
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Base-mediated homologative rearrangement of nitrogen–oxygen bonds of N-methyl-N-oxyamides†
Monika
Malik
a,
Raffaele
Senatore
a,
Thierry
Langer
a,
Wolfgang
Holzer
a and
Vittorio
Pace
 *ab
*ab
aDepartment of Pharmaceutical Sciences, Division of Pharmaceutical Chemistry, University of Vienna, Josef-Holaubek-Platz 2, 1090, Vienna, Austria. E-mail: vittorio.pace@univie.ac.at
bDepartment of Chemistry, University of Turin, Via Giuria 7, 10125 Turin, Italy
First published on 29th August 2023
Abstract
Due to the well known reactivity of C(O)–N functionalities towards canonical C1-homologating agents (e.g. carbenoids, diazomethane, ylides), resulting in the extrusion of the N-centered fragment en route to carbonyl compounds, formal C1-insertions within N–O bonds still remain obscure. Herein, we document the homologative transformation of N-methyl-N-oxyamides – with high tolerance for diverse O-substituents – into N-acyl-N,O-acetals. Under controlled basic conditions, the N-methyl group of the same starting materials acts as a competent precursor of the methylene synthon required for the homologation. The logic is levered on the formation of an electrophilic iminium ion (via N–O heterolysis) susceptible to nucleophilic attack by the alkoxide previously expulsed. The procedure documents genuine chemocontrol and flexibility, as judged by the diversity of substituents placed on both amide and nitrogen linchpins. The mechanistic rationale was validated through experiments conducted on D-labeled materials which unambiguously attributed the origin of the methylene fragment to the N-methyl group of the starting compounds.
Introduction
Homologation techniques enable the progressive insertion of a constant unit (e.g. –CH2–) within a given chemical bond.1 These operations are playing nowadays a prominent role in the assembly of complex molecular entities and exhibit a significant potential in guiding the fine-tuning of crucial physical–chemical parameters.2 Since the introduction almost one century ago by Arndt and Eistert of the archetypal homologating agent3 – diazomethane (CH2N2) –, subsequent studies converged in individuating the LG-CH2-[M] motif as the general descriptor for C1-releasing agents (Scheme 1a). Mechanistically, the transformation can be represented as the formal delivery of the C1 unit from a donor – externally added – agent to a recipient linchpin which, through the triggering of 1,2-migration-type events furnishes the homologated skeleton.4 The venerable Matteson homologation of boronates with lithium carbenoids (nucleophilic C1-agents, Scheme 1b)5 elegantly extended to iterative processes by Aggarwal,6 and Blakemore7 – well illustrates the controlled insertions into C–B bonds and served as source of inspiration for the more recent work on N–B and O–B bond homologations by Dong.8 In the course of our studies on programmable LiCH2X-mediated homologation of carbon electrophiles,9 we validated the rationale for the selective insertion of methylene units into chalcogenide bonds (S–S and Se–Se analogues) to construct geminal disulfurated methanes (dithioacetal-like – Scheme 1c).10 One intriguing aspect is whether a C1-insertion could take place selectively on a more sophisticated Z–X–Y platform, where both Z–X and X–Y bonds might undergo homologation (Scheme 1d): to the best of our knowledge, these clusters have not been previously investigated in C1-insertion sequences. In this context, N-alkoxy-substituted amides11 constitute representative models since both the C(O)–N and the N–O fragments could be subjected to the methylene insertion: as documented in recent work by our group,12 amide-type substrates undergo homologation (employing [M]-CH2-LG reagents) at the C(O)–N linkage – yielding α-substituted ketones11b,12c,13 – thus, precluding de facto the use of such agents for inserting the C1-unit within the N–O bond (Scheme 1d – up). Beside introducing the concept of N–O homologation, the prospected transformation would provide a modular and rapid approach towards challenging – yet stable14 – acyclic N-acyl-N,O-acetals (i.e. N,O-aminals).15 Noteworthy, the preparation of these analogues – i.e. RN(H)–CH2–OR1 motif – has been much less explored compared to methods for assembling substituted congeners and, still remains circumscribed to the electrochemical oxidation of aliphatic amides introduced by Linstead,16 or to the Katritzky's amidoalkylation of alcohols.17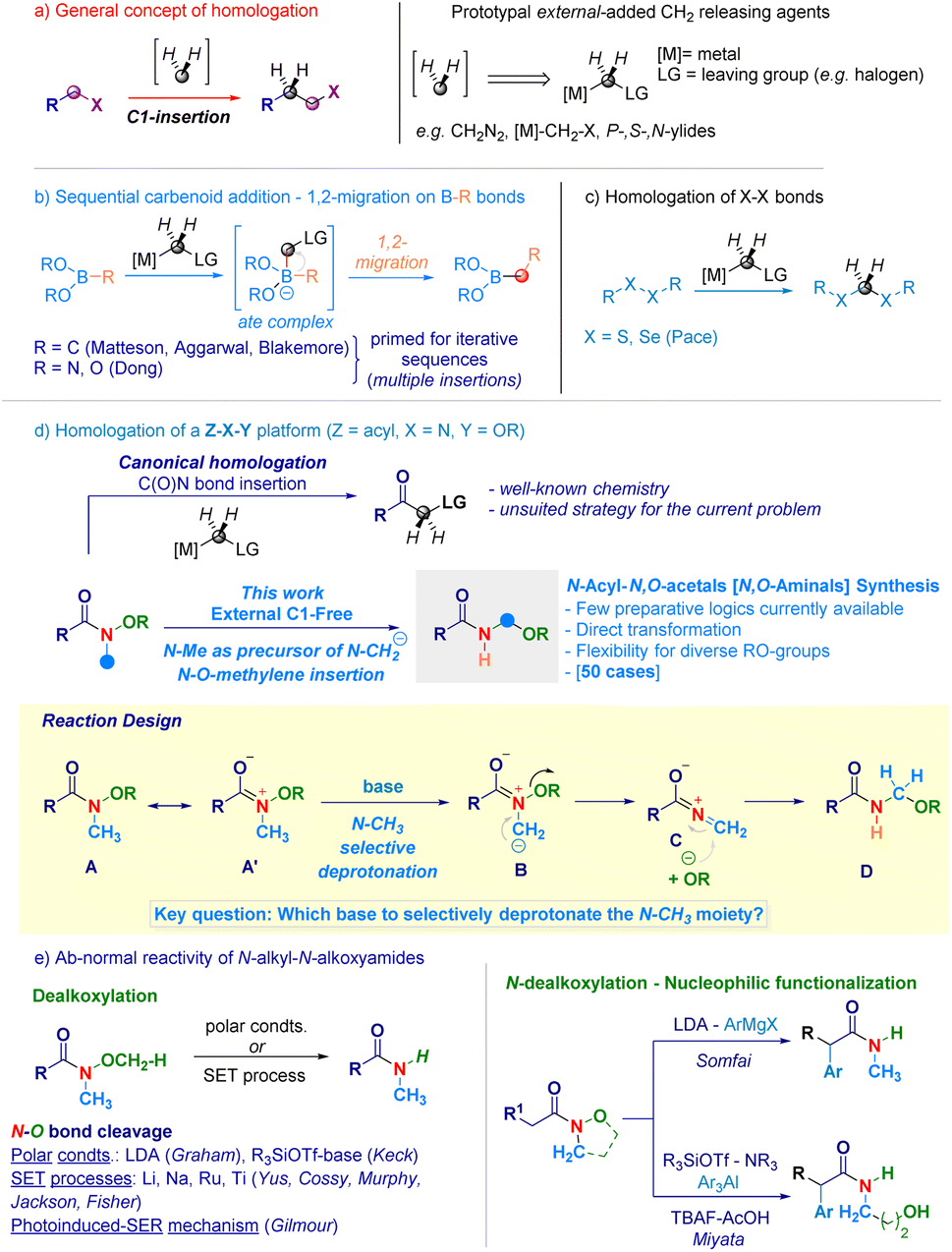 | ||
| Scheme 1 General context of the presented work. | ||
In this scenario, we wondered if the homologating element (–CH2–) could be generated from the constitutive N-methyl substituent of the N-alkoxy amide through a deprotonative event. Accordingly, a N-methyl-N-methoxyamide13aA represents a pertinent model to make productive the concept: as anticipated, it possesses – as judged by the NC(O) delocalization (A ↔ A′) – relatively acidic N-methyl protons amenable to removal with a base of adequate strength. In this circumstance, a ylide-type intermediate B would be formed prior to its collapse – via expulsion of the alkoxide – to a terminal, electrophilic iminium ion C,18 capable of intercepting the same nucleophile (RO−) and, thus yielding the homologated adduct D. A series of critical aspects required fine tuning, also in view of the hitherto explored chemistry of the O–CH3 moiety of Weinreb-like amides functional to access secondary amides (Scheme 1e). In this context, Graham pointed out that under polar conditions – the lithium amide base LDA (pKa = 36)19 selectively deprotonates the O–CH3 group giving a demethoxylated amide (via E2 mechanism),20 thus predicting a critical role of the base for generating the requested ylide.21 To further complicate this base-mediated scenario, enolizable N-alkyl-N-alkoxyamides22 can serve as manifolds for sequential N-dealkoxylation – nucleophilic functionalization to gather α-substituted amides, as documented by Somfai23 and Miyata.24 Moreover, N–O breaking operations on N-alkyl-N-alkoxyamides benefited from adopting (metal) based single electron transfer logics,25 for which Gilmour in 2022 presented an elegant organocatalytic photoinduced alternative (Scheme 1e).26
Despite these precedents on the N–O bond cleavage, the seminal work by Beak27 on the lithiation of N,N-dimethylamides supported our initial hypothesis of selectively deprotonate the N–CH3 fragment. Herein, we report the homologative assembly of N-acyl-N,O-acetals starting from N-alkoxy-N-methyl-amides through the use of the same N-methyl group of alkoxyamide as a formal C1-delivering agent. Our findings shed light on a novel reactivity – beyond classical acylation and enolate chemistry – of N-alkoxyamides in which the C(O)N bond maintains its chemical integrity in a carbanion environment while, the labile N–O linkage (55–65 kcal mol−1)26,28 undergoes homologation.
Results and discussion
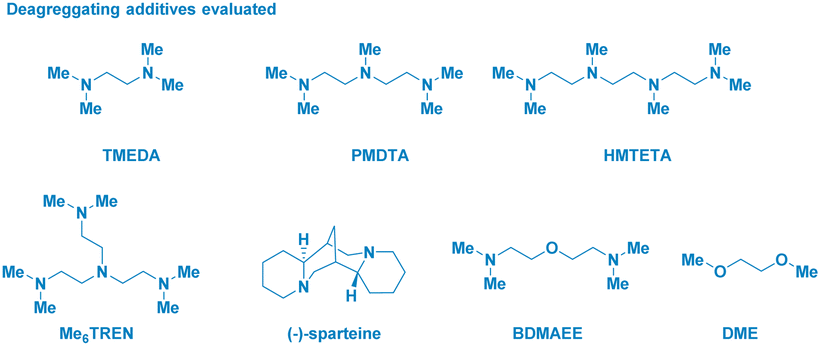
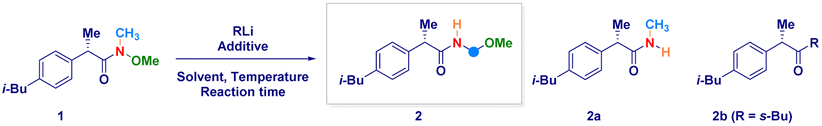
The enantiomerically pure Weinreb amide 1 generated from the common nonsteroidal anti-inflammatory drug Ibuprofen was chosen as the model substrate (Table 1). Upon treatment with s-BuLi in THF at −78 °C (entry 1), the corresponding ketone 2b – derived from classical Weinreb amide chemistry – was obtained as the unique product (with traces of the demethoxylated adduct 2a). Pleasingly, by triggering the deagreggation (and thus the basicity)29 of s-BuLi through the use of the bidentate ligand TMEDA (N,N,N′,N′-tetramethylethylendiamine), the deprotonation of the N–Me group became evident, thus furnishing the desired N-acyl-N,O-acetal 2 in a promising 29% yield, though neither formation of the Graham20a secondary amide 2a nor of s-butyl ketone 2b could be suppressed (entry 2). Running the reaction in diethyl ether (entry 3) or, increasing the loading of the additive resulted in no evident improvement (entry 4). The chemoselectivity slightly augmented in 2-methyltetrahydrofuran, probably, as a consequence of its higher stability under strongly basic conditions (entry 5).30 Considering Bauer's observation on the complete deaggregation of s-BuLi in the presence of the tridentate additive PMDTA (N,N,N′,N′′,N′′-pentamethyldiethylentriamine),31 we were pleased in noting the almost exclusive formation of the desired adduct 2, thus confirming the generation of the aminomethyl anion and the avoiding of Weinreb amides enolization phenomena (entry 6).22e The noticeable dwindling of the yield in THF is presumably ascribed to the easier α-lithiation it undergoes – compared to 2-MeTHF – (entry 7),30,32 thus suggesting a strict dependance of the N–CH3 deprotonation with the overall basicity of the reaction medium. Moreover, conducting the process in 2-MeTHF guarantees the full integrity of the stereochemical information compared to THF in which racemization phenomena became apparent (entry 6 vs. 7).32 Some additional points merit mention: (a) reactions reach completion within 2 h, as judged by lowering the reaction time to 0.5 h (entry 8); (b) keeping the stoichiometric loading of s-BuLi and PMDTA at 1.6 equiv. enables to maximize the formation of the desired product 2, thus avoiding a detrimental effect on the chemoselectivity due to less controlled lithiations (entry 9); (c) replacing PMDTA with analogous polyamine ligands such as HMTETA, Me6TREN, BDMAEE or (−)-sparteine again affected the selectivity, thus confirming the unique beneficial effect displayed by PMDTA (entries 10–14); (d) switching to alkyllithiums of distinct basicity [i.e. more basic t-BuLi (pKa = 53) and less alkaline n-BuLi (pKa = 50)]19 resulted in substantial lack of control of the transformation (entries 14–15), while the lithium amide LTMP (pKa = 35.7)19 shifted the distribution towards the demethoxylated product 2a (entry 16) in analogy to the previously used LDA;20a (e) increasing the temperature to −40 °C lowered the reaction efficiency, as a consequence of the plausible diminished stability of the generated anion (entry 17).|
|
||||
|---|---|---|---|---|
| Entry | RLi | Additive (equiv.) | Solvent | Product(s)b (%) |
a Unless otherwise stated, reactions were run at −78 °C for 2 h in the presence of 1.6 equiv. of the competent organolithium.
b Isolated yields.
c Compound 2 was obtained in 99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 er.
d Compound 2 was obtained in 95:5 er.
e Reaction time 0.5 h.
f
s-BuLi (3.2 equiv.) was used.
g Reaction run at −40 °C. :1 er.
d Compound 2 was obtained in 95:5 er.
e Reaction time 0.5 h.
f
s-BuLi (3.2 equiv.) was used.
g Reaction run at −40 °C.
|
||||
| 1 | sBuLi | — | THF | 2 (—); 2a (6); 2b (90) |
| 2 | sBuLi | TMEDA (1.6) | THF | 2 (29); 2a (17); 2b (34) |
| 3 | sBuLi | TMEDA (1.6) | Et2O | 2 (23); 2a (20); 2b (31) |
| 4 | sBuLi | TMEDA (3.2) | THF | 2 (30); 2a (24); 2b (27) |
| 5 | sBuLi | TMEDA (1.6) | MeTHF | 2 (48); 2a (11); 2b (8) |
| 6c | sBuLi | PMDTA (1.6) | MeTHF | 2 (81) |
| 7d | sBuLi | PMDTA (1.6) | THF | 2 (59); 2a (15); 2b (12) |
| 8e | sBuLi | PMDTA (1.6) | MeTHF | 2 (62) |
| 9f | sBuLi | PMDTA (3.2) | MeTHF | 2 (64); 2a (15) |
| 10 | sBuLi | HMETA (1.6) | MeTHF | 2 (43); 2a (22); 2b (30) |
| 11 | sBuLi | Me6TREN (1.6) | MeTHF | 2 (35); 2a (16); 2b (24) |
| 12 | sBuLi | (−)-Spart. (1.6) | MeTHF | 2 (51); 2a (23); 2b (18) |
| 13 | sBuLi | BDMAEE (1.6) | MeTHF | 2 (47); 2a (18); 2b (14) |
| 14 | tBuLi | PMDTA (1.6) | MeTHF | 2 (36); 2a (15); 2b (21) |
| 15 | nBuLi | PMDTA (1.6) | MeTHF | 2 (12); 2a (19); 2b (37) |
| 16 | LTMP | — | MeTHF | 2 (5); 2a (80) |
| 17g | sBuLi | PMDTA (1.6) | MeTHF | 2 (46); 2a (8); 2b (10) |
|
||||
Having established the four key elements governing the reaction (s-BuLi, PMDTA, 2-MeTHF, −78 °C), we then studied the scope of the intramolecular N-methyl-N-alkoxyamide homologation to N-acyl-N,O-acetals (Scheme 2). A series of Weinreb amides generated from aliphatic carboxylic acids were amenable substrates for the process: similarly to the ibuprofene analogue 2, also the aproxene derived species smoothly underwent the transformation (3) with full retention of the embodied stereochemical information. This latter aspect was also deducted in the case of compound 4, thus confirming that the employed strongly basic reaction conditions did not promote any racemization of the materials. Not only aliphatic Weinreb amides of different chemical hindrance [phenylethyl (5) and tert-butyl (6)] on the carbonyl carbon could be used, but also a series of alicyclic analogues of progressive ring size such as cyclopropyl (7), cyclobutyl (8), cyclopentyl (9), cyclohexyl (10) and the sterically demanding adamantyl derivative (11). Switching to aromatic Weinreb amides did not alter the validity of the general methodology. In this context, common aliphatic substituents could be conveniently placed at different positions of the phenyl ring [e.g. unsubstituted (12), p-methyl (13), m-methyl (14), o-ethyl (15), p-tert-butyl (16)]. The presence of elements modulating the electrophilicity of carbonyl carbon was tolerated, as documented in the cases of: (i) electron-donating substituents such as ethers (methoxy-, 17–19) or thioether (20) and, (ii) halogen atoms [chloro (21–23), fluoro (24–26), trifluoromethyl (27)]. The X-ray structural analysis of derivative 17 unambiguously confirmed the N-acyl-N,O-acetal cluster. Scaling up to 20 mmol scale did not influence the reaction yield, as shown in the case of compound 23. Notably, the inclusion of additional acidic functionalities [OH of a phenol (28), NH2 of an aniline residue (29) or C–H of a terminal alkyne (30)] were not detrimental for the homologative event whenever substrates were pretreated with MeLi (see ESI†). Moreover, polyaromatic [2-naphtyl (31)] and heteroaromatic [furyl (32) and thienyl (33)] systems further expanded the reaction scope. The deprotonation en route to the intermediate aminomethyl species did not suffer from the simultaneous presence of functionalities susceptible to lithiation, as deducted in the cases of the stannane (34) and the selenane (35). With much of our delight, the methodology enabled the selective transformation of a Weinreb amide featuring a carboxy-amide fragment (N,N-diethyl) into the corresponding N-acyl-N,O-acetal 36 in which no modification occurred to the functionalizing element. This is indeed an evident proof of the selective deprotonation occurring at the N-methyl group of the amides.
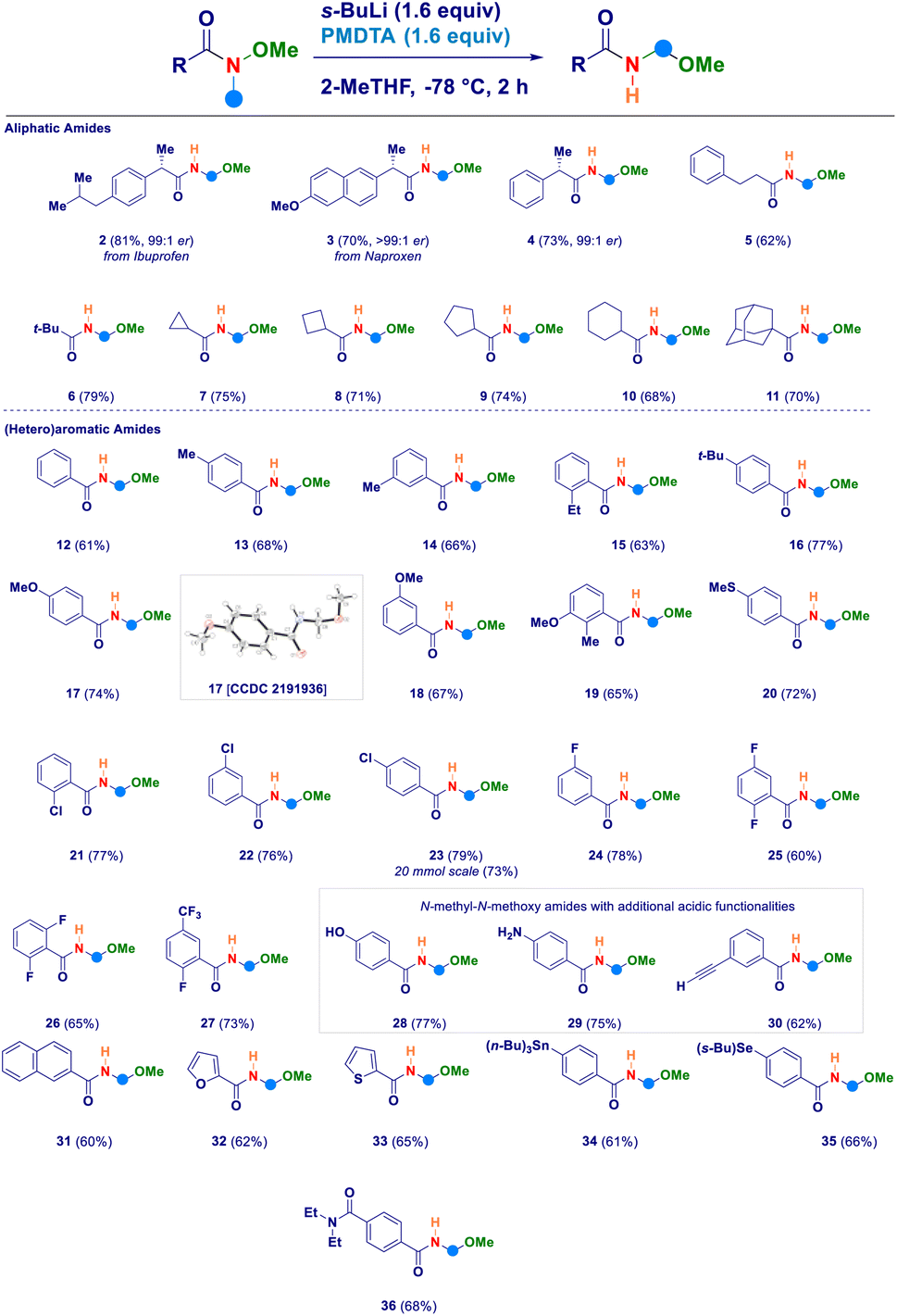 | ||
| Scheme 2 Scope of the method with N-methyl-N-methoxyamides. | ||
With the aim to uncover the full potential of the reaction, the evaluation of different O-substituted N,O-alkoxyamides was performed (Scheme 3). In this sense, the scope of Weinreb amides discussed above was efficiently implemented by engaging as reaction partners not only the O-ethyl analogues (37–39) but also the more sterically hindered O-i-propyl (40) and O-t-butyl (41) ones. The initial N–CH3 deprotonation event was successfully realized also on the corresponding O-allyl (42) and the O-propargyl (43) systems, as well as, on the O-cyclopropylmethyl (44) and the O-methoxymethyl (45) analogues. The high electrophilic reactivity of the iminium species involved, enabled the smooth attack of a less nucleophilic element as the phenolate anion, thus furnishing derivative 46. Finally, also N-methyl-O-benzyl-type alkoxyamides yielded the expected N-acyl-N,O-acetals 47 and 48 in comparable efficiency.
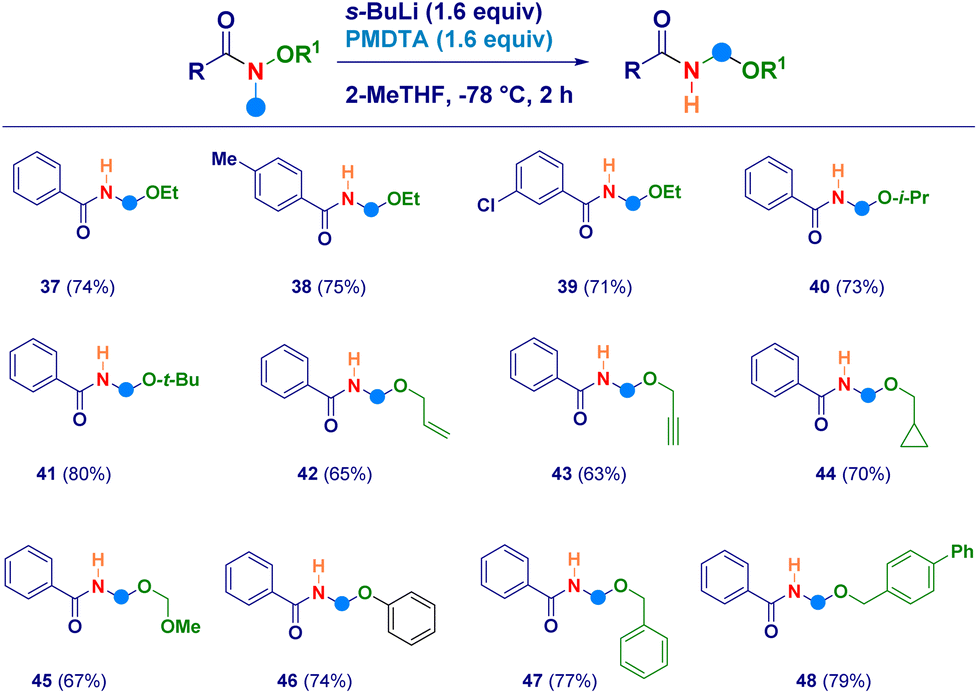 | ||
| Scheme 3 Synthesis of diverse O-substituted N-acyl-N,O-acetals. | ||
In order to confirm the proposed rationale, a series of control experiments were performed (Scheme 4). The pivotal presence of the N-methyl group as the element furnishing the active methylene unit was deducted by attempting the transformation on the N-ethyl-O-methoxy analogue 49 which simply reacted via canonical Weinreb mechanism giving ketone 50 as the unique product (path a). It is likely that N–CH2CH3 protons would exhibit lower acidity preventing the crucial deprotonation event with s-BuLi/PMDTA. Additionally, this result is also in agreement with the suitability of our conditions for preventing Graham demethoxylation.20a The critical role displayed by the acyl moiety was evidenced by employing a Weinreb amide featuring a N-methoxy-N-methyl amine fragment (51) which yielded exclusively N-acyl-N,O-acetal 52 (path b), thus suggesting the requirement for an amide functionality to promote the reaction. The unambiguous deciphering of the origin of the constitutive methylene unit of N-acyl-N,O-acetals was gathered by applying the procedure to deuterium-labeled systems. Accordingly, N-d3-methyl-N-methoxy amide (53) was converted into the d2-N-acyl-N,O-acetal 54, thus demonstrating that the effective source for the homologated N–O linkage was the N–CD3 group (path c). Ceteris paribus, N-[1H]methyl-N-d3-methoxy amide (55) was homologated to the corresponding N-acyl-N,O-[1H]-acetal 56 featuring the labeled element exclusively at the alkoxy portion (OCD3 – path d).
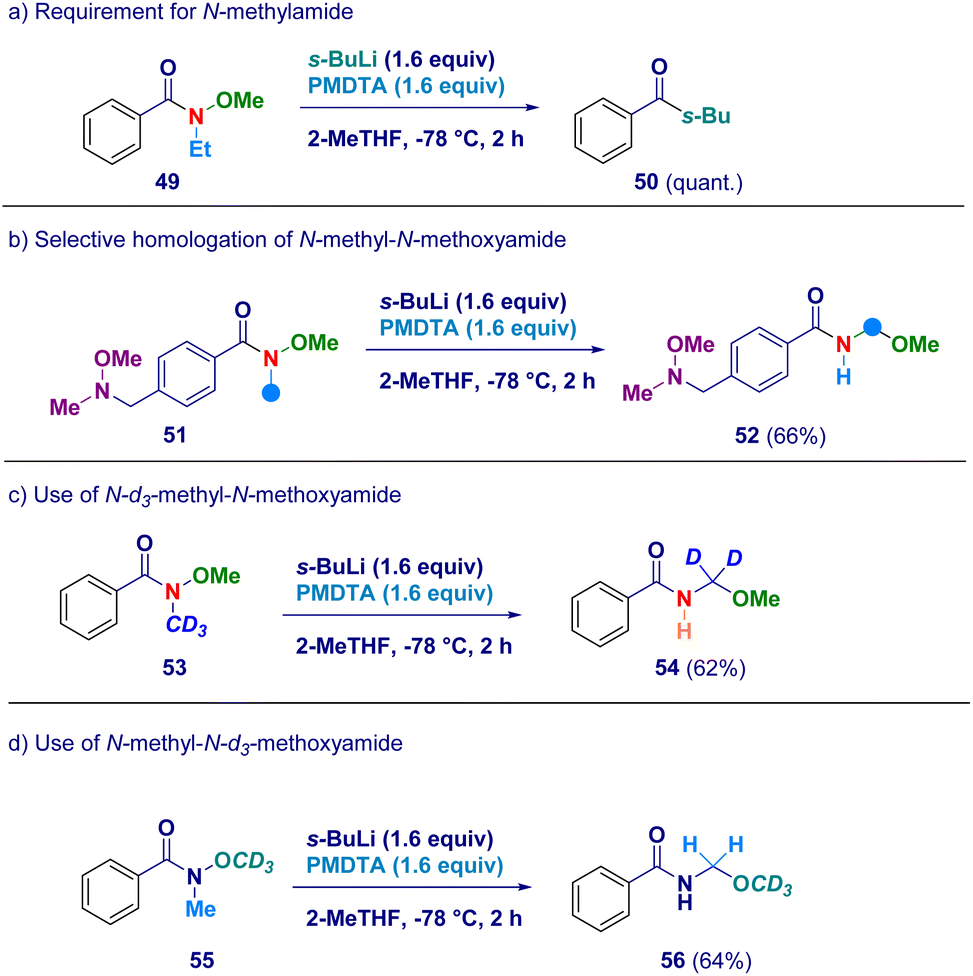 | ||
| Scheme 4 Control experiments for validating the mechanistic hypothesis. | ||
Finally, selected N-acyl-N,O-acetals were employed as starting materials for standard derivatization chemistry (Scheme 5). The stannane containing analogue 34 was used in a Stille coupling furnishing the biphenyl adduct 57 (path a);33 upon tosylation of phenol-derivative 28, followed by a Buchwald–Hartwig amination with morpholine, compound 58 was obtained (path b);34 the alkynyl-type Michael acceptor 59 reacted – in the presence of an excess of s-BuLi – giving N-acyl-N,O-acetal 60 in which the excess of s-BuLi attacked the β-position (path c); the secondary amide functionality of adduct 14 could be efficiently alkylated under basic conditions generating 61 (path d).
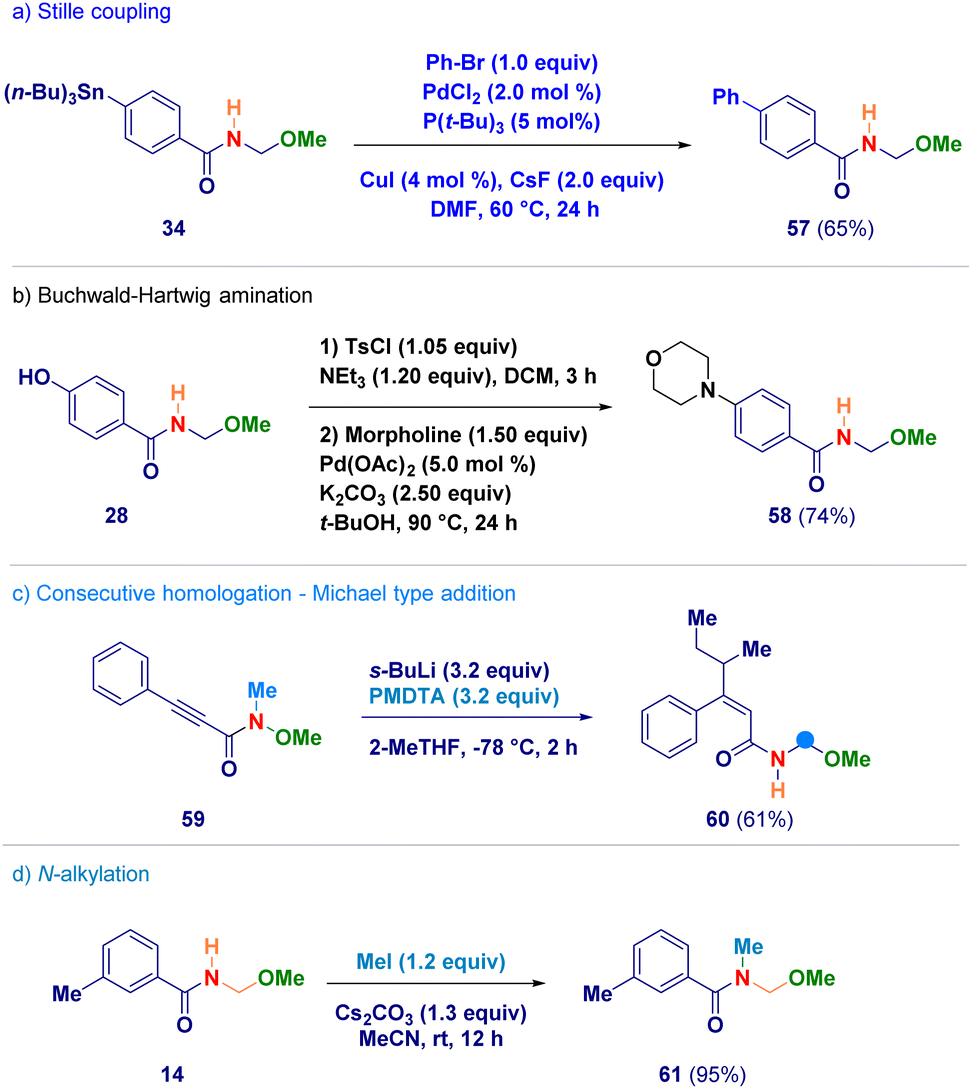 | ||
| Scheme 5 Synthetic manipulation of N-acyl-N,O-acetals. | ||
In summary, we have introduced – through the homologative transformation of N-methyl-N-oxyamides to acyclic N-acyl-N,O-acetals – the concept of C1-installation within a N–O bond. The constitutive N-methyl moiety of the starting material acted as the source of the methylene unit. Because of the well-established reactivity of N-alkoxyamides in the presence of canonical homologating agents (e.g. [M]-CH2-LG), the tactic proposed herein appears an effective tool for inserting the CH2 group into formal oxy-amines functionalities. The controlled deprotonating event, conducted with deaggregated s-BuLi (PMDTA), guarantees the tailored genesis of a α-aminomethyl carbanion which spontaneously rearranges to an electrophilic iminium intercepting the released alkoxide. Almost no restriction on the substitution pattern across the whole amide functionality – and, in particular on the N-alkoxy element – was observed, thus making flexible the access to the targeted compounds. The crucial role displayed by the N-methyl group was definitively ascertained through the application of the protocol to deuterium-labeled analogues.
Data availability
Further details of the experimental procedure, 1H, 13C, 19F, 77Se and 119Sn NMR spectra, HPLC analyses and X-ray crystallographic data for 17 are available in the ESI.†Author contributions
M. M. and R. S. performed the experiments. W. H. realized NMR analyses and elucidated the structures. V. P. conceived and directed the project and wrote the manuscript. All the authors discussed the results.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work is dedicated in memoriam of Professor José Vicente Sinisterra Gago. We thank the University of Vienna, the University of Turin and All4Labels Group (Hamburg, Germany) for generous funding. M. M. acknowledges the Austrian Academy of Sciences, Austrian UNESCO Commission and L'ORÉAL Austria for a L'ORÉAL FWIS Fellowship. The support from Project CH4.0 under MUR (Italian Ministry for the University) program “Dipartimenti di Eccellenza 2023-2027” (CUP: D13C22003520001) is acknowledged. Ing. A. Roller is gratefully appreciated for realizing X-ray analysis.Notes and references
- Homologation Reactions. Reagents, Applications and Mechanisms, ed. V. Pace, Wiley-VCH, Weinheim, 2023, vol. 1 and 2, ISBN 978-3-527-34815-2 Search PubMed.
- For a review on the modulation of biological properties via homologation processes, see: L. M. Lima, M. A. Alves and D. N. do Amaral, Curr. Top. Med. Chem., 2019, 19, 1734 CrossRef CAS PubMed.
- For seminal work, see: (a) F. Arndt, B. Eistert and W. Ender, Chem. Ber., 1929, 62, 44 CrossRef; (b) For a recent authoritative review, see: N. R. Candeias, R. Paterna and P. M. P. Gois, Chem. Rev., 2016, 116, 2937 CrossRef CAS PubMed.
- (a) A. Sidduri, M. J. Rozema and P. Knochel, J. Org. Chem., 1993, 58, 2694 CrossRef CAS; (b) S. R. Roy, D. Didier, A. Kleiner and I. Marek, Chem. Sci., 2016, 7, 5989–5994 RSC; (c) B. Dutta, N. Gilboa and I. Marek, J. Am. Chem. Soc., 2010, 132, 5588–5589 CrossRef CAS PubMed.
- For seminal work, see: (a) D. S. Matteson and D. Majumdar, J. Am. Chem. Soc., 1980, 102, 7588 CrossRef CAS; (b) For an account, see: D. S. Matteson, J. Org. Chem., 2013, 78, 10009 CrossRef CAS; (c) For the first enantioselective catalytic version, see: H. A. Sharma, J. Z. Essman and E. N. Jacobsen, Science, 2021, 374, 752 CrossRef CAS.
- (a) M. Burns, S. Essafi, J. R. Bame, S. P. Bull, M. P. Webster, S. Balieu, J. W. Dale, C. P. Butts, J. N. Harvey and V. K. Aggarwal, Nature, 2014, 513, 183 CrossRef CAS PubMed; (b) D. Leonori and V. K. Aggarwal, Acc. Chem. Res., 2014, 47, 3174 CrossRef CAS PubMed; (c) S. Balieu, G. E. Hallett, M. Burns, T. Bootwicha, J. Studley and V. K. Aggarwal, J. Am. Chem. Soc., 2015, 137, 4398 CrossRef CAS PubMed; (d) C. A. Brown and V. K. Aggarwal, Chem.–Eur. J., 2015, 21, 13900 CrossRef CAS PubMed; (e) J. Wu, P. Lorenzo, S. Zhong, M. Ali, C. P. Butts, E. L. Myers and V. K. Aggarwal, Nature, 2017, 547, 436 CrossRef CAS PubMed.
- (a) C. R. Emerson, L. N. Zakharov and P. R. Blakemore, Chem.–Eur. J., 2013, 19, 16342 CrossRef CAS PubMed; (b) P. R. Blakemore and M. S. Burge, J. Am. Chem. Soc., 2007, 129, 3068 CrossRef CAS PubMed.
- (a) Q. Xie and G. Dong, J. Am. Chem. Soc., 2022, 144, 8498 CrossRef CAS; (b) Q. Xie and G. Dong, J. Am. Chem. Soc., 2021, 143, 14422 CrossRef CAS PubMed; (c) Q. Xie, R. Zhang and G. Dong, Angew. Chem., Int. Ed., 2023, e202307118 CAS.
- (a) L. Ielo, L. Castoldi, S. Touqeer, J. Lombino, A. Roller, C. Prandi, W. Holzer and V. Pace, Angew. Chem., Int. Ed., 2020, 59, 20852 CrossRef CAS PubMed; (b) L. Ielo, S. Touqeer, A. Roller, T. Langer, W. Holzer and V. Pace, Angew. Chem., Int. Ed., 2019, 58, 2479 CrossRef CAS PubMed; (c) V. Pace, L. Castoldi, E. Mazzeo, M. Rui, T. Langer and W. Holzer, Angew. Chem., Int. Ed., 2017, 56, 12677 CrossRef CAS; (d) R. Senatore, M. Malik, T. Langer, W. Holzer and V. Pace, Angew. Chem., Int. Ed., 2021, 60, 24854 CrossRef CAS PubMed; (e) G. Parisi, M. Colella, S. Monticelli, G. Romanazzi, W. Holzer, T. Langer, L. Degennaro, V. Pace and R. Luisi, J. Am. Chem. Soc., 2017, 139, 13648 CrossRef CAS PubMed; (f) For accounts on CH2X mediated homologations, see: L. Castoldi, S. Monticelli, R. Senatore, L. Ielo and V. Pace, Chem. Commun., 2018, 54, 6692 RSC; (g) V. Pace, L. Castoldi, S. Monticelli, M. Rui and S. Collina, Synlett, 2017, 28, 879 CrossRef CAS.
- For the direct homologation of disulfides to symmetrical dithioacetals, see: (a) V. Pace, A. Pelosi, D. Antermite, O. Rosati, M. Curini and W. Holzer, Chem. Commun., 2016, 52, 2639 RSC; (b) See also for unsymmetrical analogues: L. Ielo, V. Pillari, N. Gajic, W. Holzer and V. Pace, Chem. Commun., 2020, 56, 12395 RSC; (c) The transformation could also be used for preparing O,S-acetals, see: L. Ielo, V. Pillari, M. Miele, W. Holzer and V. Pace, Adv. Synth. Catal., 2020, 362, 5444 CrossRef CAS.
- For reviews, see: (a) T. Sato and N. Chida, Org. Biomol. Chem., 2014, 12, 3147 RSC; (b) V. Pace, W. Holzer and B. Olofsson, Adv. Synth. Catal., 2014, 356, 3697 CrossRef CAS; (c) For detailed studies of the electrophilic reactivity of N-alkoxyamides, see: M. Nakajima, Y. Oda, T. Wada, R. Minamikawa, K. Shirokane, T. Sato and N. Chida, Chem.–Eur. J., 2014, 20, 17565 CrossRef CAS PubMed.
- For studies from our group on the use of Weinreb amides as acylating agents for α-substituted carbanions, see: (a) V. Pace, I. Murgia, S. Westermayer, T. Langer and W. Holzer, Chem. Commun., 2016, 52, 7584 RSC; (b) R. Senatore, L. Castoldi, L. Ielo, W. Holzer and V. Pace, Org. Lett., 2018, 20, 2685 CrossRef CAS; (c) For reviews, see: R. Senatore, L. Ielo, S. Monticelli, L. Castoldi and V. Pace, Synthesis, 2019, 51, 2792 CrossRef CAS; (d) M. Blangetti, K. de la Vega-Hernández, M. Miele and V. Pace, in Amide Bond Activation, ed. M. Szostak, Wiley-VCH, Weinheim, 2022, pp. 101–156 Search PubMed.
- (a) S. Nahm and S. M. Weinreb, Tetrahedron Lett., 1981, 22, 3815 CrossRef CAS; (b) V. Pace, L. Castoldi and W. Holzer, J. Org. Chem., 2013, 78, 7764 CrossRef CAS.
- Although much less stable, also hemiaminals generated by adding carbanion-type reagents to Weinreb amides, have been isolated. For a contribution by our group, see: L. Castoldi, W. Holzer, T. Langer and V. Pace, Chem. Commun., 2017, 53, 9498 RSC.
- For reviews, see: (a) Y.-Y. Huang, C. Cai, X. Yang, Z.-C. Lv and U. Schneider, ACS Catal., 2016, 6, 5747 CrossRef CAS; (b) X.-Y. Ma, F.-Q. Shao, X. Hu and X. Liu, Synthesis, 2022, 54, 1203 CrossRef CAS; (c) L. Duhamel, in Amino, Nitrosco and Nitro Compounds and Their Derivatives, ed. S. Patai, 1982, p. 849 Search PubMed.
- (a) R. P. Linstead, B. R. Shephard and B. C. L. Weedon, J. Chem. Soc., 1951, 2854 RSC; (b) S. D. Ross, M. Finkelstein and R. C. Petersen, J. Am. Chem. Soc., 1966, 88, 4657 CrossRef CAS.
- (a) A. R. Katritzky, W. Q. Fan, M. Black and J. Pernak, J. Org. Chem., 1992, 57, 547 CrossRef CAS; (b) For the preparation of i-propoxy analogues, see also: R. V. Hoffman and N. K. Nayyar, J. Org. Chem., 1994, 59, 3530 CrossRef CAS.
- For reviews on N-acyliminium ions, see: (a) P. Wu and T. E. Nielsen, Chem. Rev., 2017, 117, 7811 CrossRef CAS PubMed; (b) A. J. Basson and M. G. McLaughlin, Tetrahedron, 2022, 114, 132764 CrossRef CAS.
- T. L. Rathman and J. A. Schwindeman, Org. Process Res. Dev., 2014, 18, 1192 CrossRef CAS.
- (a) S. L. Graham and T. H. Scholz, Tetrahedron Lett., 1990, 31, 6269 CrossRef CAS; (b) See also for additional examples: G. E. Keck, S. F. McHardy and J. A. Murry, Tetrahedron Lett., 1993, 34, 6215 CrossRef CAS; (c) O. Labeeuw, P. Phansavath and J.-P. Genêt, Tetrahedron Lett., 2004, 45, 7107 CrossRef CAS; (d) M. P. Sibi, R. Sharma and K. L. Paulson, Tetrahedron Lett., 1992, 33, 1941 CrossRef CAS.
- For authoritative references, see: (a) M. C. Whisler, S. MacNeil, V. Snieckus and P. Beak, Angew. Chem., Int. Ed., 2004, 43, 2206 CrossRef CAS PubMed; (b) J. Clayden, Organolithiums: Selectivity for Synthesis, Elsevier, 2002 Search PubMed; (c) P. Beak and D. B. Reitz, Chem. Rev., 1978, 78, 275 CrossRef CAS; (d) V. Capriati, F. M. Perna and A. Salomone, Dalton Trans., 2014, 43, 14204 RSC; (e) Lithium Compounds in Organic Synthesis: From Fundamentals to Applications, ed. R. Luisi and V. Capriati, Wiley-VCH, Weinheim, 2014 Search PubMed.
- (a) F. A. Davis, K. R. Prasad, M. B. Nolt and Y. Wu, Org. Lett., 2003, 5, 925 CrossRef CAS PubMed; (b) F. A. Davis and M. Song, Org. Lett., 2007, 9, 2413 CrossRef CAS PubMed; (c) F. A. Davis and N. Theddu, J. Org. Chem., 2010, 75, 3814 CrossRef CAS PubMed; (d) R. De la Campa and J. Flórez, Chem.–Eur. J., 2015, 21, 1854 CrossRef CAS PubMed; (e) M. J. Houghton and D. B. Collum, J. Org. Chem., 2016, 81, 11057 CrossRef CAS.
- (a) S. Hirner, O. Panknin, M. Edefuhr and P. Somfai, Angew. Chem., Int. Ed., 2008, 47, 1907 CrossRef CAS; (b) S. Hirner, D. K. Kirchner and P. Somfai, Eur. J. Org Chem., 2008, 5583 CrossRef CAS; (c) S. Hirner and P. Somfai, J. Org. Chem., 2009, 74, 7798 CrossRef CAS PubMed.
- N. Takeda, E. Futaki, Y. Kobori, M. Ueda and O. Miyata, Angew. Chem., Int. Ed., 2017, 56, 16342 CrossRef CAS PubMed.
- (a) M. Yus, G. Radivoy and F. Alonso, Synthesis, 2001, 914 CrossRef CAS; (b) C. Taillier, V. Bellosta, C. Meyer and J. Cossy, Org. Lett., 2004, 6, 2145 CrossRef CAS PubMed; (c) J. E. Jackson, B. N. O'Brien, S. K. Kedzior, G. R. Fryz, F. S. Jalloh, A. Banisafar, M. A. Caldwell, M. B. Braun, B. M. Dunyak and J. L. Dye, Tetrahedron Lett., 2015, 56, 6227 CrossRef CAS; (d) T. You, M. Zhang, J. Chen, H. Liu and Y. Xia, Org. Chem. Front., 2021, 8, 112 RSC; (e) L. E. Fisher, J. M. Caroon, Jahangir, S. R. Stabler, S. Lundberg and J. M. Muchowski, J. Org. Chem., 1993, 58, 3643 CrossRef CAS; (f) S. P. Y. Cutulic, J. A. Murphy, H. Farwaha, S.-Z. Zhou and E. Chrystal, Synlett, 2008, 2132 CAS.
- J. Soika, C. McLaughlin, T. Neveselý, C. G. Daniliuc, J. J. Molloy and R. Gilmour, ACS Catal., 2022, 12, 10047 CrossRef CAS.
- (a) P. Beak, B. G. McKinnie and D. B. Reitz, Tetrahedron Lett., 1977, 18, 1839 CrossRef; (b) D. R. Hay, Z. Song, S. G. Smith and P. Beak, J. Am. Chem. Soc., 1988, 110, 8145 CrossRef CAS; (c) P. Beak, S. T. Kerrick and D. J. Gallagher, J. Am. Chem. Soc., 1993, 115, 10628 CrossRef CAS; (d) P. Beak and A. I. Meyers, Acc. Chem. Res., 1986, 19, 356 CrossRef CAS.
- (a) S. J. Blanksby and G. B. Ellison, Acc. Chem. Res., 2003, 36, 255 CrossRef CAS PubMed; (b) Y.-R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC Press, Boca Raton, 2007 CrossRef.
- For authoritative references, see: (a) H. J. Reich, Chem. Rev., 2013, 113, 7130 CrossRef CAS PubMed; (b) D. B. Collum, Acc. Chem. Res., 1992, 25, 448 CrossRef CAS; (c) D. Hoffmann and D. B. Collum, J. Am. Chem. Soc., 1998, 120, 5810 CrossRef CAS; (d) R. A. Gossage, J. T. B. H. Jastrzebski and G. van Koten, Angew. Chem., Int. Ed., 2005, 44, 1448 CrossRef CAS PubMed; (e) V. H. Gessner, C. Däschlein and C. Strohmann, Chem.–Eur. J., 2009, 15, 3320 CrossRef CAS PubMed; (f) The use of additives enables to finely tailor the reaction site of an organolithium. Thomas and Gutierrez just reported the selective deprotonation at C3 of THF when HMPA was present: in its absence, deprotonation at C2 is observed. See: M. P. Crockett, J. Piña, A. R. Gogoi, R. F. Lalisse, A. V. Nguyen, O. Gutierrez and A. A. Thomas, J. Am. Chem. Soc., 2023, 145, 10743 CrossRef CAS PubMed.
- For reviews on the use of 2-MeTHF in organic synthesis, see: (a) V. Pace, P. Hoyos, L. Castoldi, P. Domínguez de María and A. R. Alcántara, ChemSusChem, 2012, 5, 1369 CrossRef CAS PubMed; (b) D. F. Aycock, Org. Process Res. Dev., 2007, 11, 156 CrossRef CAS; (c) S. Monticelli, L. Castoldi, I. Murgia, R. Senatore, E. Mazzeo, J. Wackerlig, E. Urban, T. Langer and V. Pace, Monatsh. Chem., 2017, 148, 37 CrossRef CAS PubMed; (d) V. Pace, Aust. J. Chem., 2012, 65, 301 CrossRef CAS; (e) For seminal studies on the improved stability of 2-MeTHF towards strongly basic organolithiums, see: R. B. Bates, L. M. Kroposki and D. E. Potter, J. Org. Chem., 1972, 37, 560 CrossRef CAS; (f) For recent studies highlighting the beneficial application of 2-MeTHF in organolithium chemistry, see for example: M. Fairley, L. J. Bole, F. F. Mulks, L. Main, A. R. Kennedy, C. T. O'Hara, J. García-Alvarez and E. Hevia, Chem. Sci., 2020, 11, 6500 RSC; (g) V. Pace, K. de la Vega-Hernández, E. Urban and T. Langer, Org. Lett., 2016, 18, 2750 CrossRef CAS PubMed; (h) V. Pace, L. Castoldi, A. R. Alcántara and W. Holzer, Green Chem., 2012, 14, 1859 RSC; (i) G. Carbone, P. O'Brien and G. Hilmersson, J. Am. Chem. Soc., 2010, 132, 15445 CrossRef CAS PubMed.
- The deaggregating effect induced by PMDTA on s-BuLi was observed in THF, see: (a) W. Bauer, W. R. Winchester and P. v. R. Schleyer, Organometallics, 1987, 6, 2371 CrossRef CAS; (b) Klumpp reported that PMDTA (and relating polyamines) could be lithiated and, thus act as effective carbanions, instead of the same organolithium. However, polyamines deprotonations require proper thermal activation (from low temperature to 0 °C or even rt). Such conditions, however, have not been employed in the present study in which temperature was constantly kept at −78 °C. For polyamines deprotonations, see: M. Schakel, M. P. Aarnts and G. W. Klumpp, Recl. Trav. Chim. Pays-Bas, 1990, 109, 305 CrossRef CAS; (c) G. W. Klumpp, H. Luitjes, M. Schakel, F. J. J. de Kanter, R. F. Schmitz and N. J. R. v. E. Hommes, Angew. Chem., Int. Ed., 1992, 31, 633 CrossRef; (d) H. Luitjes, M. Schakel, M. P. Aarnts, R. F. Schmitz, F. J. J. de Kanter and G. W. Klumpp, Tetrahedron, 1997, 53, 9977 CrossRef CAS; (e) For more recent studies on the thermal induced deprotonation of polyamines, see: C. Strohmann and V. H. Gessner, Angew. Chem., Int. Ed., 2007, 46, 4566 CrossRef CAS PubMed; (f) Y. Wang, J. Liu, L. Huang, R. Zhu, X. Huang, R. Moir and J. Huang, Chem. Commun., 2017, 53, 4589 RSC; (g) Collectively, these literature precedents would support that under our reaction conditions, the effective deprotonating agent is the deaggregated s-BuLi. Very recently, Hevia showed the fundamental role of PMDTA during the sodiation of toluenes to generate formal NaCH2Ar. Notably, these nucleophiles were employed for acylating Weinreb amides, furnishing α-arylacetophenones. See: D. E. Anderson, A. Tortajada and E. Hevia, Angew. Chem., Int. Ed., 2023, 62, e202218498 CrossRef CAS PubMed; (h) For additional studies on the role of disaggregating agents in carbanion chemistry, see for example: V. H. Gessner and C. Strohmann, J. Am. Chem. Soc., 2008, 130, 14412 CrossRef CAS PubMed; (i) A. Harrison-Marchand and F. Mongin, Chem. Rev., 2013, 113, 7470 CrossRef CAS PubMed.
- The lower polarity of 2-MeTHF (compared to THF, as judged by dieletrics constants ε) has been often used for supporting enhanced stereoselectivities observed when reactions were run in 2-MeTHF. For recent perspectives, see: (a) J. García-Álvarez, E. Hevia and V. Capriati, Chem.–Eur. J., 2018, 24, 14854 CrossRef PubMed; (b) J. García-Álvarez, E. Hevia and V. Capriati, Eur. J. Org Chem., 2015, 2015, 6779 CrossRef; (c) M. Miele, V. Pillari, V. Pace, A. R. Alcántara and G. de Gonzalo, Molecules, 2022, 27, 6701 CrossRef CAS PubMed See also, ref. 30a–d and works cited therein..
- S. P. H. Mee, V. Lee and J. E. Baldwin, Angew. Chem., Int. Ed., 2004, 43, 1132 CrossRef CAS PubMed.
- P. A. Forero-Cortés and A. M. Haydl, Org. Process Res. Dev., 2019, 23, 1478 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2191936. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc03216g |
| This journal is © The Royal Society of Chemistry 2023 |