
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBioorthogonal dissociative rhenium(I) photosensitisers for controlled immunogenic cell death induction†
Guang-Xi
Xu
 a,
Lawrence Cho-Cheung
Lee
ab,
Peter Kam-Keung
Leung
ac,
Eunice Chiu-Lam
Mak
a,
Justin
Shum
a,
Kenneth Yin
Zhang
d,
Qiang
Zhao
d and
Kenneth Kam-Wing
Lo
*ac
a,
Lawrence Cho-Cheung
Lee
ab,
Peter Kam-Keung
Leung
ac,
Eunice Chiu-Lam
Mak
a,
Justin
Shum
a,
Kenneth Yin
Zhang
d,
Qiang
Zhao
d and
Kenneth Kam-Wing
Lo
*ac
aDepartment of Chemistry, City University of Hong Kong, Tat Chee Avenue, Kowloon, Hong Kong, P. R. China. E-mail: bhkenlo@cityu.edu.hk
bLaboratory for Synthetic Chemistry and Chemical Biology Limited, Units 1503-1511, 15/F, Building 17 W, Hong Kong Science Park, New Territories, Hong Kong, P. R. China
cState Key Laboratory of Terahertz and Millimetre Waves, City University of Hong Kong, Tat Chee Avenue, Kowloon, Hong Kong, P. R. China
dState Key Laboratory of Organic Electronics and Information Displays, Jiangsu Key Laboratory for Biosensors, Institute of Advanced Materials (IAM), Nanjing University of Posts and Telecommunications, 9 Wenyuan Road, Nanjing 210023, P. R. China
First published on 24th October 2023
Abstract
Photosensitisers for photoimmunotherapy with high spatiotemporal controllability are rare. In this work, we designed rhenium(I) polypyridine complexes modified with a tetrazine unit via a bioorthogonally activatable carbamate linker as bioorthogonally dissociative photosensitisers for the controlled induction of immunogenic cell death (ICD). The complexes displayed increased emission intensities and singlet oxygen (1O2) generation efficiencies upon reaction with trans-cyclooct-4-enol (TCO-OH) due to the separation of the quenching tetrazine unit from the rhenium(I) polypyridine core. One of the complexes containing a poly(ethylene glycol) (PEG) group exhibited negligible dark cytotoxicity but showed greatly enhanced (photo)cytotoxic activity towards TCO-OH-pretreated cells upon light irradiation. The reason is that TCO-OH allowed the synergistic release of the more cytotoxic rhenium(I) aminomethylpyridine complex and increased 1O2 generation. Importantly, the treatment induced a cascade of events, including lysosomal dysfunction, autophagy suppression and ICD. To the best of our knowledge, this is the very first example of using bioorthogonal dissociation reactions as a trigger to realise photoinduced ICD, opening up new avenues for the development of innovative photoimmunotherapeutic agents.
Introduction
Immunotherapy has emerged as a promising modality for the treatment of cancer patients. The effectiveness of immunotherapy can not only eliminate the local and metastatic tumours but also prevent the recurrence of tumours by triggering the host's immune system.1,2 Various medical approaches such as cancer vaccines,3 T-cell immunomodulators,4 checkpoint inhibitors5 and cytokine therapy6 have been utilised to stimulate the systemic immune response for immunotherapy. Although encouraging, these methods often face challenges in generating sufficient immunogenic response due to the immunosuppressive microenvironments and low immunogenicity of solid tumours, limiting their clinical applications.7,8 Recent investigations on luminescent transition metal complexes as photosensitisers for photodynamic therapy (PDT) have demonstrated their unique ability to directly damage tumours while simultaneously inducing immunogenic cell death (ICD). This process triggers the release of tumour-associated antigens leading to immune stimulation. As a result, these complexes effectively inhibit tumour growth and recurrence.9–12 These metal complexes show exceptional anticancer activity, but fine-tuning is required to resolve their intrinsic dark cytotoxicity, poor tumour accumulation behaviour and un-switchable “always-on” photosensitisation that may lead to undesired photocytotoxicity towards normal tissues.9,13 Reducing this particular side effect can aid cancer patients during recovery periods and balance the safety and efficacy of PDT agents. The development of new approaches is required to enable spatiotemporally controlled activation of the transition metal complexes with increased biocompatibility, tumour accumulation, and effective anticancer properties. We anticipate that the controlled activation can reduce photosensitivity in cancer patients, inhibit tumour growth, and provide cancer patients with long-term protective antitumour immunity.Considerable efforts have been devoted to the design of on-demand activatable reagents, aiming to restore the bioactivity of molecules within living systems through precise chemical or enzymatic reactions.14–17 One representative example is bioorthogonal ligation, which can specifically activate the pharmacological behaviour of compounds.18–21 The scope of bioorthogonal dissociation reactions has been further expanded by their ability to release therapeutic payloads in a controlled manner at targeted sites of interest.22–26 These reactions involve the specific bond cleavage through an exogenous click–reaction pair, converting the caged payload into its active form. This methodology has been applied successfully in PDT and chemotherapy,27–30 and holds great promise as a controlled stimulation approach for immunotherapy.
Our group has utilised different bioorthogonal ligation reactions to manipulate the efficiency of singlet oxygen (1O2) photosensitisation and photocytotoxic activity of transition metal complexes.31–33 By treating the tetrazine complexes with trans-cyclooct-4-enol (TCO-OH), we observed moderate changes in the 1O2 quantum yields, which was due to the ability of the dihydropyridazine products to quench the emission through photoinduced electron transfer (PET).32 Inspired by the recently reported TCO-OH-induced tetrazine elimination reaction,34 we hypothesise that modification of metal complexes with a cleavable tetrazine moiety will substantially enhance the emission intensity, 1O2 quantum yields and (photo)cytotoxicity by releasing tetrazine-free complexes. Herein, we report three rhenium(I) polypyridine complexes containing a tetrazine unit via a bioorthogonally activatable carbamate linker [Re(N^N)(CO)3(py-Tz-NHBoc)](CF3SO3) (py-Tz-NHBoc = 3-(1-(6-(4-(N-Boc-aminomethyl)phenyl)-1,2,4,5-tetrazin-3-yl)ethyloxycarbonylaminomethyl)pyridine; N^N = 1,10-phenanthroline (phen) (1), 3,4,7,8-tetramethyl-1,10-phenanthroline (Me4-phen) (2) and 4,7-diphenyl-1,10-phenanthroline (Ph2-phen) (3)) (Chart 1). Their aminomethylpyridine counterparts [Re(N^N)(CO)3(py-CH2NH2)](CF3SO3) (py-CH2NH2 = 3-(aminomethyl)pyridine; N^N = phen (1a), Me4-phen (2a) and Ph2-phen (3a)) were prepared for comparison studies (Chart 1). Additionally, to increase the solubility and biocompatibility, complex 3 with high lipophilicity and low energy emission was modified with a poly(ethylene glycol) (PEG) pendant to afford [Re(Ph2-phen)(CO)3(py-Tz-PEG5000)](CF3SO3) (4) (py-Tz-PEG5000 = 3-(1-(6-(4-(N-(3-(ω-methoxypoly(1-oxapropyl))propanoyl)aminomethyl)phenyl)-1,2,4,5-tetrazin-3-yl)ethyloxycarbonylaminomethyl)pyridine). In complex 4, the carbamate-tetrazine-PEG moiety was designed to play three roles: emission quencher, cytotoxicity regulator and bioorthogonal handle. The tetrazine suppresses the emission intensity and 1O2 generation efficiency of the complex, and the PEG unit enhances the biocompatibility. Consequently, this design strategy could minimise the undesired (photo)cytotoxicity associated with the complex. After the cycloaddition reaction of complex 4 with TCO-OH, the carbamate linker undergoes a 1,4-elimination, resulting in the detachment of the pyridazine-PEG unit and the release of the aminomethylpyridine complex 3a (Scheme 1). Without the PEG moiety, the cytotoxic activity of the rhenium(I) polypyridine complex will be restored, inhibiting the proliferation of cancer cells. Also, the removal of the tetrazine quencher enables the released complex to display increased emission intensity and 1O2 photosensitisation. Thus, the death of cancer cells could be further promoted upon photoirradiation due to the synergistic effect of the cytotoxic complex 3a and 1O2. The combination of bioorthogonal dissociation and light irradiation will also elicit potential ICD effects and stimulate immune response. Thus, with the aid of bioorthogonal dissociation reaction and light irradiation, complex 4 is expected to achieve remarkably enhanced anticancer performance in a highly controlled manner. We believe that this innovative approach will broaden the applications of bioorthogonal dissociation reactions and serve as a new strategy for the design of photoimmunotherapeutic agents.
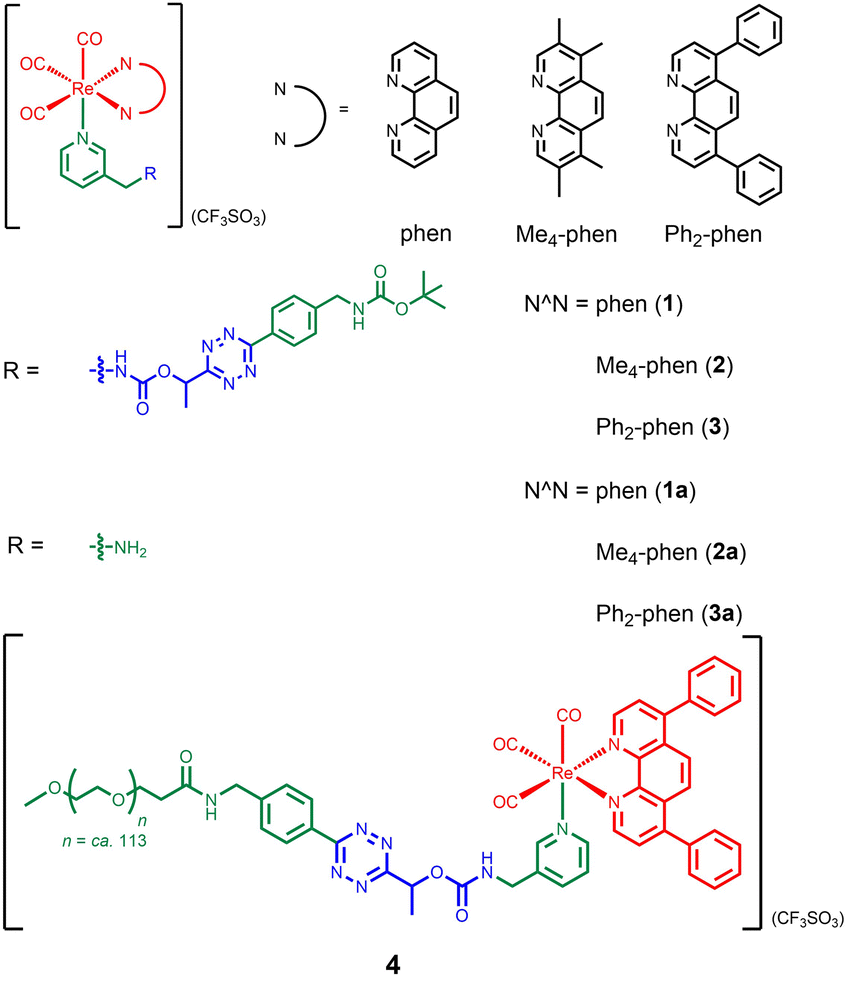 | ||
| Chart 1 Structures of the rhenium(I) complexes. | ||
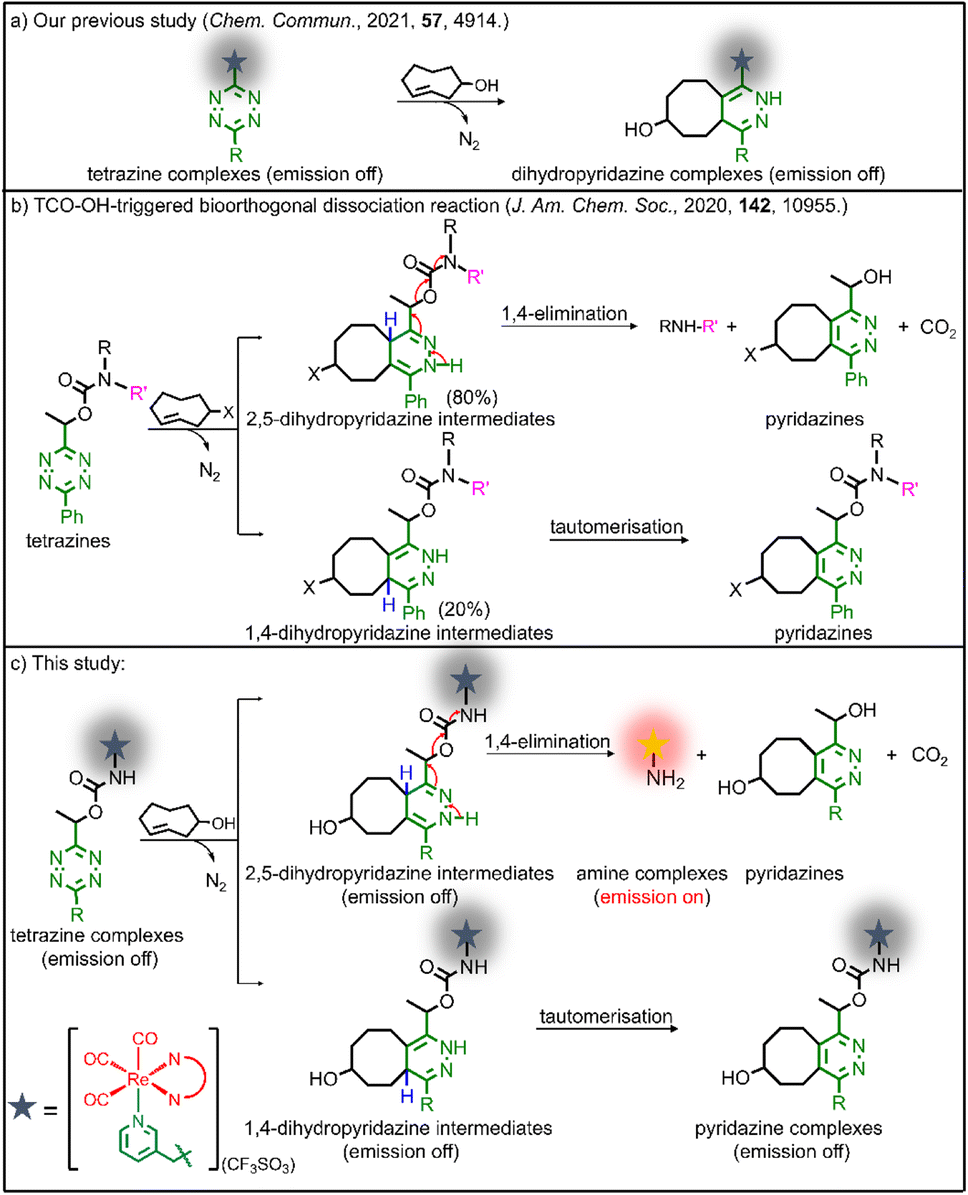 | ||
| Scheme 1 (a) Bioorthogonal cycloaddition reaction of tetrazine-modified complexes with TCO-OH. (b) Bioorthogonal dissociation reaction of tetrazine-modified compounds with TCO-OH. (c) Bioorthogonal dissociation reaction of tetrazine-modified rhenium(I) complexes with TCO-OH. | ||
Results and discussions
Synthesis and characterisation of the complexes
The ligand py-Tz-NHBoc was synthesised from the reaction of 3-(aminomethyl)pyridine with tert-butyl 4-(6-(1-(((4-nitrophenoxy)carbonyl)oxy)ethyl)-1,2,4,5-tetrazin-3-yl)benzylcarbamate in anhydrous CH2Cl2. Deprotection of the Boc and subsequent reaction with methoxy poly(ethylene glycol) succinimidyl carboxymethyl ester (mPEG5000-NHS) afforded the ligand py-Tz-PEG5000. Complexes 1–3, 1a–3a and the PEGylated complex 4 were obtained from the reaction of the precursor complexes [Re(N^N)(CO)3(CH3CN)](CF3SO3) with their respective pyridine ligands in refluxing THF (Schemes S1 and S2†). All the complexes were characterised by high-resolution ESI-MS or MALDI-TOF MS, 1H and 13C NMR and IR spectroscopy.Photophysical properties
The electronic absorption data and spectra of complexes 1–4 are shown in Table S1 and Fig. S1,† respectively. All the complexes displayed intense absorption bands at ca. 252–342 nm and weaker absorption bands/shoulders at ca. 369–397 nm, which should be attributed to the spin-allowed intraligand (1IL) (π → π*)(N^N and pyridine ligands) transitions and spin-allowed metal-to-ligand charge transfer (1MLCT) (dπ(Re) → π*(N^N)) transitions, respectively.35,36 Additionally, the weak absorption bands at ca. 530–546 nm were assigned to the n → π* transition of the tetrazine moiety.32 Upon photoexcitation, the rhenium(I) complexes displayed green to orange emission under ambient conditions (Table S2 and Fig. S2†). Complexes 1, 3 and 4 showed a broad and structureless emission band with positive solvatochromism in fluid solutions at 298 K. Their emission maxima displayed significant hypsochromic shifts upon cooling the samples to 77 K, indicative of a 3MLCT (dπ(Re) → π*(N^N)) emissive state.35,36 Complex 2 in solution exhibited less-solvent dependence, suggestive of a predominant 3IL (π → π*)(Me4-phen) emissive state.35,36 Notably, complexes 1–4 exhibited very low emission quantum yields (Φem = 0.03–0.04) compared to their aminomethylpyridine counterparts 1a–3a (Φem = 0.20–0.47)35 due to the quenching by tetrazine.31,32Reactivity and phosphorogenic properties towards TCO-OH
To study the reactivity of the tetrazine complexes towards TCO-OH, the second-order rate constants (k2) of the ligation reaction of complexes 1–4 with TCO-OH in H2O/DMSO (99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) were first determined. Complexes 1–4 showed good reaction rates with TCO-OH with k2 values of ca. 3.8–5.8 M−1 s−1 (Fig. S3†). Plots of non-zero intercepts were observed in Fig. S3,† which were ascribed to the fact that after the ligation reaction occurred, the bioorthogonal dissociation reaction also commenced, leading to the conversion of the dihydropyridazine intermediates to their aminomethylpyridine counterparts. Nevertheless, we believe that the measured second-order rate constants can serve as a benchmark to estimate how fast the ligation reaction occurs. The second-order rates of the ligation reactions (3.8–5.8 M−1 s−1) were comparable to other bioorthogonal dissociation reactions involving tetrazine and TCO as the reaction pair (3.14–15.2 M−1 s−1).34,37,38 To complement the ligation studies, the half-lives of the dihydropyridazine intermediates formed upon incubation of the tetrazine complexes with TCO-OH were studied by high-performance liquid chromatography (HPLC) to evaluate the dissociation reaction. The tetrazine complexes underwent a rapid reaction with TCO-OH and were converted to their aminomethylpyridine counterparts 1a–3a (Fig. 1a and S4†) after incubation for 12 h with the release rates (half-lives) ranging from 1.53 to 2.67 h (Fig. S5†) and elimination yields of 78–85% (Fig. 1b), respectively, comparable to a previous study.34 The formation of the aminomethylpyridine complexes was confirmed by ESI-MS (Fig. 1c and S6†). Full conversion may be impeded by the incomplete elimination of complexes 1–4 due to the formation of the non-releasing 1,4-dihydropyridazine isomer during the [4 + 2] cycloaddition process (Scheme 1b and c),34 as revealed by ESI-MS (Fig. S7†). Nevertheless, the results suggest that efficient and fast release of the payload can be achieved even for complex 4, which has a sterically demanding PEG unit. Typically, PEGylated compounds may self-assemble into micelles in aqueous solutions. Complex 4 showed promising release potential with a PEG unit, and the morphology of the complex prior to and after the TCO-OH reaction is expected to be different. Thus, the morphology and size of complex 4 with or without TCO-OH treatment were examined by transmission electron microscopy (TEM) and dynamic light scattering (DLS), respectively. TEM images demonstrated that complex 4 would form uniform spherical-shaped micelles with a size of ca. 45.0 ± 4.0 nm (Fig. 1d). Upon treatment of complex 4 with TCO-OH for 12 h, the spherical nanoparticles disappeared, and only small aggregates with diameters of 2.3 ± 1.2 nm were noticed (Fig. 1e). These observations are in line with the results of the DLS experiments, where a notable decrease in the hydrodynamic diameter from 60.1 to 4.9 nm was observed after complex 4 was treated with TCO-OH for 12 h (Fig. 1f).
:1, v/v) were first determined. Complexes 1–4 showed good reaction rates with TCO-OH with k2 values of ca. 3.8–5.8 M−1 s−1 (Fig. S3†). Plots of non-zero intercepts were observed in Fig. S3,† which were ascribed to the fact that after the ligation reaction occurred, the bioorthogonal dissociation reaction also commenced, leading to the conversion of the dihydropyridazine intermediates to their aminomethylpyridine counterparts. Nevertheless, we believe that the measured second-order rate constants can serve as a benchmark to estimate how fast the ligation reaction occurs. The second-order rates of the ligation reactions (3.8–5.8 M−1 s−1) were comparable to other bioorthogonal dissociation reactions involving tetrazine and TCO as the reaction pair (3.14–15.2 M−1 s−1).34,37,38 To complement the ligation studies, the half-lives of the dihydropyridazine intermediates formed upon incubation of the tetrazine complexes with TCO-OH were studied by high-performance liquid chromatography (HPLC) to evaluate the dissociation reaction. The tetrazine complexes underwent a rapid reaction with TCO-OH and were converted to their aminomethylpyridine counterparts 1a–3a (Fig. 1a and S4†) after incubation for 12 h with the release rates (half-lives) ranging from 1.53 to 2.67 h (Fig. S5†) and elimination yields of 78–85% (Fig. 1b), respectively, comparable to a previous study.34 The formation of the aminomethylpyridine complexes was confirmed by ESI-MS (Fig. 1c and S6†). Full conversion may be impeded by the incomplete elimination of complexes 1–4 due to the formation of the non-releasing 1,4-dihydropyridazine isomer during the [4 + 2] cycloaddition process (Scheme 1b and c),34 as revealed by ESI-MS (Fig. S7†). Nevertheless, the results suggest that efficient and fast release of the payload can be achieved even for complex 4, which has a sterically demanding PEG unit. Typically, PEGylated compounds may self-assemble into micelles in aqueous solutions. Complex 4 showed promising release potential with a PEG unit, and the morphology of the complex prior to and after the TCO-OH reaction is expected to be different. Thus, the morphology and size of complex 4 with or without TCO-OH treatment were examined by transmission electron microscopy (TEM) and dynamic light scattering (DLS), respectively. TEM images demonstrated that complex 4 would form uniform spherical-shaped micelles with a size of ca. 45.0 ± 4.0 nm (Fig. 1d). Upon treatment of complex 4 with TCO-OH for 12 h, the spherical nanoparticles disappeared, and only small aggregates with diameters of 2.3 ± 1.2 nm were noticed (Fig. 1e). These observations are in line with the results of the DLS experiments, where a notable decrease in the hydrodynamic diameter from 60.1 to 4.9 nm was observed after complex 4 was treated with TCO-OH for 12 h (Fig. 1f).
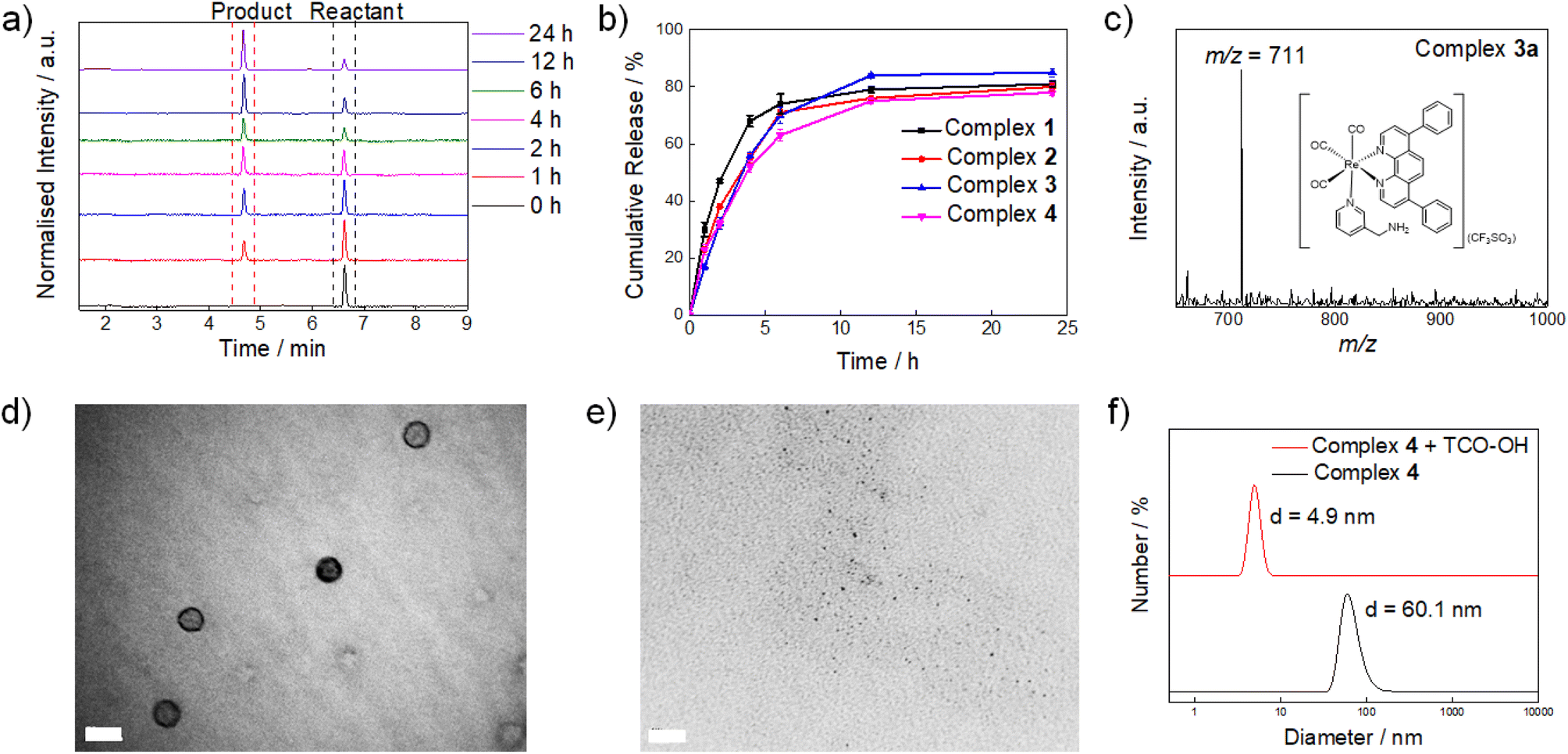 | ||
| Fig. 1 (a) HPLC traces of the reaction of complex 4 (20 μM) with TCO-OH (200 μM) in H2O/DMSO (99:1, v/v) at different time points at 298 K. (b) Release profiles of complexes 1–4 (20 μM) in the presence of TCO-OH (200 μM) in H2O/DMSO (99:1, v/v) at 298 K. (c) ESI-mass spectrum of the eluate collected at tR = 4.67 min of the reaction of complex 4 (20 μM) with TCO-OH (200 μM) in H2O/DMSO (99:1, v/v) at 298 K. (d) and (e) TEM images of complex 4 (20 μM) upon incubation without and with TCO-OH (200 μM) in H2O/DMSO (99:1, v/v) at 298 K for 12 h, respectively. Scale bar = 50 nm (d) and 20 nm (e). (f) DLS analysis of complex 4 (20 μM) upon incubation with TCO-OH (0 μM (black) and 200 μM (red)) in H2O/DMSO (99:1, v/v) at 298 K for 12 h. | ||
The possible phosphorogenic response of complexes 1–4 towards TCO-OH was examined. Treatment of the complexes with TCO-OH (100 μM) led to significant emission enhancement and lifetime extension (I/Io = 11.4–56.2, τ = 0.47–1.02 μs; Table 1 and Fig. S8†). The 1O2 generation quantum yields (ΦΔ) of complexes 1–4 and their aminomethylpyridine counterparts 1a–3a were determined in CH3CN, which is commonly used for 1O2 generation investigation.39 As shown in Table S3 and Fig. S9,† the ΦΔ values of complexes 1–3 (0.22–0.48) were comparable to those of complexes 1a–3a (0.26–0.63), most likely due to the formation of a triplet charge-separate state of the tetrazine complexes after photoexcitation.32 Notably, the 1O2 generation efficiency of the PEG complex 4 (0.27) was 2.3-fold lower than that of its aminomethylpyridine counterpart 3a (0.63). This may be attributed to the polymer shielding effect of the PEG unit, which prevents (1) oxygen molecules from approaching the excited rhenium(I) complex and (2) the escape of generated 1O2 from the PEG cage to interact with DPBF.40 The results exemplify that the release of the payload, phosphorogenic response and 1O2 generation can be finely controlled by utilising TCO-OH as an activation trigger in the bioorthogonal dissociation strategy.
:1, v/v) at 298 K
| Complex | I/Ioa | τ (μs) |
|---|---|---|
| a I o and I are the emission intensities of the complexes in the absence and presence of TCO-OH, respectively. b The emission lifetimes of the complexes after incubation with TCO-OH for 12 h. In the absence of TCO-OH, the emission lifetimes of the complexes could not be determined accurately. | ||
| 1 | 40.9 | 0.47 |
| 2 | 28.9 | 1.02 |
| 3 | 56.2 | 0.95 |
| 4 | 11.4 | 0.90 |
Intracellular imaging
The phosphorogenic response of complexes 1–4 towards TCO-OH in live cells was explored using MDA-MB-231 cells as a late-stage breast cancer model. The cells were preincubated without or with TCO-OH (100 μM) for 2 h, followed by incubation with complexes 1–4 (10 μM) for 6 h. An additional incubation time of 12 h in fresh medium was required to allow the dissociation reaction to proceed to a significant extent. Laser-scanning confocal microscopy (LSCM) images showed that strong intracellular emission intensity was noticed only in cells that were preincubated with TCO-OH (Fig. 2a), indicating the release of the strongly emissive rhenium(I) aminomethylpyridine complexes 1a–3a. We further confirmed this cleavage by ESI-MS analysis of the cell lysate (Fig. S10†). Co-localisation experiments with complex 4 using LysoTracker Deep Red revealed that the released complex was mainly distributed in the lysosomes, with a Pearson's correlation coefficient (PCC) of 0.85 (Fig. 2b), which is different from that of the free complex 3a (Fig. S11†). This result is attributed to the different uptake mechanisms exhibited by complexes 3a and 4. As shown in Fig. S12,† the intracellular emission intensity of complex 3a-treated MDA-MB-231 cells was not affected by temperature, indicating that the complex was internalised into the cells through passive diffusion. In contrast, complex 4 containing the PEG unit was much larger in size and should enter the cells through endocytosis.41 The subsequent bioorthogonal dissociation reaction with TCO-OH released the aminomethylpyridine complex 3a that can be protonated under acidic conditions, leading to its localisation in the lysosomes.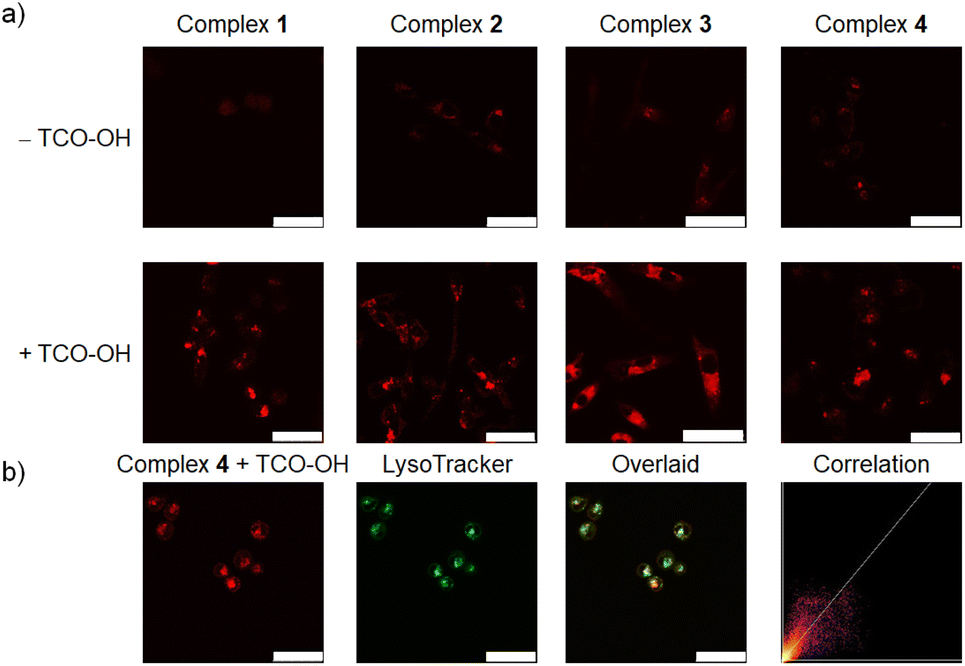 | ||
| Fig. 2 (a) LSCM images of TCO-OH-untreated (upper) or pretreated (lower, 100 μM, 2 h) MDA-MB-231 cells incubated with complexes 1–4 (10 μM, 6 h; λex = 405 nm, λem = 500–600 nm) at 37 °C, followed by incubation in fresh medium for 12 h. (b) LSCM images of TCO-OH-pretreated (100 μM, 2 h) MDA-MB-231 cells incubated with complex 4 (10 μM, 6 h; λex = 405 nm, λem = 500–600 nm), followed by incubation in fresh medium for 12 h, and then stained with LysoTracker Deep Red (100 nM, 30 min; λex = 635 nm, λem = 640–660 nm). PCC = 0.85. Scale bar = 50 μm. | ||
Cellular uptake and (photo)cytotoxicity studies
The uptake of the complexes by MDA-MB-231 cells was studied by measuring the intracellular contents of rhenium using ICP-MS. The uptake efficiencies of the complexes followed the order: 4 < 1 < 2 < 3 (Table S4†), which is in line with the increasing hydrophobic nature of the diimine ligands: phen < Me4-phen < Ph2-phen.36 Notably, the PEGylated complex 4 displayed a lower cellular uptake (Table S4†), probably due to the high hydrophilicity and large size of the PEG pendant.The (photo)cytotoxicity of the rhenium(I) complexes towards different cancer cell lines (MDA-MB-231, HepG2 and NCI-H460) was examined using the MTT assay. Without pretreatment with TCO-OH, the dark cytotoxicity of complex 4 (IC50,dark = 51.1–73.7 μM; Table 2) was much lower than that of complexes 1–3 (IC50,dark = 2.3–33.0 μM; Table 2) due to the presence of the PEG unit which may (1) prevent the complex from interacting with intracellular biomolecules42 and (2) reduce the cellular uptake. The dark cytotoxicity of the PEG-free complexes 1–3 was essentially unchanged when the cells were pretreated with TCO-OH (IC50,dark = 3.4–35.6 μM), whereas the cytotoxic activity of complex 4 significantly increased with the IC50,dark values decreasing to 15.5–20.9 μM, owing to the liberation of the cytotoxic aminomethylpyridine complex 3a. Upon irradiation at 450 nm, complexes 1–3 alone showed moderate photocytotoxicity (IC50,light = 0.7–17.7 μM), which did not significantly increase when the cells were pretreated with TCO-OH (IC50,light = 0.5–21.9 μM), since the 1O2 generation efficiencies of complexes 1–3 are comparable to those of their aminomethylpyridine counterparts 1a–3a (Table S3†). Notably, the photocytotoxicity of complex 4 before TCO-OH pretreatment (IC50,light = 3.8–13.6 μM) was markedly increased in the presence of TCO-OH (IC50,light = 0.6–4.6 μM). The enhanced photocytotoxicity of the PEGylated complex 4 is due to the synergistic release of the highly cytotoxic complex 3a and its more efficient 1O2 generation capability (ΦΔ = 0.27 for complex 4 and 0.63 for complex 3a; Table S3†).
| Complex | Treatment | MDA-MB-231 | HepG2 | NCI-H460 | |||
|---|---|---|---|---|---|---|---|
| IC50,dark (μM) | IC50,light (μM) | IC50,dark (μM) | IC50,light (μM) | IC50,dark (μM) | IC50,light (μM) | ||
| 1 | − TCO-OH | 14.4 ± 2.8 | 6.9 ± 0.4 | 16.9 ± 2.4 | 11.8 ± 0.9 | 33.0 ± 7.0 | 17.7 ± 2.1 |
| + TCO-OH | 20.4 ± 1.2 | 9.2 ± 0.5 | 17.1 ± 1.0 | 13.1 ± 2.6 | 35.4 ± 4.2 | 21.9 ± 5.7 | |
| 2 | − TCO-OH | 28.2 ± 3.8 | 12.4 ± 0.2 | 11.9 ± 1.4 | 11.2 ± 1.0 | 7.1 ± 0.4 | 1.7 ± 0.1 |
| + TCO-OH | 35.6 ± 6.7 | 5.6 ± 0.6 | 11.5 ± 0.7 | 8.7 ± 0.8 | 6.6 ± 0.2 | 1.1 ± 0.8 | |
| 3 | − TCO-OH | 8.0 ± 1.1 | 0.7 ± 0.1 | 2.3 ± 0.5 | 0.9 ± 0.1 | 4.1 ± 0.2 | 0.9 ± 0.1 |
| + TCO-OH | 5.4 ± 0.4 | 0.5 ± 0.1 | 3.4 ± 0.6 | 0.9 ± 0.1 | 4.2 ± 0.1 | 0.7 ± 0.1 | |
| 4 | − TCO-OH | 68.2 ± 1.2 | 3.8 ± 0.3 | 73.7 ± 6.5 | 4.8 ± 0.8 | 51.1 ± 1.2 | 13.6 ± 0.6 |
| + TCO-OH | 18.3 ± 1.4 | 0.6 ± 0.3 | 15.5 ± 2.0 | 3.2 ± 0.6 | 20.9 ± 0.9 | 4.6 ± 0.1 | |
Based on the promising PDT activity of complex 4, we investigated the cell viability and cell death mode with Calcein-AM/PI and Annexin V/PI assays, respectively. Cell viability studies of MDA-MB-231 cells with complex 4 (10 μM) only revealed strong green fluorescence of Calcein-AM with no red emission of the PI dye (Fig. 3a). This result indicates that the complex is innocuous to the cancer cells. When the cells were treated with TCO-OH and complex 4, or complex 4 and light, the Calcein-AM fluorescence decreased, and the emission of PI increased in MDA-MB-231 cells. The combined treatment of TCO-OH, complex 4 and light resulted in the highest loss of cell viability with minimal green fluorescence and strong red fluorescence. A similar observation was noted in 3D multicellular tumour spheroids (Fig. 3b), demonstrating that complex 4 efficiently damaged both monolayer cells and 3D multicellular tumour spheroids upon the combined treatments. Cell death mode studies with Annexin V/PI disclosed that treatment of cells with complex 4 and TCO-OH slightly increased the population of necrotic cells to 17.0% (Fig. 3c). However, treatment of MDA-MB-231 cells with TCO-OH and complex 4 upon photoirradiation increased the necrotic cell population to 41.8% (Fig. 3c), illustrating that necrosis is the primary cell death mode in MDA-MB-231 cells. Taken together, these data demonstrated that the (photo)cytotoxic activity of complex 4 can be finely modulated through the combination of bioorthogonal dissociation reaction and photoirradiation, highlighting the great promise of using complex 4 as a bioorthogonally controllable therapeutic for precise anticancer therapy.
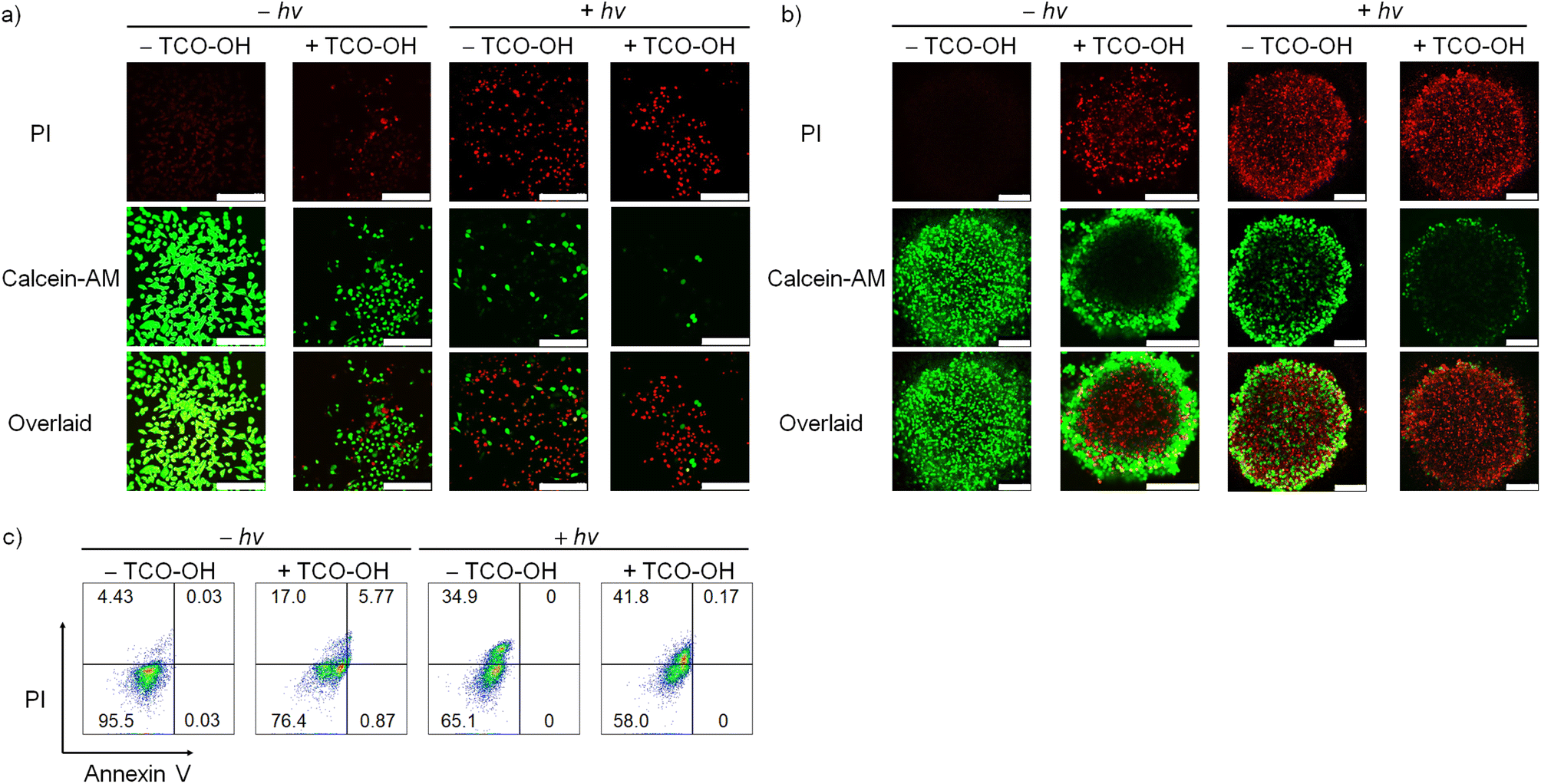 | ||
| Fig. 3 Live/dead cell staining of (a) monolayer MDA-MB-231 cells and (b) 3D multicellular tumour spheroids of MDA-MB-231 cells under different treatments. The cells or tumour spheroids were first pretreated without or with TCO-OH (100 μM) for 2 h, then incubated with complex 4 (10 μM) for 6 h, left in fresh medium for 12 h, remained in the dark or were irradiated at 450 nm (20 mW cm−2) for 20 min and subsequently incubated in the dark for 24 h. All the cells and tumour spheroids were stained with Calcein-AM (1 μM; λex = 488 nm, λem = 500–520 nm) and PI (10 μM; λex = 532 nm, λem = 600–650 nm) for 1 h prior to imaging experiments. Scale bar = 50 μm (a) and 200 μm (b). (c) Flow cytometric analysis of MDA-MB-231 cells under the same treatments as described above. After the treatments, the cells were then stained with PI (2 μL, 100 μg mL−1; λex = 561 nm) and Alexa Fluor 647-Annexin V conjugate (5 μL, standard conc; λex = 638 nm) for 15 min and analysed by flow cytometry. | ||
Lysosome disruption and autophagy inhibition
Given the potent anticancer performance, the cell death mechanism associated with complex 4 was investigated. The intracellular ROS levels in MDA-MB-231 cells under different treatments were examined using CM-H2DCFDA as the ROS indicator (Fig. 4a). No emission was observed for cells incubated with complex 4 only in the dark. Only very weak emission signals were detected in cells treated with TCO-OH and complex 4 in the dark, and complex 4 only with light. Importantly, substantial intracellular emission was observed in cells treated with TCO-OH, complex 4 and light irradiation, indicating the increased capacity for intracellular 1O2 generation.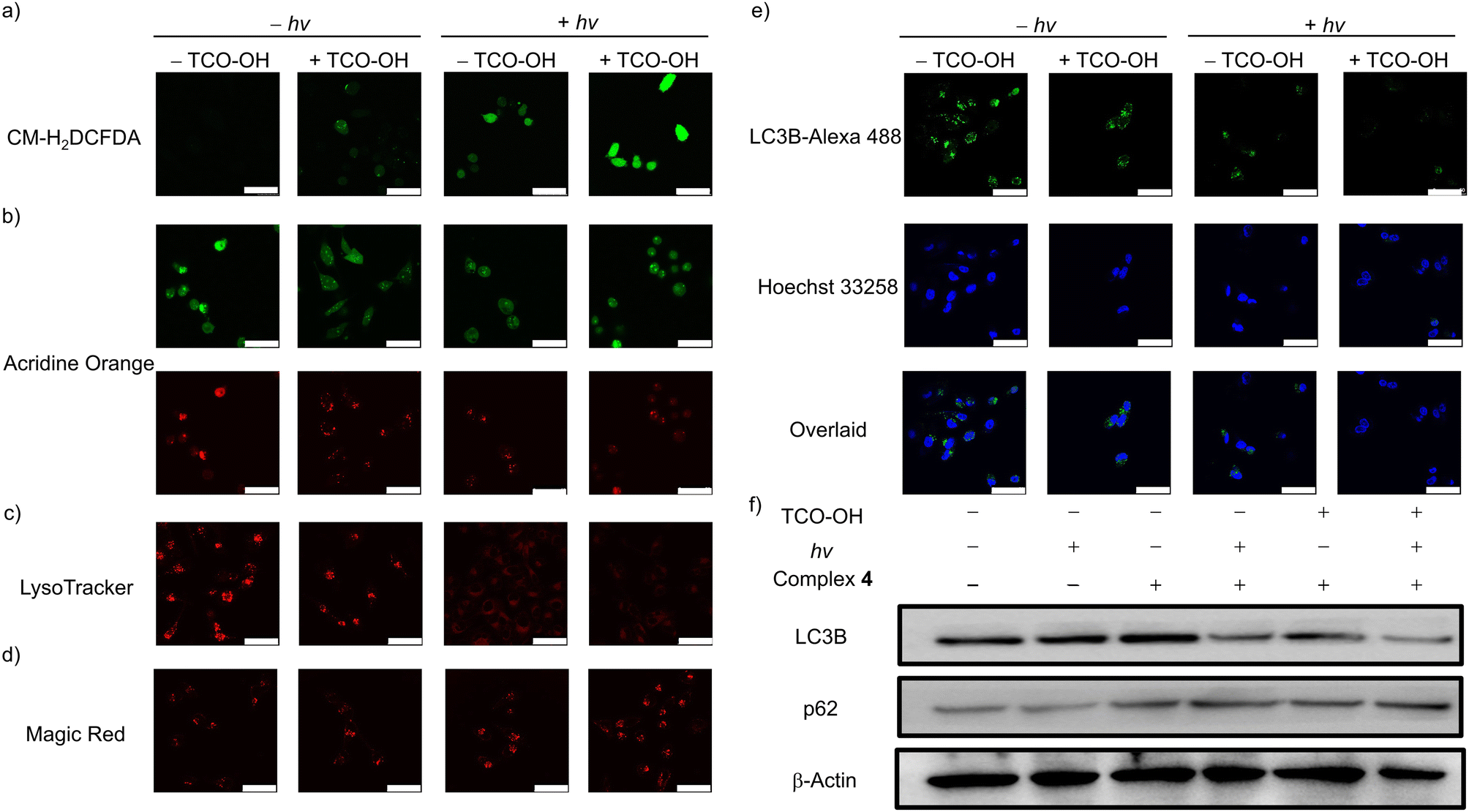 | ||
| Fig. 4 LSCM images of MDA-MB-231 cells under different conditions: (a) intracellular ROS generation, (b)–(d) lysosome disruption and (e) LC3B protein expression. The cells were first pretreated without or with TCO-OH (100 μM) for 2 h, then incubated with complex 4 (10 μM) for 6 h, left in fresh medium for 12 h, remained in the dark or irradiated at 450 nm (20 mW cm−2) for 20 min and subsequently incubated in the dark for 2 h. The samples were then stained with CM-H2DCFDA (10 μM, 20 min; λex = 488 nm, λem = 500–520 nm) or AO (10 μM, 30 min; λex = 488 nm, λem = 500–520 nm (green), λem = 610–640 nm (red)) or LysoTracker Deep Red (100 nM, 15 min; λex = 635 nm, λem = 640–660 nm) or Magic Red (10 μM, 1:100 solution, 1 h; λex = 532 nm, λem = 630–650 nm). The immunofluorescence staining of LC3B (LC3B-Alexa 488 (10 μM, 1:200 solution, 2 h; λex = 488 nm, λem = 500–520 nm); Hoechst 33258 (10 μM, 1 μg mL−1, 10 min; λex = 405 nm, λem = 420–450 nm)) was performed prior to imaging experiments. Scale bar = 50 μm. (f) Western blot analysis of autophagy protein markers LC3B and p62 in MDA-MB-231 cells. | ||
Hence, complex 4 may induce lysosomal damage through the released cytotoxic aminomethylpyridine complex 3a and the in situ-generated 1O2. The lysosome integrity, acidic microenvironment and the distribution of cathepsin B were studied in MDA-MB-231 cells using acridine orange (AO), LysoTracker Deep Red and Magic Red MR-(RR)2, respectively, to assess the integrity of lysosomes. The AO exhibits dual emission in the nuclei and cytoplasm (green) and lysosomes (red), and a decrease in red fluorescence intensity indicates that the lysosomal membrane is disrupted. As depicted in Fig. 4b, the intense red emission of AO decreased after the cells were treated with TCO-OH (in the dark) or treated with complex 4 only (with irradiation). Remarkably, the red emission intensity was greatly reduced after the combined treatments, indicative of potent disruption to the lysosomal membranes. The acidic microenvironment of the lysosomes was monitored by the pH-sensitive LysoTracker Deep Red (Fig. 4c). No emission was observed with the combined treatments, showing that the luminal pH regulation of the lysosomes was impacted. Besides, lysosomal membrane permeabilisation could induce the release of lysosomal hydrolases, as such, the release of cathepsin B was monitored using Magic Red MR-(RR)2 as the indicator. The combined treatment of TCO-OH, complex 4 and light irradiation led to obvious dispersive red fluorescence in the cytoplasm (Fig. 4d), indicating the release of cathepsin B.
Cancer cells exhibit high protein turnover rates, and impairment to cellular degradation processes, such as autophagy, will impede cell survival.43 Thus, the autophagic inhibition effects of complex 4 were explored by monitoring the expression levels of intracellular LC3B protein (an autophagy-related protein) using an immunofluorescence staining assay (Fig. 4e). MDA-MB-231 cells treated with complex 4 only in the dark showed typical LC3 punctate immunofluorescence. Upon pretreatment of MDA-MB-231 cells with TCO-OH, a slight reduction in the emission intensity was revealed, which may result from the released cytotoxic aminomethylpyridine complex 3a after bioorthogonal dissociation. No emission could be detected after treating MDA-MB-231 cells with the combined treatments, indicating strong inhibition of the autophagic process. A similar trend was noted for MDC staining assay in MDA-MB-231 cells upon the combined treatments (Fig. S13†). The absence of MDC fluorescence suggests the reduction in autophagic vacuoles and suppression of autophagosome formation in cells. To further validate the autophagic inhibition, the expression of LC3B and p62 was investigated (Fig. 4f). Western blot analysis of MDA-MB-231 cells treated with complex 4 indicates that autophagic inhibition did not arise from the complex. Pretreatment of cells with TCO-OH in the dark or treatment of cells with complex 4 alone and upon light irradiation revealed similar results to the immunofluorescence experiments (Fig. 4e), with a slight decrease in LC3B expression. A significant reduction of LC3B expression was noted in the combined treatments. The protein p62 is a substrate degraded in the autolysosomes, and higher expression levels indicate inhibition of lysosomal degradation.44 Upon combined treatments, the expression level of p62 was remarkably upregulated compared to the control groups. Collectively, the activation of complex 4 with TCO-OH followed by light irradiation can efficiently inhibit autophagosome formation and lysosomal degradation, owing to the release of the more cytotoxic complex 3a, which also acts as an efficient 1O2 photosensitiser. The cascade of events blocks the autophagic process, resulting in necrotic cell death.
Lysosome-induced immunogenic cell death
The induction of ROS in lysosomes has been linked to lysosome-induced immunogenic cell death (LIICD), as ROS-induced lysosomal membrane permeabilisation is known to lead to the release of lysosomal hydrolases, thereby stimulating ICD.45,46 Based on the successful intracellular 1O2 generation of the combined bioorthogonal dissociation approach and light irradiation in the lysosomes, the induction of LIICD was investigated by monitoring three representative ICD-related biomarkers: calreticulin (CRT), high mobility group box 1 (HMGB1) and adenosine triphosphate (ATP). As revealed in Fig. 5a, dark conditions without or with TCO-OH pretreatment did not induce any CRT translocation. In contrast, light irradiation without TCO-OH led to a slight increase in emission intensity for the CRT-antibody dye, and the immunofluorescence intensity was the highest for the combined condition, indicative of translocation of CRT from the cytoplasm to the membrane. This trend was reciprocal for HMGB1, with no detectable immunofluorescence for the combined treatments (Fig. 5b), suggesting the migration of HMGB1 from the nucleus to the extracellular space. ATP secretion from the treated cells was measured by an ATP determination assay. The extracellular ATP concentration was greatly increased after the cells were pretreated with TCO-OH, followed by complex 4 and light irradiation compared to the other three conditions (Fig. 5c). These results provide evidence that the bioorthogonally dissociative complex 4 can successfully evoke ICD through the 1O2-induced lysosomal damage in a highly controlled manner. Similar observations were also noted in cells that were treated with the ER-targeted free complex 3a and light irradiation (Fig. S14 and S15†), which should be associated with 1O2-induced ER dysfunction.47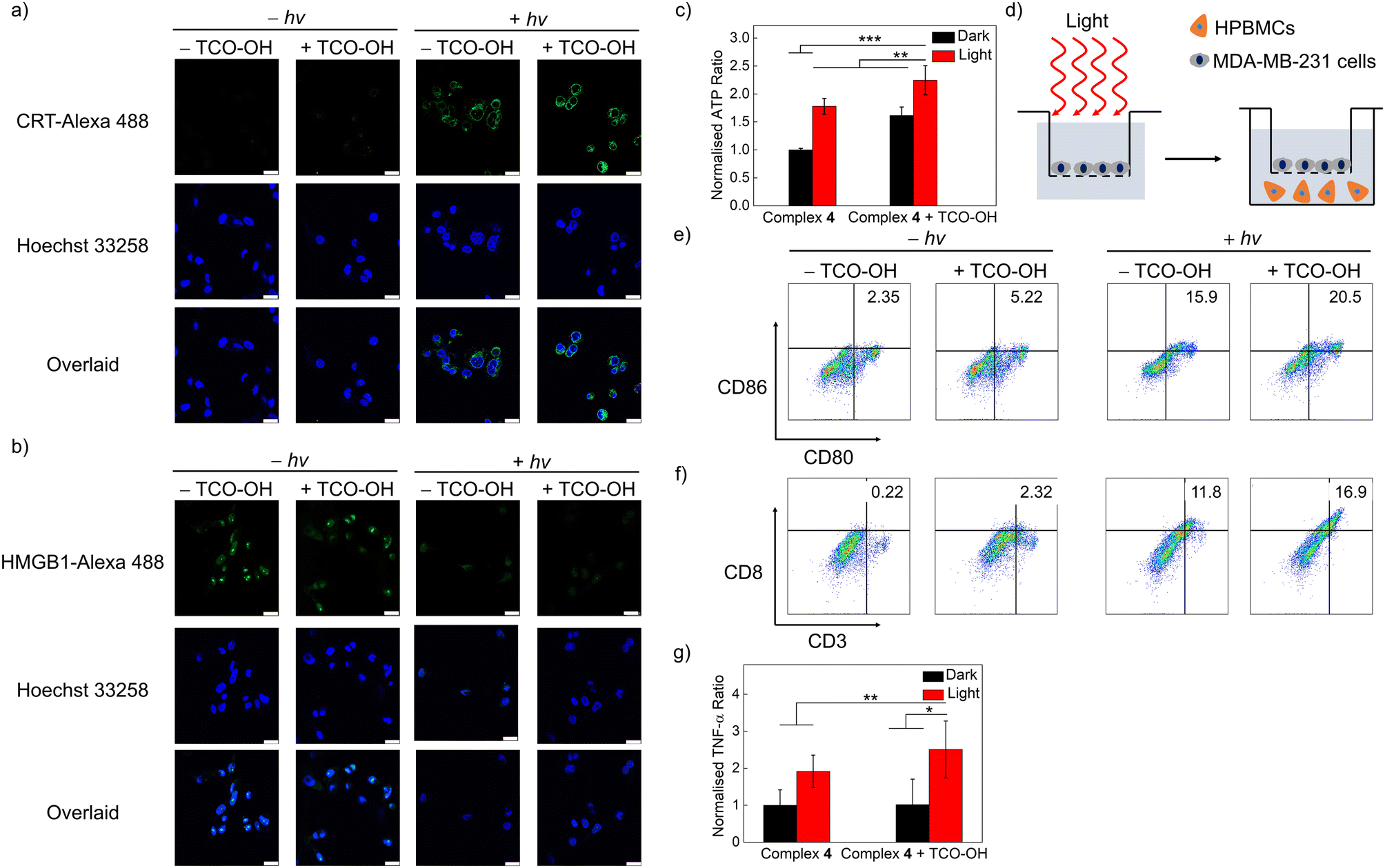 | ||
| Fig. 5 LSCM images of (a) CRT (CRT-Alexa 488 (10 μM, 1:200 solution, 2 h; λex = 488 nm, λem = 500–520 nm); Hoechst 33258 (10 μM, 1 μg mL−1, 10 min; λex = 405 nm, λem = 420–450 nm)) and (b) HMGB1 (HMGB1-Alexa 488 (10 μM, 1:200 solution, 2 h; λex = 488 nm, λem = 500–520 nm); Hoechst 33258 (10 μM, 1 μg mL−1, 10 min; λex = 405 nm, λem = 420–450 nm)). The cells were first pretreated without or with TCO-OH (100 μM) for 2 h, incubated with complex 4 (10 μM) for 6 h, left in fresh medium for 12 h, remained in the dark or were irradiated at 450 nm (20 mW cm−2) for 20 min and subsequently incubated in the dark for 2 h. (c) Extracellular ATP concentrations of MDA-MB-231 cells. (d) Schematic illustration of the transwell system for immune cell response studies using HPBMCs. MDA-MB-231 cells were first pretreated without or with TCO-OH (100 μM) for 2 h, incubated with complex 4 (10 μM) for 6 h, left in fresh medium for 12 h, remained in the dark or were irradiated at 450 nm (20 mW cm−2) for 20 min and co-cultured with HPBMCs for 24 h. HPBMCs were cultured in the lower chamber; the treated MDA-MB-231 cells were added to the upper chambers. Flow cytometry analysis of (e) mature DCs (CD80+CD86+) and (f) cytotoxic T cells (CD3+CD8+). (g) Levels of TNF-α secreted by HPBMCs. *p < 0.1, **p < 0.01, ***p < 0.001. | ||
The released HMGB1 and ATP from dead cells act as “danger” and “find me” signals, respectively, to trigger the maturation of dendritic cells (DCs) (CD80+CD86+), which further promote the activation of effector T cells, such as cytotoxic T cells to stimulate the systemic immune response.48 Thus, the immune activation efficiency of complex 4 was assessed by measuring the immune cell proliferation of human peripheral blood mononuclear cells (HPBMCs) through co-incubation with treated-MDA-MB-231 cells using a transwell system for 24 h (Fig. 5d). The combined treatment of TCO-OH, complex 4 and light irradiation upregulated both the population of mature DCs (CD80+CD86+) and cytotoxic T cells (CD3+CD8+) compared to the other three groups (Fig. 5e and f), with the percentage values increased from 2.35 to 20.5% and from 0.22 to 16.9%, respectively. Additionally, the expression levels of tumour necrosis factor (TNF-α), a proinflammatory cytokine, were enhanced by 2.5-fold after the same treatments (Fig. 5g). These promising results exemplify that the synergistic biorthogonal dissociation and light irradiation approach can effectively trigger the maturation of DCs and activation of effector T cells, suggesting that complex 4 can serve as a bioorthogonally activatable and controllable photosensitiser for photoimmunotherapy, which is, to the best of our knowledge, the first of its kind.
Conclusions
In summary, we developed a series of rhenium(I) polypyridine complexes as bioorthogonally dissociative phototherapeutic agents for the controllable induction of ICD by conjugating a tetrazine moiety to the metal complexes using a bioorthogonally activatable carbamate linker. Upon reaction with TCO-OH, all the complexes showed significantly increased emission intensities and 1O2 generation due to the TCO-OH-triggered tetrazine elimination. Notably, the PEGylated complex 4 showed increased cytotoxicity after reaction with TCO-OH because of the release of the PEG unit to yield a more cytotoxic aminomethylpyridine complex 3a with enhanced anticancer performance upon photoirradiation. Cellular studies revealed that the intracellular bioorthogonal dissociation reaction of complex 4 with TCO-OH followed by light irradiation induced lysosomal dysfunction, autophagy suppression and activation of ICD. The activation of ICD using a bioorthogonal dissociation reaction and photoirradiation is unprecedented, and this strategy can enhance therapeutic efficacy along with long-term protective antitumour immunity in cancer patients to prevent the recurrence of tumours. Future studies will be performed to investigate whether complex 4 can elicit systemic immune response and inhibit the growth of established tumours in vivo after treatment with TCO-OH and light.Data availability
Data available in the ESI: instrumentation, methods, synthetic details, characterisation, analytical and photophysical and kinetic data and results of cellular studies, microscopy experiments and bioassays.†Author contributions
G.-X. X.: conceptualization, data curation, formal analysis, investigation, writing – original draft, writing – review & editing; L. C.-C. L.: data curation, formal analysis, writing – original draft, writing – review & editing; P. K.-K. L.: data curation, formal analysis, writing – original draft, writing – review & editing; E. C.-L. M.: data curation, writing – original draft, writing – review & editing; J. S.: writing – original draft, writing – review & editing; K. Y. Z.: funding acquisition, project administration, writing – review & editing; Q. Z.: funding acquisition, project administration, supervision, writing – review & editing; K. K.-W. L.: conceptualization, funding acquisition, project administration, resources, supervision, writing – original draft, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Hong Kong Research Grants Council (Project no. CityU 11302820, CityU 11301121, CityU 11317022, C6014-20W and C7075-21GF) and the Hong Kong Research Grants Council, National Natural Science Foundation of China (Project no. N_CityU104/21). We also thank the funding support from “Laboratory for Synthetic Chemistry and Chemical Biology” under the Health@InnoHK Program launched by Innovation and Technology Commission, The Government of Hong Kong SAR, P. R. China. G.-X. X. acknowledges the receipt of a Postgraduate Studentship and Research Tuition Scholarship, both administered by City University of Hong Kong.References
- M. McNutt, Science, 2013, 342, 1417 CrossRef CAS PubMed.
- A. D. Waldman, J. M. Fritz and M. J. Lenardo, Nat. Rev. Immunol., 2020, 20, 651 CrossRef CAS PubMed.
- C. Liu, X. Liu, X. Xiang, X. Pang, S. Chen, Y. Zhang, E. Ren, L. Zhang, X. Liu, P. Lv, X. Wang, W. Luo, N. Xia, X. Chen and G. Liu, Nat. Nanotechnol., 2022, 17, 531 CrossRef CAS PubMed.
- J. Maher, R. J. Brentjens, G. Gunset, I. Riviere and M. Sadelain, Nat. Biotechnol., 2002, 20, 70 CrossRef CAS PubMed.
- W. Wu, Y. Pu, B. Zhou, Y. Shen, S. Gao, M. Zhou and J. Shi, J. Am. Chem. Soc., 2022, 144, 19038 CrossRef CAS PubMed.
- B. Uricoli, L. A. Birnbaum, P. Do, J. M. Kelvin, J. Jain, E. Costanza, A. Chyong, C. C. Porter, S. Rafiq and E. C. Dreaden, Adv. Healthcare Mater., 2021, 10, e2002214 CrossRef PubMed.
- T. F. Gajewski, H. Schreiber and Y.-X. Fu, Nat. Immunol., 2013, 14, 1014 CrossRef CAS PubMed.
- F. S. Hodi, S. J. O'Day, D. F. McDermott, R. W. Weber, J. A. Sosman, J. B. Haanen, R. Gonzalez, C. Robert, D. Schadendorf, J. C. Hassel, W. Akerley, A. J. M. van den Eertwegh, J. Lutzky, P. Lorigan, J. M. Vaubel, G. P. Linette, D. Hogg, C. H. Ottensmeier, C. Lebbé, C. Peschel, I. Quirt, J. I. Clark, J. D. Wolchok, J. S. Weber, J. Tian, M. J. Yellin, G. M. Nichol, A. Hoos and W. J. Urba, N. Engl. J. Med., 2010, 363, 711 CrossRef CAS PubMed.
- X. Su, W.-J. Wang, Q. Cao, H. Zhang, B. Liu, Y. Ling, X. Zhou and Z.-W. Mao, Angew. Chem., Int. Ed., 2022, 61, e202115800 CrossRef CAS PubMed.
- L. Wang, R. Guan, L. Xie, X. Liao, K. Xiong, T. W. Rees, Y. Chen, L. Ji and H. Chao, Angew. Chem., Int. Ed., 2021, 60, 4657 CrossRef CAS PubMed.
- B. Wang, D. Tang, J. Karges, M. Cui and H. Xiao, Adv. Funct. Mater., 2023, 33, 2214824 CrossRef CAS.
- L. Zhang, N. Montesdeoca, J. Karges and H. Xiao, Angew. Chem., Int. Ed., 2023, 62, e202300662 CrossRef CAS PubMed.
- S. Monro, K. L. Colón, H. Yin, J. Roque III, P. Konda, S. Gujar, R. P. Thummel, L. Lilge, C. G. Cameron and S. A. McFarland, Chem. Rev., 2019, 119, 797 CrossRef CAS PubMed.
- X. Ji, Z. Pan, B. Yu, L. K. De La Cruz, Y. Zheng, B. Ke and B. Wang, Chem. Soc. Rev., 2019, 48, 1077 RSC.
- G. Saravanakumar, J. Kim and W. J. Kim, Adv. Sci., 2017, 4, 1600124 CrossRef PubMed.
- M. H. Lee, J. L. Sessler and J. S. Kim, Acc. Chem. Res., 2015, 48, 2935 CrossRef CAS PubMed.
- M. Rooseboom, J. N. M. Commandeur and N. P. E. Vermeulen, Pharmacol. Rev., 2004, 56, 53 CrossRef CAS PubMed.
- E. Kozma, M. Bojtár and P. Kele, Angew. Chem., Int. Ed., 2023, 62, e202303198 CrossRef CAS PubMed.
- Y. You, F. Cao, Y. Zhao, Q. Deng, Y. Sang, Y. Li, K. Dong, J. Ren and X. Qu, ACS Nano, 2020, 14, 4178 CrossRef CAS PubMed.
- K. K.-W. Lo, Acc. Chem. Res., 2020, 53, 32 CrossRef CAS PubMed.
- N. K. Devaraj and R. Weissleder, Acc. Chem. Res., 2011, 44, 816 CrossRef CAS PubMed.
- J. Wang, X. Wang, X. Fan and P. R. Chen, ACS Cent. Sci., 2021, 7, 929 CrossRef CAS PubMed.
- J. Li and P. R. Cheng, Nat. Chem. Biol., 2016, 12, 129 CrossRef CAS PubMed.
- L. Lin, L. Jiang, E. Ren and G. Liu, Front. Chem. Sci. Eng., 2021, 17, 483 CrossRef.
- X. Xie, B. Li, J. Wang, C. Zhan, Y. Huang, F. Zeng and S. Wu, ACS Mater. Lett., 2019, 1, 549 CrossRef CAS.
- Q. Yao, F. Lin, X. Fan, Y. Wang, Y. Liu, Z. Liu, X. Jiang, P. R. Chen and Y. Gao, Nat. Commun., 2018, 9, 5032 CrossRef PubMed.
- R. M. Versteegen, R. Rossin, W. ten Hoeve, H. M. Janssen and M. S. Robillard, Angew. Chem., Int. Ed., 2013, 52, 14362 CrossRef.
- J. Tu, M. Xu, S. Parvez, R. T. Peterson and R. M. Franzini, J. Am. Chem. Soc., 2018, 140, 8410 CrossRef CAS PubMed.
- K. Porte, B. Renoux, E. Péraudeau, J. Clarhaut, B. Eddhif, P. Poinot, E. Gravel, E. Doris, A. Wijkhuisen, D. Audisio, S. Papot and F. Taran, Angew. Chem., Int. Ed., 2019, 58, 6366 CrossRef CAS PubMed.
- J. Xiong, E. Y. Xue, Q. Wu, P.-C. Lo and D. K. P. Ng, J. Control. Release, 2023, 353, 663 CrossRef CAS PubMed.
- A. M.-H. Yip, C. K.-H. Lai, K. S.-M. Yiu and K. K.-W. Lo, Angew. Chem., Int. Ed., 2022, 61, e202116078 CrossRef CAS PubMed.
- P. K.-K. Leung, L. C.-C. Lee, H. H.-Y. Yeung, K.-W. Io and K. K.-W. Lo, Chem. Commun., 2021, 57, 4914 RSC.
- J. Shum, L. C.-C. Lee, M. W.-L. Chiang, Y.-W. Lam and K. K.-W. Lo, Angew. Chem., Int. Ed., 2023, 135, e202303931 CrossRef.
- A. H. A. M. van Onzen, R. M. Versteegen, F. J. M. Hoeben, I. A. W. Filot, R. Rossin, T. Zhu, J. Wu, P. J. Hudson, H. M. Janssen, W. ten Hoeve and M. S. Robillard, J. Am. Chem. Soc., 2020, 142, 10955 CrossRef CAS PubMed.
- G.-X. Xu, L. C.-C. Lee, C. W.-C. Kwok, P. K.-K. Leung, J.-H. Zhu and K. K.-W. Lo, Eur. J. Inorg. Chem., 2021, 2021, 3432 CrossRef CAS.
- P. K.-K. Leung, L. C.-C. Lee, T. K.-Y. Ip, H.-W. Liu, S.-M. Yiu, N. P. Lee and K. K.-W. Lo, Chem. Commun., 2021, 57, 11256 RSC.
- R. M. Versteegen, W. Ten Hoeve, R. Rossin, M. A. R. de Geus, H. M. Janssen and M. S. Robillard, Angew. Chem., Int. Ed., 2018, 57, 10494 CrossRef CAS PubMed.
- M. Wilkovitsch, M. Haider, B. Sohr, B. Herrmann, J. Klubnick, R. Weisslede, J. C. T. Carlson and H. A. Mikula, J. Am. Chem. Soc., 2020, 142, 19132 CrossRef CAS PubMed.
- H. D. Cole, J. A. Roque III, G. Shi, L. M. Lifshits, E. Ramasamy, P. C. Barrett, R. O. Hodges, C. G. Cameron and S. A. McFarland, J. Am. Chem. Soc., 2022, 144, 9543 CrossRef CAS PubMed.
- S. P.-Y. Li, C. T.-S. Lau, M.-W. Louie, Y.-W. Lam, S. H. Cheng and K. K.-W. Lo, Biomaterials, 2013, 34, 7519 CrossRef CAS PubMed.
- J.-H. Zhu, G.-X. Xu, J. Shum, L. C.-C. Lee and K. K.-W. Lo, Chem. Commun., 2021, 57, 12008 RSC.
- J. Nicolas, S. Mura, D. Brambilla, N. Mackiewicz and P. Couvreur, Chem. Soc. Rev., 2013, 42, 1147 RSC.
- G. Kroemer and M. Jäättelä, Nat. Rev. Cancer, 2005, 5, 886 CrossRef CAS PubMed.
- X. Liang, Y. Ji, Y. Zhou, S. Wang, L. B. Vong and N. Li, Chem. Eng. J., 2022, 443, 136379 CrossRef CAS.
- T. Iulianna, N. Kuldeep and F. Eric, Cell Death Dis., 2022, 13, 509 CrossRef CAS PubMed.
- E. Fossel and I. Taritsa, J. Immunol. Res. Infect. Dis., 2022, 2, 1 Search PubMed.
- D. V. Krysko, A. D. Garg, A. Kaczmarek, O. Krysko, P. Agostinis and P. Vandenabeele, Nat. Rev. Cancer, 2012, 12, 860 CrossRef CAS PubMed.
- F. Sun, Q. Zhu, T. Li, M. Saeed, Z. Xu, F. Zhong, R. Song, M. Huai, M. Zheng, C. Xie, L. Xu and H. Yu, Adv. Sci., 2021, 8, 2002746 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc04903e |
| This journal is © The Royal Society of Chemistry 2023 |