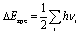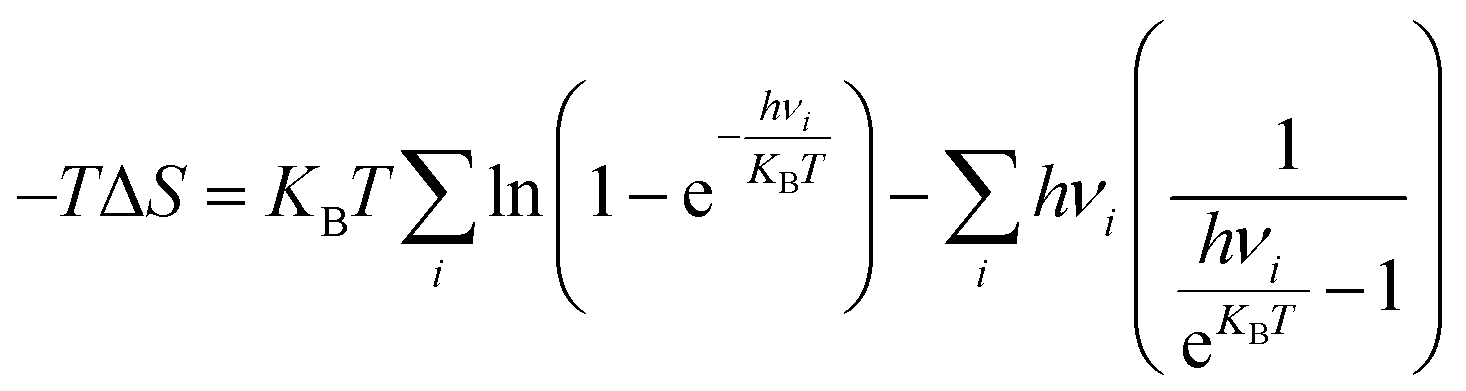DOI:
10.1039/D3SC03474G
(Edge Article)
Chem. Sci., 2023,
14, 13518-13529
Synergistic coupling of interface ohmic contact and LSPR effects over Au/Bi24O31Br10 nanosheets for visible-light-driven photocatalytic CO2 reduction to CO†
Received
7th July 2023
, Accepted 26th October 2023
First published on 27th October 2023
Abstract
The challenge of synergistically optimizing different mechanisms limits the further improvement of plasmon-mediated photocatalytic activities. In this work, an Au/Bi24O31Br10 composite, combining an interface ohmic contact and localized surface plasmon resonance (LSPR), is prepared by a thermal reduction method. The LSPR effect induces the local resonance energy transfer effect and the local electric field enhancement effect, while the interface ohmic contact forms a stronger interface electric field. The novel synergistic interaction between the interface ohmic contact and LSPR drives effective charge separation and provides more active sites for the adsorption and activation of CO2 with improved photocatalytic efficiency. The optimized 0.6 wt% Au (5.7 nm) over Bi24O31Br10 nanosheets showed an apparently improved photocatalytic activity without any sacrificial reagents, specifically CO and O2 yields of 44.92 and 17.83 μmol g−1 h−1, and demonstrated superior stability (only lost 6%) after continuous reaction for 48 h, nearly 5-fold enhanced compared to Bi24O31Br10 and a great advantage compared with other bismuth-based photocatalysts.
1. Introduction
Since the second industrial revolution, human beings have been in a state of over-use of fossil fuels, resulting in an energy crisis and severe environmental pollution problems.1 So far, a variety of solutions have been proposed. Among them, the photocatalytic CO2 reduction/conversion process can be regarded as the most promising one, and the driving force of this reaction is inexhaustible light energy, so it has attracted the interest of a large number of researchers.2 To date, many kinds of photocatalysts for CO2 reduction have been reported, such as transition metal oxides, transition metal sulfides, and metal–organic frameworks. The factors that affect the photocatalytic performance include: the spectral response range;3 the separation and migration of photogenerated charges;4 surface chemical reaction.5 Therefore, the construction of photocatalysts with high photoenergy efficiency, strong photogenerated charge transfer ability and a large specific surface area is of great significance for the photoreduction of CO2.
Among the reported photocatalysts, two-dimensional (2D) ultrathin nanosheets have attracted much attention because of their excellent photoinduced charge separation and transfer efficiency, large specific surface area, lots of surface-active sites and better substrate interaction. Therefore, it is of great research value to construct a highly efficient composite based on a 2D photocatalyst. Among many photocatalysts, BiOX (X= Cl, Br and I) easily form a nanosheet structure due to their unique “sandwich configuration” (X−–[Bi2O2]2+–X−).6 And their band gap is relatively narrow, and the utilization rate of sunlight is relatively high, which has aroused widespread concern. However, bismuth-based photocatalytic materials have a high photogenerated charge recombination rate and easy photo-corrosion, which limits their applications in practical applications. Cui et al. summarized three key strategies for improving photocatalytic efficiency through extensive experiments, such as composition adjustment, vacancy engineering and preparation of heterostructures.7 As a commonly used method to improve photocatalytic activity, the bismuth-rich strategy has constructed a series of semiconductors,8 such as Bi5O7Br,9 Bi24O31Cl10 (ref. 10) and Bi24O31Br10 (ref. 11), which show satisfactory photocatalytic performance under visible light. Among them, Bi24O31Br10 (Eg = 2.51 eV), as a new bismuth halide compound, has a smaller band gap and better performance than BiOBr (Eg = 2.75 eV). Its crystal structure is composed of BiO5 polyhedrons and Br− ions.12 Recently, Bi24O31Br10 has been used in some photocatalytic systems.13–15 For example, Xiao et al. used Bi24O31Br10 to realize the high selective conversion of benzyl alcohol to benzaldehyde.11 Wang et al. used Bi24O31Br10 as a catalyst to realize the efficient degradation of tetracycline hydrochloride.4 However, the weak photocatalytic reduction ability and high photogenerated charge recombination rate hinder the further development of Bi24O31Br10 in the field of photocatalysis. To overcome these shortcomings, it is necessary to further develop Bi24O31Br10.
One of the most attractive characteristics of noble metals (e.g., Au, Ag, etc.) is the nanoscale local surface plasmon resonance (LSPR) effect, which is produced by conducting electrons in response to the collective oscillation of incident light under resonance conditions. Due to the LSPR effect, plasma metal is a kind of highly sensitive material for photothermal catalysis. Once exposed to light, they promote the activity of chemical reactions in the following ways: (1) local electric field enhancement (LEFE),16 (2) local resonance energy transfer (LRET),17 and (3) photothermal effect (PE).18 The LEFE can observably improve the band transition rate of the adjacent semiconductor, if the resonance energy of the plasma metal surpasses the band gap of the semiconductor.16,19 Furthermore, the plasmonic oscillation energy can be delivered via LRET to the semiconductor photocatalyst by local electric field or non-radiative dipole–dipole coupling, which requires spectral overlap between the LSPR peak and semiconductor light absorption range.17,20 The surface plasmon can improve the generation and separation of e−/h+ pairs through these two mechanisms. Ma et al. prepared core–shell (Au/AgAu)@CdS hybrid nanostructures with strong multi-interface plasmon coupling. Strong plasmon absorption and near-field enhancement caused by multi-interface plasmon coupling provide more plasmon energy, which significantly promotes the generation and separation of e−/h+ pairs in catalysts.21 Wang et al. have developed a simple surfactant-free nano-processing method, which limits Au nanoparticles to N-doped TiO2 nano-bowl arrays. N-doping extends the optical absorption of TiO2 to the visible region, which better overlaps the plasmon bands of Au nanoparticles. This design cooperates well with the Schottky junction and the plasmon LRET process, thus significantly improving the performance of photocatalytic hydrogen production in the full spectrum range.22 In addition, due to different work functions, the noble metal and the base photocatalyst will form Schottky contacts22,23 and ohmic contacts24,25 at the coupling interface, which will lead to energy band bending, which is beneficial to the migration of photogenerated electrons, thus improving the separation efficiency of charges. As far as we know, few people have studied the synergistic effect between the LSPR effect and interface contact on photocatalytic performance, and the internal mechanism is not clear.
In the work, 0D/2D Au/Bi24O31Br10 (Au/BOB) was synthesized by a thermal reduction method, and the impact of the synergy between the LSPR of Au NPs and ohmic contact between interfaces on the photocatalytic reduction of CO2 was investigated. Surprisingly, the CO yield of the composite can reach 44.92 μmol g−1 h−1, and the yield of CO only decreased 6% after continuous reaction for 48 h in the absence of sacrificial agents and photosensitizers. Emphatically, since the plasma band of Au NPs was mainly located in the visible band, the coupling between Au and BOB can effectively improve the utilization of solar energy through the LRET mechanism. Therefore, the optical absorption of BOB was no longer restricted by the width of the bandgap. Multi-physical field coupling analysis (COMSOL) proved that the LEFE effect can significantly promote the generation and separation of photogenerated charges. In addition, the interface ohmic contact induces a strong interface electric field, which can directly improve the photogenerated charge transfer process and drive the photoelectron transfer from BOB to Au NPs, which is confirmed by DFT calculation. The synergistic effect of the interface ohmic contact and LSPR effect makes it possible to further optimize the photocatalyst, prolong the light absorption wavelength and improve the charge separation.
2. Experimental section
2.1. Chemicals and reagents
All reagents were used directly without further purification. Bismuth nitrate pentahydrate (Bi(NO3)3·5H2O, AR), cetyl trimethyl ammonium bromide (CTAB, AR), sodium hydroxide (NaOH, AR), and methanol (99.5%) were purchased from Xi long Chemical Co., Ltd. Chlorauric acid tetrahydrate (HAuCl4·4H2O, AR) and silicon dioxide (SiO2) were purchased from Aladdin Reagent (Shanghai) Co., Ltd. Sodium borohydride (NaBH4, 96%) and anhydrous ethanol (AR) were purchased from Sinopec Chemical Reagent Co., Ltd. Deionized water was used throughout the whole experiment.
2.2. Synthesis of 2D Bi24O31Br10 (BOB) ultrathin nanosheets
Firstly, 0.243 g of bismuth nitrate pentahydrate and 0.182 g of cetyl trimethyl ammonium bromide (CTAB) were added into 20 mL of deionized water. The suspension was ultrasonically treated for 10 min, and then stirred for 15 min until it was dissolved. Then NaOH aqueous solution (6 M) was added to adjust the pH value of the obtained solution to about 9. Then the uniform mixture was transferred to a Teflon lined stainless steel autoclave, which was held in a heated oven at 160 °C for 16 h. After naturally cooling down to room temperature, the solid was filtered, and washed several times with ethanol, and vacuum dried at 60 °C overnight to obtain (BOB) ultrathin nanosheets.
2.3. Synthesis of the Au NPs/Bi24O31Br10 (Au/BOB) composite
100 mg of BOB prepared in the above experiment was added to 30 mL of ethanol solution (v(H2O)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :v(ethanol) = 1:1) and stirred at room temperature for 10 min to obtain a uniform suspension. Then, 12 mL of HAuCl4 (50 mg L−1) and NaOH (1 M) were added in turn and stirred for 30 min in the dark to ensure uniform dispersion. Then, 0.05 mL of NaBH4 (112 mM) was dropwise added under vigorous stirring, and the reaction was carried out at room temperature for 3 h. The product was washed with ethanol and ultrapure water three times and then heated in a vacuum drying oven at 60 °C for 12 hours. Various mass ratios of Au NPs were investigated for optimization experiments, including 0.3, 0.6 and 1.2 wt%, and the samples are abbreviated as XAu/BOB, where X = 0.3, 0.6 and 1.2.
:v(ethanol) = 1:1) and stirred at room temperature for 10 min to obtain a uniform suspension. Then, 12 mL of HAuCl4 (50 mg L−1) and NaOH (1 M) were added in turn and stirred for 30 min in the dark to ensure uniform dispersion. Then, 0.05 mL of NaBH4 (112 mM) was dropwise added under vigorous stirring, and the reaction was carried out at room temperature for 3 h. The product was washed with ethanol and ultrapure water three times and then heated in a vacuum drying oven at 60 °C for 12 hours. Various mass ratios of Au NPs were investigated for optimization experiments, including 0.3, 0.6 and 1.2 wt%, and the samples are abbreviated as XAu/BOB, where X = 0.3, 0.6 and 1.2.
2.4. Photocatalytic CO2 production experiments
The photocatalytic reduction CO2 experiment was carried out in a 500 mL sealed quartz reactor, which can be irradiated vertically. The light source was a 300 W xenon lamp equipped with a 420 nm cut-off filter (CEL-HXF300-T3). Briefly, 50 mg photocatalyst powder was added to 100 mL aqueous solution. Prior to illumination, the reactor was degassed to completely remove air and dissolved oxygen by continuously blowing high-concentration CO2 gas. During illumination, the system temperature was maintained at about 5 °C by circulating condensed water. The photocatalytic reduction CO2 reaction lasted for 4 h, and the gas sample was collected every 1 h. Finally, the product was quantified using a gas chromatograph (Agilent 8890 GC) equipped with a FID detector, TCD detector and column (TDX-01). The product 13CO obtained from 13CO2 reduction was confirmed by using a dual instrument of gas chromatography and mass spectrometry (GC-MS, Agilent 7890A GC and Agilent 5975C MS).
2.5. Characterization
The surface morphology of the samples was examined by using a field emission scanning electron microscope (FESEM) (NovaNaNoSEM450, operation voltage 10 kV). The microstructure and elemental distribution were characterized by field emission transmission electron microscopy (FETEM, FEI Talos F200X), high-resolution TEM (HRTEM), high-angle annular dark field scanning TEM (HAADF-STEM) and selected area energy dispersive X-ray spectroscopy (EDX) mapping. The thickness and surface potential difference of the nanosheets were measured by atomic force microscopy (AFM, NT-MDT, NTEGRA). The operating conditions were an acceleration voltage 200 kV, a LaB6 filament, and a resolution of 0.235 nm. The optical properties of the samples were studied by using a UV-vis DIFFUSE reflectance spectrometer (UV-vis DRS, Shimadzu UV-2450) with BaSO4 as a reference. The surface chemical composition was analyzed by X-ray photoelectron spectroscopy (XPS, Al Ka, 150 W, and C 1s 284.8 eV). The separation and migration rate of photogenerated charges were analyzed by using photoluminescence (PL) and the fluorescence lifetime spectrum (TRPL) obtained with a fluorescence spectrometer (Hitachif-7000, 420 nm excitation). The Brunauer–Emmett–Teller (BET) method was employed to measure the surface area and pore size distribution of the samples using a NDVA2000e instrument with N2 as the adsorbate at 77 K. The CO2 adsorption capacity of the sample was evaluated by using a NDVA2000e instrument. The temperature programmed desorption data of CO2 were recorded by using a TCD detector on an AutoChem II 2920 analyzer (measured flow rate: 1.339 mmol min−1). In situ infrared spectroscopy (In situ FTIR, Perkin Elmer-3100) was used to determine the change trend of intermediates on the sample surface with time during photocatalytic CO2 reduction. Electron paramagnetic resonance (EPR) data were recorded by using a Bruker A300 spectrometer (state frequency 9.54 GHz, modulation amplitude 2 G, locking frequency 100 kHz, and attenuation 30 dB). Multi-physical coupling analysis software (COMSOL Multiphysics) was used to analyze the local electric field of the sample surface.
2.6. Photoelectrochemical and electrochemical measurements
To study the photochemical properties of all samples, photocurrent was measured in a three-electrode system, where a platinum wire, Ag/AgCl (saturated KCl) and FTO substrate coated with the photocatalyst served as the counter electrode, reference electrode, and working electrode, respectively. The specific experiment was carried out using an electrochemical working station (CH2660C). The working electrode area was 2 cm2 (1 cm × 2 cm) and the electrolyte was 0.1 M Na2SO4 solution. A 300 W xenon lamp (CEL-HXF300-T3) was the light source. The lamp was switched on and off to measure photocurrent under light irradiation and in the dark. Electrochemical impedance spectroscopy (EIS) was performed using the same working station. The Mott–Schottky curve (M–S) was scanned in 0.1 M Na2SO4 solution with a frequency range of 0.1 Hz to 100 kHz. The test amplitude of AC voltage was set at 1 V. In addition, the photoelectrochemical properties of the samples were studied in a three-electrode system, in which carbon rods, calomel electrodes and carbon cloth substrates coated with photocatalysts were used as counter electrodes, reference electrodes and working electrodes, respectively. The specific experiments were carried out on an electrochemical workbench (CH2660C). The working electrode area was 1 cm2 (1 cm × 1 cm) and the electrolyte was 1 M KOH solution.
2.7. DFT calculation
In the density functional theory calculations, we built a planar model of Bi24O31Br10 (002) and Bi24O31Br10 (002) loading Au NPs. The atom number of Au NPs is 20. The vacuum layer along the Z direction was set to be 15 Å to avoid the interactions between images. Structural optimization was performed by using the Vienna Ab initio Simulation Package (VASP)26 with the projector augmented wave (PAW) method.27 The Perdew–Burke–Ernzerhof (PBE)28 exchange–correlation functional with the generalized gradient approximation (GGA)29 was adopted to describe the electronic exchange–correlation energy. The cut-off energy of the plane-wave basis was set at 450 eV. For the optimization of both geometry and lattice size, the Brillouin zone integration is performed with 2 × 2 × 1 Gamma k-point sampling.30 The self-consistent calculations applied a convergence energy threshold of 10−5 eV. The equilibrium geometries and lattice constants were optimized with maximum stress on each atom within 0.01 eV Å−1. Dipole correction was adopted along the Z direction in the calculation of the work function to describe the unsymmetrical surface. The isosurface level of the charge density difference was set to be 5 × 10−4 eÅ−3.
The Gibbs free energy of intermediates was calculated as ΔG = ΔE + ΔEzpe − TΔS, where ΔE is the electronic energy difference directly obtained by DFT calculation, and ΔEzpe and ΔS indicate the zero-point energy and entropy of the surface adsorbing H atom, respectively. The entropies of the free molecules of H2 and H2O were referenced from the NIST database.31 Besides, the Kelvin temperature T was set to be 298.15 K. The reaction energy in photocatalytic CO2 reduction is the reaction free energy of the potential limiting step. The ΔEzpe and (−TΔS) of all intermediates in the CO2 reduction on Au/BOB are calculated by the following equation:
| | 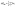 | (1) |
| | 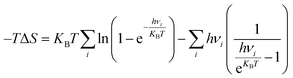 | (2) |
where
h,
ν and
KB represent Planck's constant, vibrational frequencies, and Boltzmann's constant, respectively. The reduction pathway from CO
2 to CO was calculated according to:
| | | *CO2 + H+ + e− → *COOH | (4) |
| | | *COOH + H+ + e− → *CO + H2O | (5) |
3. Results and discussion
3.1. Structure characterization of the photocatalysts
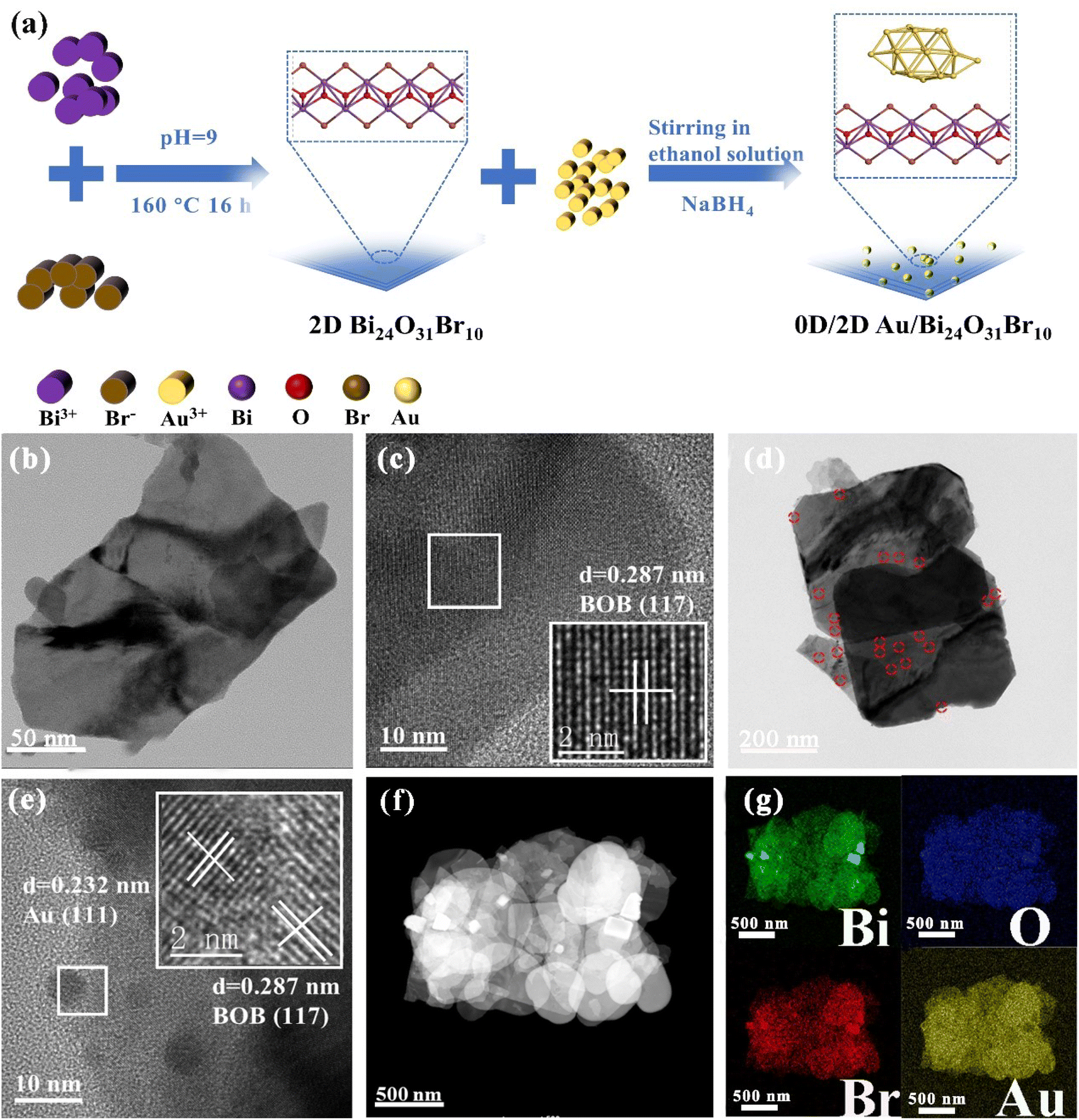
Fig. 1a shows that Au NPs were in situ grown on the surface of 2D BOB nanosheets by impregnation reduction, and the details and particular characterization of the modified 0D/2D Au/BOB composite are displayed in the ESI.† The prepared samples are proved to be monoclinic BOB by XRD (JCPDS No. 75-0888),32 while no obvious diffraction peaks belonging to Au NPs are observed on 0.6Au/BOB, proving the high degree of fine dispersion of Au NPs (Fig. S1†). The content of Au NPs in 0.6Au/BOB was measured using an inductively coupled plasma analyzer. The results are consistent with expectations, indicating that Au NPs were successfully recombined in Au/BOB (Table S1†). Besides, the chemical structure information of BOB and 0.6Au/BOB is compared by FT-IR (Fig. S2†). The results show that the 0.6Au/BOB composite shows a new characteristic peak at 661 cm−1, corresponding to the Au–Bi–O vibrational dynamics, which further proves the successful loading of Au NPs.33
 |
| | Fig. 1 (a) Synthesis schematic diagram of 0D/2D Au/BOB; TEM and HRTEM images of (b and c) BOB and (d and e) 0.6Au/BOB; HAADF-STEM image (f) and corresponding elemental mappings (g) of 0.6Au/BOB. | |
In order to intuitively observe the loading of Au NPs on the surface of BOB nanosheets, the microstructure of BOB and 0.6Au/BOB was obtained by SEM and TEM. As shown in Fig. S3a and b,† both BOB and 0.6Au/BOB exist in the form of irregular nanosheets. In addition, atomic force microscopy (AFM) further confirmed the nanosheet structure of BOB and 0.6Au/BOB (Fig. S4a and b†). The results show that BOB and 0.6Au/BOB are both ultrathin nanosheet structures with thickness close to 6.6 nm, which indicate that the loading of Au NPs will not change the morphology and size of the substrate. TEM further confirmed the results of SEM and AFM. As shown in Fig. 1b and c, BOB exists in the form of irregular nanosheets with no excess longitudinal clutter, which is conducive to the exposure of the active surface and the loading of Au NPs. There are a large number of Au NPs in the TEM image of 0.6Au/BOB (Fig. 1d and e). In addition, through the statistical distribution, the diameter of Au NPs is calculated to be about 5.7 nm (Fig. S3c†). From the HRTEM spectra of 0.6Au/BOB (Fig. 1e), it can be clearly seen that the crystal plane spacings of 0.287 nm and 0.232 nm correspond to the (117) and (111) crystal planes of BOB and Au NPs, respectively. HRTEM indirectly shows that Au NPs were successfully loaded on BOB nanosheets. In addition, as shown in Fig. 1f and g, HAADF-STEM and EDX mappings on the selected areas indicate that the distributions of Bi, O and Br elements are uniform. And also, the uniform distribution of Au elements on the nanosheets can also be observed.34 The above results show that Au NPs are very finely dispersed on BOB, which explains why the specific surface area of the composites increases with the loading of Au NPs (Fig. S5a and b, Table S2†). As a contrast, the same content of Au NPs (0.6Au/SiO2) is loaded on the surface of commercial SiO2, and the results show that the size of Au NPs has not changed significantly (Fig. S6†), which provides more reference data for the proof of the synergy effect and reduces the error of external experiments.
To further verify the chemical interaction between Au NPs and BOB, 0.6Au/BOB was tested by XPS and compared with BOB. As shown in Fig. S7a,† the 0.6Au/BOB composite determines the presence of Bi 4f, Br 3d, O 1s and Au 4f elements, which proves the successful loading of Au NPs. Fig. S7b† shows that BOB has two symmetry peaks at 159.20 and 164.45 eV, which are related to the characteristics of Bi 4f7/2 and Bi 4f5/2 of Bi3+. The characteristic peak of the 0.6Au/BOB composite shifts 0.1 eV to a lower value. As shown in Fig. S7c and d,† compared with BOB, the characteristic peaks of O 1s and Br 3d in the 0.6Au/BOB composite shift negatively and the binding energy decreases. The result further confirms that there is a strong interfacial interaction between Au NPs and BOB. In addition, the XPS spectrum of Au 4f, as shown in Fig. S7e,† is located at the peaks of 87.45 and 83.74 eV, corresponding to Au 4f5/2 and Au 4f7/2, respectively, which is consistent with the results in the literature, and confirms that the Au element exists in the composite in the form of Au0.35
3.2. Photophysical and electrochemical measurements
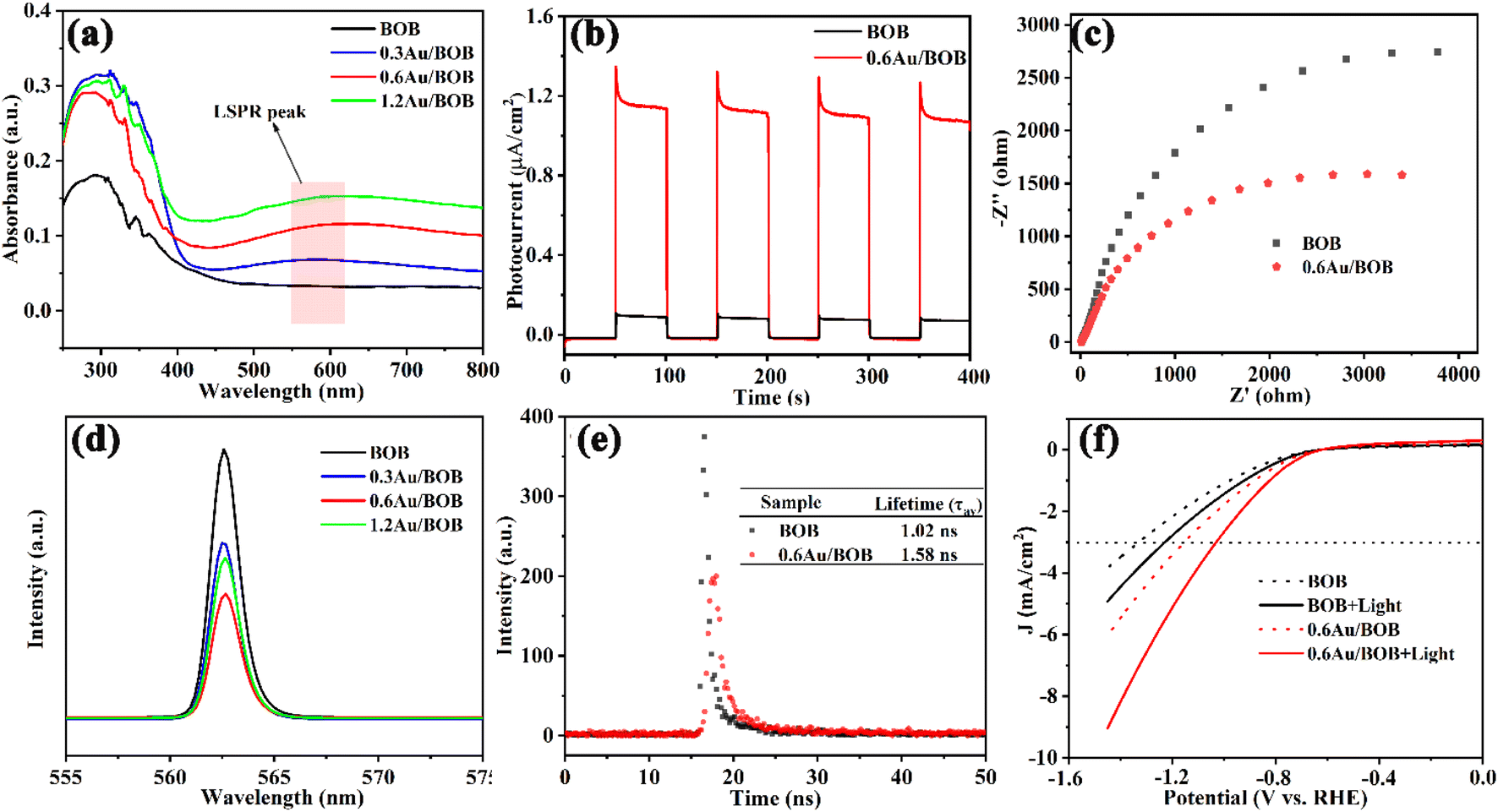
The optical properties of BOB and Au/BOB were determined using UV-vis DRS. As shown in Fig. 2a, pristine BOB exhibits a visible light response, and the edge is about 470 nm. The corresponding band gap is 2.54 eV (Fig. S8†), which is consistent with a previous report.36 Interestingly, Au/BOB has an obvious wide peak near 570 nm, which may be caused by the LSPR effect of Au NPs,17,37 assigned to quadrupole and dipole plasmon resonance signals. In addition, Fig. 2a shows a broadened and enhanced light absorption edge of Au/BOB compared with BOB, indicating that the Au NPs form impurity energy levels within the band gap, effectively reduce the band gap, and improve the absorption of visible light. The DOS curve proves the correctness of the results (Fig. S9†).38 Moreover, if the visible absorption spectrum of BOB partially overlaps with the LSPR peak of Au NPs, this may lead to the formation of a local resonance energy transfer (LRET) mechanism, similar to that of hot electrons at the interface.17,39 Therefore, we investigated the dependence of CO yield on light intensity at λ > 570 nm, and the results are shown in Fig. S10.† As observed, the rate of CO generation was linearly correlated with light intensity, indicating the presence of the LRET mechanism between the interfaces. Thus, the energy acquisition of the BOB CB can be extended beyond the spectral response range, which enhances visible light absorption, photogenerated charge generation and effective charge separation.
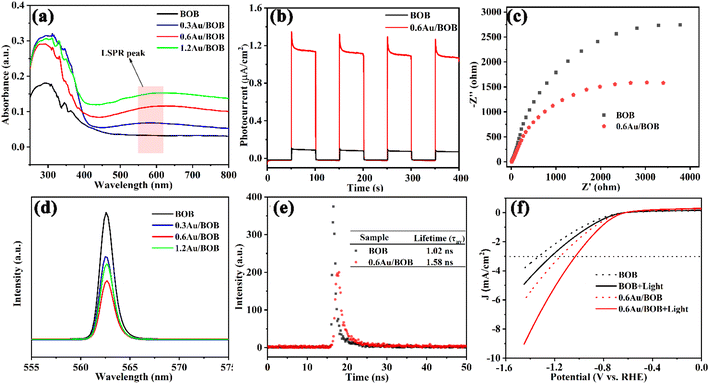 |
| | Fig. 2 (a) UV-vis diffuse reflection spectra of samples; (b) transient photocurrent responses (λ > 420 nm) and (c) EIS of BOB and 0.6Au/BOB; (d) PL spectra; (e) time-resolved PL (TRPL) spectra; (f) LSV of BOB and 0.6Au/BOB under visible light or not. | |
The photocatalytic properties of BOB and 0.6Au/BOB were studied by transient photocurrent response and electrochemical impedance spectroscopy (EIS). As shown in Fig. 2b, the photocurrent response of pure BOB is very weak, which is due to its fast charge recombination. The optical current intensity of the Au/BOB composite has been greatly improved. Among them, the average optical current intensity of 0.6Au/BOB is 1.12 μA cm−2, which is about 11 times that of pure BOB. Therefore, the composite structure of 0.6Au/BOB can effectively separate the photogenerated charge, thus improving the photocatalytic performance. In addition, the near-field electromagnetic coupling of Au NPs also plays an important role, which shows that the yield of photogenerated charges in semiconductor BOB is improved in the vicinity of Au NPs with the LSPR effect. At the same time, electrochemical impedance spectroscopy (EIS) measurement shows that the radius of 0.6Au/BOB is smaller than that of BOB (Fig. 2c), implying less interface resistance for carrier transfer due to the improvement of electron mobility, which is consistent with the photocurrent results. All these data together determine the fast electron transfer in 0.6Au/BOB, which means that the formation of composites promotes charge separation. To confirm this improved charge separation, photoluminescence spectra (PL) and time-resolved PL decay (TRPL) spectra of BOB and 0.6Au/BOB were recorded.40 As shown in Fig. 2d and e, 0.6Au/BOB has the lowest PL intensity and longer lifetime (1.58 ns, Table S3†), proving effective suppression of charge recombination, which is consistent with the results of photocurrent response and EIS. The LSV results in Fig. 2f further proved that to reach the same cathodic current density (e.g. −3 mA cm−2), the lowest potential (−0.95 V vs. RHE) was needed for 0.6Au/BOB, while the highest potential (−1.25 V vs. RHE) was required for BOB.41 Therefore, the composite structure and LRET mechanism can effectively promote the separation and transport of photogenerated charges and inhibit their recombination.
3.3. DFT calculation evidence for the formation of the ohmic contact
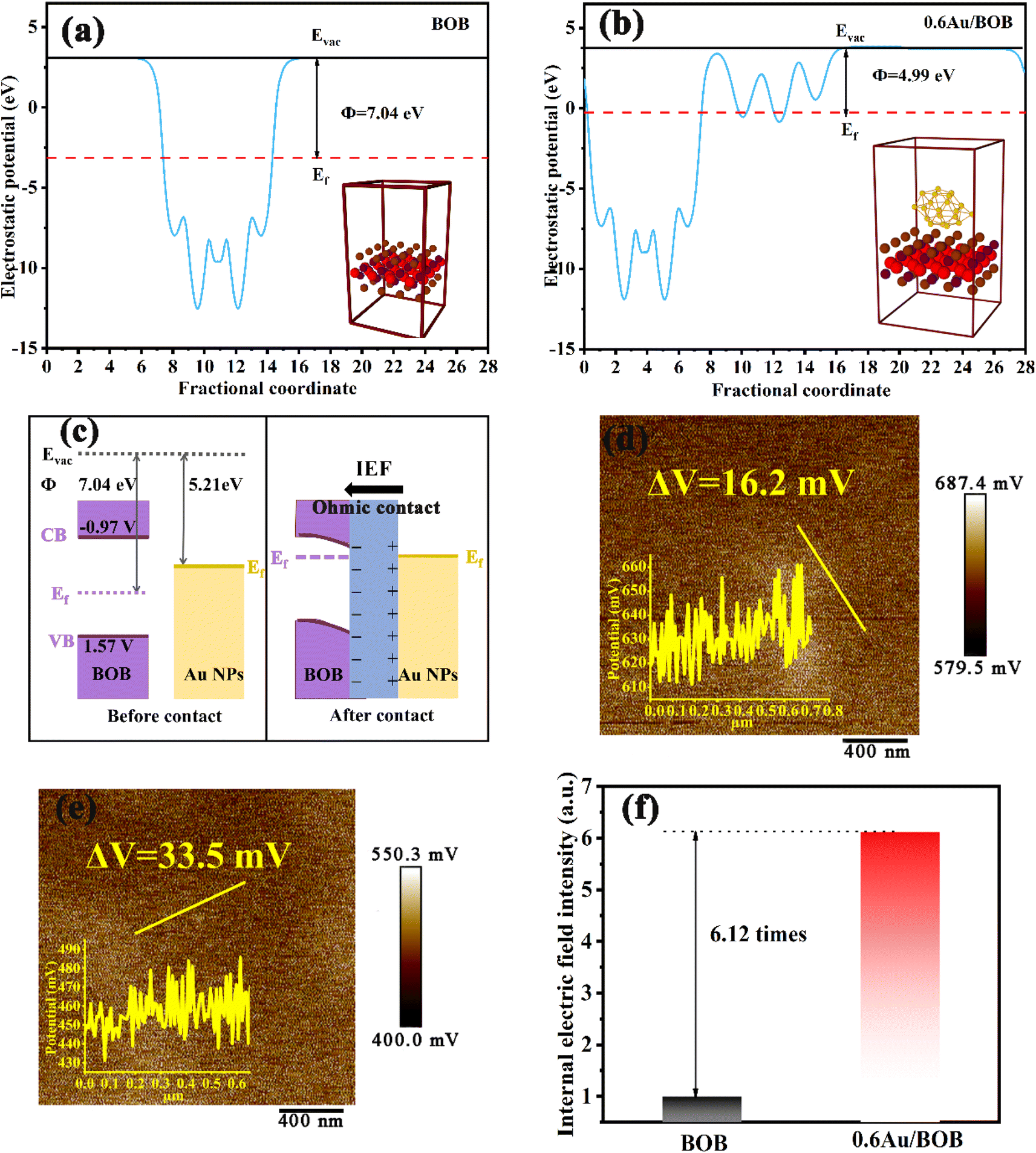
To clarify the charge separation and transfer behaviors of the Au/BOB composites, Density Function Theory (DFT) calculations were employed to investigate the work function and spatial charge density difference of the as-prepared materials. As shown in Fig. 3a and b, the crystal structures of BOB and Au/BOB are simulated by DFT, and the work functions (Φ) of BOB and Au/BOB are 7.04 and 4.99 eV, respectively. Obviously, the Au/BOB composite has a smaller work function, indicating easier overflowing electrons and higher separation efficiency of photogenerated charges. The work function of Au is 5.21 eV,42,43 which is much smaller than that of BOB (7.04 eV), implying that a unique ohmic contact will be formed between Au NPs and BOB (as mentioned by Tang et al.).44 When BOB and Au NPs are in contact (Fig. 3c), the energy band of BOB will bend downward and form an electron accumulation region. Due to electron diffusion, Au NPs will form a hole accumulation area at the interface. Theoretically, this charge distribution at the interface of the composite will form an internal electric field (IEF), and the direction of the IEF is from Au NPs to BOB. The differential charge distribution of Au/BOB confirms this conclusion. As shown in Fig. S11,† the electron consumption occurs on the Au NP side, while charge accumulation occurs on the BOB side.45,46 This result also confirms the electron enrichment trend of BOB in the dark, which is consistent with the results of XPS. Different from Schott contact, the downward bending of the band caused by ohm contact can reduce the transfer barrier of BOB conduction band electrons to Au NPs and greatly increases the separation efficiency of photogenerated charge under the action of the IEF.
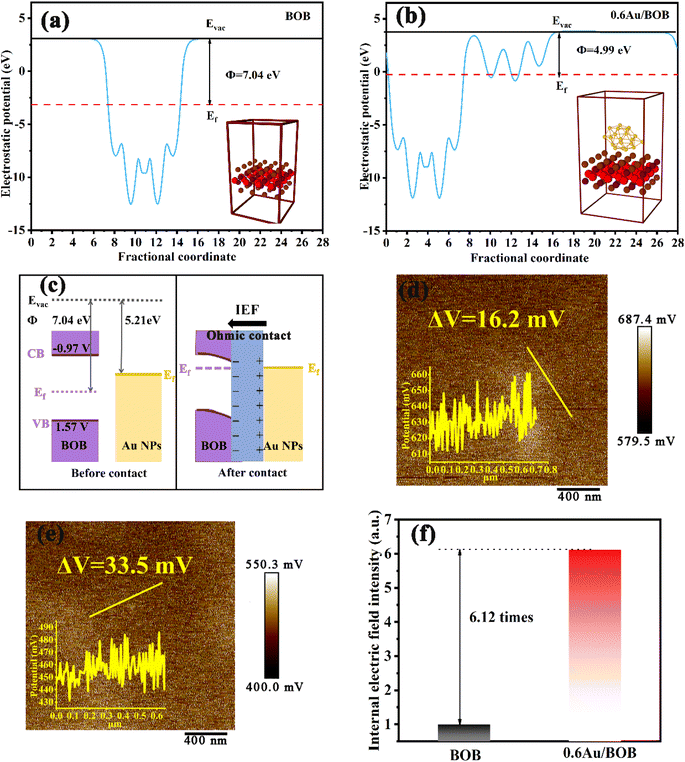 |
| | Fig. 3 Work functions of (a) BOB and (b) Au/BOB; (c) schematic diagram of the energy band structure of the ohmic contact; KPFM images of (d) BOB and (e) 0.6Au/BOB; internal electrical field intensities (f) of BOB and 0.6Au/BOB. | |
Generally, the interface IEF takes on the dynamic driving force for the separation and migration of photogenerated charges. And the intensity of the IEF determines the efficiency of charge separation and migration. The IEF intensities of BOB and Au/BOB composites were calculated. Firstly, the intensity of the IEF is directly proportional to the surface potential of the catalyst, and a Kelvin probe force microscopy (KPFM) test was conducted.47,48 As shown in Fig. 3d and e, the surface potential of BOB is only 16.2 mV, and while the surface potential of the Au/BOB composite reaches 33.5 mV. Secondly, an experimental test was conducted based on the model proposed by Kanata et al.49 (Fig. S12†). According to this model, the square roots of the products of surface voltage (Vs) and surface charge density (ρ) are proportional to IEF intensities. As shown in Fig. 3f, the IEF intensity of Au/BOB increases by 6.12 times compared to that of BOB. The above results show that the ohmic contact between Au NPs and BOB can effectively improve the IEF intensity and stimulate the separation and migration of photogenerated charges.
3.4. CO2 photoreduction performance measurements
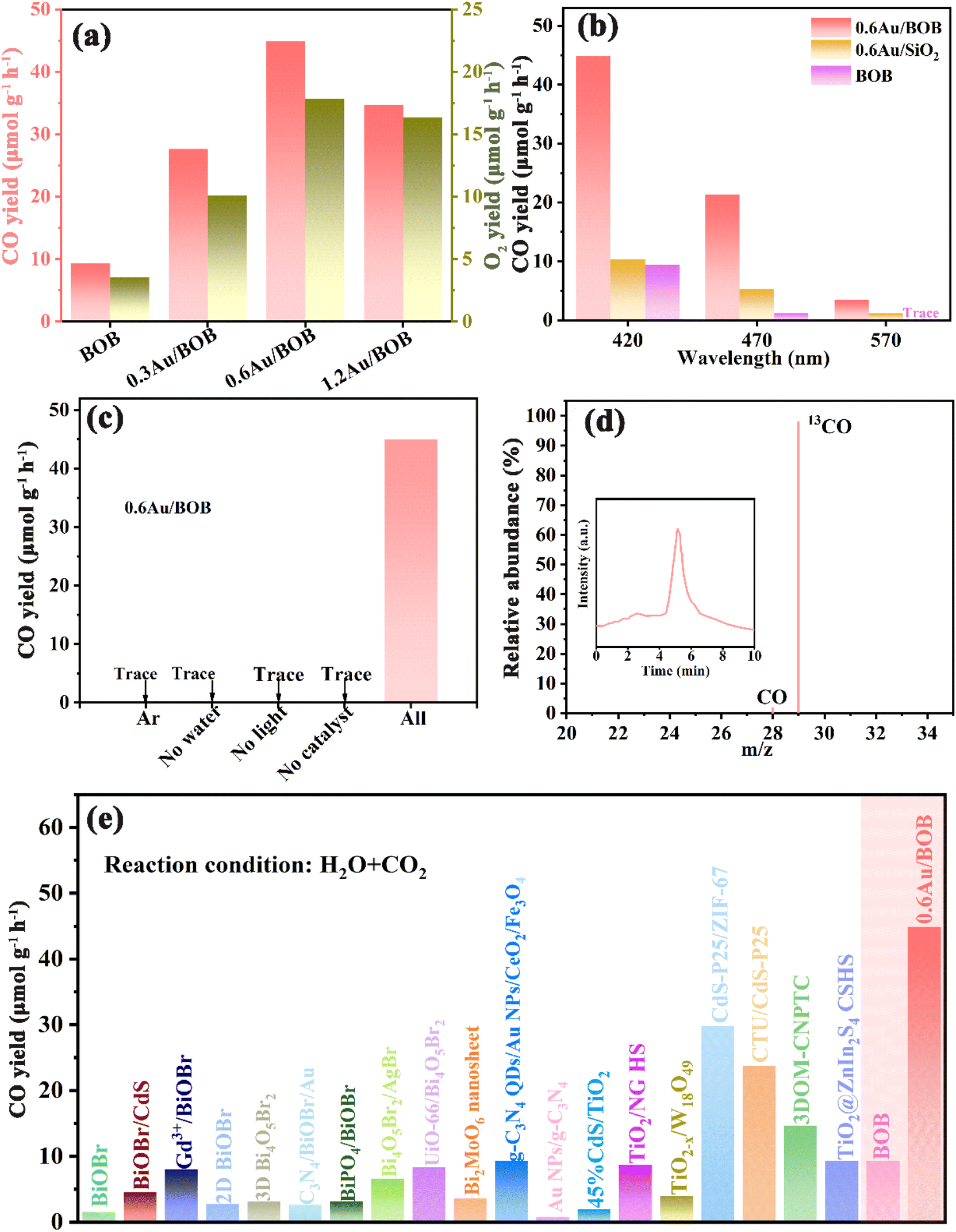
Photocatalytic performance is evaluated to reveal the advantages of the catalytic system. Here, the photocatalytic performance of BOB and different Au/BOB composites was studied. The photocatalytic reduction of CO2 with H2O (no sacrificial agent) was evaluated under simulated sunlight (λ > 420 nm). CO was detected as the major CO2 reduction product with a small amount of CH4 but no other C2–C4 hydrocarbon was detected. O2 was also generated from H2O oxidation. As shown in Fig. 4a, the CO yield of BOB is only 9.36 μmol g−1 h−1. In contrast, the CO yield of 0.6Au/BOB is the highest, reaching 44.92 μmol g−1 h−1. With the loading of Au NPs, the changing trend of properties is 0.6Au/BOB (44.92 μmol g−1 h−1) > 1.2Au/BOB (34.68 μmol g−1 h−1) > 0.3Au/BOB (27.62 μmol g−1 h−1). Obviously, the performance of O2 also has the same trend, and the performance of 0.6Au/BOB is the highest, which can reach 17.83 μmol g−1 h−1. Although the performance of the Au/BOB composite did not increase linearly with the increase of the Au NP content, it was improved. In addition, the performance of 1.2Au/BOB does not increase but decreases compared with that of 0.6Au/BOB, which can be attributed to the fact that superfluous Au NPs will shield the incident light, thus hindering the generation of photogenerated carriers and becoming the recombination center.
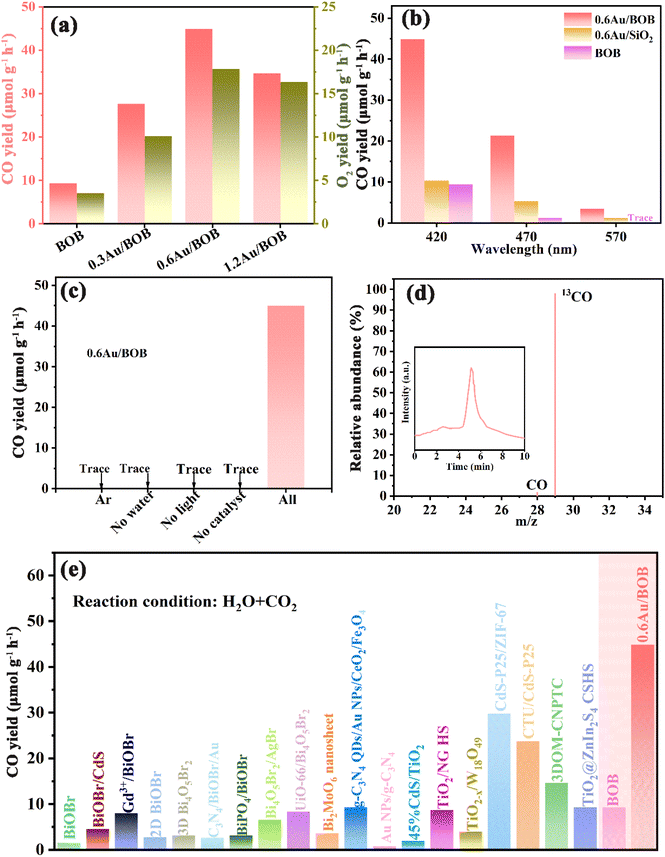 |
| | Fig. 4 (a) Photocatalytic CO yield and O2 yield; (b) CO yield of BOB and 0.6Au/BOB under different wavelengths of light irradiation; (c) CO yield on 0.6Au/BOB under different reaction conditions; (d) mass spectrometry spectra of 13CO2 isotope experiments in the presence of 0.6Au/BOB (inset: gas chromatography spectra); (e) comparison of the CO yield of 0.6Au/BOB and some representative photocatalysts under similar conditions. | |
The excellent CO yield of 0.6Au/BOB can be attributed to the following aspects. Firstly, the IEF at the interface between Au NPs and BOB greatly improves the migration rate of photogenerated charges. Secondly, Au NPs formed a strong LSPR effect. Among them, the LEFE mechanism further enhances the charge transfer of the IEF. The LRET mechanism enhances the generation of photogenerated charges. The photocatalytic performance of 0.6Au/BOB composites at different wavelengths was investigated to confirm the positive influence of the LSPR effect of Au NPs on the photocatalytic performance (Fig. 4b). The best photocatalytic performance was obtained under simulated sunlight (λ > 420 nm) because of more photon energy excited by its wide spectral range, which was further proved by the significant reduction of photocatalytic CO yield under the irradiation of incident light wavelengths λ > 470 nm and λ > 570 nm (ref. 42). Of course, this also proved that the LSPR effect of Au NPs can indeed broaden the spectral response range of the photocatalyst, and then increase the yield of photogenerated charges. In addition, to verify the synergy effect of the IEF and LSPR effect on the improvement of photocatalytic performance, the activities of BOB and 0.6Au/SiO2 at different wavelengths were studied. It can be found that when the incident wavelength was greater than 420 nm, the photocatalytic performance of 0.6Au/BOB was 5 times and 6 times that of BOB and 0.6Au/SiO2, respectively. When the incident wavelength was greater than 570 nm, the photocatalytic CO yields of 0.6Au/BOB and 0.6 Au/SiO2 are 3.44 μmol g−1 h−1 and 0.98 μmol g−1 h−1, respectively, and BOB has almost no light response. These data prove that it was the synergy effect of the IEF and LSPR effect that improved the photocatalytic CO yield of the 0.6Au/BOB composite, and both of them were indispensable.
Fig. 4c shows the effect of different experimental conditions on the photocatalytic reduction of CO2 with 0.6Au/BOB. No CO is detected in the Ar atmosphere. Similarly, CO is not detected under no water, no light or no catalyst conditions, indicating that the integrity of reaction conditions is a prerequisite for the success of an experiment. These results also confirm that the added CO2 is the main source of CO, which is further confirmed by the isotope labeling experiment of 13CO2 (Fig. 4d). Using 13CO2 as the isotope-labeled raw material, the m/z values of 13CO and CO were observed to be 29 and 28, respectively, in the GC-MS dual instrument, and the abundance ratio is close to 49:1, which confirms the accuracy of the conjecture.50 Furthermore, Table S4† shows the wavelength dependent CO yield of 0.6Au/BOB, except that a bandpass filter is equipped to obtain monochromatic incident light (λ = 380, 420, 470 and 570 nm). The optical power of the different monochromatic lights is given in Table S4.† Based on this, the apparent quantum yield (AQY) of the 0.6Au/BOB photocatalyst from CO2 to CO can be determined (see the ESI† for the detailed calculation process). The action spectrum is presented in Fig. S13a.† The UV-visible absorption spectrum and action spectrum of the 0.6Au/BOB photocatalyst were found to be congruent, evincing its commendable light absorption and utilization capacities.51 In addition, the positive correlation trend between the obtained intensity and AQY at λ = 420 nm further supports this result (Fig. S13b†).52 Considering that the 0.6Au/BOB photocatalyst was prepared by thermal reduction, it is reasonable to suspect that crystal defects were formed in 0.6Au/BOB. As shown in Fig. S14,† unsurprisingly, strong oxygen vacancies did exist in 0.6Au/BOB and positively enhanced the photocatalytic CO2 reduction performance.53 Moreover, recycling stability is a key factor for the practical application of photocatalysts, in addition to excellent photocatalytic performance. As shown in Fig. S15,† the photocatalytic activity of 0.6Au/BOB only decreased by 6% after 48 h of continuous reaction, and the activity showed a stable trend in the later stage of the reaction, indicating that loading Au NPs could not only improve the photocatalytic activity of BOB but also greatly improve its stability. In addition, the XRD, TEM, XPS and FTIR spectra of 0.6Au/BOB remained almost unchanged before and after the reaction, which also proved its excellent structural stability (Fig. S16 and S17†). For comparison, Fig. 4e and Table S5† summarize some important experiments in recent years, showing that loading Au NPs based on a bismuth-rich strategy has an advantage in photocatalytic CO2 reduction.
3.5. Local electric field enhancement effect analysis
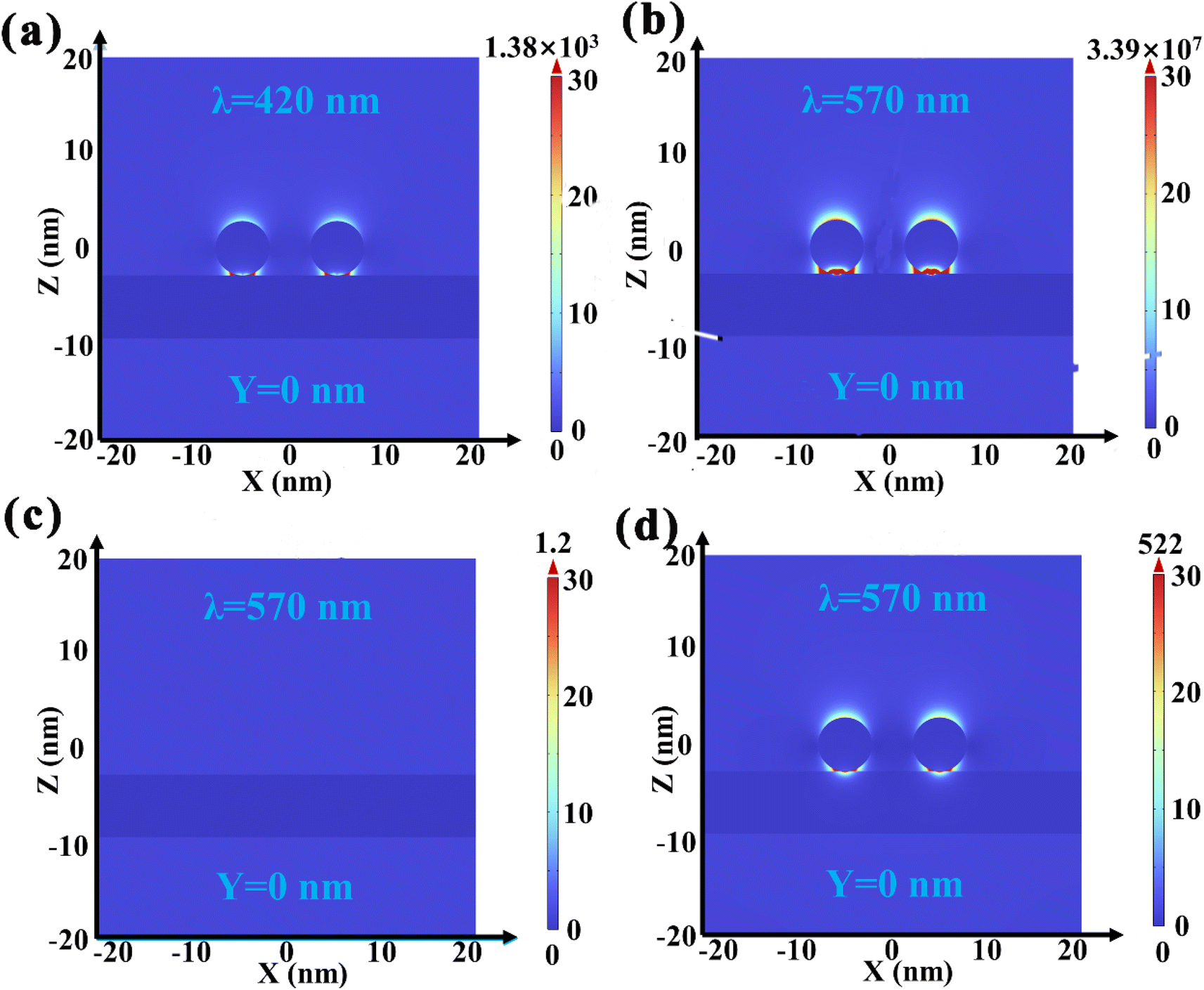
The inherent mechanism of the effect of Au NP LSPR on the photocatalytic performance of Au/BOB is surveyed by the multi-physical field coupling analysis (COMSOL) simulation method. Under the excitation of polarized 420 nm light along the Y axis (Fig. 5a), a hot spot representing the focused electromagnetic field is found at the interface.35 The electromagnetic intensity of the local hot spot is much higher than that of the far-field excited light, and the electromagnetic field intensity of the far-field excited light is at least one order of magnitude higher than that of the far-field excited light. The appearance of a local hot spot means that the photogenerated electrons will migrate from BOB to Au NPs driven by the IEF. As shown in Fig. 5b, with the increase of the incident light wavelength, the electromagnetic intensity corresponding to the local hot spot increases by four orders of magnitude, which means that the local electric field enhancement effect (LEFE) can promote the migration of photogenerated charge.54,55 At the same time, the enhancement of the local electric field of 0.6Au/BOB can further explain the synergy effect of the LEFE and interfacial ohmic contact on the effective separation and migration of photogenerated charges, which can be confirmed by the lower local electric field of Au NPs and BOB (Fig. 5c and d).
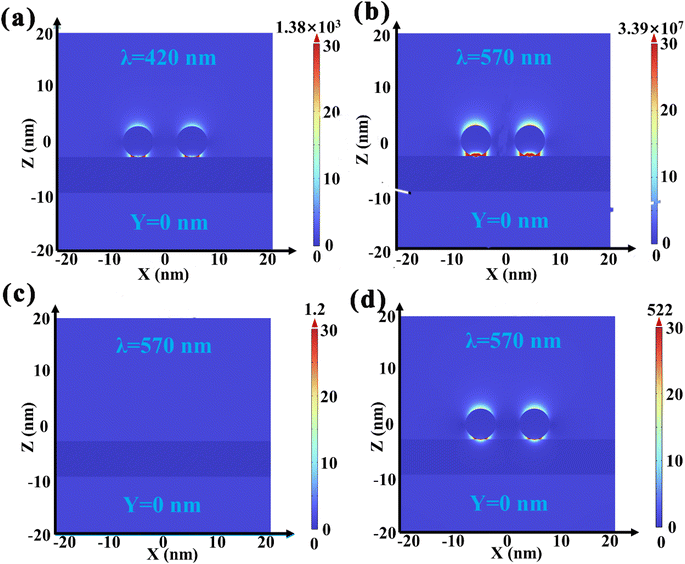 |
| | Fig. 5 COMSOL simulated the local electric field distribution of (a) 0.6Au/BOB at 420 nm and (b) 0.6Au/BOB, (c) BOB and (d) 0.6Au/SiO2 under 570 nm monochromatic light excitation, respectively. | |
3.6. Photocatalytic mechanism
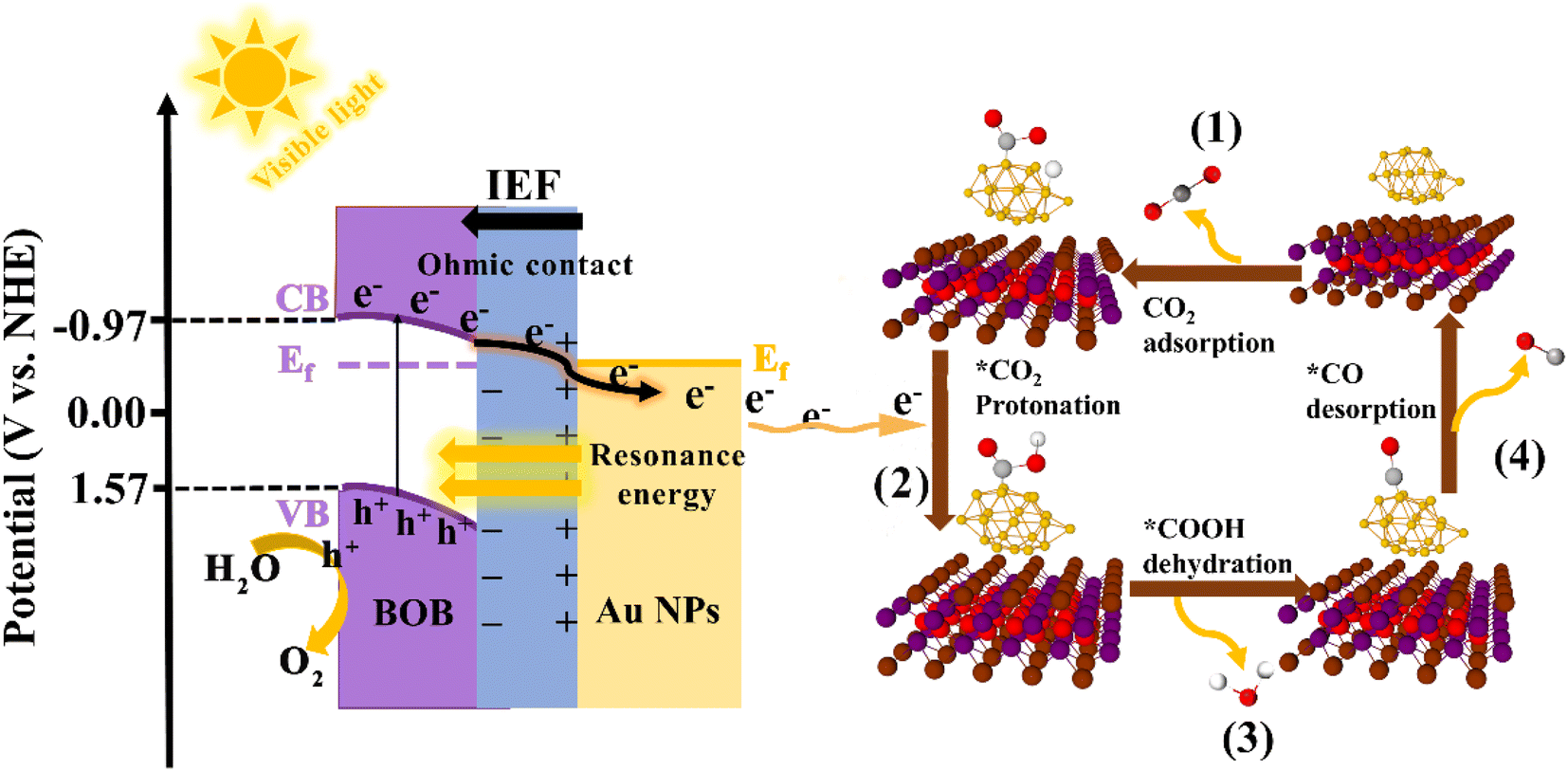
Based on the above analysis, we proposed a feasible mechanism of Au/BOB to enhance photocatalytic CO2 reduction (Fig. 6). Due to the different work functions (Φ), the contact between BOB and Au NPs leads to a charge redistribution with the BOB side negatively charged and the Au NPs positively charged. This charge distribution caused the energy band near the BOB side to bend downward, forming an internal electric field (IEF) from Au NPs to BOB. An ohmic contact was formed at the heterogeneous interface to realize the directional migration of carriers. Under the irradiation of visible light, the photogenerated electrons and photogenerated holes were separated. Driven by the IEF, the photogenerated electrons transferred to the BOB CB were quickly guided and migrated to the Au NP surface in one direction. At the same time, the LSPR effect of Au NPs promotes this process. On the one hand, the local resonance energy transfer (LRET) effect was induced by the LSPR effect, which broadened the spectral response range of BOB and increased the photocharge yield efficiency. On the other hand, the LSPR effect induced the local electric field enhancement effect (LEFE), which led to a strong local electric field on the Au NPs to further accelerate the transfer of photogenerated charge. Under the double synergistic action of the interface ohmic contact and LSPR effect, more photogenerated electrons migrated to the Au NP surface along the bending direction of the BOB energy band and participated in the reduction reaction of CO2 to CO. Surely, the significantly increased CO2 adsorption capacity and the appearance of double CO2-TPD desorption peaks in the 0.6Au/BOB composite further proved that the synergistic effect promoted the adsorption and activation of CO2 (Fig. S18†), which was consistent with its performance results. In addition, we also characterized the process of photocatalytic reduction of CO2 to CO by in situ FTIR and Gibbs free energy, and the specific reaction process is shown in Fig. S19.† Noticeably, due to no sacrificial agent in the reaction system, the holes left by the BOB VB would react directly with H2O to form O2, as shown in Fig. 6.
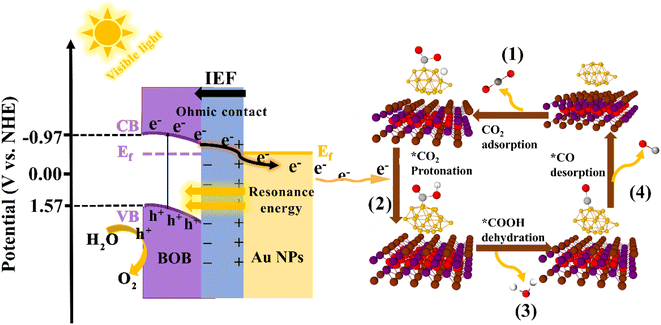 |
| | Fig. 6 The photocatalytic mechanism of Au/BOB and the detailed steps of reducing CO2 to CO. | |
4. Conclusion
In summary, Au NPs with a dual function emphasized in the Au/Bi24O31Br10 (Au/BOB) composite, and the ultrafast transfer of photogenerated charges from BOB to Au NPs was realized through the synergistic effect of the interfacial ohmic contact and LSPR effect. Here, the introduction of Au NPs could not only collect more electrons for CO2 reduction as an electron trap, but also induced the local electric field enhancement effect (LEFE) to enhance the driving force of the internal electric field (IEF) for photogenerated charge transfer. In addition, the ultrahigh CO yield and selectivity were significantly attributed to the reduced energy barrier and strong CO2 adsorption and activation. Under visible light irradiation, the optimal 0.6Au/BOB exhibited nearly 5 times higher activity than BOB (9.36 μmol g−1 h−1) without any sacrificial reagents. More importantly, 0.6Au/BOB demonstrated superior stability (only lost 6%) after continuous reaction for 48 h without obvious deactivation.
Data availability
The data is available in ESI.†
Author contributions
Jie Liu: conceptualization, methodology, software, investigation; Yu Xie: supervision, project administration, funding acquisition; Yiqiao Wang: writing – review & editing; Kai Yang, Shuping Su, Yun Ling and Pinghua Chen: writing – review & editing, investigation.
Conflicts of interest
The authors declare no competing interests.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 22272070 and 22066017), the First Training-class High-end Talents Projects of Science and Technology Innovation in Jiangxi Province (No. CK202002473), the Key Laboratory of Photochemical Conversion and Optoelectronic Materials, TIPC, CSA (No. PCOM201906), the Jiangxi Province “double thousand plan” project (jxsq2019201007, jxsq2020102027) and the Open Fund of Jiangxi Province Key Laboratory of Synthetic Chemistry (No. JXSC202003).
References
- K. Maeda, Adv. Mater., 2019, 31, e1808205 CrossRef PubMed.
- L. Liu, H. Huang, Z. Chen, H. Yu, K. Wang, J. Huang, H. Yu and Y. Zhang, Angew. Chem., Int. Ed., 2021, 60, 18303–18308 CrossRef CAS PubMed.
- D. Zhang, Y. Guo and Z. Zhao, Appl. Catal., B, 2018, 226, 1–9 CrossRef CAS.
- C. Y. Wang, X. Zhang, H. Bin Qiu, G. X. Huang and H. Q. Yu, Appl. Catal., B, 2017, 205, 615–623 CrossRef CAS.
- J. Wan, Y. Zhang, R. Wang, L. Liu, E. Liu, J. Fan and F. Fu, J. Hazard. Mater., 2018, 384, 121484 CrossRef PubMed.
- K. Li, B. Peng and T. Peng, ACS Catal., 2016, 6, 7485–7527 CrossRef CAS.
- D. Cui, L. Wang, Y. Du, W. C. Hao and J. Chen, ACS Sustainable Chem. Eng., 2018, 6, 15936–15953 CrossRef CAS.
- M. Ji, Y. Liu, J. Di, R. Chen, Z. Chen, J. Xia and H. Li, Appl. Catal., B, 2018, 237, 1033–1043 CrossRef CAS.
- X. Chen, M. Y. Qi, Y. H. Li, Z. R. Tang and Y. J. Xu, Chin. J. Catal., 2021, 42, 2020–2026 CrossRef CAS.
- X. Jin, C. Lv, X. Zhou, C. Zhang, B. Zhang, H. Su and G. Chen, J. Mater. Chem. A, 2018, 6, 24350–24357 RSC.
- X. Xiao, C. Zheng, M. Lu, L. Zhang, F. Liu, X. Zuo and J. Nan, Appl. Catal., B, 2018, 228, 142–151 CrossRef CAS.
- R. L. Tang, C. L. Hu, B. L. Wu, Z. Fang, Y. Chen and J. G. Mao, Angew. Chem., Int. Ed., 2019, 58, 15358–15361 CrossRef CAS PubMed.
- Y. Liu, C. Miao, P. Yang, Y. He, J. Feng and D. Li, Appl. Catal., B, 2019, 244, 919–930 CrossRef CAS.
- Y. Dai, C. Li, Y. Shen, S. Zhu, M. S. Hvid, L. C. Wu, J. Skibsted, Y. Li, J. W. H. Niemantsverdriet, F. Besenbacher, N. Lock and R. Su, J. Am. Chem. Soc., 2018, 140, 16711–16719 CrossRef CAS PubMed.
- C. Zhao, Z. Wang, X. Li, X. Yi, H. Chu, X. Chen and C. C. Wang, Chem. Eng. J., 2020, 389, 123431 CrossRef CAS.
- R. Verma, R. Belgamwar and V. Polshettiwar, ACS Mater. Lett., 2021, 3, 574–598 CrossRef CAS.
- X. Li, H. Jiang, C. Ma, Z. Zhu, X. Song, H. Wang, P. Huo and X. Li, Appl. Catal., B, 2021, 283, 119638 CrossRef CAS.
- P. Zhang, L. jun Wu, W. guo Pan, S. cheng Bai and R. tang Guo, Appl. Catal., B, 2021, 289, 120040 CrossRef CAS.
- L. Y. Lin, S. Kavadiya, X. He, W. N. Wang, B. B. Karakocak, Y. C. Lin, M. Y. Berezin and P. Biswas, Chem. Eng. J., 2020, 389, 123450 CrossRef CAS.
- W. Hou and S. B. Cronin, Adv. Funct. Mater., 2013, 23, 1612–1619 CrossRef CAS.
- L. Ma, Y. L. Chen, D. J. Yang, H. X. Li, S. J. Ding, L. Xiong, P. L. Qin and X. B. Chen, Nanoscale, 2020, 12, 4383–4392 RSC.
- X. Wang, R. Long, D. Liu, D. Yang, C. Wang and Y. Xiong, Nano Energy, 2016, 24, 87–93 CrossRef CAS.
- C. Cheng, J. Zhang, R. Zeng, F. Xing and C. Huang, Appl. Catal., B, 2022, 310, 121321 CrossRef CAS.
- H. Ren, T. Dittrich, H. Ma, J. N. Hart, S. Fengler, S. Chen, Y. Li, Y. Wang, F. Cao, M. Schieda, Y. H. Ng, Z. Xie, X. Bo, P. Koshy, L. R. Sheppard, C. Zhao and C. C. Sorrell, Adv. Mater., 2019, 31, 1–9 Search PubMed.
- C. Feng, L. Tang, Y. Deng, J. Wang, Y. Liu, X. Ouyang, Z. Chen, H. Yang, J. Yu and J. Wang, Appl. Catal., B, 2020, 276, 119167 CrossRef CAS.
- P. Mazierski, W. Lisowski, T. Grzyb, M. J. Winiarski, T. Klimczuk, A. Mikołajczyk, J. Flisikowski, A. Hirsch, A. Kołakowska, T. Puzyn, A. Zaleska-Medynska and J. Nadolna, Appl. Catal., B, 2017, 205, 376–385 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- A. S. Rosen, V. Fung, P. Huck, C. T. O'Donnell, M. K. Horton, D. G. Truhlar, K. A. Persson, J. M. Notestein and R. Q. Snurr, npj Comput. Mater., 2022, 8, 1–10 CrossRef.
- K. Hu, M. Wu, S. Hinokuma, T. Ohto, M. Wakisaka, J. I. Fujita and Y. Ito, J. Mater. Chem. A, 2019, 7, 2156–2164 RSC.
- J. Haslmayr and T. Renger, J. Chem. Phys., 2013, 139, 044103 CrossRef PubMed.
- Z. W. Wang, Q. Wan, Y. Z. Shi, H. Wang, Y. Y. Kang, S. Y. Zhu, S. Lin and L. Wu, Appl. Catal., B, 2021, 288, 120000 CrossRef CAS.
- S. G. Fard, M. Haghighi and M. Shabani, Appl. Catal., B, 2019, 248, 320–331 CrossRef CAS.
- Y. Zhou, C. Zhang, D. Huang, W. Wang, Y. Zhai, Q. Liang, Y. Yang, S. Tian, H. Luo and D. Qin, Appl. Catal., B, 2022, 301, 120749 CrossRef CAS.
- G. Yu, J. Qian, P. Zhang, B. Zhang, W. Zhang, W. Yan and G. Liu, Nat. Commun., 2019, 10, 1–8 CrossRef CAS PubMed.
- J. Guo, X. Liao, M. H. Lee, G. Hyett, C. C. Huang, D. W. Hewak, S. Mailis, W. Zhou and Z. Jiang, Appl. Catal., B, 2019, 243, 502–512 CrossRef CAS.
- Y. Tong, L. Chen, S. Ning, N. Tong, Z. Zhang, H. Lin, F. Li and X. Wang, Appl. Catal., B, 2017, 203, 725–730 CrossRef CAS.
- X. Cai, L. Mao, S. Yang, K. Han and J. Zhang, ACS Energy Lett., 2018, 3, 932–939 CrossRef CAS.
- R. Verma, R. Belgamwar, P. Chatterjee, R. Bericat-Vadell, J. Sa and V. Polshettiwar, ACS Nano, 2023, 17, 4526–4538 CrossRef CAS PubMed.
- Y. B. Kim, J. S. Kim, S. K. Baek, Y. D. Yun, S. W. Cho, S. H. Jung and H. K. Cho, Energy Environ. Sci., 2018, 11, 2540–2549 RSC.
- Y. Yoon, B. Yan and Y. Surendranath, J. Am. Chem. Soc., 2018, 140, 2397–2400 CrossRef CAS PubMed.
- H. Zhu, X. Yuan, Q. Yao and J. Xie, Nano Energy, 2021, 88, 106306 CrossRef CAS.
- T. Han, X. Cao, K. Sun, Q. Peng, C. Ye, A. Huang, W. C. Cheong, Z. Chen, R. Lin, D. Zhao, X. Tan, Z. Zhuang, C. Chen, D. Wang and Y. Li, Nat. Commun., 2021, 12, 1–11 CrossRef PubMed.
- M. Tang, Y. Ao, C. Wang and P. Wang, Appl. Catal., B, 2020, 270, 118918 CrossRef CAS.
- Z. Miao, Q. Wang, Y. Zhang, L. Meng and X. Wang, Appl. Catal., B, 2022, 301, 120802 CrossRef CAS.
- Z. Wu, J. Jing, K. Zhang, W. Li, J. Yang, J. Shen, S. Zhang, K. Xu, S. Zhang and Y. Zhu, Appl. Catal., B, 2022, 307, 121153 CrossRef CAS.
- J. Xu, Z. Ju, W. Zhang, Y. Pan, J. Zhu, J. Mao, X. Zheng, H. Fu, M. Yuan, H. Chen and R. Li, Angew. Chem., Int. Ed., 2021, 60, 8705–8709 CrossRef CAS PubMed.
- J. Li, L. Cai, J. Shang, Y. Yu and L. Zhang, Adv. Mater., 2016, 28, 4059–4064 CrossRef CAS PubMed.
- T. Kanata, M. Matsunaga, H. Takakura, Y. Hamakawa and T. Nishino, J. Appl. Phys., 1990, 68, 5309–5313 CrossRef CAS.
- Z. Yu, K. Yang, C. Yu, K. Lu, W. Huang, L. Xu, L. Zou, S. Wang, Z. Chen, J. Hu, Y. Hou and Y. Zhu, Adv. Funct. Mater., 2022, 1–10 Search PubMed.
- X. Wang, X. Wang, J. Huang, S. Li, A. Meng and Z. Li, Nat. Commun., 2021, 12, 1–11 CrossRef.
- S. Singh, R. Verma, N. Kaul, J. Sa, A. Punjal, S. Prabhu and V. Polshettiwar, Nat. Commun., 2023, 14, 2551 CrossRef CAS PubMed.
- Z. Miao, Q. Wang, Y. Zhang, L. Meng and X. Wang, Appl. Catal., B, 2022, 301, 120802 CrossRef CAS.
- M. Liu, Q. Kang, Z. Xie, L. Lu, K. Dai and G. Dawson, J. Phys. D Appl. Phys., 2022, 55, 043002 CrossRef CAS.
- M. Fang, X. Tan, Z. Liu, B. Hu and X. Wang, Research, 2021, 2021, 9794329 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Yiqiao
Wang
,
Kai
Yang
*,
Yiqiao
Wang
,
Kai
Yang