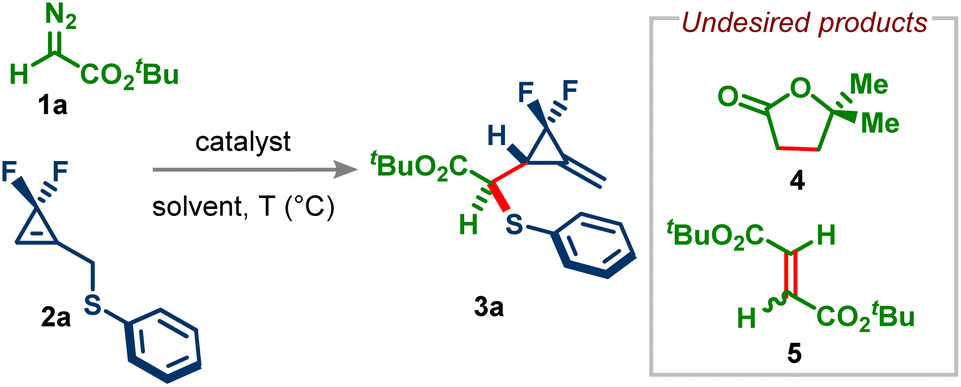
With the optimized conditions in hand, we examined the reaction scope of α-diazo esters (Scheme 1). 2-Adamantyl and benzyl diazoacetates provided the corresponding F2MCPs 3b and 3c in excellent yields and selectivities. Using simple ethyl diazoacetate, the desired product 3d was obtained in moderate yield but with a high diastereoselectivity. The low yield is due to the formation of a significant amount of dimerization products. Aryl diazo esters of different steric bulks reacted well and afforded the desired products (3e–3g) in good yields and selectivities. Pleasingly, diazo Weinreb amide and diazo phosphonate were also viable substrates in this reaction, giving products 3h and 3i. Diazo esters bearing heterocycles such as furan and pyridine also participated in this reaction (products 3j and 3k). Alkyl-substituted diazo compounds are less explored in the Doyle–Kirmse reaction than the simple acceptor diazo compounds. Therefore, we subjected tert-butyl α-diazo propionate to standard conditions. To our delight, the desired F2MCP 3l with a quaternary center was obtained in high yield and selectivity.
 | ||
| Scheme 1 Scope of diazo compounds and difluorocyclopropenyl methyl sulfanes. Standard conditions: 0.3 mmol of 2, 2.0 equiv. of 1, 5.0 mol% of Cu(acac)2, 3.0 mL of dry DCM, 40 °C, 12 h. Yields are of isolated products. dr was determined by 19F-NMR from a crude reaction mixture. aRh2(OAc)4 was used instead of Cu(acac)2. bRh2(TFA)4 was used instead of Cu(acac)2. cThere was no stereochemical induction from menthol, borneol, and cholesterol. d0.15 mmol scale. | ||
Donor–acceptor diazo compounds also underwent the desired transformation, giving thioethers 3m–3p albeit with low diastereoselectivity because such diazo compounds are known to provide metal-unbound ylides with allyl sulfides (vide infra), thus resulting in poor selectivity over the rearrangement step. Finally, acceptor–acceptor diazo compounds 1q and 1r were tested in our reaction; unfortunately, under the standard conditions, the desired products 3q and 3r were obtained in very poor yield. However, using more reactive Rh2(TFA)4, the yield of 3q and 3r could be improved to 88% and 67% yields, respectively. Modification of bioactive natural products may help enhance their biological activity. Therefore, our method was applied to diazo derivatives of natural products such as menthol, borneol, and cholesterol. Pleasingly, the desired products 3s–3u were obtained in good yields with excellent selectivities. Tosylhydrazone derived from methyl 2-oxo-2-phenylacetate didn't provide the desired product under the standard conditions. However, simple light irradiation delivered the desired F2MCP 3o in 35% yield (See the ESI† for more details).
We then turned our attention to the scope of difluorocyclopropenyl methyl sulfides. p-Methyl and p-methoxy groups were well tolerated on the aryl ring of sulfides, giving products 3v and 3w in good yields with excellent diastereoselectivities. These sulfides also reacted well with disubstituted diazo compounds, leading to quaternary-carbon-containing products 3x and 3y. Simple benzyl sulfide also participated well in this reaction (product 3z). Notably, methyl and phenyl-substituted difluorocyclopropenes underwent the desired transformation successfully, giving products 3aa and 3ab possessing an all-carbon quaternary stereocenter. Methyl substitution α to the sulfur was also tolerable, affording alkylidene difluorocyclopropanes 3ac and 3ad with good Z/E selectivity.15
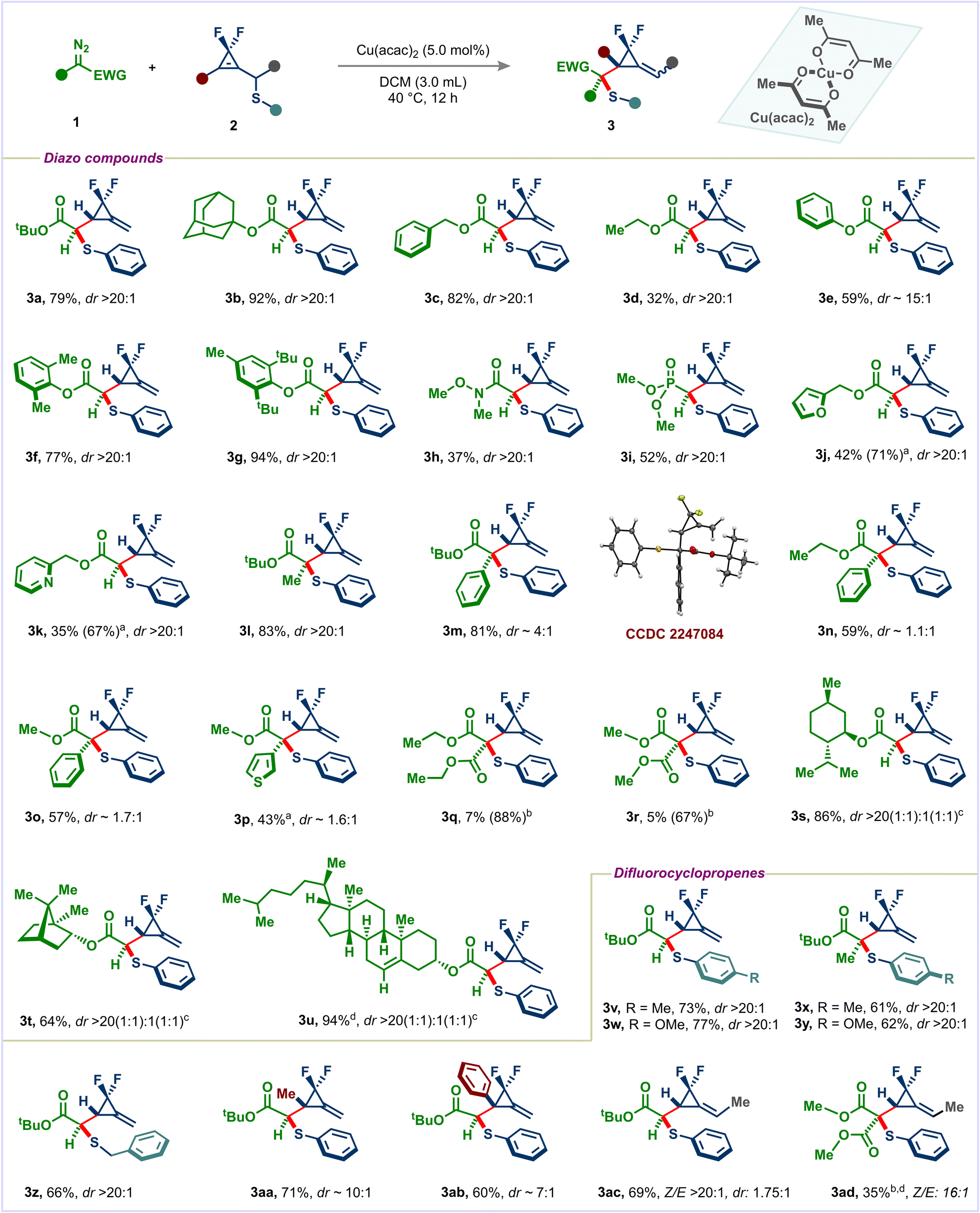
Organoselenium compounds are not only present in several bioactive compounds but also useful synthetic handles for introducing highly valuable functional groups. The [2,3]-sigmatropic rearrangement of allylic selenides with diazo esters is considerably less explored than the lighter group VI elements.16 Therefore, we have extended our methodology to difluorocyclopropenyl selenium analogues. When we subjected (3,3-difluorocycloprop-1-en-1-yl)methyl)(phenyl)selane (4) to our standard conditions, the desired F2MCP 5a was obtained in 31% yield with excellent diastereoselectivity (Scheme 2). Switching to Rh2(OAc)4, this reaction successfully provided the desired product 5a in a much improved 65% yield. Benzyl and dimethyl phenyl diazo esters were also viable substrates in this reaction giving products 5b and 5c. The relative stereochemistry of the major diastereomer 5c was determined by X-ray analysis (CCDC 2250643). The transformation was not limited to α-diazo esters: both diazo Weinreb amide and diazo phosphonate gave the desired products 5d and 5e in 58% and 44% yields with high selectivities. tert-Butyl α-diazo propionate also participated well in this reaction providing selenoether 5f with a quaternary stereocenter in 64% yield.
 | ||
| Scheme 2 Scope of diazo compounds with difluorocyclopropenyl methyl selane. Standard conditions: 0.15 mmol of 4, 2.0 equiv. of 1, 5.0 mol% of Cu(acac)2/2.0 mol% of Rh2(OAc)4, 1.0 mL of dry DCM, 30 °C, 12 h. Yields are of isolated products. dr was determined by 19F-NMR of the crude reaction mixture. | ||
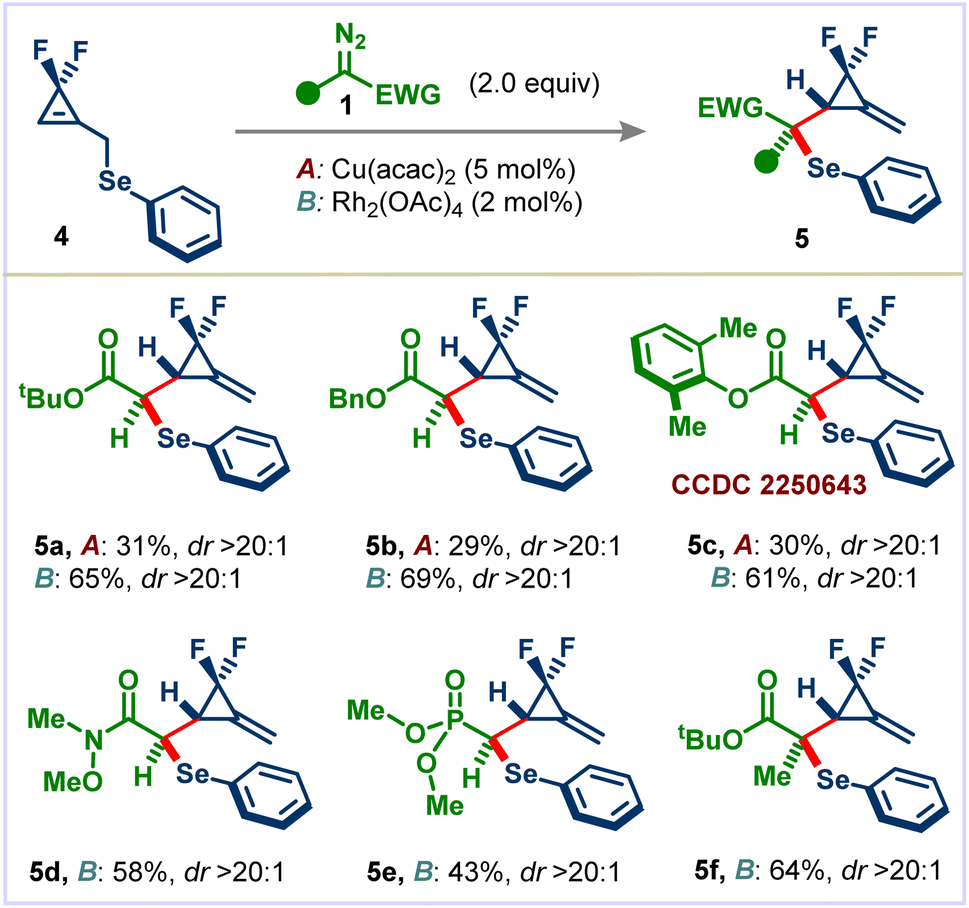
To illustrate the practicality of this method, we performed the reaction on a 1.0 mmol scale using 1a and 2a. The desired product 3a was obtained in 60% yield (Scheme 3A). Next, we carried out several experiments to highlight the potential synthetic utility of the obtained products (Scheme 3B). A [3 + 2] cycloaddition reaction between F2MCP 3a and nitrone afforded spirocycle 6 in 67% yield with excellent diastereoselectivity. Diels–Alder cycloaddition of 3a with furan in toluene at 140 °C delivered spiro-bridged heterocyclic adduct 7 in 58% yield. Oxidation of 3a using m-CPBA led to sulfone 8 in excellent yield, which is not possible to synthesize using the Doyle–Kirmse reaction from the corresponding difluorocyclopropenyl methyl sulfone. Bromination of F2MCP 3a provided the corresponding highly functionalized difluorocyclopropane derivative 9 in 89% yield with 1.5:1 dr. Skipped dienes are core frameworks in a myriad of products, including primary fatty acid metabolites and alkaloids.17 Many methods have been reported for the synthesis of these privileged molecules.18 However, reports on synthesizing fluorinated skipped dienes are very scarce; only a single example has been reported.19 To our delight, treating 3a with bromine followed by sodium hydride delivered highly functionalized skipped diene 10 in 85% yield with an excellent E/Z selectivity. Furthermore, irradiation of 3a using 20 mol% Ph2Se2 gave the skipped diene 11 in good yield and selectivity (see the ESI† for the mechanism).
 | ||
| Scheme 3 (A) Scale-up. (B) Synthetic utility of the obtained products. Yields are of isolated products. dr was determined by 19F-NMR of the crude reaction mixture. Reactions were carried out on a 0.1 mmol scale. | ||
Based on the literature reports9a,20 and our observations, we proposed a plausible mechanism in Scheme 4A. The copper catalyst first reacts with diazo compound 1a to form copper–carbene intermediate I, which reacts with difluorocyclopropenyl methyl sulfide 2a, generating sulfonium ylide II. The subsequent [2,3]-sigmatropic rearrangement furnishes the desired product 3a. We have proposed a stereochemical model to account for the observed stereochemistry, which was confirmed by X-ray analysis of products 3m and 5c. The rearrangement would be expected to proceed via a five-membered envelope-like transition state, in which the ester group occupies a pseudo-equatorial position where the A1,3 strain is minimalized (TS-A) (Scheme 4B). This transition state would give the observed major diastereomer. Next, we sought to investigate why the selectivity was very low in the case of donor–acceptor diazo compounds 1m–1p (see products 3m–3p). In 2021, Tantillo and co-workers proposed that the rhodium catalyst dissociates from the ylide that is generated from rhodium–carbene and donor–acceptor diazo compounds before the [2,3]-sigmatropic rearrangement,21 thus resulting in poor selectivity. If a similar free ylide mechanism were operative in our copper catalyst system, one would expect that irradiation of diazo 1m, known to give free-carbene,22 and 2awould give the product 3m with the same diastereoselectivity (4:1 dr). To this end, when we irradiated the diazo compound 1m and 2a, the product 3m was obtained in 40% yield with 4:1 dr. Furthermore, irradiation of the diazo compound 1n and 2a also gave similar results. These results suggest a free ylide mechanism for donor–acceptor diazo compounds. Control experiments with cyclic trisubstituted alkene 12 and cinnamyl(phenyl)sulfane (14)9j,16a,23 under the standard conditions gave the corresponding products 13 and 15 in 19% and 65% yields with poor selectivities. These results suggest that the high reactivity and selectivity observed with difluorocyclopropene derivatives could be due to the strain release.
 | ||
| Scheme 4 (A) Plausible mechanism. (B) Stereochemical model. (C) Control experiments. | ||
Conclusion
In summary, we have developed a catalytic highly diastereoselective strain-release Doyle–Kirmse reaction. It is worth mentioning that this is the first catalytic stereoselective method for the synthesis of functionalized F2MCPs. The transformation showed excellent compatibility with various functionalities on diazo compounds and difluorocyclopropenyl methyl sulfanes. Furthermore, it was successfully extended to difluorocyclopropenyl methyl selanes. The diverse synthetic modifications of the obtained products, such as spiroheterocycles, difluorocyclopropanes, sulfones, and skipped dienes, further demonstrate the potential of this method. We are currently exploring its application to the synthesis of complex molecules.Data availability
General information, experimental procedures, characterization data for all new compounds, and NMR spectra are in the ESI.† Data for the crystal structure reported in this paper have been deposited at the Cambridge Crystallographic Data Centre (CCDC) under the deposition number CCDC 2247084 and 2250643.Author contributions
S. M. and D. P. H. conceived and designed the project. S. M. carried out optimization, substrate scope, and mechanistic studies. D. P. H. and S. M. wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Science and Engineering Research Board (SERB), Government of India (File CRG/2022/007372) is gratefully acknowledged. D. P. H. thanks the Indian Institute of Science (IISc) Bangalore for the start-up grant and infrastructure. S. M. thanks the Indian Institute of Science (IISc) Bangalore for doctoral fellowships. We thank Dineshchakravarthy Senthurpandi for solving the X-ray crystal structures.Notes and references
- A. Brandi, S. Cicchi, F. M. Cordero and A. Goti, Chem. Rev., 2014, 114, 7317–7420 CrossRef CAS PubMed
.
-
(a) Y. Wang, M. E. Muratore, Z. Rong and A. M. Echavarren, Angew. Chem., Int. Ed., 2014, 53, 14022–14026 CrossRef CAS PubMed
-
(a) J. T. Pechacek, T. M. Bargar and M. R. Sabol, Bioorg. Med. Chem. Lett., 1997, 7, 2665–2668 CrossRef CAS
-
(a) W. R. Dolbier Jr, M. Seabury, D. Daly and B. E. Smart, J. Org. Chem., 1986, 51, 974–979 CrossRef
- A. Shibuya, M. Okada, Y. Nakamura, M. Kibashi, H. Horikawa and T. Taguchi, Tetrahedron, 1999, 55, 10325–10340 CrossRef CAS
- T. Taguchi, M. Kurishita and A. Shibuya, J. Fluorine Chem., 1999, 97, 157–159 CrossRef CAS
- Z.-L. Cheng, J.-C. Xiao, C. Liu and Q.-Y. Chen, Eur. J. Org Chem., 2006, 5581–5587 CrossRef CAS
- G. Ernouf, J.-L. Brayer, B. Folléas, J.-P. Demoute, C. Meyer and J. Cossy, J. Org. Chem., 2017, 82, 3965–3975 CrossRef CAS PubMed
-
(a) T. H. West, S. S. M. Spoehrle, K. Kasten, J. E. Taylor and A. D. Smith, ACS Catal., 2015, 5, 7446–7479 CrossRef CAS
-
(a) D. S. Carter and D. L. Van Vranken, Org. Lett., 2000, 2, 1303–1305 CrossRef CAS PubMed
-
(a) M. A. A. Walczak, T. Krainz and P. Wipf, Acc. Chem. Res., 2015, 48, 1149–1158 CrossRef CAS PubMed
-
(a) K. B. Wiberg, Angew. Chem., Int. Ed., 1986, 25, 312–322 CrossRef
-
(a) F. Wang, T. Luo, J. Hu, Y. Wang, H. S. Krishnan, P. V. Jog, S. K. Ganesh, G. K. S. Prakash and G. A. Olah, Angew. Chem., Int. Ed., 2011, 50, 7153–7157 CrossRef CAS PubMed
- S. J. Hwang, S. H. Cho and S. Chang, Pure Appl. Chem., 2008, 80, 873–879 CrossRef CAS
-
(a) D. A. Evans, G. C. Andrews, T. T. Fujimoto and D. Wells, Tetrahedron Lett., 1973, 14, 1385–1388 CrossRef
-
(a) Y. Nishibayashi, K. Ohe and S. Uemura, J. Chem. Soc., Chem. Commun., 1995, 1245–1246, 10.1039/C39950001245
- G. Petruncio, Z. Shellnutt, S. Elahi-Mohassel, S. Alishetty and M. Paige, Nat. Prod. Rep., 2021, 38, 2187–2213 RSC
- T. Sato, T. Suto, Y. Nagashima, S. Mukai and N. Chida, Asian J. Org. Chem., 2021, 10, 2486–2502 CrossRef CAS
- A. L. Henne and E. G. DeWitt, J. Am. Chem. Soc., 1948, 70, 1548–1550 CrossRef CAS
- Z. Li, B. T. Parr and H. M. L. Davies, J. Am. Chem. Soc., 2012, 134, 10942–10946 CrossRef CAS PubMed
- C. J. Laconsay and D. J. Tantillo, ACS Catal., 2021, 11, 829–839 CrossRef CAS
-
(a) R. Hommelsheim, Y. Guo, Z. Yang, C. Empel and R. M. Koenigs, Angew. Chem., Int. Ed., 2019, 58, 1203–1207 CrossRef CAS PubMed
-
(a) V. K. Aggarwal, M. Ferrara, R. Hainz and S. E. Spey, Tetrahedron Lett., 1999, 40, 8923–8927 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2247084 and 2250643. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc04749k |
| This journal is © The Royal Society of Chemistry 2023 |