
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModulating the folding and binding of peptides using a stimuli-responsive molecular tweezer†
Sooho
Ko‡
a,
Joo-Young
Kim‡
b,
Jung Yeon
Park
c,
You-jin
Jung
 a,
Min-Jae
Choi
d,
Kyeong Sik
Jin
e,
Yongju
Kim
c,
Yong-beom
Lim
*a and
Woo-jin
Jeong
*bf
a,
Min-Jae
Choi
d,
Kyeong Sik
Jin
e,
Yongju
Kim
c,
Yong-beom
Lim
*a and
Woo-jin
Jeong
*bf
aDepartment of Materials Science and Engineering, Yonsei University, Seoul 03722, Republic of Korea
bDepartment of Biological Sciences and Bioengineering, Inha University, Incheon, 22212, Republic of Korea. E-mail: wjjeong@inha.ac.kr
cKU-KIST Graduate School of Converging Science and Technology, Department of Integrative Energy Engineering, Korea University, Seoul 02841, Republic of Korea
dDepartment of Chemical & Biochemical Engineering, Dongguk University, Seoul, 06420, Republic of Korea
ePohang Accelerator Laboratory, Pohang University of Science and Technology, Pohang 790-784, Republic of Korea
fDepartment of Biological Engineering, Inha University, Incheon, 22212, Republic of Korea
First published on 1st September 2023
Abstract
This study presents the development of a β-hairpin (tryptophan zipper, Trpzip)-based molecular tweezer (MT) that can control the folding and binding of α-helical peptides. When an α-helix isolated from the p53 protein was conjugated with Trpzip in an optimized macrocyclic structure, the folded β-hairpin stabilized the helix conformation through the side chain-to-side chain stapling strategy, which notably enhanced target (hDM2) affinity of the peptide. On the other hand, the helicity and binding affinity were significantly reduced when the hairpin was unfolded by a redox stimulus. This stimulus-responsive property was translated into the effective capture and release of model multivalent biomaterials, hDM2-gold nanoparticle conjugates. Since numerous protein interactions are mediated by α-helical peptides, these results suggest that the β-hairpin-based MT holds great potential to be utilized in various biomedical applications, such as protein interaction inhibition and cancer biomarker (e.g., circulating tumor cells and exosomes) detection.
Introduction
Various molecular machines have been developed by mimicking the working mechanisms of macroscopic machines.1 In particular, molecular tweezers (MTs) have attracted considerable interest in many fields, as controlling the capture-and-release process allows effective manipulation of nano- and microscale substances.2 Armed with two interaction sites bridged by a linker, MTs can exhibit enhanced target affinity compared to their monovalent counterparts.3 By introducing stimulus-responsive properties, it is also possible to develop MTs that switch open and closed conformations, thereby delivering and releasing guest molecules in a spatiotemporally controlled manner.4Peptides are promising materials for the development of MTs for biomedical applications,5 as (1) they are biocompatible and bioactive; many protein interactions are mediated by small peptide moieties,6 and (2) their amino acid compositions and macromolecular topologies are easily controllable using the solid-phase peptide synthesis (SPPS) method.7 Interestingly, β-hairpin peptides, which contain two β-strands connected by a turn structure, are morphologically similar to tweezers. In addition, they generally show a reversible and two-state folding behavior,8 which is suggestive of the pinching action of tweezers. However, the development of β-hairpin-based MTs has been significantly limited, likely because the formation of a stable β-hairpin in a short peptide is challenging due to their low conformational stability.9
Herein, to overcome this limitation, we employed the tryptophan zipper peptide (GEWTWDDATKTWTWTE, Trpzip), which was found to distinctively maintain a stable β-hairpin conformation.10 An α-helical peptide was grafted to the Trpzip sequence as the target binding motif of the MT in this study (Fig. 1a), considering that numerous protein–protein interactions (PPIs) are mediated by α-helical peptides. Since binding properties of peptides are closely related to their folding structures, α-helix stabilization in a short peptide has been one of important issues in the peptide research field.12 Among various strategies, the peptide stapling technique recently enabled an α-helix-based PPI inhibitor to demonstrate its clinical potential.13 In this approach, a synthetic brace stabilizes an α-helix by covalently connecting two remotely located amino acid residues (side chain-to-side chain stapling, Fig. 1b).
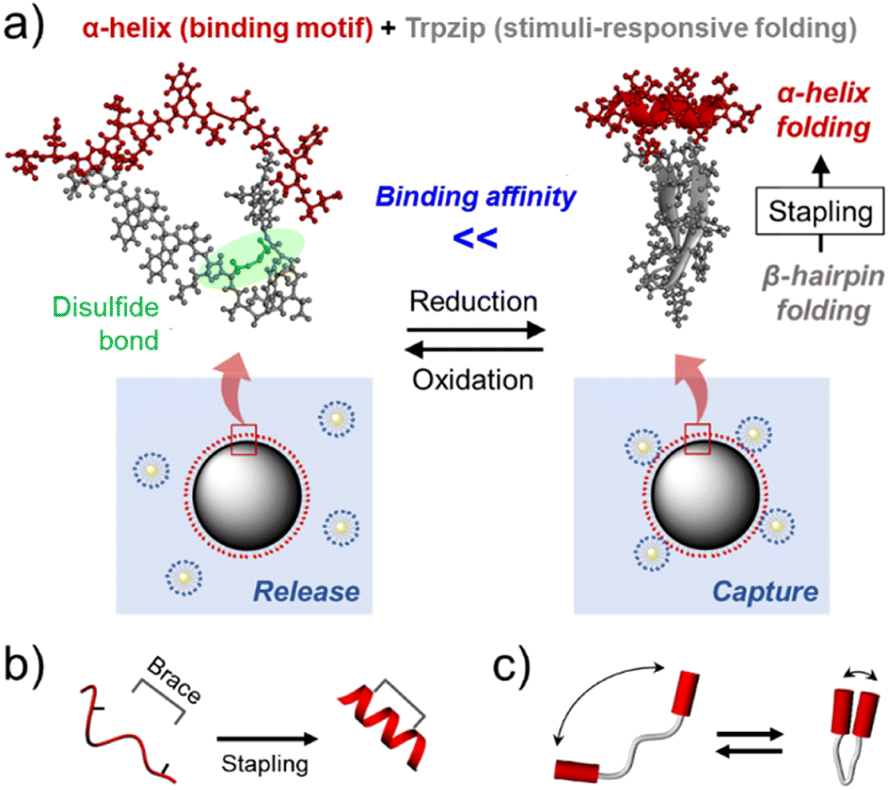 | ||
| Fig. 1 (a) A schematic illustration of the stimulus-responsive folding and binding of an α-helix/Trpzip-based molecular tweezer, which enabled effective capture and release of model multivalent biomaterials (protein-gold nanoparticle conjugates). Schematic illustrations of (b) the side chain-to-side chain stapling strategy for α-helix stabilization, (c) the folding behavior of molecular tweezers. | ||
As β-hairpin peptides are also able to confine guest molecules in a small space when folded (Fig. 1c), it was assumed that Trpzip could play the same role as the brace for α-helix stabilization. Previous studies have shown that β-hairpin folding can be controlled by introducing stimulus-responsive properties to the peptide.14 Hence, we also hypothesized that it would be possible to regulate the hairpin folding-mediated α-helix stabilization and, in turn, enable the helix to exhibit stimulus-responsive binding properties (Fig. 1a). To examine these assumptions, the trans-activation domain of p53 (residues 15–29, SQETFSDLWKLLPEN) to hDM2 was employed as a model α-helical peptide (p53α).15 The β-hairpin folding was controlled using an intrapeptide disulfide bond; two amino acid residues in the Trpzip sequence were substituted by cysteine residues, which were expected to prevent hairpin formation when oxidized. This redox-responsive behavior is opposite to that of typical disulfide stapling, which stabilizes α-helix with the disulfide bond formation (Fig. S5†). Thus, this α-helix/Trpzip-based molecular tweezer (αTMT) can be used as a unique tool for the control of α-helix-mediated protein interactions.
Results and discussion
For effective control of α-helix folding, the conformation of TMT should be distinctly changed in response to redox stimuli. We thus prepared three engineered Trpzip peptides with different cysteine substitutions and compared their folding behaviors (Trpzip-1, Trpzip-2, and Trpzip-3, Fig. 2a). Note that threonine residues were selected as the substitution sites, because they are abundant in the sequence and have physicochemical properties analogous to those of cysteine.16 Without the disulfide bond, all the engineered peptides formed stable β-hairpins, as indicated by the signature circular dichroism (CD) signal of Trpzip, which presented a strong negative band at 215 nm (β-sheet) and a positive band at 228 nm (indole–indole interactions of Trp pairs, Fig. 2b).10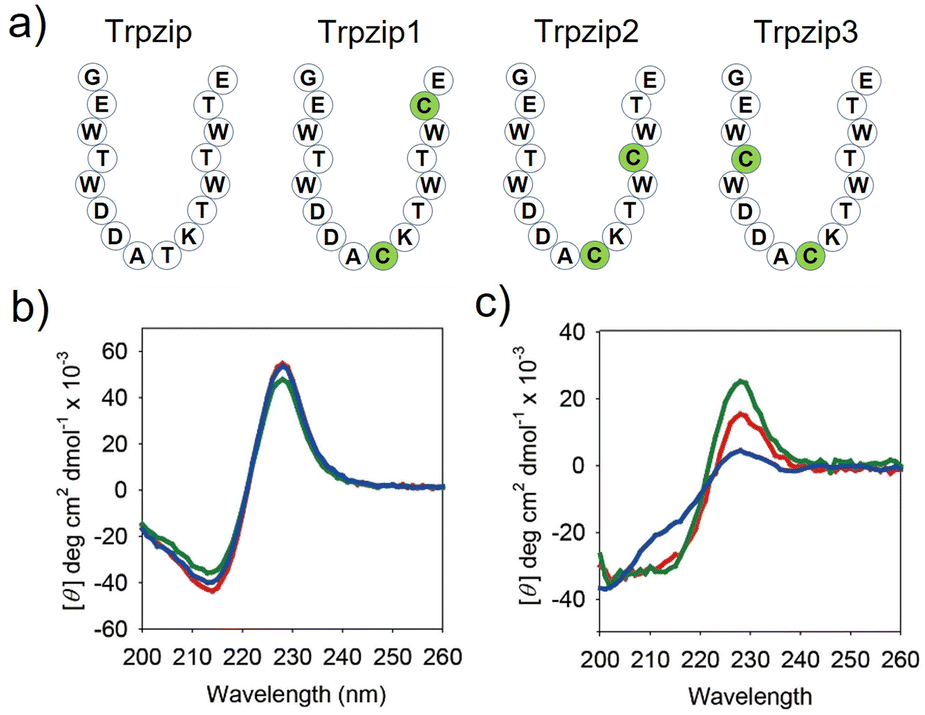 | ||
| Fig. 2 Sequence modification of the Trpzip peptide to exhibit redox stimuli-responsive folding behavior. (a) Schematic structures of Trpzip, TMT1, TMT2, and TMT3. CD spectra of Trpzip1 (red), Trpzip2 (green), and Trpzip3 (blue) in PBS (pH 7.0, 20 mM, 150 mM NaCl) at room temperature, (b) before and (c) after oxidation. [peptide] = 20 μM. | ||
However, after incubation for one day in an oxidizing solution (10% DMSO, 45% water, 45% acetonitrile) at room temperature, the CD spectrum of Trpzip-3 revealed destabilization of the hairpin structure with a substantially reduced positive band at 228 nm, a weakened negative band at 215 nm, and an enhanced negative band at approximately 200 nm (random coil, Fig. 2c). On the other hand, the CD spectra of Trpzip-1 and Trpzip-2 showed much less change. It is noted that the formation of disulfide bonds was confirmed by (1) observing HPLC peak shift (Fig. S6†) and (2) coupling n-methoxycarbonyl maleimide to the peptides (thiol-maleimide chemistry) and monitoring the molecular weight change using matrix-assisted laser desorption/ionization (MALDI) mass spectrometry (Fig. S7†). Hence, Trpzip-3 was selected as the MT sequence, showing the largest conformational change in response to the stimulus.
Next, the Trpzip-3 sequence was hybridized with p53α within a macrocyclic structure (αTMT). The helix stapling efficiency is known to be highly influenced by the length and geometry of the brace.11b,17 We thus prepared three αTMT peptides with different stapling sites (i,i + 3, i,i + 7, and i,i + 10 staplings for αTMT1, αTMT2, αTMT3, respectively) to identify a configuration that fulfills the structural requirements for effective helix stabilization (Fig. 3a). As shown in Fig. 3b, CD analysis revealed that αTMT2 had more α-helix propensity than other peptides, which was indicated by more intense negative CD bands at 208 and 222 nm (α-helix).18 Subtraction of CD spectrum of αTMT1 from that of αTMT2 resulted in a typical α-helix CD signal, displaying negative bands at 208 and 222 nm and positive signal at the lowest wavelength (inset in Fig. 3b). Although a positive maximum at 190 nm is another band characteristic of α-helices, significant far-UV absorption of the buffer solution limited the CD measurements below 200 nm. The results of Fourier transform infrared (FT-IR) analysis were also consistent with the CD data (Fig. 3c). Deconvolution of the FT-IR spectrum revealed the coexistence of a β-hairpin and an α-helix in the αTMT2 peptides, which was indicated by the bands at 1630 cm−1 (β-sheet), 1653 cm−1 (α-helix), and 1674 cm−1 (turn).18
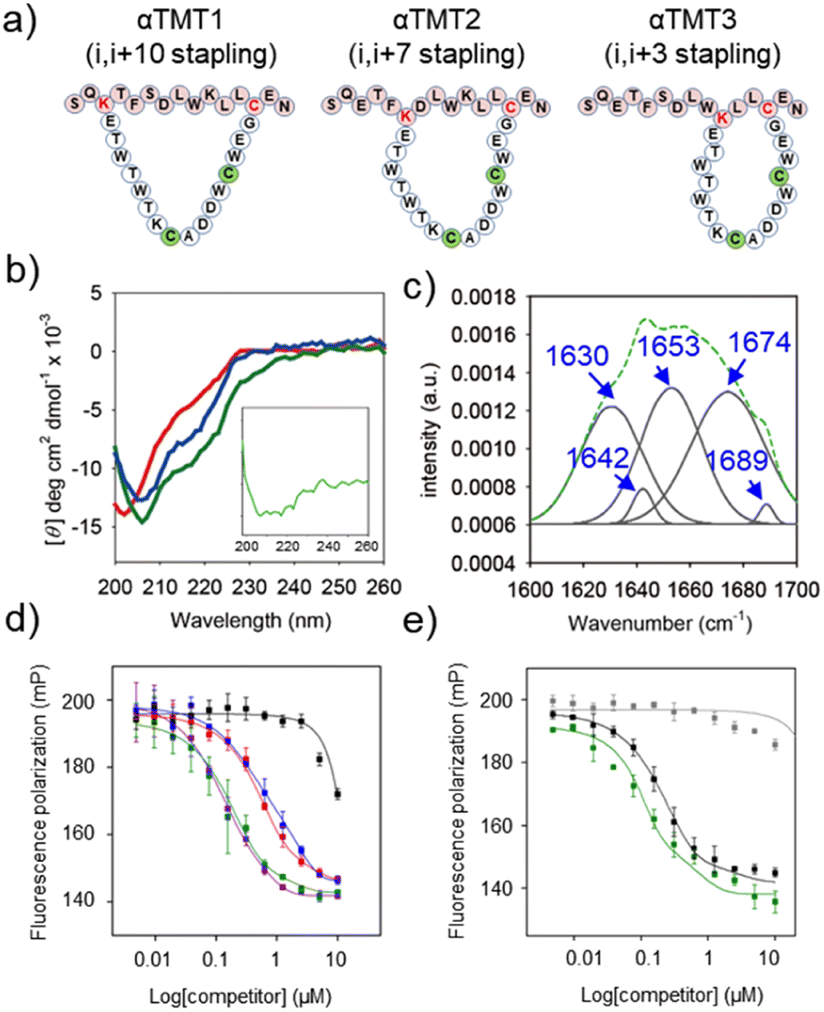 | ||
| Fig. 3 Hybridization of the TMT3 and p53α sequences in a macrocyclic structure. (a) Schematic structures of αTMT1, αTMT2, and αTMT3. (b) CD spectra of αTMT1 (red), αTMT2 (green), and αTMT3 (blue) in PBS (pH 7.0, 20 mM, 150 mM NaCl) at room temperature. [peptide] = 20 μM. Inset: CD spectrum obtained by subtracting the CD spectrum of αTMT1 from that of αTMT2. (c) FTIR data of αTMT2. Original spectrum (green) and Fourier self-deconvolution (gray). FP competition experiments: titration of (d) αTMT1 (red), αTMT2 (green), αTMT3 (blue), Nutlin-3a (purple), αTMT2A (black), (e) αTMT2 (green), stapled p53α (black), and native p53α (gray) against the p53f/hDM2 (100 nM/4 μM) complexes in Tris–HCl buffer solution (pH 8.0, 20 mM, 10 mM imidazole, 150 mM NaCl, 10% glycerol) at room temperature. The filter sets were 485/15 excitation and 535/25 emission. | ||
To further examine the stapling effect, the αTMT peptides were subjected to fluorescence polarization (FP) analysis, based on the fact that the hDM2 binding efficiency of the p53α peptides is correlated to their helicity.19 The fluorescein-conjugated p53α peptides (p53f) were synthesized (Fig. S1, S3, and S4†) and employed as a fluorescent probe in the FP assays. Fig. S8† shows the change in FP intensity of the p53f as a function of hDM2 concentration, which reflects peptide–protein complex formation. FP competition assays were then carried out to determine which peptide most effectively competed with p53f for hDM2 binding. As shown in Fig. 3d and S9,† αTMT2 exhibited a significantly lower EC50 value than other peptides (0.547 μM, 0.149 μM, and 0.655 μM for αTMT1, αTMT2, and αTMT3, respectively), which was comparable to that of a well-known small molecule inhibitor, Nutlin-3a (0.154 μM). The substitution of a phenylalanine (one of the essential amino acids for hDM2 binding)19b with an alanine (αTMT2A) reduced the competition efficiency approximately 30 times (EC50 value of 4.584 μM), confirming the selectivity of the interactions. In addition, the inhibitory effect of αTMT2 was compared to the control peptides, namely the native and stapled p53α peptides (Fig. S1, S3, S4, and S10†). Consequently, the inhibitory effect of αTMT2 was similar to that of stapled p53α peptides and considerably higher than that of native unstapled p53α peptides (Fig. 3e).
We then explored if β-hairpin-mediated α-helix folding could be controlled by the redox state (Fig. 4a). Note that the αTMT2 peptides formed no disulfide bond without the oxidants for 24 h at least (Fig. S11†). As shown in Fig. 4b and S12,† disulfide bond formation (αTMT2-ox) resulted in a blueshift of the CD spectrum of αTMT2, which indicated a partial transition from an ordered structure to a disordered random coil conformation.
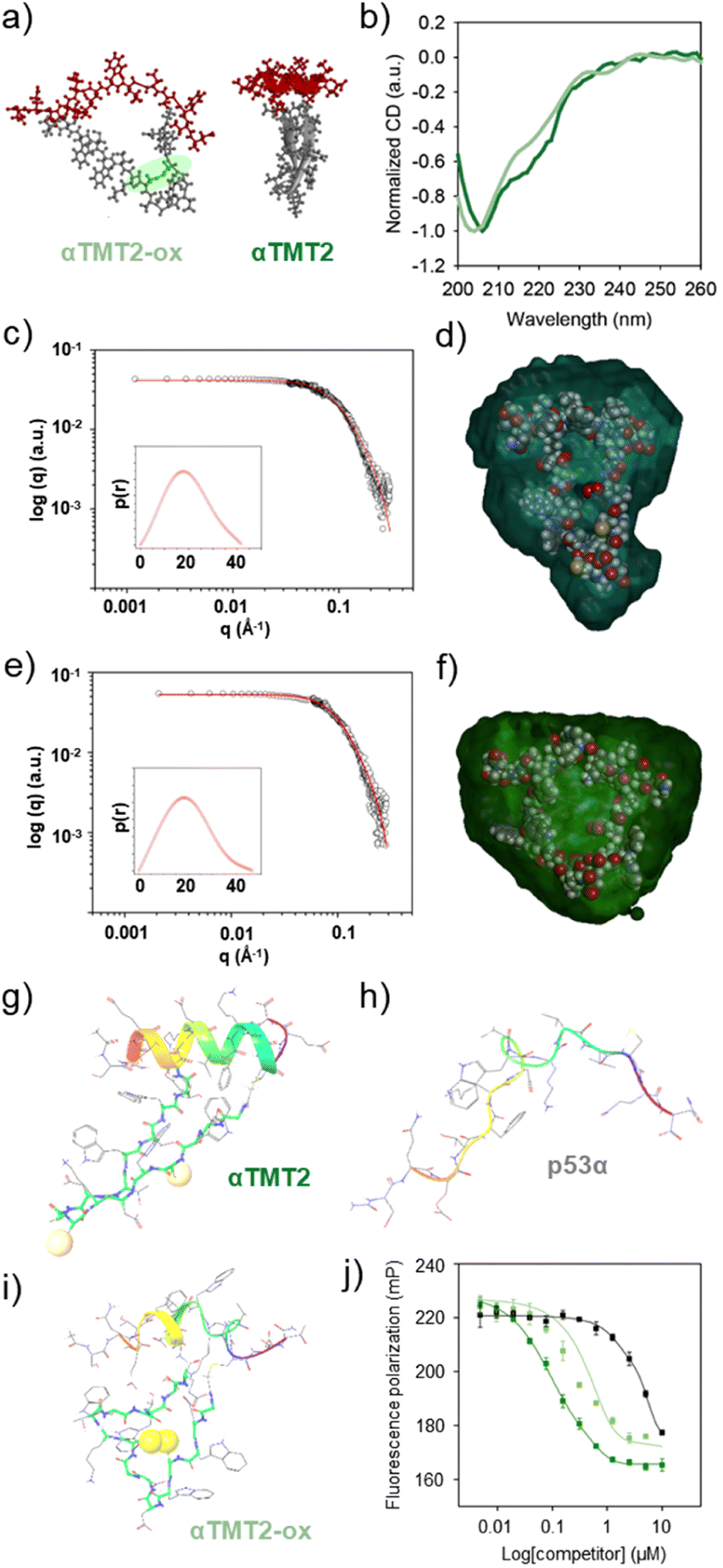 | ||
| Fig. 4 Redox stimuli-responsive folding and binding of αTMT2. (a) Molecular models of αTMT2-ox and αTMT2. (b) Normalized CD spectra of αTMT2 (green) and αTMT2-ox (light green) in PBS (pH 7.0, 20 mM, 150 mM NaCl) at room temperature. [peptide] = 20 μM. Small-angle X-ray scattering (SAXS) profiles of (c) αTMT2 and (e) αTMT2-ox in water at room temperature. Insets: pair distance distribution function of the peptides based on the scattering profiles. Superimposition of the molecular structures of (d) αTMT2 and (f) αTMT2-ox with the molecular envelope derived from the SAXS data. MD simulation results of folding behaviors of (g) αTMT2, (h) p53α, and (i) αTMT2-ox. (j) FP competition experiments: titration of competitors (i.e., αTMT2, green; αTMT2-ox, light green; and αTMT2A, black) against the p53f/hDM2 (100 nM/4 μM) complexes in Tris–HCl buffer solution (pH 8.0, 20 mM, 10 mM imidazole, 150 mM NaCl, 10% glycerol) at room temperature. The filter sets were 485/15 excitation and 535/25 emission. | ||
Small-angle X-ray scattering (SAXS) was also employed to investigate structural changes of the peptides upon oxidation. The shape and size information for αTMT2 and αTMT2-ox derived from their scattering profiles using a pair-distance distribution function P(r), which represents the profiles in real space (Fig. 4c and e). The calculated radius of gyration (Rg,p(r)) and maximum dimension (Dmax) of αTMT2 were 1.47 ± 0.014 nm and 4.19 nm, respectively, whereas the values of αTMT2-ox were 1.63 ± 0.18 nm and 4.61 nm. In addition, the reconstructed structural models, which fit well with the molecular models of the peptides, also demonstrated the structural difference between αTMT2 and αTMT2-ox (Fig. 4d and f).
This result was again supported by molecular dynamics (MD) simulations, in which peptide folding behaviors were commonly observed for 100 ns from the initially folded p53α. The simulation results revealed that the α-helix structure was maintained when coupled with the folded hairpin (Fig. 4g and S13†), whereas the native p53α and αTMT2-ox peptides showed significantly distorted helical conformations (Fig. 4h and i). Moreover, αTMT2-ox peptides have a lower number of hydrogen bonds than that of αTMT2, indicating weaker maintenance of the helical conformers (Fig. S14†). Furthermore, the FP competition assay revealed that αTMT2-ox exhibited approximately fivefold lower competition efficiency than the peptides without the disulfide bond (EC50 values of 0.111 μM and 0.569 μM for αTMT2 and αTMT2-ox, respectively, Fig. 4j). Therefore, the collective results demonstrated that αTMT2 has the most effective molecular configuration to exhibit stimulus-responsive folding and binding properties.
Next, we tested whether the redox-induced secondary structure change was reversible. As shown in Fig. S15a and S15b,† the CD spectra of the peptides exhibited negligible change throughout at least six cycles of oxidation–reduction reactions. In addition, the molecular weight of αTMT2 also remained constant following the repeated redox processes (Fig. S15c†). When αTMT2-ox was introduced into the p53f/hDM2 complex solution with reducing agents (1 mM of tris(2-carboxyethyl)phosphine hydrochloride, TCEP), we observed a time-dependent decrease in FP intensity (approximately a 90% decrease at 2 hours), which confirmed the recovery of the target-binding ability of the peptides due to the reduction-mediated α-helix folding (Fig. S16†). Therefore, we have verified that the stimulus-responsive folding and binding of αTMT2 are reversible.
With the optimized sequence in hand, we finally asked whether αTMT2 could capture and release target substances, which is a fundamental function of tweezers. The peptides were conjugated to the PEG linkers (500 Da) on the surface of magnetic bead substrates (αTMT2-bead) for a facile control of the environmental redox condition (Fig. 5a and S17†). Because this system was developed to interact with biomaterials, biocompatible oxidative condition (micromolar concentrations of hydrogen peroxide)20 was used for disulfide bond formation in the release step. The process of the capture-and-release (CAR) experiment in summary was as follows (Fig. 5a): (1) αTMT2-beads displaying the stabilized α-helices were incubated with the target materials for 1 h (capture step). (2) The buffer solution was changed to a hydrogen peroxide-containing solution with the aid of a magnet. (3) After incubation under the oxidative condition (release step), the released materials were separated from αTMT2-bead using the magnetic force and subjected to quantification.
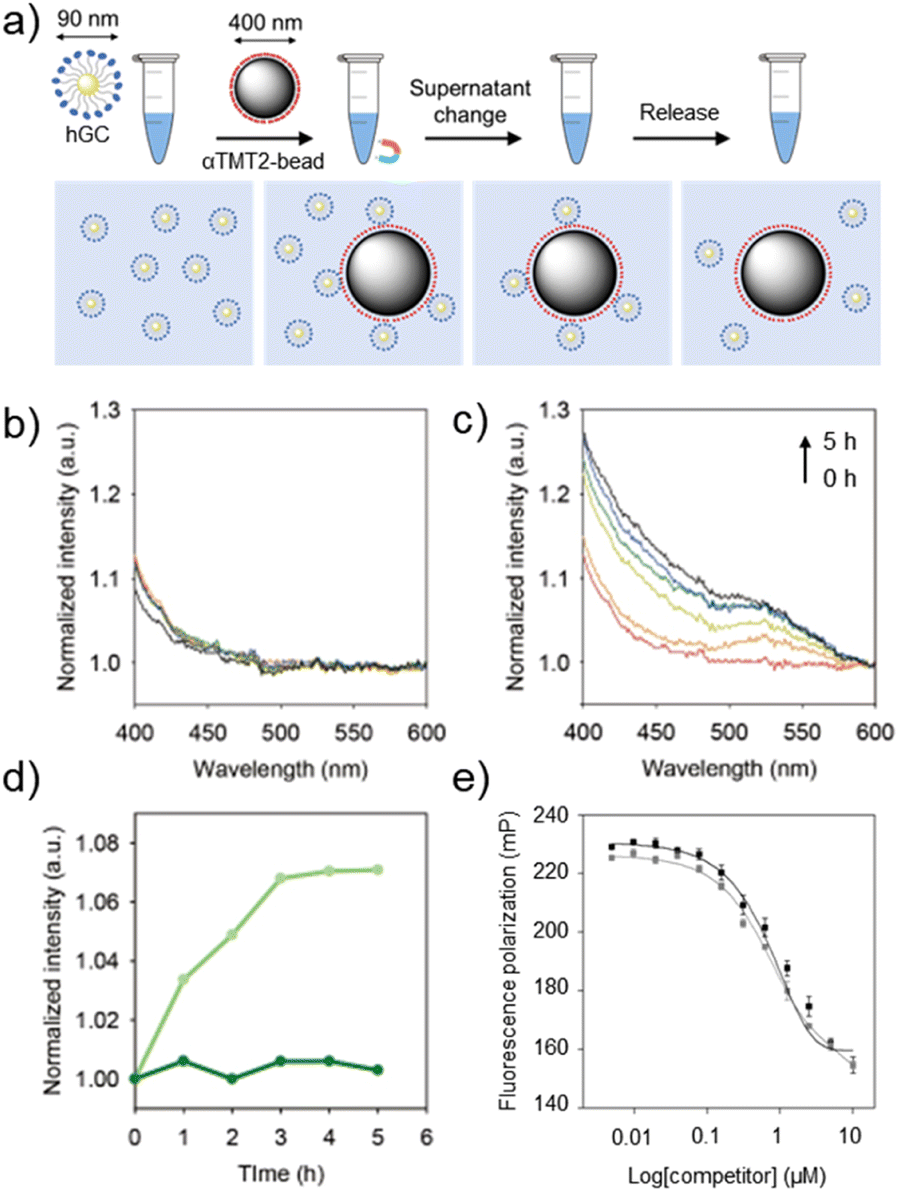 | ||
| Fig. 5 Controlled capture and release of hDM2-gold nanoparticle conjugates (hGCs). (a) An illustration of the capture-and-release (CAR) experiment. Time-dependent UV-vis absorption spectra of released hGCs after incubation in (b) PBS and (c) H2O2-containing PBS at different time intervals (0–5 h). (d) The release profiles of hGCs from αTMT2-bead in PBS (green) and H2O2-containing PBS (light green). (e) FP competition assays on the stapled p53α peptides against p53f/hDM2 (100 nM/4 μM) complexes in Tris–HCl buffer solution (pH 8.0, 20 mM, 10 mM imidazole, 150 mM NaCl, 10% glycerol) at room temperature, with (gray) and without (black) the oxidizing agents (H2O2). The filter sets were 485/15 excitation and 535/25 emission. | ||
Numerous interactions of biomaterials are mediated by multivalent binding, which is generally much stronger than monovalent interactions.21 In addition, the separation of multivalent objects (e.g., exosomes and tumor cells) from biological fluids has been one of important issues in recent biomedical research.22 We thus prepared hDM2-gold nanoparticle conjugates (hGCs) as a model multivalent target to test whether the αTMT system can be used in such applications. The GNP substrates also facilitated simple hGC quantification using their characteristic optical property, plasmon-induced visible light (400–800 nm) absorption.23 As shown in Fig. 5b, αTMT2-bead-bound hGCs were merely released without the redox stimulus in the CAR experiment. On the other hand, the peptides released hGCs in a time-dependent manner when exposed to the oxidative condition (Fig. 5c). Note that no hGCs were detected in the experiment with the PEG-magnetic bead conjugates without the peptides (Fig. S18†). The plots of absorbance intensity change at 520 nm illustrated a clear difference in the release behavior between αTMT2 and αTMT2-ox (Fig. 5d). This significant discrepancy was likely due to (1) the hairpin unfolding-mediated helix structure distortion (Fig. S13†), and (2) intensified hDM2-affinity difference between the oxidized and reduced peptides by the multivalent binding.24 Since disulfide bonds are essential for the structural integrity of numerous proteins, the protein elution under the mild oxidative condition can be a promising approach, which is unattainable with general disulfide-mediated α-helix stapling approaches (Fig. S5†). As demonstrated in previous research, we believe that the CAR efficiency could be improved through optimization of the orientation, density, arrangement of the peptides on the magnetic bead surface, and the length of the linker.21 In addition, the control of environmental parameters, such as pH and temperature, could also be another important factors in enhancing CAR efficiency. Lastly, to probe the influence of the oxidizing agents on the CAR experiment, the stapled p53α peptides were added to the p53f/hDM2 complex solution with and without hydrogen peroxide (Fig. 5e). As a result, the stapled p53α peptides exhibited little change in the hDM2 binding efficiency regardless of the presence of the oxidizing agents.
Conclusions
In conclusion, we have demonstrated that the β-hairpin-based MT can regulate the folding behavior of α-helical peptides, which enabled them to exhibit controlled target affinity. This stimulus-responsive binding property was translated into effective capture-and-release of model multivalent biomaterials. As various α-helix sequences and stimulus-responsive properties can be introduced to αTMT, our strategy provides a powerful platform to sense and modulate biomolecular interactions. For instance, one can potentially develop αTMT-based PPI inhibitors that selectively operate responding to cancer-related stimuli (e.g., hypoxia and reducing intracellular environment). The conjugation of bioactive ligands to the termini is another promising approach to utilize the β-hairpin-based MT. Therefore, this novel and versatile MT system holds great potential for use in a wide range of biomedical applications.Materials and methods
Peptide synthesis
The peptides were chemically synthesized on Rink amide MBHA resin LL (Novabiochem, Germany), based on the general fluorenylmethyloxycarbonyl, Mmt: 4-methoxytrityl (Fmoc) solid-phase peptide synthesis (SPPS) protocol. Except for Fmoc-Cys(Mmt)-OH and Fmoc-Lys(1-(4,4-dimethyl-2,6-dioxocyclohexylidene)ethyl, allyl ester; Dde)-OH, standard amino acid protecting groups were used.In α,N,N-trimethyltryptamine (αTMT) peptides, the tryptophan zipper residue is connected to the p53 α-helix through the ε-amino group on the lysine and thiol groups of cysteine. Two milliliters of 2% (v/v) hydrazine/dimethylformamide (DMF) were added to the resin (20 μmol) four times to remove the Dde-protecting group.
For cyclization, bromoacetic acid was coupled to the N-terminus of the tryptophan zipper residue. Bromoacetic acid (28 mg, 200 μmol) and N,N′-diisopropylcarbodiimide (DIC, 15.6 μL, 100 μmol) were mixed and incubated for 10 min in N-methyl-2-pyrrolidone (NMP, 1 mL) and added to the resin. After shaking for 1 h at room temperature, the resin was washed five times with NMP and dichloromethane (DCM) (1 mL each time). The cleavage cocktail for the Mmt protecting group contained DCM![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :triisopropylane:trifluoroacetic acid (TFA) in a ratio of 94:5:1. The cocktail was applied 10 times for 1 min each application. The resin was then washed several times with DCM and NMP. The intermolecular cyclization reaction was performed in 3 mL of 1% (v/v) N,N-diisopropylethylamine (DIPEA) in NMP for 2 days with shaking at room temperature. The resin was then successively washed with NMP and DMF.
:triisopropylane:trifluoroacetic acid (TFA) in a ratio of 94:5:1. The cocktail was applied 10 times for 1 min each application. The resin was then washed several times with DCM and NMP. The intermolecular cyclization reaction was performed in 3 mL of 1% (v/v) N,N-diisopropylethylamine (DIPEA) in NMP for 2 days with shaking at room temperature. The resin was then successively washed with NMP and DMF.
For stapling of the p53α peptide, the Mmt protecting group was cleaved. Next, 200 μmol of N,N′-1,3-phenylene bismaleimide in 2 mL of 5% (v/v) DIPEA/DMF was added to 10 μmol resin for 24 h with shaking. The resin was then washed several times with DMF.
For deprotection of all protecting groups and cleavage from the resin, the resin was treated with a cleavage cocktail consisting of TFA:1,2-ethanedithiol:thioanisole (95:2.5:2.5) for 3 h. The solution was triturated with excess tert-butyl methyl ether. The cleaved peptides were purified by reverse-phase high-performance liquid chromatography (RP-HPLC). RP-HPLC was performed on a Vydac C18 column (10 × 250 mm, 300 Å) using a gradient of 30% to 50% MeCN with 0.1% TFA. The flow rate was set to 2 ml min−1, and samples were detected at 280 nm. The molecular weight and purity of the peptides were confirmed by matrix-assisted laser desorption/ionization-time of flight mass spectrometry (MALDI-TOF MS) with α-cyano-4-hydroxycinnamic acid (CHCA) matrix.
Disulfide bond oxidation and reduction
Unless otherwise noted, the peptides were oxidized in 10% dimethyl sulfoxide (DMSO), 45% water, and 45% ACN solution, or 20 mM potassium phosphate buffered saline (KPBS) buffer solution (pH 7.4) containing 10% DMSO. After a 1 day incubation at room temperature, the oxidized peptides were purified using RP-HPLC. The reduction of disulfide bonds of oxidized peptides was performed in 1 mM 2-mercaptoethanol in 20 mM KPBS (pH 7.4) overnight at room temperature. Oxidation and reduction of disulfide bonds were determined by RP-HPLC and MALDI-TOF MS after the reaction with maleimide compounds.Circular dichroism (CD) spectroscopy
Structural changes caused by disulfide bond formation in peptides were observed by CD using a Chirascan circular dichroism spectrometer equipped with a Peltier temperature controller (Applied Photophysics, Ltd, UK). CD spectra for the peptides were recorded from 260 to 190 nm using a 2 mm pathlength cuvette. All sample peptides were prepared at a concentration of 20 μM in PBS (pH 7.0, 20 mM, 150 mM NaCl) at room temperature. Scans were repeated three times and averaged.Fourier transform infrared spectroscopy (FT-IR)
For FT-IR measurements, 200 μL of solution (100 μM sample peptide in deionized water) was loaded onto the zinc selenide window. Three thousand scans were acquired using a VERTEX 70 FT-IR spectrometer (Bruker, USA) at room temperature.Small-angle X-ray scattering (SAXS)
SAXS analysis was performed at the 4C SAXS II beamline of the Pohang Accelerator Laboratory (Korea). The beam source was in-vacuum undulator 20 of the Pohang Light Source II storage ring, and it was concentrated with a vertical toroidal focusing mirror to deliver an X-ray beam of 0.675 Å. The 50 μL of sample solution were flowed along the tube for 1 minute and were irradiated for a total of 10 times at room temperature. The concentrations of peptides were 1 mM. X-Ray scatting patterns were collected by using a charge-coupled detector (Mar USA, Inc.) positioned 1, 4 m from the samples. The data were analyzed using SAS Data Analysis.Protein expression and purification
Recombinant human Mdm2 protein (residues 17–125) was cloned into the pET28a(+) vector containing a histone and TEV cleavage site. The recombinant protein was overexpressed in Escherichia coli BL-21-Codon Plus (DE3)-RIPL expressing cells. Cells were grown at 37 °C until the optical density at 600 nm (OD600) reached 0.8. The cells were induced by the addition of 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG). After 8 h of incubation at 25 °C, cells were harvested by centrifugation at 4000×g for 20 min and resuspended in lysis buffer (20 mM Tris–HCl pH 8.0, 200 mM NaCl, 5 mM imidazole, and 10% glycerol). The cells were lysed by sonication. After centrifugation at 10000×g to remove cell debris, the recombinant hDM2 protein was purified using Ni-NTA agarose resin following the standard purification protocol provided by the vendor (QIAGEN, Germany). To cleave the histone residue from the recombinant hDM2 protein, thrombin was added to the purified protein in 20 mM Tris–HCl (pH 7.5, containing 150 mM NaCl, 2.5 mM CaCl2 and 10% glycerol at 4 °C). Protein purification and cleavage reactions were monitored by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The purity and identity of the proteins were confirmed using SDS-PAGE and MALDI-TOF MS.
Binding and competition assays
The peptide affinity for hDM2 was measured by a fluorescence polarization (FP) assay. FP binding and competition assays were performed in 384-well plates using a Victor X5 multilabel plate reader (PerkinElmer, USA). All FP experiments were performed in a buffer containing 20 mM Tris–HCl pH 8.0, 500 mM NaCl, 10 mM imidazole, and 10% glycerol at room temperature. A binding assay between the fluorescein-conjugated p53 α-helix peptide (p53f) and hDM2 was conducted to calculate the dissociation constant (Kd). The Kd value was calculated by fitting the experimental data to the following single-site binding model-based equation:A = measured fluorescence anisotropy, Al = fluorescence anisotropy of the ligand at an infinite concentration of hDM2, A0 = fluorescence anisotropy in the absence of hDM2, [hDM2] = concentration of the hDM2 protein, [L] = concentration of ligand, p53f.
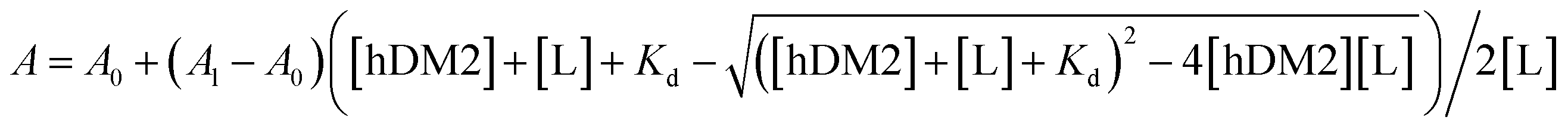
The interaction Kd value of hDM2 with 100 nM p53f peptide and binary concentration was 1.6 μM. The FP competitive binding assay used sample peptides as inhibitors, based on the binding assay between p53f and hDM2. Ten microliters of a mixture of 200 mM p53f and 10 μM hDM2 protein was incubated for 2 h and then added to each well containing 10 μL of sample peptides (0–20 μM). After additional incubation for 2 h, the FP value was measured in millipolarization (mp) units. The p53α peptide without tryptophan zipper residue was used to determine the effect of hydrogen peroxide (H2O2) on the p53-hDM2 interaction. Under the same conditions as those in the previous experiment, H2O2 was added 10 times to the peptide.
In the competition assay, the dissociation constant and half maximal effective concentration (EC50) were calculated by fitting the experimental data to the following explicit equation:
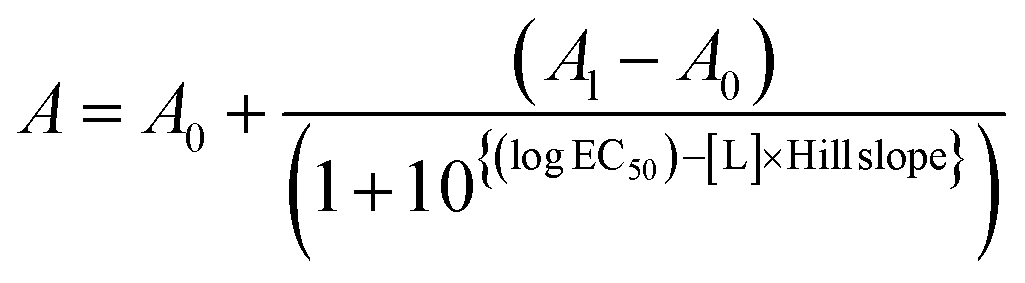
The data were analyzed and fitted using SigmaPlot, Origin and GraphPad Prism9 software.
Molecular dynamics (MD) simulations
The molecular dynamics simulations were performed to measure the β-hairpin-mediated α-helix folding by the redox state via the Desmond module in Schrödinger Suites with the following parameters-force field: OPLS4; solvent model: TIP5P; ion placement: chloride; boundary conditions: orthorhombic box shape, box size calculation method (buffer); simulation time: 100 ns; approximate number of frames: 1000; ensemble class: NPT; temperature: 300 K; pressure: 1.01325 bar; thermostat method: Nose–Hoover chain; coulombic interaction cutoff radius: 9.0 Å. Hydrogen bonds in the helical backbone of oxidized peptide and reduced peptide were analyzed per 100 ps.Peptide-magnetic bead conjugation
COOH magnetic nanobeads were purchased from BIONEER (Daejeon, Republic of Korea), and NH2-PEG-COOH (500 Da) was obtained from Biochempeg (Watertown, MA, USA). Phosphate buffered saline (PBS, pH 7.4) was purchased from WELGENE (Daegu, Republic of Korea). All other reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA). The amine groups of NH2-PEG-COOH and carboxyl groups of magnetic beads were conjugated by N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC)/N-hydroxysuccinimide (NHS) chemistry.Magnetic nanobeads (0.85 mg) dissolved in 1 mL PBS were activated using EDC and NHS (100-fold molar excess of the carboxyl group of magnetic nanobeads) at room temperature for 30 min.
Excess reagents were removed using a magnetic rack, and the magnetic nanobeads were washed three times with PBS. NH2-PEG-COOH (10-fold molar excess of the carboxyl group of magnetic nanobeads) dissolved in 1 mL PBS was added, and the reaction proceeded at room temperature overnight. Excess reagents were removed and the magnetic nanobeads were washed and activated as just described.
After activation, excess reagents were removed using a magnetic rack, and the magnetic nanobeads were washed three times with PBS. The nanobeads were mixed with αTMT2 and αTMT2A (equimolar with the carboxyl group of magnetic nanobeads) at room temperature overnight. Finally, the supernatant was replaced with PBS. The number of peptides conjugated to the magnetic beads was estimated to decrease by comparing the fluorescence of the peptide before conjugation with the peptide fluorescence of the supernatant at 280/260 nm after conjugation. We confirmed that 8.16 nmol of the peptide was conjugated to magnetic nanobeads per 8.782 fmol of αTMT2-beads.
hDM2-gold nanoparticle conjugation
Thirty-nanometer NHS-Activated Gold Nanoparticle Conjugation kit was purchased from Cytodiagnostics (Burlington, Ontario, Canada). A total of 60 μL of reaction buffer and 48 μL of hDM2 (20 μg mL−1) were mixed. Lyophilized NHS-gold nanoparticles (NHS-AuNPs) were dissolved in 90 μL of the mixture and incubated at room temperature for 3 h. Next, 10 μL of the quencher buffer was added and incubated for 10 min to inactivate excess NHS. The supernatant was replaced by PBS by centrifugation at 2500×g for 30 min. The final concentration of the gold nanoparticles (AuNPs) was quantified using a plate reader using molar extinction coefficients for 30 nm AuNPs at 526 nm.Capture-and-release experiment
In the binding step, 4.58 fmol of αTMT2-beads and hGCs with one hDM2 for every 288 αTMT2 on the magnetic bead surface were mixed in 620 μL of PBS buffer for 1 h. Three hundred microliters of the αTMT2-bead mixture were collected twice. The αTMT2-beads were washed three times with PBS and deionized distilled water, and the buffer was replaced by PBS containing 35 μM H2O2 (approximately 17-fold molar excess of αTMT2). A total of 105 μL of the sample was collected after 0.5, 1, 2, 3, 4, 5, and 6 h. After removing the magnetic particles using a magnetic rack, the absorbance of 100 μL of each sample was measured at 400–600 nm with a plate reader.In the αTMT2-bead and αTMT2A-bead binding tests, the bead types were individually mixed with 264.39 pM hGCs in 300 μL of PBS at a concentration of 3.02 pM respectively and shaken for 1 h to bind hGCs. The supernatants were replaced by 400 μL of 35 μM H2O2 using a magnetic rack, and the αTMT2-bead and αTMT2A-bead mixtures were shaken for 3 h. Two hundred microliters of the supernatant were measured at 400–600 nm.
Data availability
The data related to this study are available from the corresponding author upon reasonable request.Author contributions
S. K., J.-Y. K., Y.-B. L., and W.-J. J. designed research; S. K., J. K., Y.-J. P., and J. Y. P. performed research; S. K., J.-Y. K., J. Y. P., M.-J. C., K. S. J., Y. K., Y.-B. L., and W.-J. J. analyzed data; and S. K., J. K., J. Y. P., Y. K., Y.-B. L., and W.-J. J. wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Ministry of Science and ICT (2021R1C1C1009912, 2021R1A4A3024237, 2020R1A2C2007578, 2022M3E5F1016877); the Technology Innovation Program (20015577) funded by the Ministry of Trade, Industry & Energy (MOTIE, Korea); the Technology development Program(S3040004) funded by the Ministry of SMEs and Startups (MSS, Korea); and Inha University Research Grant.Notes and references
- (a) S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Chem. Rev., 2015, 115, 10081–10206 CrossRef CAS PubMed; (b) C. Cheng, et al. , Nat. Nanotechnol., 2015, 10, 547–553 CrossRef CAS PubMed; (c) L. Zhang, V. Marcos and D. A. Leigh, Proc. Natl. Acad. Sci. U.S.A., 2018, 115, 9397–9404 CrossRef CAS PubMed.
- (a) J. Leblond and A. Petitjean, Chemphyschem, 2011, 12, 1043–1051 CrossRef CAS PubMed; (b) X. G. Han, Z. H. Zhou, F. Yang and Z. X. Deng, J. Am. Chem. Soc., 2008, 130, 14414 CrossRef CAS PubMed; (c) T. Bartelmann, F. Gnannt, M. Zitzmann, P. Mayer and H. Dube, Chem. Sci., 2021, 12, 3651–3659 RSC; (d) J. Leblond, H. Gao, A. Petitjean and J. C. Leroux, J. Am. Chem. Soc., 2010, 132, 8544–8545 CrossRef CAS PubMed.
- (a) S. Kim, et al. , Angew. Chem., Int. Ed. Engl., 2012, 51, 1890–1894 CrossRef CAS PubMed; (b) H. Y. Kuchelmeister, S. Karczewski, A. Gutschmidt, S. Knauer and C. Schmuck, Angew. Chem., Int. Ed. Engl., 2013, 52, 14016–14020 CrossRef CAS PubMed; (c) M. Takeda, et al. , J. Am. Chem. Soc., 2018, 140, 6336–6342 CrossRef CAS PubMed.
- (a) J. C. Lou, X. Y. Zhang and M. D. Best, Chem. –Eur. J., 2019, 25, 20–25 CrossRef CAS PubMed; (b) W. Viricel, et al. , Nanoscale, 2017, 9, 31–36 RSC.
- J. Y. Kim, et al. , Pept. Sci., 2022, 114, e24233 CrossRef CAS.
- V. Azzarito, K. Long, N. S. Murphy and A. J. Wilson, Nat. Chem., 2013, 5, 161–173 CrossRef CAS PubMed.
- (a) W. J. Jeong, S. H. Kwon and Y. B. Lim, Adv. Funct. Mater., 2018, 28, 1803114 CrossRef; (b) W. J. Jeong, S. H. Choi, H. S. Lee and Y. B. Lim, Sens. Actuators, B, 2019, 290, 93–99 CrossRef CAS; (c) H. Neitz, et al. , Chem. Sci., 2022, 13, 9079–9090 RSC.
- K. A. Olsen, R. M. Fesinmeyer, J. M. Stewart and N. H. Andersen, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 15483–15487 CrossRef CAS PubMed.
- (a) M. Pelay-Gimeno, A. Glas, O. Koch and T. N. Grossmann, Angew. Chem., Int. Ed. Engl., 2015, 54, 8896–8927 CrossRef CAS PubMed; (b) W. J. Jeong, et al. , J. Am. Chem. Soc., 2020, 142, 1832–1837 CrossRef CAS PubMed; (c) J. Bu, et al. , Biosens. Bioelectron., 2022, 213, 114445 CrossRef CAS PubMed.
- (a) A. G. Cochran, N. J. Skelton and M. A. Starovasnik, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 5578–5583 CrossRef CAS PubMed; (b) T. L. Pham, et al. , Chembiochem, 2023, 24, e202200588 CrossRef CAS PubMed.
- (a) T. A. Hill, N. E. Shepherd, F. Diness and D. P. Fairlie, Angew. Chem., Int. Ed. Engl., 2014, 53, 13020–13041 CrossRef CAS PubMed; (b) A. D. de Araujo, et al. , RSC Chem. Biol., 2022, 3, 895–904 RSC.
- D. A. Guarracino, et al. , Chem. Rev., 2019, 119, 9915–9949 CrossRef CAS PubMed.
- (a) M. N. Saleh, et al. , Clin. Cancer Res., 2022, 28, 429 CrossRef PubMed; (b) Y. H. Lau, et al. , Chem. Sci., 2014, 5, 1804–1809 RSC.
- (a) S. Lou, X. Wang, Z. Yu and L. Shi, Adv. Sci., 2019, 6, 1802043 CrossRef PubMed; (b) T. E. Schrader, et al. , Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 15729–15734 CrossRef CAS PubMed; (c) M. C. Branco, D. J. Pochan, N. J. Wagner and J. P. Schneider, Biomaterials, 2009, 30, 1339–1347 CrossRef CAS PubMed.
- (a) J. H. Lee, et al. , J. Am. Chem. Soc., 2011, 133, 676–679 CrossRef CAS PubMed; (b) A. Czarna, et al. , Cell Cycle, 2009, 8, 1176–1184 CrossRef CAS PubMed.
- J. C. Biro, Theor. Biol. Med., 2006, 3, 15 CrossRef CAS PubMed.
- J. Ceballos, E. Grinhagena, G. Sangouard, C. Heinis and J. Waser, Angew. Chem., Int. Ed. Engl., 2021, 60, 9022–9031 CrossRef CAS PubMed.
- W. J. Jeong, M. S. Lee and Y. B. Lim, Biomacromolecules, 2013, 14, 2684–2689 CrossRef CAS PubMed.
- (a) W. Borcherds, et al. , Nat. Chem. Biol., 2014, 10, 1000–1002 CrossRef CAS PubMed; (b) F. Bernal, A. F. Tyler, S. J. Korsmeyer, L. D. Walensky and G. L. Verdine, J. Am. Chem. Soc., 2007, 129, 2456–2457 CrossRef CAS PubMed.
- (a) Q. Chen, et al. , Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 5343–5348 CrossRef CAS PubMed; (b) M. Gulden, A. Jess, J. Kammann, E. Maser and H. Seibert, Free Radical Biol. Med., 2010, 49, 1298–1305 CrossRef PubMed.
- W. J. Jeong, et al. , Adv. Sci., 2022, 9, e2103098 CrossRef PubMed.
- (a) Y. Song, et al. , Angew. Chem., Int. Ed. Engl., 2019, 58, 2236–2240 CrossRef CAS PubMed; (b) L. L. Wu, et al. , Small Methods, 2019, 3, 1800544 CrossRef.
- M. A. C. Potenza, et al. , Nanoscale, 2017, 9, 2778–2784 RSC.
- (a) H. Bila, K. Paloja, V. Caroprese, A. Kononenko and M. M. C. Bastings, J. Am. Chem. Soc., 2022, 144, 21576–21586 CrossRef CAS PubMed; (b) M. R. W. Scheepers, L. J. van IJzendoorn and M. W. J. Prins, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 22690–22697 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc03758d |
| ‡ These two authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |