
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Towards a prebiotic chemoton – nucleotide precursor synthesis driven by the autocatalytic formose reaction†
Quoc Phuong
Tran
ab,
Ruiqin
Yi
 d and
Albert C.
Fahrenbach
*abc
d and
Albert C.
Fahrenbach
*abc
aSchool of Chemistry, University of New South Wales, Sydney, NSW 2052, Australia. E-mail: a.fahrenbach@unsw.edu.au
bAustralian Centre for Astrobiology, University of New South Wales, Sydney, NSW 2052, Australia
cUNSW RNA Institute, University of New South Wales, Sydney, NSW 2052, Australia
dEarth-Life Science Institute, Tokyo Institute of Technology, Tokyo 152-8550, Japan
First published on 29th August 2023
Abstract
The formose reaction is often cited as a prebiotic source of sugars and remains one of the most plausible forms of autocatalysis on the early Earth. Herein, we investigated how cyanamide and 2-aminooxazole, molecules proposed to be present on early Earth and precursors for nonenzymatic ribonucleotide synthesis, mediate the formose reaction using HPLC, LC-MS and 1H NMR spectroscopy. Cyanamide was shown to delay the exponential phase of the formose reaction by reacting with formose sugars to form 2-aminooxazole and 2-aminooxazolines thereby diverting some of these sugars from the autocatalytic cycle, which nonetheless remains intact. Masses for tetrose and pentose aminooxazolines, precursors for nucleotide synthesis including TNA and RNA, were also observed. The results of this work in the context of the chemoton model are further discussed. Additionally, we highlight other prebiotically plausible molecules that could have mediated the formose reaction and alternative prebiotic autocatalytic systems.
Introduction
Tibor Gánti's theoretical model of the chemoton1 describes a minimal compartmentalized system capable of growth, division, and evolution that serves as a heuristic model for the generation of a minimal cell. At the heart of the chemoton is a self-sustaining autocatalytic system capable of providing the substrates for the other two required autocatalytic cycles, which afford the template-directed synthesis of a genetic polymer and the production of components for the compartment, e.g., lipids. While these autocatalytic systems have been investigated independently2,3 in the context of prebiotic chemistry, for example, nonenzymatic template-directed nucleotide replication from activated mononucleotides,4–9 studies for how these autocatalytic systems could be integrated together10 remain few and far between. The present work herein demonstrates an example of how an autocatalytic reaction network, i.e., the formose reaction, can fuel the synthesis of nucleotide precursors, and that the autoamplification kinetics of the former are intertwined with the kinetics of the latter.The formose reaction, discovered by Butlerow,11 remains one of the most plausible autocatalytic reactions on early Earth as a result of the proposed abundance of the necessary feedstocks and reaction conditions.12–15 The formose reaction is often cited as a source of ribose for the prebiotic synthesis of ribonucleotides, a key ingredient for the RNA world,16–20 although not without criticism.21,22 As “messy chemistry” and “metabolism-first” approaches23–27 receive increasingly more attention within the prebiotic chemistry community, the diversity of formose products and the robustness of the reaction under different conditions have become renewed topics of interest. For example, Trapp and coworkers28,29 demonstrated a nonaqueous (solid state) formose reaction accelerated by mechanochemical processes. The authors also showed that the minerals incorporated as catalysts during the milling process lead to changes in product distributions. Recent report from the Huck group30,31 used a continuously stirred-tank reactor to elucidate how different combinations of potentially relevant environmental factors (e.g., formaldehyde concentrations, initiator sugar identity, and NaOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CaCl2 ratios) activate different reaction mechanisms and pathways.
:CaCl2 ratios) activate different reaction mechanisms and pathways.
The autocatalytic nature of the formose reaction was described by Breslow32 (Scheme 1A, black arrows) and starts with the aldol addition between glycolaldehyde and formaldehyde (CH2O) to form glyceraldehyde. This C3 aldose isomerises to the ketose, dihydroxyacetone. A subsequent aldol addition with formaldehyde produces C4 ketotetroses. These ketotetroses isomerise into aldose stereoisomers, which undergo retroaldol fragmentation30,33–35 into two molecules of glycolaldehyde. While the complexity of the formose reaction pathways and product distributions have been extensively elaborated upon,30,34,36–44 Breslow's model32 serves as a simple demonstration of its autocatalytic nature, although more complex cycles containing higher-order sugars are also possible.30,33,34,38,39
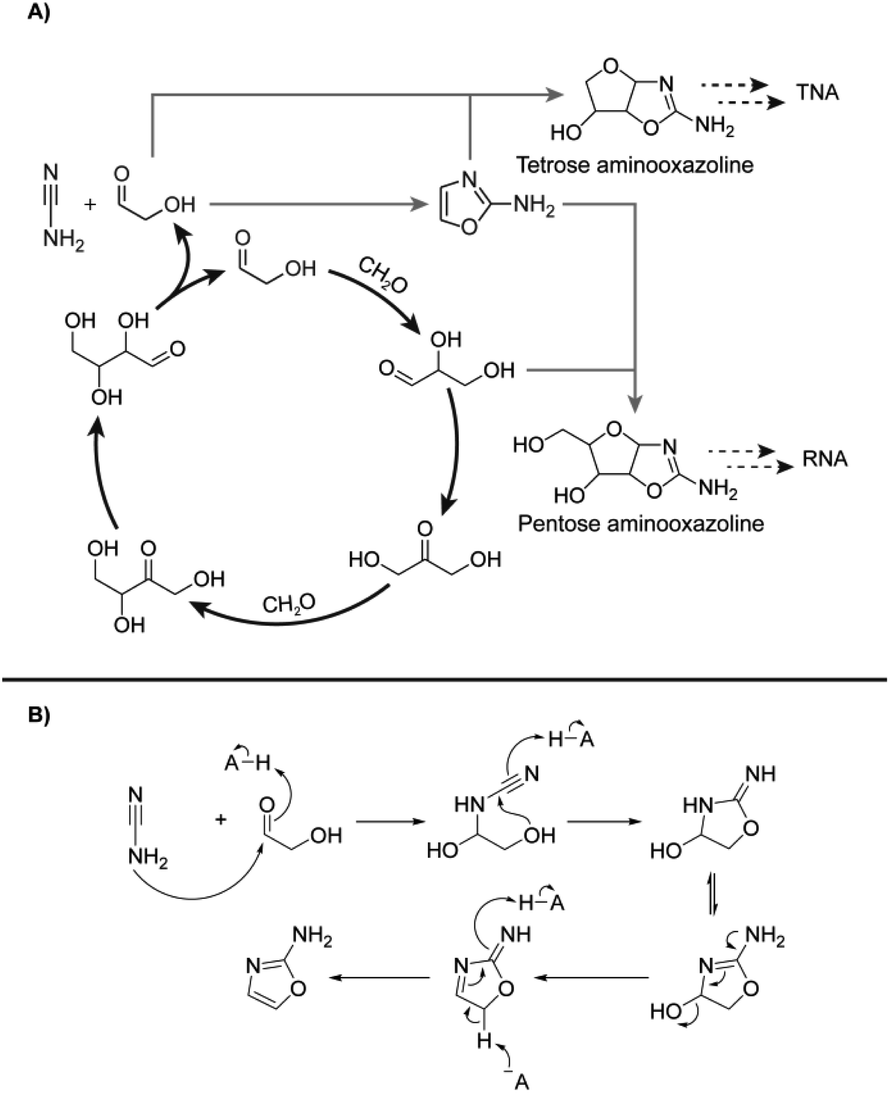 | ||
| Scheme 1 (A) Initially proposed reaction scheme involving Breslow's autocatalytic cycle32 (black) coupling with RNA and TNA nucleotide synthesis proceeding through 2-aminooxazole (2-NH2Ox) previously demonstrated by Sutherland and coworkers51 (grey dashed). (B) Mechanism of 2-NH2Ox synthesis from glycolaldehyde and cyanamide under general-base catalysed conditions.51 | ||
The kinetics of the formose reaction can be divided into three phases. The lag phase,22 characterised by a period of little formaldehyde consumption, ends once glycolaldehyde and higher-order sugars have accumulated past a critical concentration. At the onset of the exponential phase, formaldehyde is rapidly consumed by aldol additions to produce higher-order sugars.37 These sugars then undergo retroaldol reaction, forming two additional aldol nucleophiles for every one fragmentation, creating a positive feedback loop. When formaldehyde is depleted, also known as the yellowing point,22 the reaction transitions to the degradation phase, which is characterised by the development of a yellow colour and a complex mixture of oligomeric products.
It is unlikely, however, that formaldehyde and other simple sugars existed as neat mixtures on early Earth. Such mixtures likely co-existed with other plausibly abundant prebiotic molecules. Yet, little is known how such molecules affect formose reaction kinetics and product distributions. For example, cyanamide (NH2CN) is one such simple molecule derivable prebiotically from hydrogen cyanide,45,46 and is known as a reagent for prebiotic chemical activation,47,48 production of 2-aminoimidazole24,46,49 and nucleotide precursor synthesis50,51 as well as the synthesis of arginine.52
Herein, the addition of cyanamide was shown to commandeer the formose reaction for nucleotide precursor synthesis, observable through changes in kinetics and product distributions. Cyanamide was investigated herein due to its established relevance to threo- and ribonucleotide synthesis51,53–57 and prebiotic plausibility.24,45,46 The effects of cyanamide and 2-aminooxazole (2-NH2Ox), its cyclisation product with glycolaldehyde, on formose reaction kinetics were determined via time course experiments monitored by high-performance liquid chromatography (HPLC), 1D proton nuclear magnetic resonance (1H NMR) spectroscopy and liquid chromatography-high resolution (QToF) mass spectrometry (LC-MS). It was observed that cyanamide has an inhibitory effect on formose reaction kinetics while 2-NH2Ox does not, and that autocatalytic sugar synthesis can be effectively commandeered for nucleotide precursor production.
Results
The reaction of cyanamide with glycolaldehyde to form 2-NH2Ox, an intermediate in the Powner-Sutherland pathway for nucleotide synthesis51 has been well-characterised51,53 (see Scheme 1B). We initially hypothesised that cyanamide would react with glycolaldehyde, as well as other Breslow cycle intermediates, thereby disrupting the autocatalytic cycle, inhibiting the onset of the exponential phase, while synthesising 2-NH2Ox and perhaps other higher-order derivatives (Scheme 1A, grey arrows), e.g., tetrose and pentose aminooxazolines.Formose reaction mixtures containing 100 mM CH2O, 1 mM glycolaldehyde, with various concentrations of cyanamide (0, 7, and 9 mM) were heated at 50 °C and monitored for 22 minutes by HPLC (Fig. 1A). Samples were collected every two minutes and placed on ice to stop the reaction. The concentrations of formaldehyde and glycolaldehyde were determined by 2,4-dinitrophenylhydrazine (DNPH) derivatisation followed by HPLC analysis monitored by UV-absorbance (190–450 nm). Although initial cyanamide concentrations >9 mM were also tested, it was difficult to achieve a consistent yellowing point between repeats, which varied from ∼1 hour to no yellowing point at all. It is hypothesised that the formose reaction, being autocatalytic, becomes highly sensitive to the initial conditions at these higher cyanamide concentrations, and so the variation in the yellowing point becomes large. One possible contributing factor lies in the competing Cannizarro reaction,33,34,37,44 which gives rise to a constant decrease in pH, further inhibiting the formose reaction, especially at these longer time scales.
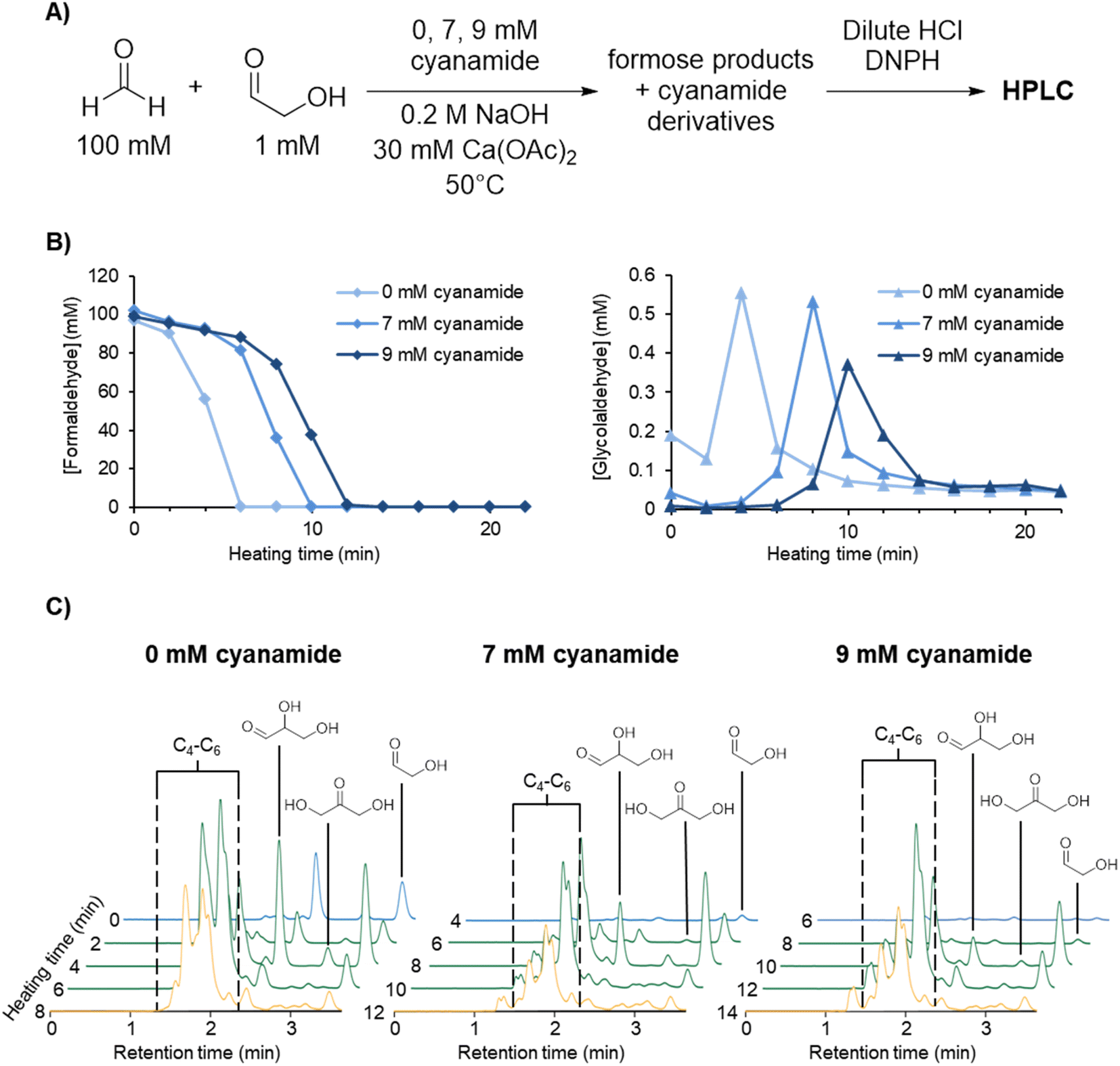 | ||
| Fig. 1 (A) Experimental scheme for formose reaction experiments containing 0, 7, and 9 mM initial cyanamide analysed by HPLC (see Methods for more details). (B) [CH2O] and [glycolaldehyde] over time in the formose reaction experiments outlined above based on the average of triplicates (see ESI Fig. S9–S11†). (C) Extracted HPLC chromatograms at 360 nm absorbance at selected timepoints for formose reaction experiments containing 0, 7, and 9 mM initial cyanamide (see ESI Fig. S12–S14† for all timepoints). Hemiaminal formation may lower the aldehyde concentrations during the reaction (see ESI Fig. S36 and S37† and Discussion). Chromatograms in the lag phase are coloured blue, exponential phase green, and degradation phase yellow. The intensity of the sugar peaks have a general tendency to decrease with increasing cyanamide concentrations included initially, suggesting that cyanamide is serving as a sink for formose sugars, hence inhibiting the positive feedback. | ||
Fig. 1B depicts the CH2O (left graph) and glycolaldehyde (right graph) concentration changes over time in formose reaction mixtures containing initial concentrations of 0, 7, and 9 mM cyanamide. The point at which CH2O consumption deviates from a linear decrease was used to determine the onset of the exponential phase. During this phase, glycolaldehyde production as measured by concentration reached its highest point, while CH2O concentration plummeted. This depletion of CH2O transitions the reaction from the exponential phase to the degradation phase.
The simultaneous consumption of formaldehyde and glycolaldehyde production followed by glycolaldehyde degradation after the yellowing point is consistent with trends observed in literature.37,58 The discrepancy between the theoretical glycolaldehyde concentration before heating (1 mM) and the observed values (0.01–0.2 mM) can be explained by glycolaldehyde participating in aldol additions, e.g., with CH2O, as well as reaction with cyanamide immediately after initial preparation of the solution prior to heating.
It was observed that the onset of the exponential phase is delayed with increasing cyanamide concentrations. At an initial concentration of 9 mM, the maximum concentration of glycolaldehyde detected during the exponential phase was also noticeably lower than reactions initially containing 7 mM or no cyanamide. This extension of the lag phase is attributed to cyanamide serving as a sink for glycolaldehyde and other sugars in the formose reaction (see Fig. 1C and ESI Fig. S15 and S16†). By preventing glycolaldehyde and larger carbohydrates from participating in autocatalytic cycles, cyanamide provides an inhibitory mechanism. The products of this inhibition are 2-NH2Ox alongside other oxazole and oxazoline derivatives yielded by reaction of cyanamide with other formose intermediates (e.g., glyceraldehyde, tetroses, and pentoses), which were observed by LC-MS (see ESI Fig. S22–S29†). The formation of 2-NH2Ox at the end of the experiment (22 minutes) was also confirmed with 1H NMR spectroscopy (see Fig. 2 and ESI Fig. S17–S19†).
 | ||
| Fig. 2 Partial 1H NMR spectrum of the formose reaction containing 7 mM initial cyanamide after 22 minutes of heating recorded in 10% D2O at pH 6.7 with phosphate buffer (see ESI Fig. S17–S19† for comparison with standards). | ||
To understand whether 2-NH2Ox was synthesised from the initial glycolaldehyde or that produced by the formose reaction, the experiment with 7 mM cyanamide was repeated with 13C-labelled CH2O and analysed using LC-MS (ESI Fig. S20 and S21†). Before heating, only unlabelled 2-NH2Ox is observed. After heating started, the concentration of doubly-13C-labelled 2-NH2Ox quickly increased. The rate of production is highest during the exponential phase (6–10 minutes) and slowed down as formaldehyde is depleted (10 minutes). By the end of the observation window (22 minutes), the concentration of the doubly-13C-labelled species (m/zobs 87.0456) was ∼1 mM (∼14% yield with respect to cyanamide), approximately four times that of the unlabelled species (m/zobs 85.0396, ∼3.4% yield with respect to cyanamide). This observation indicates that the majority of 2-NH2Ox was made from formose-produced glycolaldehyde, which became the dominant product after the exponential phase started. Singly-13C-labelled 2-NH2Ox was also observed but only in trace amounts.
To provide secondary confirmation that the majority of 2-NH2Ox is produced from formose-derived glycolaldehyde, the formose reaction with 7 mM cyanamide was repeated but with 1,2-13C2-glycolaldehyde and unlabelled CH2O (Fig. 3A); LC-MS was used to monitor the carbohydrate-cyanamide adducts (Fig. 3B and ESI Fig. S22†). Here, the trend previously observed for 2-NH2Ox was reversed, i.e., the concentration of unlabelled 2-NH2Ox (m/zobs 85.0388) overtook the doubly-13C-labelled species (m/zobs 87.0458) after the onset of the exponential phase, the concentration of which did not increase considerably throughout the experiment. The LC-MS data was also monitored for the 2-NH2Ox hydrate (Fig. 3B and ESI Fig. S23†), the intermediate in 2-NH2Ox synthesis (Scheme 1B), the growth and decrease of which is consistent with the synthetic mechanism. The concentration of the doubly-13C-labelled (m/zobs 105.0561) 2-NH2Ox hydrate was highest at the start of the reaction and decreased significantly as the reaction progressed. Meanwhile, the concentration of the unlabelled 2-NH2Ox hydrate (m/zobs 103.0493), which was not detected before heating, began to rise at the same time as the increased glycolaldehyde production during the exponential phase and subsequently dropped at the end of it. The reaction mechanism between cyanamide and glycolaldehyde, which first yields the 2-NH2Ox hydrate that then undergoes dehydration to yield 2-NH2Ox, is consistent with these LC-MS results.
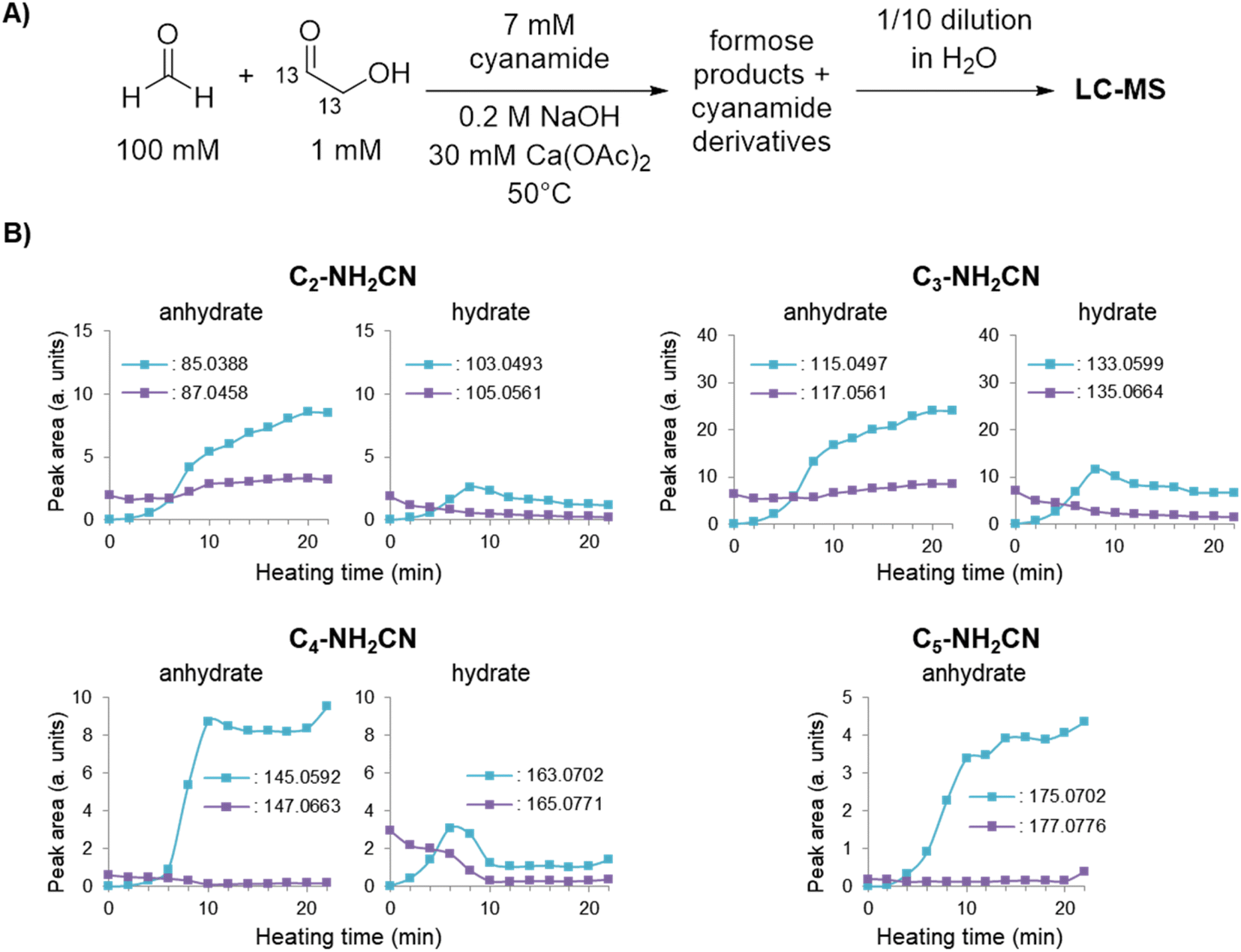 | ||
| Fig. 3 (A) Experimental scheme for formose reaction experiments containing 7 mM cyanamide analysed by LC-MS (see Methods for more details). (B) Peak areas of cyanamide adducts with C2–5 sugars containing 0 or 2 13C isotopes based on the average of extracted ion chromatograms (EICs) taken in triplicates over time (see ESI Fig. S22–S28† for EICs). | ||
To demonstrate that the mechanism by which cyanamide delays the exponential phase is not limited to the removal of glycolaldehyde but also other intermediates in the Breslow cycle, we also looked for the products of cyanamide reacting with C3–5 sugars. As shown in Fig. 3B (see also ESI Fig. S24–S28†), the changes in concentration of cyanamide adducts with C3 and C4 sugars resembled that of the 2-NH2Ox hydrate, i.e., the concentrations of the doubly-13C-labelled species (m/zobs 135.0664 for C3, and 165.0771 for C4) exhibited gradual decreases while the unlabelled species (m/zobs 133.0599 for C3 and 163.0702 for C4) started undetectable, reached their maximum during the exponential phase, and decreased afterwards. The concentrations of dehydrated cyanamide adducts with C3–5 sugars also followed similar patterns to that of 2-NH2Ox, i.e., the doubly-13C-labelled species (m/zobs 117.0561, 147.0663, 177.0776) did not show appreciable change, while the unlabelled species (m/zobs 115.0497, 145.0592, and 175.0702) displayed significant growth, especially at the onset of the exponential phase. The proposed pathways for the production of these adducts are summarised in Scheme 2. It is important to note, the m/zobs signals of 145.0592 and 175.0702 are consistent with those of tetrose and pentose aminooxazolines, threo-, ribo- and arabino-isomers of which serve as precursors for prebiotic TNA and RNA nucleotides.51,53–57 However, the extracted ion chromatograms for these m/z signals showed multiple peaks, suggesting a complex mixture of isomers. Collision-induced fragmentation (MS/MS) was carried out to test whether the m/zobs = 175.0702 signals arise from pentose aminooxazolines. The tandem mass spectrum recorded on this ion at the end of the observation window (22 minutes) showed the same fragments in comparison to the MS/MS recorded for synthesised arabino-, ribo- and xylo-aminooxazoline standards (ESI Fig. S6–S8†), suggesting the formation of one or more of these pentose aminooxazoline stereoisomers in the formose reaction (ESI Fig. S29†).
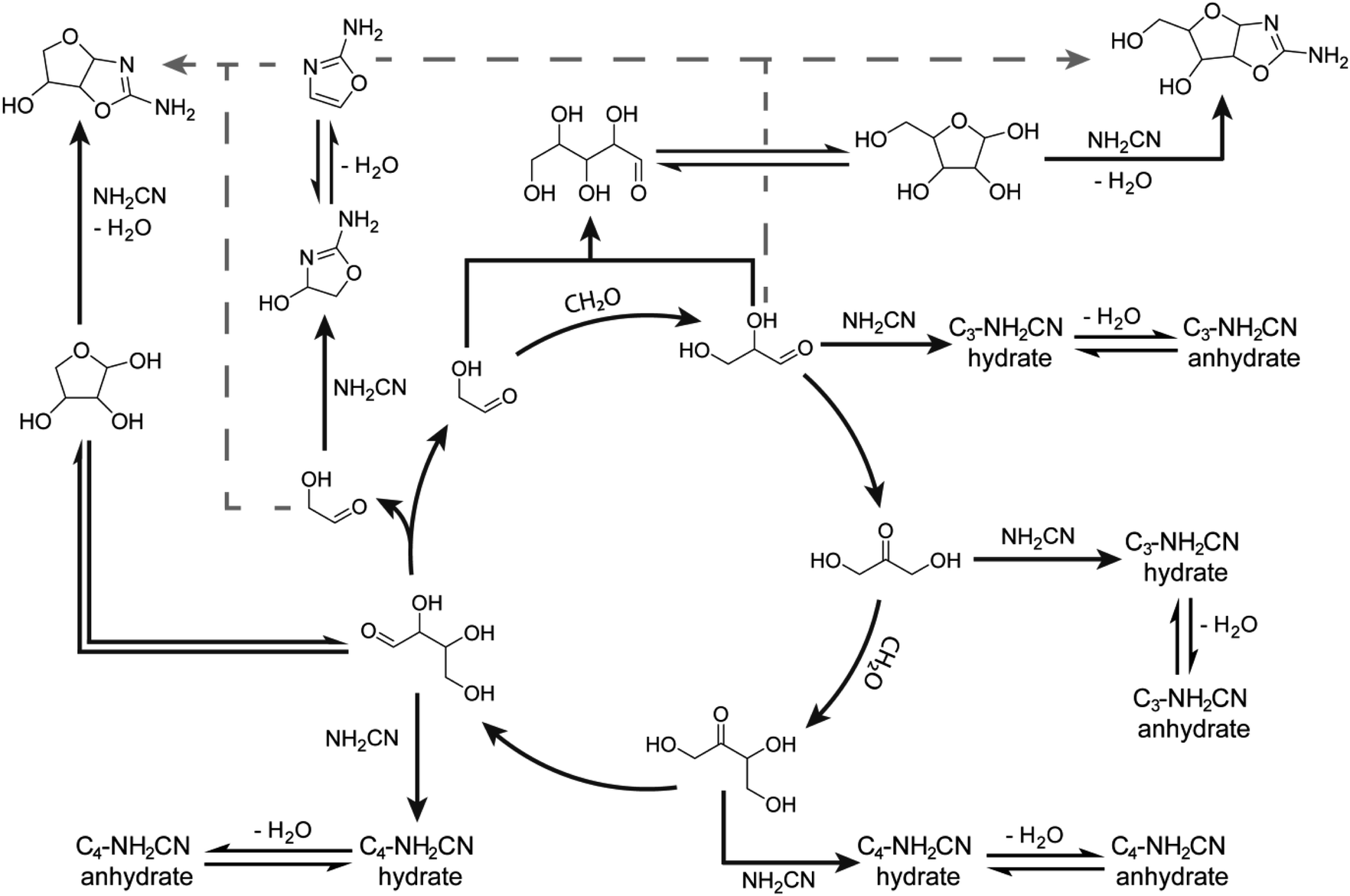 | ||
| Scheme 2 Some pathways for the proposed reaction network of formose intermediates and cyanamide based on the autocatalytic cycle proposed by Breslow32 and experiments herein. It is hypothesised that the formation of tetrose and pentose aminooxazolines via the addition of 2-NH2Ox with glycolaldehyde and glyceraldehyde (gray dashed arrows), respectively, is likely not the dominant pathway as originally hypothesised in Scheme 1. See ESI Table S3† for more proposed structures. Note, that although the Breslow autocatalytic cycle is shown for simplicity, the full reaction network is much more complex; see ref. 30, 41 and 42. | ||
To test the impact of 2-NH2Ox as a potential inhibitor, the formose reaction was repeated, replacing cyanamide with 4 mM 2-NH2Ox (Fig. 4A), which is ∼4 times the 2-NH2Ox concentration yielded at the end of the formose reaction with 7 mM cyanamide (ESI Fig. S20†). 2-NH2Ox was hypothesised to also be capable of extending the lag phase due to its reaction as a carbon-centered nucleophile with sugars to form aminooxazoline derivatives among other products.51,53 Similar to previous experiments, the timepoints were analysed using HPLC and LC-MS (Fig. 4B–D). The changes in CH2O and glycolaldehyde concentrations displayed in Fig. 4B resemble that of the formose reaction without cyanamide (Fig. 1B). This observation suggests that 2-NH2Ox does not have a significant effect on the kinetics of the reaction, while the HPLC chromatograms displayed in Fig. 4C also reveal that the product distributions with and without 2-NH2Ox (Fig. 1C, the 0 mM cyanamide plot) are comparable.
 | ||
| Fig. 4 (A) Experimental procedure for formose reaction experiments containing 4 mM initial 2-NH2Ox analysed by HPLC and LC-MS (see Methods for more details). (B) [CH2O] and [glycolaldehyde] over time in formose reaction experiments outlined above based on the average of triplicates of the HPLC data (see ESI Fig. S30†). (C) Extracted HPLC chromatograms at 360 nm absorbance at selected timepoints (see ESI Fig. S31† for all timepoints). Hemiaminal formation may lower the aldehyde concentrations during the reaction (see ESI Fig. S36 and S37† and Discussion). Chromatograms in the lag phase are coloured blue, exponential phase yellow, and degradation phase green. (D) Peak areas of 2-NH2Ox and 2-NH2Ox adducts with CH2O and glycolaldehyde are based on the average of LC-MS EICs taken in triplicate over time (see ESI Fig. S32–S34† for EICs). | ||
To gain insight about the mechanism of aminooxazoline production, we also monitored m/z values consistent with tetrose and pentose aminooxazolines in the LC-MS data. It was initially expected for these signals to increase as a result of the excess 2-NH2Ox included initially, which ought to react with glycolaldehyde and glyceraldehyde to produce their respective aminooxazoline derivatives. Contrary to these expectations, the signal for m/zobs 145.0592 was very weak (Fig. 4D and ESI Fig. S33†) while m/zobs 175.0702 was negligible (Fig. 4D and ESI Fig. S34†). It was previously observed by Sutherland and coworkers that glyceraldehyde is unstable in basic pH, which prevents the assembly of pentose aminooxazolines.53 This finding suggests that the signals at m/zobs 145.0592 and 175.0702 previously detected in the 7 mM initial cyanamide experiment arise from tetrose and pentose aminooxazolines albeit an alternative pathway, i.e., the one demonstrated by Orgel and coworkers50 where the tetro- and pentofuranose sugars react with cyanamide to form their respective aminooxazoline products (Scheme 2).
Discussion
The results herein demonstrate that the addition of cyanamide integrates nucleotide precursor synthesis with the autocatalytic formose reaction. Furthermore, the two processes do not simply co-exist, but rather, HPLC and LC-MS data (Fig. 1 and 2) revealed them to be kinetically coupled, a characteristic shared by many of life's chemical processes.59 As shown in Scheme 2, the onset of the formose exponential phase increases the production of cyanamide adducts with formose intermediates, including 2-NH2Ox, tetrose- and pentose-aminooxazolines. At the end of the exponential phase, production starts to plateau and the concentrations of transient hydrated species subsequently drop, likely as they convert to their corresponding anhydrates. In essence, cyanamide is capable of “peeling off” excess sugars produced by the formose reaction without completely shutting down autocatalysis. In light of these results, this work serves as a useful example of a prebiotically plausible autocatalytic cycle that fuels the synthesis of intermediates required for the production of genetic material. While the highly alkaline conditions employed herein are incompatible with nucleic acid base-pairing and hydrolytic stability, it has been shown that the formose reaction can occur under milder pH conditions,58 although at higher temperatures (200 °C) and pressures (100 bar).While providing useful insights, several challenges still need to be addressed in order to fully realize the chemoton model in this context. First is the selectivity, or lack thereof, of cyanamide reactivity with other formose intermediates. In the context of prebiotic nucleotide synthesis, the cyanamide-formose products observed, excluding 2-NH2Ox, threo-, ribo- and arabino-aminooxazolines, might be considered as waste products which may interfere with further downstream reactions. However, it has also been proposed that the waste of one autocatalytic system can activate and/or serve as “food” for another, giving rise to networks of sequentially triggered autocatalytic subsystems.60 The activation or deactivation of autocatalytic motifs in such a network could give rise to different heritable states and provide the basis for chemical evolution, however, the prediction of (coupled) autocatalytic networks is a notoriously difficult problem, and their emergence in a prebiotic context has been criticised as improbable.61 Further research is required.
Alternatively, encapsulation of the reaction network within a selectively permeable membrane that could allow for the accumulation of useful intermediates while removing unwanted waste is also a potential solution. For example, Sacerdote and Szostak62 showed that a range of model fatty acid or phospholipid membranes exhibit enhanced permeability for ribose over other sugars of similar size. The authors noted that the preference for ribose assimilation may have potentially contributed to the emergence of the RNA world.62
Another method to accumulate useful products is to phosphorylate them to prevent their diffusion through the membrane. The addition of a negatively charged phosphate group is how modern cells trap glucose in the first step of glycolysis. A potential mechanism for aqueous prebiotic phosphorylation involves the use of diamidophosphate (DAP), a di-nitrogenous analogue of orthophosphate which could have formed on the early Earth, e.g., by reacting aqueous ammonia with iron phosphide or phosphorus(V) oxide.63 DAP has been shown to phosphorylate a wide variety of biological building blocks including nucleo(s/t)ides, amino acids, and lipid precursors under mild conditions.64–67
Another issue is the fact that that under the experimental conditions tested herein, 2-NH2Ox does not contribute significantly to tetrose and pentose aminooxazoline synthesis. This observation was attributed mechanistically to the relative instability of glycolaldehyde and glyceraldehyde, which more rapidly undergo aldol additions instead of cycloaddition with 2-NH2Ox under alkaline pH. We propose that regular fluctuations in pH between alkaline and neutral conditions may afford periods of autocatalytic sugar and 2-NH2Ox production followed by 2-NH2Ox-dependent synthesis of tetrose and pentose aminooxazolines, respectively. Such fluctuating conditions of pH could have been realized in a terrestrial hydrothermal field where hot springs of highly varied conditions of pH exist in close proximity and frequently mix with one another.68,69
A crucial feature of the chemoton model implied in this discussion is the complete synthesis of (activated) nucleotides that can be eventually exploited in autocatalytic (template-directed) genetic polymer replication. Following the production of aminooxazolines, the next step in the Powner-Sutherland nucleoside synthesis involves the cycloaddition of cyanoacetylene with tetrose or pentose aminooxazolines to form anhydronucleosides, which serve as common intermediates for both purine and pyrimidine nucleo(s/t)ides.51,54–57 Arabino- and threo-anhydronucleosides can be converted to pyrimidine nucleotides via urea-mediated phosphorylation.51,57 Ribo-anhydronucleosides can undergo addition with 8-mercaptoadenine followed by photoreduction in the presence of sulfite/bisulfite to form purine ribo- and deoxyribonucleosides.55,56 Given sufficient activation chemistry, the resulting racemic mixture of DNA, RNA, and TNA nucleotides may give rise to genetic polymers with heterogenous sugar backbones70 that could potentially serve as the basis for nonenzymatic template-directed replication in the chemoton. While all these prebiotic nucleotide syntheses have been reported, whether or not such chemistry can be effectively coupled to the autocatalytic formose reaction and encapsulated in the context of a chemoton model is yet to be determined.
In this work, we have demonstrated the effect of cyanamide on the formose reaction, however, other prebiotically plausible molecules that could mediate the formose reaction in interesting ways deserve highlight. Racemic alanine was previously demonstrated to catalyse aldol additions in slightly acidic conditions, forming not only higher order aldoses and ketoses but also pyruvate, pyruvaldehyde, and glyoxal.71 The reaction between cyanide with aldehydes and ketones form cyanohydrins, which can be highly stable (e.g., for glycolonitrile, K = 106 M−1).72,73 It has been observed that cyanohydrins such as glycolonitrile can accelerate HCN oligomerisation, potentially contributing to prebiotic nucleobase synthesis.74,75 Given that CH2O is in large excess, we supposed that formaldehyde would quickly sequester any cyanide, leaving the autocatalytic cycle of the formose reaction and its kinetics relatively unaffected. Fig. S35† shows 0.1 eq of sodium cyanide has little to no effect on the timing of the formose reaction yellowing point. The produced cyanohydrins, primarily glycolonitrile, may accelerate HCN oligomerisation in a neighbouring environment, leading to nucleobase formation.
However, free HCN and/or its derivatives are necessary for cyanamide synthesis.46,52,76 There may be potential prebiotic scenarios that can afford cyanamide synthesis yet prevent HCN from hindering the formose reaction. For example, in one scenario, a transiently reduced atmosphere caused by exogenous delivery and impact of reduced materials leads to the atmospheric synthesis of cyanamide.46,76 Precipitation onto the surface introduces cyanamide to bodies of water which previously accumulated formaldehyde and other formose intermediates while the atmosphere was neutral.77 Another scenario involves gamma irradiation which has been previously demonstrated by Yi et al.24 Here, hydrogen cyanide serves as the feedstock for the continuous production of cyanamide as well as formaldehyde- and glycolaldehyde-derived cyanohydrins. Addition of excess CH2O produced through other geochemical processes13,14,76 may liberate enough glycolaldehyde to kickstart the formose reaction in the presence of cyanamide, assuming sufficiently basic pH and catalytic Ca2+ concentrations are present.
At basic pH, borate is known to complex with formose intermediates that possess syn-1,2-diols, which stabilises higher-order sugars (C4 and above) from undergoing retroaldol fragmentation, a feature which also essentially inhibits the positive feedback mechanism needed for autocatalysis.40,42,43,78 If these sugar–borate complexes were to undergo pH fluctuations to neutral and borate subsequently sequestered, the released sugars may be able to initiate the formose reaction once the pH increases. Hence, although the “messy” early Earth could have hosted an array of potential prebiotic molecules capable of interfering with the formose reaction, we have identified some cases where such interference could trigger downstream reaction cycles and give rise to further and potentially beneficial complexity.
Alternatives for prebiotic sugar synthesis can be found in Eschenmoser's glyoxylate scenario79 and the recently proposed glyoxylose reaction.80 The glyoxylate scenario79 relies on the glyoxylic acid dimer, dihydroxyfumarate, which reacts with its constituents and other compounds to produce a range of classes of biomolecules, including sugars, amino acids and pyrimidines. The glyoxylose reaction proposed by Krishnamurthy and Liotta80 relies on aldol additions involving glyoxylate to produce sugars and sugar acids. Although the potential for retroaldol fragmentation in both reactions suggests the possibility for autocatalysis, this has yet to be confirmed experimentally. Nevertheless, the ability to produce sugars, among other classes of molecules, and the potential for autocatalysis hint that these proposed reactions could serve the role of the central metabolic cycle in the chemoton model.
Outside sugar synthesis, other autocatalytic cycles capable of fuelling membrane synthesis and self-replication have also been investigated, most notably the reductive TCA (rTCA) cycle. The appeal of the rTCA cycle involves the production of five universal metabolites including the biological precursors for lipids and nucleotides, molecules that make up the membrane and self-replication mechanism.59 While the nonenzymatic rTCA cycle is autocatalytic in theory, its unfavourable kinetics prevents the cycle from being of practical use,61 especially in “messy” environments often associated with early Earth. Although much progress81–85 has been made in elucidating the prebiotic conditions necessary for various steps in the cycle, a complete cycle of sufficient turnover has not yet been demonstrated. As such, its relevance to the chemoton model remains uncertain.
Conclusions
Herein, we demonstrate the synthesis of nucleotide precursors fuelled by an autocatalytic cycle as proposed by Tibor Gánti's chemoton model1 that is consistent with prebiotic chemistry. The addition of cyanamide was observed to commandeer the autocatalytic formose reaction for the production of intermediates in the nucleotide synthesis pathways proposed by Orgel et al.50 and Sutherland et al.51,53 Reaction of cyanamide and formose-derived glycolaldehyde led to the production – primarily during the exponential phase – of 2-NH2Ox, an intermediate in nonenzymatic nucleotide synthesis.51,53 Masses consistent with tetrose and pentose aminooxazolines, certain stereoisomers of which serve as the next intermediates in TNA and RNA nucleoside synthesis,53 respectively, were also detected in the cyanamide-mediated formose reactions. Moreover, LC-MS data revealed that the synthesis of these precursors is kinetically coupled to the production of sugars and are not simply linear chemical reactions between cyanamide and isolated formose-derived sugars. With this work, we aim to contribute to experimental studies investigating the chemoton model in the context of prebiotic chemistry as well as integrating nonlinear dynamics and prebiotic synthesis.Experimental
Materials and methods
:1 ratio by volume (6.8 mL each) in a 15 mL centrifuge tube and mixed immediately via shaking and vortexing. The combined mixture was subsequently divided into 12 × 1 mL aliquots which were placed into 1.5 mL centrifuge tubes containing magnetic stirrer bars. 11 of the 12 sample tubes were continuously stirred and incubated at 50 °C while the remaining tube was placed on ice to quench the reaction as an initial timepoint. To monitor the reaction over time, every 2 minutes one of the 1 mL aliquot samples being heated at 50 °C was removed from heat and placed on ice in order to stop the reaction.
HPLC analysis was carried out using a Shimadzu Nexera 40 Series UPLC system with PDA detector (Kyoto, Japan). An aliquot of 1 μL of each derivatised sample was injected into the HPLC and eluted with a 1 mL min−1 isocratic flow of 50:50 aqueous 0.1% formic acid (solvent A) and ACN with 0.1% formic acid (solvent B) for 20 minutes. Following each sample run was a binary gradient wash. The gradient started at 50% solvent B and stayed at 50% for 9 minutes. Solvent B was then ramped up to 90% in 3 minutes after which the concentration remained at 90% for 4 minutes. The concentration of B then returned to 50% in 30 seconds and remained at 50% for the rest of the run. The stationary phase was a Shimadzu Shim-pak GIST C18 column (5 μm particle size, 4.6 mm I.D. and 150 mm length) with the oven temperature maintained at 25 °C.
LC-MS analysis was carried out using a Shimadzu Nexera 40 Series UPLC system connected to a Shimadzu LC-MS-9030 quadruple-time-of-flight mass spectrometer (Kyoto, Japan). A Thermo Scientific Hypercarb UPLC column (3 μm particle size, 2.1 mm I.D., 50 mm length) was used with the oven temperature maintained at 40 °C. The diluted formose samples were injected at 1 μL aliquots and eluted at 0.2 mL min−1 using a binary gradient made up from two solvents: (A) water + 0.1% formic acid and (B) acetonitrile + 0.1% formic acid. Each run started with 0% acetonitrile, which was maintained for 2 minutes, then increased from 0% to 50% over 4 minutes and maintained at 50% for 1.5 minutes. Solvent B was then ramped down from 50% back to 0% in 1 minute and maintained at 0% for 2.5 minutes to complete the gradient. Between each analysis, Milli-Q water (10 μL) was injected and the column was washed using the same gradient. Analytes with retention time 0.4–5 min were analysed using positive-mode MS analysis. Ions with m/z 175.0719 were further analysed via collision-induced disassociation MS/MS with collision energy set to 35 V.
NMR spectroscopy was performed using a Bruker Avance III HD 600 (600.16 MHz, 1H) spectrometer. 1D 1H NMR spectra were collected with water suppression using a presaturation pulse program (zgcpgppr). The number of scans was set to 32.
Data availability
All the supporting experimental data is a part of ESI.†Author contributions
Q. P. T, R. Y., and A. C. F. designed research; Q. P. T. performed research; Q. P. T. and A. C. F. analysed data; Q. P. T. and A. C. F. wrote the paper.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by Japan Society for the Promotion of Science (JSPS) Grant-in-aid 21K14029 by the WPI-funded Earth-Life Science institute at Tokyo institute of Technology. A. C. F acknowledges support from the University of New South Wales Strategic Hires and Retention Pathways (SHARP) program, the Australian Research Council Discovery Project Grant DP210102133 and a Future Fellowship, FT220100757. The authors thank Professor Ram Krishnamurthy and Professor Charles Liotta for their constructive feedback on the manuscript.References
- T. Gánti, The Principles of Life, Oxford University Press, 2003 Search PubMed.
- P. A. Bachmann, P. L. Luisi and J. Lang, Nature, 1992, 357, 57–59 CrossRef CAS.
- L. Liu, Y. Zou, A. Bhattacharya, D. Zhang, S. Q. Lang, K. N. Houk and N. K. Devaraj, Nat. Chem., 2020, 12, 1029–1034 CrossRef CAS PubMed.
- B. J. Weimann, R. Lohrmann, L. E. Orgel, H. Schneider-Bernloehr and J. E. Sulston, Science, 1968, 161, 387 CrossRef CAS PubMed.
- G. F. Joyce, T. Inoue and L. E. Orgel, J. Mol. Biol., 1984, 176, 279–306 CrossRef CAS PubMed.
- N. Prywes, J. C. Blain, F. Del Frate and J. W. Szostak, eLife, 2016, 5, e17756 CrossRef PubMed.
- E. Hänle and C. Richert, Angew. Chem., Int. Ed., 2018, 57, 8911–8915 CrossRef PubMed.
- T. Walton, L. Pazienza and J. W. Szostak, Biochemistry, 2019, 58, 755–762 CrossRef CAS PubMed.
- G. Leveau, D. Pfeffer, B. Altaner, E. Kervio, F. Welsch, U. Gerland and C. Richert, Angew. Chem., Int. Ed., 2022, 61, e202203067 CrossRef CAS PubMed.
- S. S. Mansy, J. P. Schrum, M. Krishnamurthy, S. Tobé, D. A. Treco and J. W. Szostak, Nature, 2008, 454, 122–125 CrossRef CAS PubMed.
- A. Butlerow, C. R. Acad. Sci., 1861, 53, 145–147 Search PubMed.
- S. L. Miller and H. C. Urey, Science, 1959, 130, 245–251 CrossRef CAS PubMed.
- A. Bar-nun and H. Hartman, Origins Life, 1978, 9, 93–101 CrossRef CAS PubMed.
- J. P. Pinto, G. R. Gladstone and Y. L. Yung, Science, 1980, 210, 183–185 CrossRef CAS PubMed.
- N. E. B. Zellner, V. P. McCaffrey and J. H. E. Butler, Astrobiology, 2020, 20, 1377–1388 CrossRef CAS PubMed.
- L. E. Orgel, J. Mol. Biol., 1968, 38, 381–393 CrossRef CAS PubMed.
- W. Gilbert, Nature, 1986, 319, 618 CrossRef.
- R. F. Gesteland and J. F. Atkins, The RNA world: The Nature of Modern RNA Suggests A Prebiotic RNA World, Cold Spring Harbor Laboratory Press, 1993 Search PubMed.
- L. E. Orgel, Crit. Rev. Biochem. Mol. Biol., 2004, 39, 99–123 CrossRef CAS PubMed.
- M. Yadav, R. Kumar and R. Krishnamurthy, Chem. Rev., 2020, 120, 4766–4805 CrossRef CAS PubMed.
- C. Reid and L. E. Orgel, Nature, 1967, 216, 455 CrossRef CAS PubMed.
- R. Shapiro, Origins Life Evol. Biospheres, 1988, 18, 71–85 CrossRef CAS PubMed.
- K. B. Muchowska, S. J. Varma and J. Moran, Nature, 2019, 569, 104–107 CrossRef CAS PubMed.
- R. Yi, Q. P. Tran, S. Ali, I. Yoda, Z. R. Adam, H. J. Cleaves II and A. C. Fahrenbach, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 13267–13274 CrossRef CAS PubMed.
- Q. P. Tran, Z. R. Adam and A. C. Fahrenbach, Life, 2020, 10, 352 CrossRef CAS PubMed.
- G. Springsteen, J. R. Yerabolu, J. Nelson, C. J. Rhea and R. Krishnamurthy, Nat. Commun., 2018, 9, 91 CrossRef PubMed.
- R. T. Stubbs, M. Yadav, R. Krishnamurthy and G. Springsteen, Nat. Chem., 2020, 12, 1016–1022 CrossRef CAS PubMed.
- S. Lamour, S. Pallmann, M. Haas and O. Trapp, Life, 2019, 9, 52 CrossRef CAS PubMed.
- M. Haas, S. Lamour, S. B. Christ and O. Trapp, Commun. Chem., 2020, 3, 140 CrossRef CAS PubMed.
- W. E. Robinson, E. Daines, P. van Duppen, T. de Jong and W. T. S. Huck, Nat. Chem., 2022, 14, 623–631 CrossRef CAS PubMed.
- P. van Duppen, E. Daines, W. E. Robinson and W. T. S. Huck, J. Am. Chem. Soc., 2023, 145, 7559–7568 CrossRef CAS PubMed.
- R. Breslow, Tetrahedron Lett., 1959, 1, 22–26 CrossRef.
- R. F. Socha, A. H. Weiss and M. M. Sakharov, J. Catal., 1981, 67, 207–217 CrossRef CAS.
- A. N. Simonov, O. P. Pestunova, L. G. Matvienko and V. N. Parmon, Kinet. Catal., 2007, 48, 245–254 CrossRef CAS.
- R. Yi, R. Kern, P. Pollet, H. Lin, R. Krishnamurthy and C. L. Liotta, Chem.–Eur. J., 2023, 29, e202202816 CrossRef CAS PubMed.
- C. Appayee and R. Breslow, J. Am. Chem. Soc., 2014, 136, 3720–3723 CrossRef CAS PubMed.
- T. Mizuno and A. H. Weiss, Adv. Carbohydr. Chem. Biochem., 1974, 29, 173–227 CrossRef CAS.
- G. Harsch, M. Harsch, H. Bauer and W. Voelter, Z. Naturforsch. B, 1983, 38, 1269–1280 CrossRef.
- W. P. Huskey and I. R. Epstein, J. Am. Chem. Soc., 1989, 111, 3157–3163 CrossRef CAS.
- A. Ricardo, M. A. Carrigan, A. N. Olcott and S. A. Benner, Science, 2004, 303, 196 CrossRef CAS PubMed.
- A. Ricardo, F. Frye, M. A. Carrigan, J. D. Tipton, D. H. Powell and S. A. Benner, J. Org. Chem., 2006, 71, 9503–9505 CrossRef CAS PubMed.
- H.-J. Kim, A. Ricardo, H. I. Illangkoon, M. J. Kim, M. A. Carrigan, F. Frye and S. A. Benner, J. Am. Chem. Soc., 2011, 133, 9457–9468 CrossRef CAS PubMed.
- S. A. Benner, H.-J. Kim and M. A. Carrigan, Acc. Chem. Res., 2012, 45, 2025–2034 CrossRef CAS PubMed.
- I. V. Delidovich, A. N. Simonov, O. P. Taran and V. N. Parmon, ChemSusChem, 2014, 7, 1833–1846 CrossRef CAS PubMed.
- R. Yi, Y. Hongo, I. Yoda, Z. R. Adam and A. C. Fahrenbach, ChemistrySelect, 2018, 3, 10169–10174 CrossRef CAS.
- Z. Liu, L.-F. Wu, A. D. Bond and J. D. Sutherland, Chem. Commun., 2020, 56, 13563–13566 RSC.
- R. Lohrmann and L. E. Orgel, Science, 1968, 161, 64–66 CrossRef CAS PubMed.
- M. Tsanakopoulou and J. D. Sutherland, Chem. Commun., 2017, 53, 11893–11896 RSC.
- A. C. Fahrenbach, C. Giurgiu, C. P. Tam, L. Li, Y. Hongo, M. Aono and J. W. Szostak, J. Am. Chem. Soc., 2017, 139, 8780–8783 CrossRef CAS PubMed.
- R. A. Sanchez and L. E. Orgel, J. Mol. Biol., 1970, 47, 531–543 CrossRef CAS PubMed.
- M. W. Powner, B. Gerland and J. D. Sutherland, Nature, 2009, 459, 239–242 CrossRef CAS PubMed.
- B. H. Patel, C. Percivalle, D. J. Ritson, C. D. Duffy and J. D. Sutherland, Nat. Chem., 2015, 7, 301–307 CrossRef CAS PubMed.
- S. Islam, J. A. Aguilar, M. W. Powner, M. Nilsson, G. A. Morris and J. D. Sutherland, Chem.–Eur. J., 2013, 19, 4586–4595 CrossRef CAS PubMed.
- J. Xu, N. J. Green, C. Gibard, R. Krishnamurthy and J. D. Sutherland, Nat. Chem., 2019, 11, 457–462 CrossRef CAS PubMed.
- J. Xu, V. Chmela, N. J. Green, D. A. Russell, M. J. Janicki, R. W. Góra, R. Szabla, A. D. Bond and J. D. Sutherland, Nature, 2020, 582, 60–66 CrossRef CAS PubMed.
- J. Xu, N. J. Green, D. A. Russell, Z. Liu and J. D. Sutherland, J. Am. Chem. Soc., 2021, 143, 14482–14486 CrossRef CAS PubMed.
- B. W. F. Colville and M. W. Powner, Angew. Chem., Int. Ed., 2021, 60, 10526–10530 CrossRef CAS PubMed.
- D. Kopetzki and M. Antonietti, New J. Chem., 2011, 35, 1787–1794 RSC.
- K. B. Muchowska, S. J. Varma and J. Moran, Chem. Rev., 2020, 120, 7708–7744 CrossRef CAS PubMed.
- Z. Peng, J. Linderoth and D. A. Baum, PLoS Comput. Biol., 2022, 18, e1010498 CrossRef CAS PubMed.
- L. E. Orgel, PLoS Biol., 2008, 6, e18 CrossRef PubMed.
- M. G. Sacerdote and J. W. Szostak, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 6004–6008 CrossRef CAS PubMed.
- C. Gibard, I. B. Gorrell, E. I. Jiménez, T. P. Kee, M. A. Pasek and R. Krishnamurthy, Angew. Chem., Int. Ed., 2019, 58, 8151–8155 CrossRef CAS PubMed.
- R. Krishnamurthy, S. Guntha and A. Eschenmoser, Angew. Chem., Int. Ed., 2000, 39, 2281–2285 CrossRef CAS PubMed.
- C. Gibard, S. Bhowmik, M. Karki, E.-K. Kim and R. Krishnamurthy, Nat. Chem., 2018, 10, 212–217 CrossRef CAS PubMed.
- A. Osumah and R. Krishnamurthy, ChemBioChem, 2021, 22, 3001–3009 CrossRef CAS PubMed.
- H. Lin, L. J. Leman and R. Krishnamurthy, Chem. Sci., 2022, 13, 13741–13747 RSC.
- B. Damer and D. Deamer, Astrobiology, 2020, 20, 429–452 CrossRef PubMed.
- M. J. Van Kranendonk, R. Baumgartner, T. Djokic, T. Ota, L. Steller, U. Garbe and E. Nakamura, Astrobiology, 2021, 21, 39–59 CrossRef CAS PubMed.
- S. Bhowmik and R. Krishnamurthy, Nat. Chem., 2019, 11, 1009–1018 CrossRef CAS PubMed.
- A. L. Weber, Origins Life Evol. Biospheres, 2001, 31, 71–86 CrossRef CAS PubMed.
- T. Arrhenius, G. Arrhenius and W. Paplawsky, Origins Life Evol. Biospheres, 1994, 24, 1–17 CrossRef CAS PubMed.
- G. Arrhenius, K. K. Baldridge, S. Richards-Gross and J. S. Siegel, J. Org. Chem., 1997, 62, 5522–5525 CrossRef CAS PubMed.
- A. W. Schwartz, H. Joosten and A. B. Voet, Biosystems, 1982, 15, 191–193 CrossRef CAS PubMed.
- A. W. Schwartz and M. Goverde, J. Mol. Evol., 1982, 18, 351–353 CrossRef CAS PubMed.
- K. J. Zahnle, R. Lupu, D. C. Catling and N. Wogan, Planet. Sci. J., 2020, 1, 11 CrossRef.
- S. A. Benner, H.-J. Kim and E. Biondi, Life, 2019, 9, 84 CrossRef CAS PubMed.
- A. Franco, J. R. Ascenso, L. Ilharco, H. P. Diogo, V. André and J. A. L. da Silva, J. Mol. Struct., 2019, 1184, 281–288 CrossRef CAS.
- A. Eschenmoser, Tetrahedron, 2007, 63, 12821–12844 CrossRef CAS.
- R. Krishnamurthy and C. L. Liotta, Chem, 2023, 9, 784–797 CAS.
- G. D. Cody, N. Z. Boctor, R. M. Hazen, J. A. Brandes, H. J. Morowitz and H. S. Yoder Jr, Geochim. Cosmochim. Acta, 2001, 65, 3557–3576 CrossRef CAS.
- X. V. Zhang and S. T. Martin, J. Am. Chem. Soc., 2006, 128, 16032–16033 CrossRef CAS PubMed.
- K. B. Muchowska, S. J. Varma, E. Chevallot-Beroux, L. Lethuillier-Karl, G. Li and J. Moran, Nat. Ecol. Evol., 2017, 1, 1716–1721 CrossRef PubMed.
- S. A. Rauscher and J. Moran, Angew. Chem., Int. Ed., 2022, 61, e202212932 CrossRef CAS PubMed.
- N. Kitadai, R. Nakamura, M. Yamamoto, K. Takai, N. Yoshida and Y. Oono, Sci. Adv., 2019, 5, eaav7848 CrossRef PubMed.
- Cold Spring Harbor Protocols, Sodium Phosphate, 2006, DOI:10.1101/pdb.rec8303.
- Cold Spring Harbor Protocols, 37% Formaldehyde, 2011, DOI:10.1101/pdb.rec067645.
- M. Haas, S. Lamour and O. Trapp, J. Chromatogr. A, 2018, 1568, 160–167 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and data analysis details, HPLC chromatograms, LC-MS, MS/MS, EICs, and structures of potential products mentioned in the text. See DOI: https://doi.org/10.1039/d3sc03185c |
| This journal is © The Royal Society of Chemistry 2023 |