
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ferrous and ferric complexes with cyclometalating N-heterocyclic carbene ligands: a case of dual emission revisited†
Catherine Ellen
Johnson
 a,
Jesper
Schwarz
b,
Mawuli
Deegbey
c,
Om
Prakash
b,
Kumkum
Sharma
b,
Ping
Huang
a,
Tore
Ericsson
d,
Lennart
Häggström
d,
Jesper
Bendix
e,
Arvind Kumar
Gupta
b,
Elena
Jakubikova
*c,
Kenneth
Wärnmark‡
*b and
Reiner
Lomoth‡
*a
a,
Jesper
Schwarz
b,
Mawuli
Deegbey
c,
Om
Prakash
b,
Kumkum
Sharma
b,
Ping
Huang
a,
Tore
Ericsson
d,
Lennart
Häggström
d,
Jesper
Bendix
e,
Arvind Kumar
Gupta
b,
Elena
Jakubikova
*c,
Kenneth
Wärnmark‡
*b and
Reiner
Lomoth‡
*a
aDepartment of Chemistry –Ångström Laboratory, Uppsala University, Box 523, SE-75120 Uppsala, Sweden. E-mail: reiner.lomoth@kemi.uu.se
bCentre for Analysis and Synthesis, Department of Chemistry, Lund University, Box 124, SE-22100 Lund, Sweden
cDepartment of Chemistry, North Carolina State University, Raleigh, North Carolina 27695, USA
dDepartment of Physics – Ångström Laboratory, Uppsala University, Box 523, SE-751 20 Uppsala, Sweden
eDepartment of Chemistry, University of Copenhagen, Universitetsparken 5, DK-2100 Copenhagen, Denmark
First published on 29th August 2023
Abstract
Iron N-heterocyclic carbene (FeNHC) complexes with long-lived charge transfer states are emerging as a promising class of photoactive materials. We have synthesized [FeII(ImP)2] (ImP = bis(2,6-bis(3-methylimidazol-2-ylidene-1-yl)phenylene)) that combines carbene ligands with cyclometalation for additionally improved ligand field strength. The 9 ps lifetime of its 3MLCT (metal-to-ligand charge transfer) state however reveals no benefit from cyclometalation compared to Fe(II) complexes with NHC/pyridine or pure NHC ligand sets. In acetonitrile solution, the Fe(II) complex forms a photoproduct that features emission characteristics (450 nm, 5.1 ns) that were previously attributed to a higher (2MLCT) state of its Fe(III) analogue [FeIII(ImP)2]+, which led to a claim of dual (MLCT and LMCT) emission. Revisiting the photophysics of [FeIII(ImP)2]+, we confirmed however that higher (2MLCT) states of [FeIII(ImP)2]+ are short-lived (<10 ps) and therefore, in contrast to the previous interpretation, cannot give rise to emission on the nanosecond timescale. Accordingly, pristine [FeIII(ImP)2]+ prepared by us only shows red emission from its lower 2LMCT state (740 nm, 240 ps). The long-lived, higher energy emission previously reported for [FeIII(ImP)2]+ is instead attributed to an impurity, most probably a photoproduct of the Fe(II) precursor. The previously reported emission quenching on the nanosecond time scale hence does not support any excited state reactivity of [FeIII(ImP)2]+ itself.
Introduction
The past decade has seen remarkable progress towards photoactive first-row transition metal complexes1–3 by ligand design that has to an increasing extent overcome the problem of ultrafast deactivation of charge transfer states via low-lying metal-centered states.4 As a result, Earth-abundant metal complexes showing luminescence and excited-state reactivity have become available. They enable the substitution of precious metal photosensitizers and luminophores for the conversion of radiant-to-electric energy or vice versa in e.g. solar energy conversion and photoredox catalysis5–8 or OLEDs.9,10 Iron complexes as analogues of the archetypal ruthenium polypyridyl photosensitizers11 have since long attracted particular interest12–20 and the more recent progress with N-heterocyclic carbene (NHC) ligands21–28 has eventually led to ferrous and ferric complexes with demonstrated excited-state electron transfer reactivity of their triplet metal-to-ligand charge transfer (3MLCT)29–35 and doublet ligand-to-metal charge transfer (2LMCT)36–42 states, respectively. Furthermore, with lifetimes up to nanoseconds and larger oscillator strengths of the spin-allowed transition between the 2LMCT and the doublet ground state, Fe(III)NHCs can also feature significant photoluminescence quantum yields.29,43,44 Emission from the 3MLCT states of the Fe(II)NHCs remains, however, to be demonstrated as it is precluded by current sub-nanosecond lifetimes in combination with the spin-forbidden radiative decay. 2MLCT states of Fe(III) complexes are on the other hand energetically above the 2LMCT states and are expected to undergo rapid non-radiative deactivation that should typically preclude any emission from these higher states. We were hence intrigued by the remarkable photophysics of an Fe(III) complex with the tridentate ligand bis(2,6-bis(3-methylimidazol-2-ylidene-1-yl))phenylene-1-yl (ImP),45–48 containing two N-heterocyclic carbenes in conjugation with one cyclometalating arene moiety. The previously described complex [FeIII(ImP)2]+,45 was recently reported to feature emission from a higher energy 2MLCT state next to the more expected emission from the lowest energy 2LMCT state as a first example of dual emission from an FeNHC complex.49 Regarding the nanosecond lifetime of the blue emission attributed to the 2MLCT state, we were concerned about the absence of an excited state with a corresponding lifetime in the reported transient absorption data of [FeIII(ImP)2]+. Since we have previously found that FeNHC complexes can combine remarkable excited-state properties in both Fe(III) and Fe(II) oxidation states of the same complex,41 we were enticed to compare the notable photophysical properties reported for [FeIII(ImP)2]+ to those of its hitherto unexplored Fe(II) analogue.Here we report on the synthesis, characterization and photophysics of the [FeII(ImP)2] complex and have revisited the photophysics of its Fe(III) analogue. Based on our data, [FeIII(ImP)2]+ lacks the previously reported dual emission while its non-luminescent, photolabile Fe(II) precursor was identified as a source of a blue-emitting photoproduct.
Results and discussion
Synthesis and characterization
Pristine [FeIII(ImP)2]+ (ImP = bis(2,6-bis(3-methylimidazol-2-ylidene-1-yl)phenylene)) was synthesized (see ESI for details†) from the ligand precursor 1,1′-(1,3-phenylene)bis(3-methyl-1-imidazolium) dibromide (HImPBr2) which was reacted with tetrakis(dimethylamido)zirconium in tetrahydrofuran (THF) under inert atmosphere and in the dark (Fig. 1a).45 Transmetalation with FeBr2 resulted in the formation of the bis-tridentate complex. Work-up under air was enough to fully oxidize the complex to the Fe(III) state. The synthesis is similar to that originally described by Hollis and Webster,45 and later adapted by Bauer and coworkers,49 however differing in several aspects. Our synthesis used the bromide salt instead of the iodide salt of the ligand precursor, [HImP]2+, introducing another purification step of the ligand precursor. Similar to Bauer, we used FeIIBr2, instead of FeIIICl3 as Hollis and Webster. We ensured to run the reaction in the dark, but some exposure to ambient light during setup and work-up was allowed. The complex was finally purified by size exclusion chromatography (BioBeads S-X1) to obtain pristine [FeIII(ImP)2]PF6 (see ESI for experimental details†).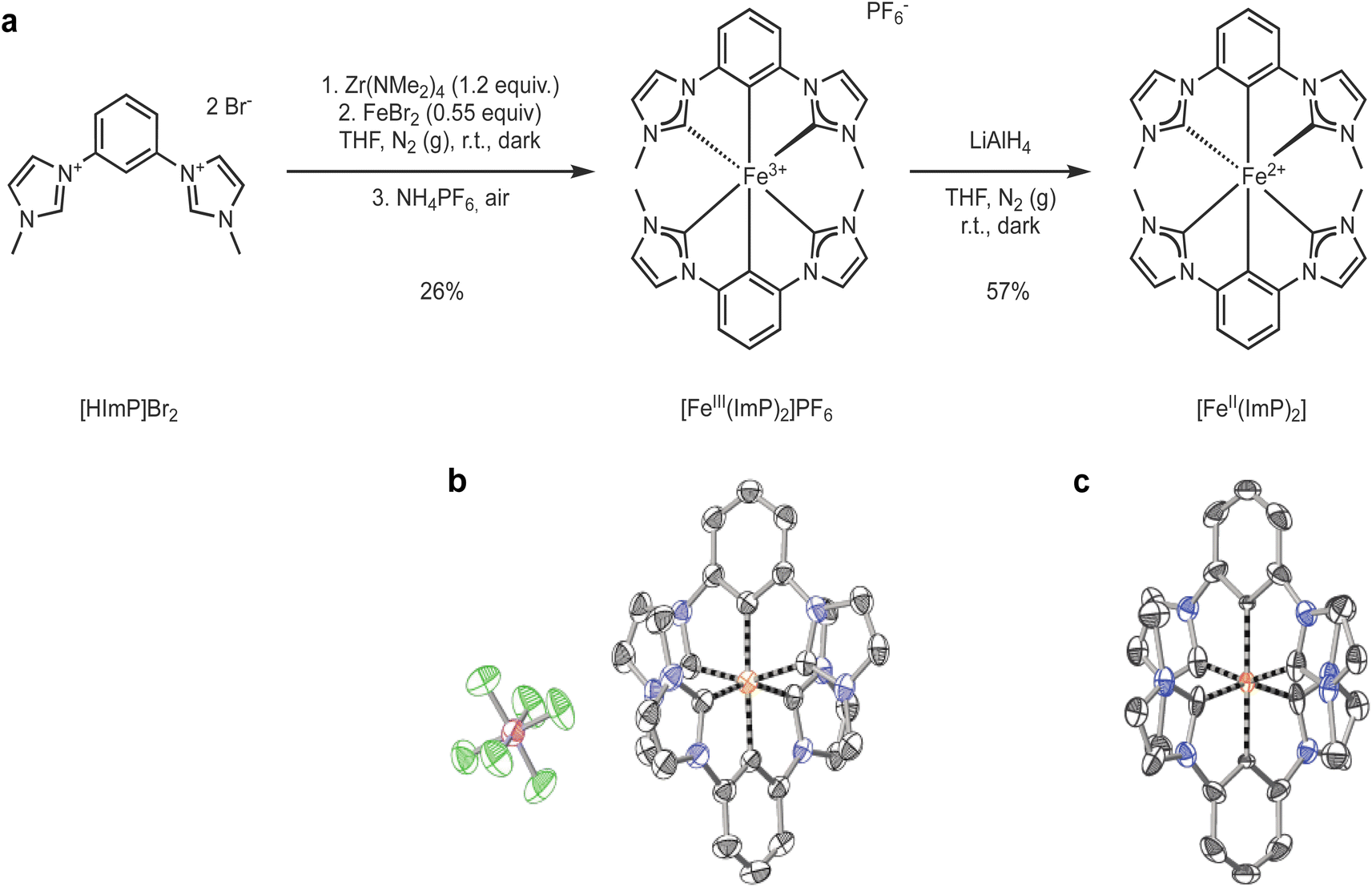 | ||
| Fig. 1 (a) Synthesis of [FeIII(ImP)2]PF6 and [FeII(ImP)2] from the ligand precursor 1,1′-(1,3-phenylene)bis(3-methyl-1-imidazolium) dibromide, [HImp]Br2. (b) X-ray diffraction structure of [FeIII(ImP)2]PF6 with C in black, F in green, Fe in orange, N in blue, and P in purple. Hydrogen atoms are omitted for clarity. (c) X-ray diffraction structure of [FeII(ImP)2]. | ||
[FeIII(ImP)2]PF6 was characterized by NMR, HRMS, elemental analysis and single crystal X-ray diffraction, all consistent with the structure and composition of the complex (Fig. 1b and ESI†). The NMR showed large paramagnetic shifts, with 1H-NMR shifts ranging from +25 to −36 ppm and 13C-NMR shifts from +520 to −200 ppm. Despite several attempts, it was not possible to detect the carbon resonances directly bound to the iron, presumably due to fast relaxation aided by the paramagnetic iron. The spin state of the complex was further characterized by magnetic susceptibility measurements in solution, solid-state magnetization, and Mößbauer measurements (see ESI†). The paramagnetic susceptibility of [FeIII(ImP)2]+ in acetonitrile was determined by Evans' NMR method to be 2.09 ± 0.04 Bohr magneton (μB). In the solid state, the magnetization (M) of [FeIII(ImP)2]PF6 shows a variation with the reduced field (BT−1) consistent with the solution data, indicating a saturation only slightly above 1 μB (Fig. S3†). The isothermally measured magnetization curves are superimposable in the (BT−1) plot, demonstrating the absence of zero field splitting as required for the low spin (LS) t52g ground state. Mößbauer spectra of [FeIII(ImP)2]PF6 at 295 K and 85 K reveal a quadrupole split doublet structure with slight asymmetry, especially for the 85 K spectrum (Fig. S4†). These measurements are all consistent with a d5 LS electronic configuration (t52g, S = 1/2) enforced by the strong field carbene ligands.
[FeII(ImP)2] was obtained by reduction of its Fe(III) precursor [FeIII(ImP)2]+ using LiAlH4 in THF solution. The change in oxidation state was accompanied by a drastic color change from dark blue to bright orange. The sensitive product could be isolated and characterized as a diamagnetic (LS d6) FeII-complex by NMR, elemental analysis, and single crystal X-ray diffraction (Fig. 1c and ESI†). The FeII structure is very similar to the FeIII structure, in accordance with what has been observed for other iron NHC complexes isolated in the two oxidation states.43,50 The Fe–C distances of 1.93 Å are very similar to those in the related [Fe(pbmi)2](PF6)2 (pbmi = 1,1′-(pyridine-2,6-diyl)bis(methylimidazol-2-ylidene)) complex containing a central pyridine moiety instead of a phenyl anion.24
Photophysics of [FeIII(ImP)2]+
Recognizing that the very oxidation-sensitive [FeII(ImP)2] might potentially contain some [FeIII(ImP)2]+ (see below), it was necessary to reassess the photophysical properties of the Fe(III) complex for reference. The electronic absorption spectrum of [FeIII(ImP)2]+ in acetonitrile (Fig. 2a and S9†) is in good agreement with published data and the previous assignment of the absorption bands at about 600 nm and 350 nm to transitions with strong LMCT and MLCT character, respectively,49 is supported by our computational data (Fig. S19†). Photoexcitation of [FeIII(ImP)2]+ into its lowest-energy absorption band (LMCT, λex = 585 nm) reproduced the reported red emission peaking at around 750 nm with its excitation spectrum matching well with the ground state absorption spectrum (Fig. 2a). Notably, upon excitation of [FeIII(ImP)2]+ into its higher-energy absorption band (MLCT, λex = 350 nm, Fig. 2a), we observed only a very minor blue emission. The intensity ratio between the blue and red emission bands is very different from the reported data with the dominating blue emission.49 Hence, it was evident that the two emission bands cannot be attributed to a dual emission of [FeIII(ImP)2]+, indicating that the blue emission must instead originate from a different species.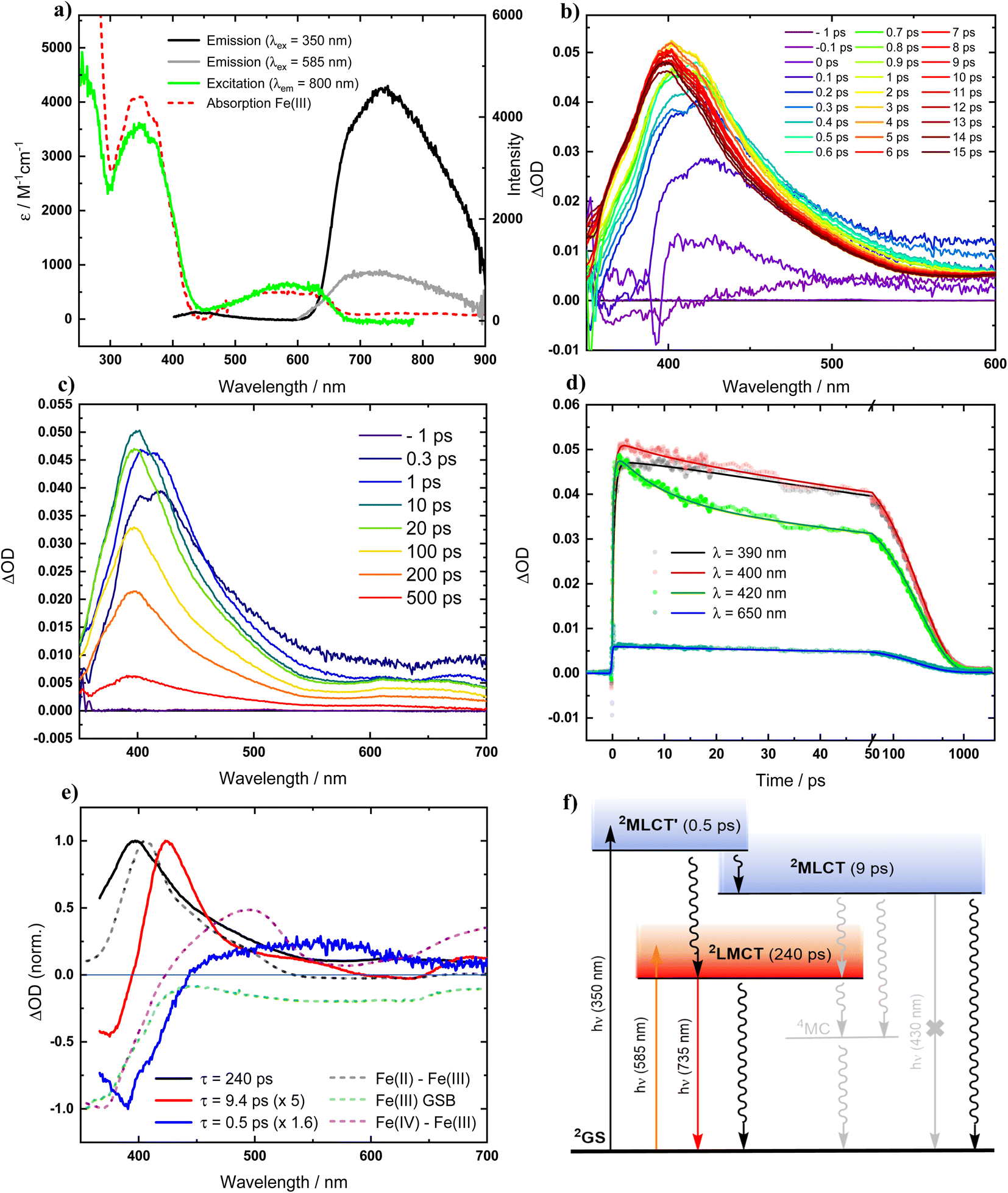 | ||
| Fig. 2 Photophysics of [FeIII(ImP)2]+ in deaerated acetonitrile solution (at room temperature). (a) Absorption, emission and excitation spectra. (b) and (c) Transient absorption spectra at indicated time delays after photoexcitation (λex = 350 nm, 140 fs). (d) Transient absorption kinetics at indicated wavelengths, fitted by global analysis. (e) Normalized decay-associated spectra (DAS) from global analysis of transient absorption spectra along with the differential absorption spectra for the metal centered reduction and oxidation of the complex and the expected ground-state bleach (GSB). (f) Schematic state energy diagram. | ||
In view of the above discrepancy, we also reexamined the excited-state dynamics of [FeIII(ImP)2]+ by fs-transient absorption spectroscopy (fs-TA) in acetonitrile. TA spectra upon excitation of [FeIII(ImP)2]+ in its lowest energy absorption band (λex = 585 nm) are in good agreement with the reported data49 confirming the formation of a 2LMCT state with a pronounced 400 nm absorption band characteristic of its Fe(II) nature and a lifetime of 230 ps (Fig. S12†). The same transient absorption spectrum is observed upon excitation of [FeIII(ImP)2]+ into its higher energy (MLCT) absorption band (λex = 350 nm). In this case, the 2LMCT state is not formed instantaneously but emerges within about a picosecond (Fig. 2b–d) before it decays to the GS with the established lifetime of 240 ps.49 For the initially populated excited state which eventually gave rise to the 2LMCT state, global analysis returns a lifetime of 0.5 ps. The corresponding decay associated spectrum (DAS) shown in Fig. 2e would be consistent with an excited state of MLCT character that we denote as 2MLCT′. The decay of this state not only gives rise to the 2LMCT state but leads in parallel to the formation of a third transient as indicated by the 420 nm peak that can be seen next to the 400 nm peak of the 2LMCT state during the first few picoseconds. Global analysis returned a lifetime of 9 ps and a DAS (Fig. 2e) that combines features expected for Fe(III) to Fe(IV) oxidation and a 420 nm peak that we tentatively attribute to the superimposed absorption of a ligand radical anion in an electronically and vibrationally relaxed MLCT state here denoted as 2MLCT. Overall, our TA data on [FeIII(ImP)2]+ essentially reproduced the observations presented in the previous report, with a relatively minor difference (6 ps vs. 9 ps) for one of the time constants. However, our interpretation of the data, as summarized in the schematic state energy diagram (Fig. 2f), disagrees strongly with the interpretation put forward in the previous report. While it is undebatable that photoexcitation of [FeIII(ImP)2]+ in acetonitrile eventually leads to a relatively long-lived (240 ps) 2LMCT state responsible for the red emission, the TA data obviously does not support the existence of any excited state with a lifetime that matches the 4.6 ns decay time of the blue emission described in a previous report.49 Instead, the two higher excited states populated upon UV excitation, here formally denoted as 2MLCT′ and 2MLCT, decay with time constants of 0.5 ps and 9 ps, respectively. The reported assignment of these time constants (0.5 ps and 6 ps in the previous report49) to parallel decay paths of the initially excited state leading to the 2LMCT state and a long-lived (4.6 ns) 2MLCT state disagrees with the TA data and the underlying kinetic scheme. Instead, the decay of the common precursor as well as the parallel formation of the 2MLCT and 2LMCT product states are described with a single time constant τ = 1/∑ki where ki are the individual rate constants for all decay pathways of the precursor; i.e. with the observed 0.5 ps. With the 9 ps (6 ps) decay of the 2MLCT state, the assignment of the 4.6 ns emission to this state is evidently inconsistent with the TA data. The TA results hence corroborate our conclusion that the blue emission cannot be attributed to any excited state of [FeIII(ImP)2]+ itself but originates instead from some impurity. Minor quantities of an impurity would readily escape detection by transient absorption spectroscopy but could be easily detectable by its emission. The appealing notion of an emissive Fe(II) impurity in the form of [FeII(ImP)2] is at odds with the relatively high energy of the emission band compared to the lowest energy absorption band of [FeII(ImP)2] (Fig. S9† and 3a). Instead, its photophysical characterization (see below) revealed the formation of an emissive photoproduct from the Fe(II) complex that can contaminate [FeIII(ImP)2]+ prepared from its Fe(II) precursor and thereby give rise to the blue emission. It is therefore important to note that the reported quenching of the blue nanosecond emission attributed to the 2MLCT state of [FeIII(ImP)2]+ has instead to be ascribed to the quenching of emission from an impurity. Reactions involving the non-emissive 2MLCT states of [FeIII(ImP)2]+ cannot be monitored by emission quenching and would be limited to conditions eliminating the need for diffusional approach of the reactants during the very short (<10 ps) lifetime at extremely high quencher concentrations or in molecular assemblies. It is only the 2LMCT state of [FeIII(ImP)2]+ that provides a lifetime (240 ps) that might be sufficient for reasonably efficient diffusional reactions, but still too short to account for the reported emission quenching on the ns timescale. Despite its lower excited state energy (1.89 eV), it is in theory still rather strongly oxidizing (0.73 V vs. Fc0/+) and reducing (−1.85 V vs. Fc0/+) but offers yet no advantages over e.g. the 2LMCT state of [FeIII(phtmeimb)2]+ (phtmeimb = [phenyltris(3-methyl-imidazolin-2-ylidene-1-yl)borate]−) (2 ns, 2.12 eV, 0.97 V, −1.88 V), the Fe(III)NHC complex having the longest excited state lifetime to date.29 Furthermore, as far as reductive quenching cycles of [FeIII(ImP)2]+ are concerned, such applications would need to avoid coordinating solvents in combination with exposure to light with wavelengths <550 nm to safely circumvent degradation of the photolabile Fe(II) state (see below).
Photophysics of [FeII(ImP)2]
Absorption spectra of [FeII(ImP)2] in acetonitrile obtained by spectroelectrochemistry and by chemical synthesis and isolation, respectively (Fig. 3a and S9†), are in good agreement with each other, but differ significantly from the published spectrum.49 Specifically, our spectra unambiguously disprove any absorption above 550 nm and reveal a much more pronounced 400 nm peak. It is hence the latter band with its low-energy shoulder(s) that represent the lowest-energy absorptions of [FeII(ImP)2]. MLCT excitation with energies of about 3 eV would be in line with the electrochemical data and computational results corroborating the MLCT assignment. The calculated absorption spectrum shows two absorption bands attributed to MLCT transitions (Fig. 4). The lowest-energy absorption band consists of nearly degenerate MLCT transitions that populate the lowest unoccupied molecular orbital (LUMO) and LUMO+1 orbitals. In contrast, the higher-energy transitions predominantly populate LUMO+3 and LUMO+4 orbitals with distinctly different nodal structures (Fig. S23† for MO pictures), which is also reflected in particle states represented by the natural transition orbitals (NTO) (Fig. 4).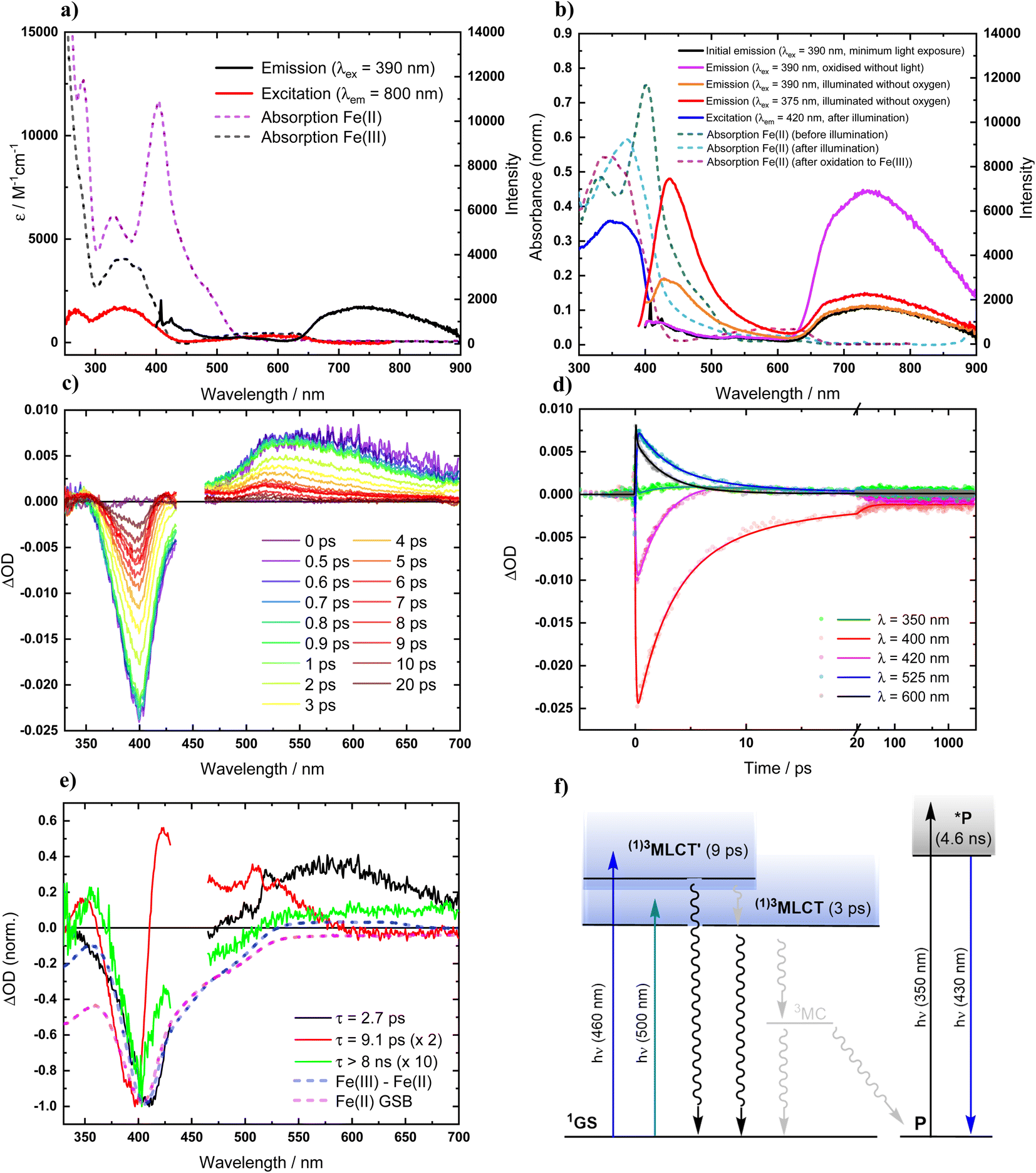 | ||
| Fig. 3 Photophysics of [FeII(ImP)2] in deaerated acetonitrile solution (at room temperature). (a) Absorption, emission and excitation spectra. (b) Effects of oxidation (atmospheric O2) and light exposure (illumination with white light lamp). (c) Transient absorption spectra at indicated time delays after photoexcitation (λex = 450 nm, 140 fs). (d) Transient absorption kinetics at indicated wavelengths, fitted by global analysis. (e) Normalized decay-associated spectra (DAS) from global analysis of transient absorption spectra, along with the differential absorption spectra for the metal centered reduction of the complex and the expected ground-state bleach (GSB). (f) Schematic state energy diagram. | ||
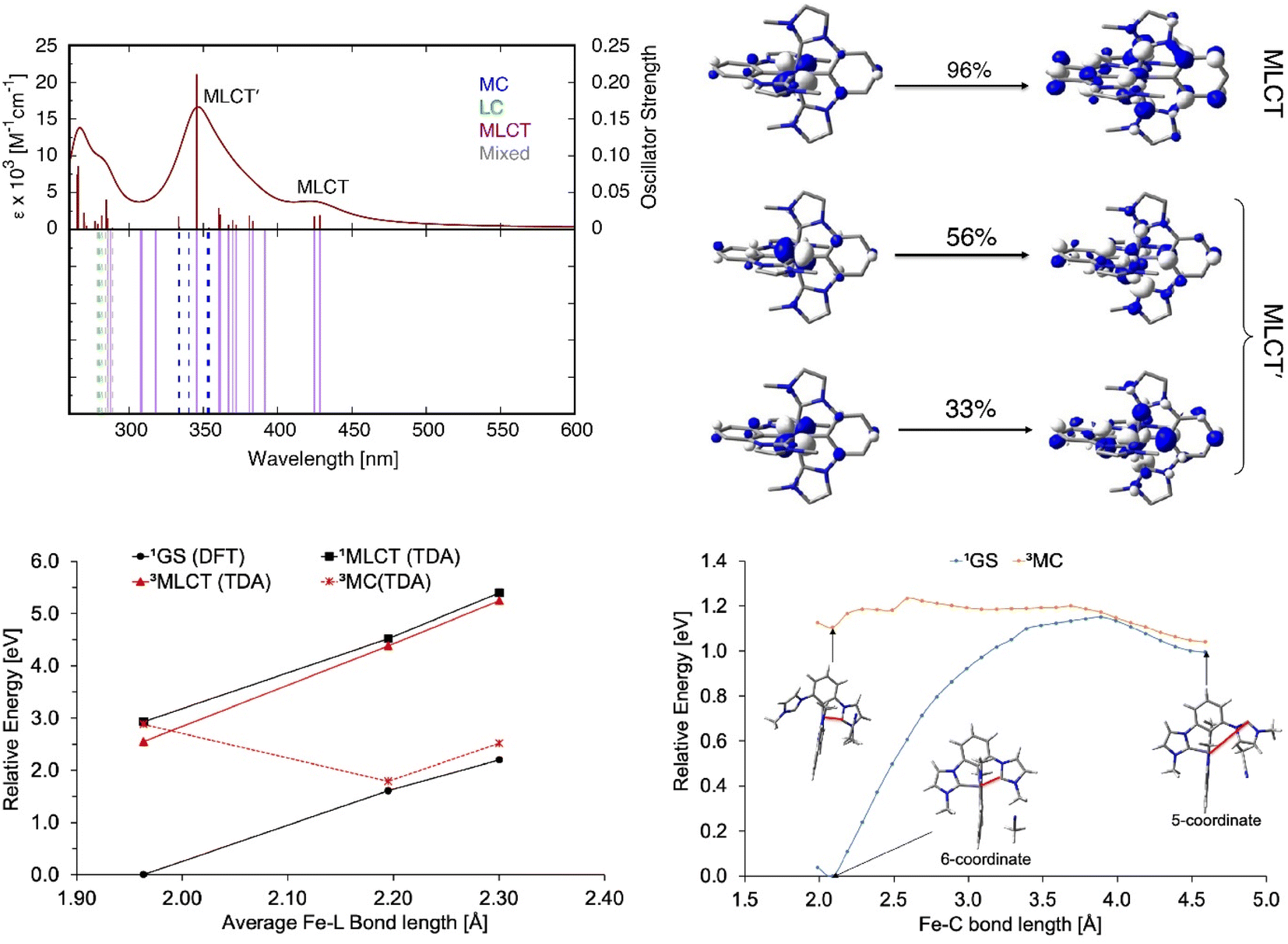 | ||
| Fig. 4 Calculated electronic absorption spectra of [FeII(ImP)2]. Top left: UV-Vis spectra of Fe(II); top right: NTOs for MLCT and MLCT′ transition; bottom left: PECs for Fe(II); bottom right: PECs for the ligand detachment in 1GS and 3MC state. Percent contribution of the NTO pairs to the total excitation is given above the arrow. MLCT denotes metal-to-ligand charge transfer, MC metal-centered, LC ligand-centered. The red line denotes the scanned Fe–C bond length. | ||
Excitation of [FeII(ImP)2] in acetonitrile in the UV region (350 nm) resulted in the emission spectrum shown in Fig. 3a. The broad red emission band suggests some contamination of the highly oxygen-sensitive Fe(II) complex with its Fe(III) congener [FeIII(ImP)2]+. This assignment was further confirmed with the excitation spectrum of the red emission band matching the absorbance spectrum of Fe(III) and the increase of this emission band upon exposure to oxygen (Fig. 3b). The additional band peaking at about 430 nm matches the blue emission band previously attributed to emission from a higher excited state of [FeIII(ImP)2]+ and also the emission lifetime of 5.1 ns (Fig. S10†) agrees well with the reported value of the reported blue emission from [FeIII(ImP)2]+.49 The intensity of the blue emission, however, does not increase together with the red emission upon oxidation (Fig. 3b), supporting our conclusion that [FeIII(ImP)2]+ does not feature any emission in the blue. Instead, the blue emission was found to increase in intensity upon exposure of [FeII(ImP)2] to light indicating that the emissive species is a product of a photoreaction (Fig. 3b). Corresponding absorption changes can also be observed, with a well-defined product absorption peak in the UV (370 nm) that agrees with the excitation spectrum for the blue emission (Fig. 3b and 5a). Control experiments in the dark only showed a slow oxidation to the Fe(III) state (Fig. S5†). While samples of [FeII(ImP)2] inevitably contain some [FeIII(ImP)2]+, any contribution of the latter to the formation of photoproduct(s) can be excluded based on the pronounced photostability of the Fe(III) complex (Fig. S6†). The detailed structure of the emissive species remains unclear at the current stage. However, similar emission (410 nm, 1.7 ns, 4.6 ns) and absorption (365 nm) characteristics were found for the ImP ligand precursor [HImp]2+ (Fig. S11†) suggesting that the emissive species might be some ligand derivative. Furthermore, solutions of [FeII(ImP)2] in THF showed weaker signs of photobleaching without any discernible product absorption (Fig. 5b) indicating some involvement of the solvent in the photodegradation.
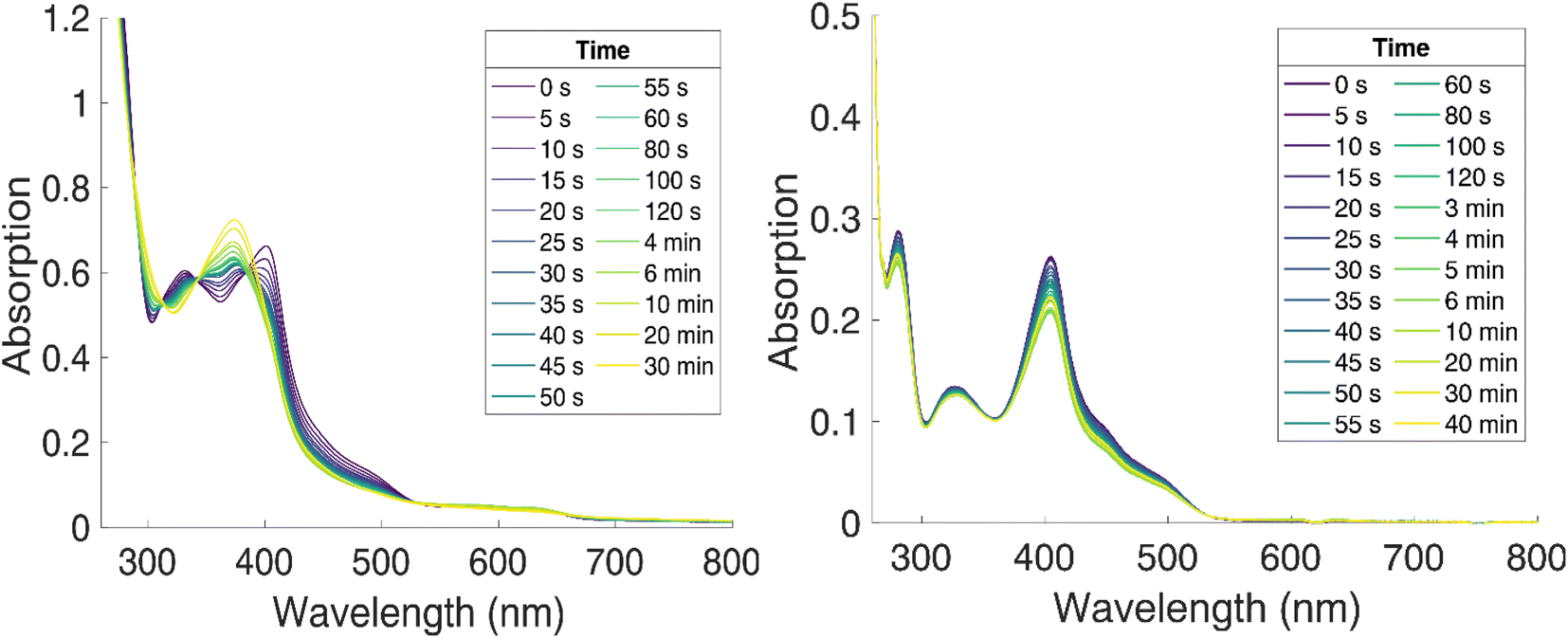 | ||
| Fig. 5 Photostability of [FeII(ImP)2] in acetonitrile (left) and THF (right) under N2 (g), blue light irradiation (λ = 450 nm) at room temperature, monitored by UV-vis absorption spectroscopy. | ||
The potential energy curves (PECs) for the relevant electronic states of the [FeII(ImP)2] complex were also computed (Fig. S27†), revealing the 3MLCT to be the lowest-energy excited state, with the 3MC (metal-centred) and 5MC states 0.33 and 1.82 eV higher in energy, respectively, at the ground-state optimized geometry. At the relaxed geometry of the 3MC, it lies below the 3MLCT, suggesting a possibility of internal conversion. Structures of the 3,5MC states optimized with the density functional theory (DFT) show a partial ligand detachment, suggesting a possibility of the solvent (acetonitrile) coordination in the excited state (Fig. S28–S31 and ESI for further details†). To further examine the partial ImP ligand detachment, we looked at interactions between [FeII(ImP)2] and acetonitrile, in which a single molecule of acetonitrile coordinates to the Fe center upon the partial ligand detachment (“head-on” coordination) or inserts itself into the pocket created by the detached ligand, weakly interacting with the 5-coordinate iron complex (“van der Waals” coordination, see Fig. S28†). We found that although both head-on and van der Waals coordination are thermodynamically uphill for 1GS, in the 3MC and 5MC states the 5-coordinate complex with a partially detached ligand is stabilized through van der Waals coordination of the acetonitrile solvent molecule (Table S10†). The one-dimensional PEC scans connecting the 6-coordinate and 5-coordinate [FeII(ImP)2]-acetonitrile structures show that the partial ligand detachment and acetonitrile coordination are unfavorable in the ground state (the 5-coordinate structure is ∼1 eV or 23 kcal mol−1 higher in energy than the 6-coordinate structure, with a transition state of ∼1.1 eV or 25 kcal mol−1). In contrast to this, the optimized 6-coordinate structure of the 3MC state already features one lengthened Fe–C bond at ∼3.2 Å, and van der Waals coordination of acetonitrile is favored and easily accessible (Fig. 4). Moreover, the 1GS and 3MC states lie near each other in energy at the 5-coordinate geometry, suggesting a possibility of a nonradiative decay from 3MC to 1GS at this geometry, with a subsequent conversion back to a 6-coordinate [FeII(ImP)2] complex (an estimated transition state for this conversion is ∼0.1 eV or 2 kcal mol−1, see Fig. 4), reversing the partial ligand detachment occurring upon irradiation with light. The formation of the 5-coordinate complex could also represent the first step in the formation of a different photoproduct (see above) and more investigation would be necessary to fully explore this issue.
The excited state dynamics of [FeII(ImP)2] in acetonitrile were studied with excitation at 450 nm to avoid excitation of any [FeIII(ImP)2]+ potentially present in the sample. A moving sample stage was employed to limit any effects of irreversible photobleaching or accumulating photoproducts. Good agreement with the TA data obtained in THF solution (Fig. S13–S15†) where [FeII(ImP)2] is more photostable confirmed that with this approach, the TA data collected in acetonitrile can also be attributed to intact [FeII(ImP)2]. The initial transient absorption spectra show a ground state bleach signal at 400 nm and broad transient absorption peaking between 500 nm and 600 nm, which would be consistent with the bleach expected for the Fe(II) to Fe(III) oxidation together with the ligand anion radical absorption of an MLCT state (Fig. 3c and d). Global analysis reveals two major components with lifetimes of 2.7 and 9.1 ps respectively and their normalized DAS are shown in Fig. 3e. The former component is responsible for the faster decay of the absorption >550 nm, while the latter describes the somewhat more long-lived TA below 550 nm, including the more slowly recovering portion on the blue side of the ground state bleach. These features are attributed to two MLCT states with probably not only different degrees of vibrational and solvent relaxation but also different electronic structures in agreement with the computational data. As intersystem crossing can be expected to occur within the instrument response function (IRF) of the TA experiments (<140 fs) and hence would not be resolved,51,52 these states are denoted as 3MLCT′ and 3MLCT, respectively (Fig. 3f). Their short lifetimes established by TA spectroscopy confirm that intact [FeII(ImP)2] is not responsible for the blue nanosecond emission. The kinetics at 420 nm indicate that the 3MLCT forms at least partly concomitant with the decay of 3MLCT′ while a portion of the two states may be formed in parallel during the IRF. Notably, the ligand radical contributions of both 3MLCT states resembles the corresponding spectral features of the two 2MLCT states of the Fe(III) complex where they appear next to the Fe(III) to Fe(IV) oxidation features (Fig. 2e and 3e). A minor part of the ground state bleach after excitation of [FeII(ImP)2] does not recover within the 8 ns time window of the TA experiments. The remaining TA spectrum is consistent with a bleaching of the ground state absorption or alternatively oxidation to the Fe(IV) state and therefore attributed to some irreversible photochemistry. Correspondingly, these irreversible changes were even less prominent in THF where otherwise very similar TA data was obtained with essentially identical spectra and only minor variations in the time constants (4.9 vs. 2.7 ps and 8.4 vs. 9.1 ps) (Fig. S13–S15†). In THF solution, TA data was also collected with 500 nm excitation of [FeII(ImP)2], i.e., at the red edge of its lowest energy absorption band. Under these conditions the TA data greatly simplifies as only the characteristics of a 3MLCT state with a lifetime of 5.4 ps are observed next to the very minor irreversible ground state bleach. A 3MLCT excited state with a lifetime of a few ps is comparable to most Fe(II) complexes with NHC ligands that have excited state energies close to or equal to 2 eV. Together with the low potential of its Fe(III/II) couple, [FeII(ImP)2] is a particularly strong excited state reductant (EIII/II1/2* < −3 V) that might even reduce the acetonitrile solvent but applications will be hampered by the rather short 3MLCT lifetime even if the photolability of the complex can be circumvented in suitable media such as THF. Substantially longer lifetimes of pure Fe 3MLCT states of Fe(II) complexes have so far only been observed for a hexa-NHC complex [Fe(btz)3](PF6)2 (0.5 ns),50 a double cyclometalated phenanthroline complex (1 ns)53 and benzannulated diarylamido complex (3 ns).54 The latter complexes are however characterized by rather low energies of their 3MLCT states (ca. 1–1.2 eV and 1.2 eV) which might be a major factor behind their slower deactivation via MC states.
Conclusions
We have synthesized and characterized the Fe(II) and Fe(III) complexes [FeII(ImP)2] and [FeIII(ImP)2]+ with cyclometalating NHC ligands and have investigated the photophysical properties of the Fe(II) complex and revisited those recently reported for its Fe(III) analogue [FeIII(ImP)2]+ by experimental and computational techniques. For the Fe(II) complex, the moderate lifetime of its 3MLCT state (9 ps in acetonitrile) indicates that the cyclometalating ligand in this case does not provide the expected advantage over previously used NHC/pyridine or pure NHC ligand sets when it comes to the destabilization of metal-centered states. Notably, our results for [FeII(ImP)2] revealed the formation of a luminescent product from the photodegradation of the intrinsically non-luminescent Fe(II) complex. The spectral characteristics and 5.1 ns lifetime of the emission from this product are near identical to the blue emission previously attributed to the emission from a higher (2MLCT) state of its Fe(III) analogue [FeIII(ImP)2]+. For the Fe(III) complex itself, our results confirm however that its higher (2MLCT) excited states are short-lived (<10 ps) and therefore cannot give rise to the reported blue emission with its nanosecond lifetime. Accordingly, pristine [FeIII(ImP)2]+, synthesized by our protocol that eliminates contamination with photoproducts of the Fe(II) precursor, only shows red emission from its lower (2LMCT) state. Regarding applications of [FeIII(ImP)2]+ as a photosensitizer, the previously reported quenching of the nanosecond emission does not demonstrate any excited state reactivity of [FeIII(ImP)2]+ itself but has to be instead attributed to the quenching of the impurity's emission. Generally, these results illustrate the challenges with the assignment of the sometimes rather weak emission from coordination compounds, particularly in the blue part of the spectrum where already minor impurities in the form of e.g., simple organics can give rise to significant emission upon UV excitation.Data availability
Crystallographic data for [FeII(ImP)2] and [FeIII(ImP)2]PF6 have been deposited as CCDC 2254082 and 2254083. Additional experimental details and data are provided in the ESI,† including synthesis, single-crystal XRD, NMR, HRMS, Mößbauer and electronic absorption spectra, electrochemistry and spectroelectrochemistry, magnetic susceptibility and magnetization, TCSPC and fs-TA, stability measurements and DFT calculations.Author contributions
J. S., O. P and K. S. carried out the synthesis of the compounds. J. S. and O. P. did the synthesis characterization of the compounds. C. J. conducted the steady state and time-resolved photophysical measurements, carried out the electrochemical measurements, and analyzed the data. M. D. carried out the quantum chemical investigations and analyzed the data. J. S., P. H. and J. B. carried out the measurements related to the magnetic characterizations and analyzed the data. T. E. and L. H. recorded and analyzed the 57Mößbauer data. A. G. recorded and analyzed the single-crystal X-ray diffraction data for the title compounds. J. S. planned and carried out the (photo)degradation studies and analyzed the results. E. J. planned the quantum chemical investigations and analyzed the results. K. W. conceived, planned the research, and designed the title compounds. R. L. planned and guided the photophysical studies and drafted the manuscript together with C. J. C. J., J. S., M. D., E. J., K. W. and R. L. wrote the manuscript. All authors read and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Sofia Essén for HRMS measurements and Göran Carlström for assistance with NMR measurements. The Swedish Strategic Research Foundation (SSF, EM16- 0067) and the Knut and Alice Wallenberg (KAW, 2018.0074) Foundation are gratefully acknowledged for their support. K. W. acknowledges support from the Swedish Research Council (VR, 2020-03207), the Swedish Energy Agency (Energimyndigheten, P48747-1), the LMK Foundation, and the Sten K Johnson Foundation. J. S. acknowledges support from the Royal Physiographic Society of Lund. R. L. is grateful to the Swedish Research Council for financial support (VR, 2020-05058). E. J. and M. D. gratefully acknowledge the support from the National Science Foundation (CHE-1554855) and the computing resources provided by North Carolina State University High Performance Computing Services Core Facility (RRID: SCR 022168).Notes and references
- O. S. Wenger, Photoactive Complexes with Earth-Abundant Metals, J. Am. Chem. Soc., 2018, 140, 13522–13533 CrossRef CAS PubMed.
- C. Förster and K. Heinze, Photophysics and photochemistry with Earth-abundant metals - fundamentals and concepts, Chem. Soc. Rev., 2020, 49, 1057–1070 RSC.
- C. Wegeberg and O. S. Wenger, Luminescent First-Row Transition Metal Complexes, JACS Au, 2021, 1, 1860–1876 CrossRef CAS PubMed.
- J. K. McCusker, Electronic structure in the transition metal block and its implications for light harvesting, Science, 2019, 363, 484–488 CrossRef CAS PubMed.
- C. B. Larsen and O. S. Wenger, Photoredox Catalysis with Metal Complexes Made from Earth-Abundant Elements, Chemistry, 2018, 24, 2039–2058 CrossRef CAS PubMed.
- L. A. Büldt and O. S. Wenger, Chromium(0), Molybdenum(0), and Tungsten(0) Isocyanide Complexes as Luminophores and Photosensitizers with Long-Lived Excited States, Angew. Chem., Int. Ed., 2017, 56, 5676–5682 CrossRef PubMed.
- B. Bozic-Weber, E. C. Constable and C. E. Housecroft, Light harvesting with Earth abundant d-block metals: Development of sensitizers in dye-sensitized solar cells (DSCs), Coord. Chem. Rev., 2013, 257, 3089–3106 CrossRef CAS.
- L. H. M. de Groot, A. Ilic, J. Schwarz and K. Wärnmark, Iron Photoredox Catalysis-Past, Present, and Future, J. Am. Chem. Soc., 2023, 145, 9369–9388 CrossRef CAS PubMed.
- R. Hamze, J. L. Peltier, D. Sylvinson, M. Jung, J. Cardenas, R. Haiges, M. Soleilhavoup, R. Jazzar, P. I. Djurovich, G. Bertrand and M. E. Thompson, Eliminating nonradiative decay in Cu(I) emitters: > 99% quantum efficiency and microsecond lifetime, Science, 2019, 363, 601–606 CrossRef CAS PubMed.
- M. J. Leitl, V. A. Krylova, P. I. Djurovich, M. E. Thompson and H. Yersin, Phosphorescence versus Thermally Activated Delayed Fluorescence. Controlling Singlet-Triplet Splitting in Brightly Emitting and Sublimable Cu(I) Compounds, J. Am. Chem. Soc., 2014, 136, 16032–16038 CrossRef CAS PubMed.
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. von Zelewsky, Ru(II) Polypyridine Complexes - Photophysics, Photochemistry, Electrochemistry, and Chemi-Luminescence, Coord. Chem. Rev., 1988, 84, 85–277 CrossRef CAS.
- S. Ferrere and B. A. Gregg, Photosensitization of TiO2 by [Fe-II(2,2'-bipyridine-4,4'-dicarboxylic acid)(2)(CN)(2)]: Band selective electron injection from ultra-short-lived excited states, J. Am. Chem. Soc., 1998, 120, 843–844 CrossRef CAS.
- J. E. Monat and J. K. McCusker, Femtosecond Excited-State Dynamics of an Iron(II) Polypyridyl Solar Cell Sensitizer Model, J. Am. Chem. Soc., 2000, 122, 4092–4097 CrossRef CAS.
- W. Zhang, K. S. Kjær, R. Alonso-Mori, U. Bergmann, M. Chollet, L. A. Fredin, R. G. Hadt, R. W. Hartsock, T. Harlang, T. Kroll, K. Kubiček, H. T. Lemke, H. W. Liang, Y. Liu, M. M. Nielsen, P. Persson, J. S. Robinson, E. I. Solomon, Z. Sun, D. Sokaras, T. B. van Driel, T.-C. Weng, D. Zhu, K. Wärnmark, V. Sundström and K. J. Gaffney, Manipulating charge transfer excited state relaxation and spin crossover in iron coordination complexes with ligand substitution, Chem. Sci., 2017, 8, 515–523 RSC.
- L. L. Jamula, A. M. Brown, D. Guo and J. K. McCusker, Synthesis and characterization of a high-symmetry ferrous polypyridyl complex: approaching the 5T2/3T1 crossing point for Fe(II), Inorg. Chem., 2014, 53, 15–17 CrossRef CAS PubMed.
- A. K. C. Mengel, C. Forster, A. Breivogel, K. Mack, J. R. Ochsmann, F. Laquai, V. Ksenofontov and K. Heinze, A Heteroleptic Push-Pull Substituted Iron(II) Bis(tridentate) Complex with Low-Energy Charge-Transfer States, Chem. - Eur. J., 2015, 21, 704–714 CrossRef CAS PubMed.
- A. Breivogel, C. Förster and K. Heinze, A Heteroleptic Bis(tridentate)ruthenium(II) Polypyridine Complex with Improved Photophysical Properties and Integrated Functionalizability, Inorg. Chem., 2010, 49, 7052–7056 CrossRef CAS.
- S. M. Fatur, S. G. Shepard, R. F. Higgins, M. P. Shores and N. H. Damrauer, A Synthetically Tunable System To Control MLCT Excited-State Lifetimes and Spin States in Iron(II) Polypyridines, J. Am. Chem. Soc., 2017, 139, 4493–4505 CrossRef CAS PubMed.
- S. Mukherjee, D. N. Bowman and E. Jakubikova, Cyclometalated Fe(II) Complexes as Sensitizers in Dye-Sensitized Solar Cells, Inorg. Chem., 2015, 54, 560–569 CrossRef CAS PubMed.
- I. M. Dixon, F. Alary, M. Boggio-Pasqua and J. L. Heully, Reversing the relative 3MLCT-3MC order in Fe(II) complexes using cyclometallating ligands: a computational study aiming at luminescent Fe(II) complexes, Dalton Trans., 2015, 44, 13498–13503 RSC.
- P. Chabera, L. Lindh, N. W. Rosemann, O. Prakash, J. Uhlig, A. Yartsev, K. Wärnmark, V. Sundström and P. Persson, Photofunctionality of iron(III) N-heterocyclic carbenes and related d(5) transition metal complexes, Coord. Chem. Rev., 2021, 426, 213517 CrossRef CAS.
- L. Lindh, P. Chabera, N. W. Rosemann, J. Uhlig, K. Wärnmark, A. Yartsev, V. Sundström and P. Persson, Photophysics and Photochemistry of Iron Carbene Complexes for Solar Energy Conversion and Photocatalysis, Catalysts, 2020, 10, 315 CrossRef CAS.
- O. S. Wenger, Is Iron the New Ruthenium?, Chem. - Eur. J., 2019, 25, 6043–6052 CrossRef CAS PubMed.
- Y. Liu, T. Harlang, S. E. Canton, P. Chabera, K. Suarez-Alcantara, A. Fleckhaus, D. A. Vithanage, E. Göransson, A. Corani, R. Lomoth, V. Sundström and K. Wärnmark, Towards longer-lived metal-to-ligand charge transfer states of iron(II) complexes: an N-heterocyclic carbene approach, Chem. Commun., 2013, 49, 6412–6414 RSC.
- Y. Liu, P. Persson, V. Sundström and K. Wärnmark, Fe N-Heterocyclic Carbene Complexes as Promising Photosensitizers, Acc. Chem. Res., 2016, 49, 1477–1485 CrossRef CAS PubMed.
- S. Kaufhold and K. Wärnmark, Design and Synthesis of Photoactive Iron N-Heterocyclic Carbene Complexes, Catalysts, 2020, 10, 132 CrossRef CAS.
- C. Cebrian, M. Pastore, A. Monari, X. Assfeld, P. C. Gros and S. Haacke, Ultrafast Spectroscopy of Fe(II) Complexes Designed for Solar-Energy Conversion: Current Status and Open Questions, Chemphyschem, 2022, 23, e202100659 CrossRef CAS PubMed.
- A. Francés-Monerris, P. C. Gros, X. Assfeld, A. Monari and M. Pastore, Toward Luminescent Iron Complexes: Unravelling the Photophysics by Computing Potential Energy Surfaces, ChemPhotoChem, 2019, 3, 666–683 CrossRef.
- K. S. Kjaer, N. Kaul, O. Prakash, P. Chabera, N. W. Rosemann, A. Honarfar, O. Gordivska, L. A. Fredin, K. E. Bergquist, L. Haggstrom, T. Ericsson, L. Lindh, A. Yartsev, S. Styring, P. Huang, J. Uhlig, J. Bendix, D. Strand, V. Sundström, P. Persson, R. Lomoth and K. Wärnmark, Luminescence and reactivity of a charge-transfer excited iron complex with nanosecond lifetime, Science, 2019, 363, 249–253 CrossRef CAS PubMed.
- N. W. Rosemann, P. Chabera, O. Prakash, S. Kaufhold, K. Wärnmark, A. Yartsev and P. Persson, Tracing the Full Bimolecular Photocycle of Iron(III)-Carbene Light Harvesters in Electron-Donating Solvents, J. Am. Chem. Soc., 2020, 142, 8565–8569 CrossRef CAS PubMed.
- N. Kaul and R. Lomoth, The Carbene Cannibal: Photoinduced Symmetry-Breaking Charge Separation in an Fe(III) N-Heterocyclic Carbene, J. Am. Chem. Soc., 2021, 143, 10816–10821 CrossRef CAS PubMed.
- J. Schwarz, A. Ilic, C. Johnson, R. Lomoth and K. Wärnmark, High turnover photocatalytic hydrogen formation with an Fe(iii) N-heterocyclic carbene photosensitiser, Chem. Commun., 2022, 58, 5351–5354 RSC.
- A. Aydogan, R. E. Bangle, A. Cadranel, M. D. Turlington, D. T. Conroy, E. Cauët, M. L. Singleton, G. J. Meyer, R. N. Sampaio, B. Elias and L. Troian-Gautier, Accessing Photoredox Transformations with an Iron(III) Photosensitizer and Green Light, J. Am. Chem. Soc., 2021, 143, 15661–15673 CrossRef CAS PubMed.
- A. Aydogan, R. E. Bangle, S. De Kreijger, J. C. Dickenson, M. L. Singleton, E. Cauët, A. Cadranel, G. J. Meyer, B. Elias, R. N. Sampaio and L. Troian-Gautier, Mechanistic investigation of a visible light mediated dehalogenation/cyclisation reaction using iron(III), iridium(III) and ruthenium(II) photosensitizers, Catal. Sci. Technol., 2021, 11, 8037–8051 RSC.
- T. Duchanois, T. Etienne, C. Cebrián, L. Liu, A. Monari, M. Beley, X. Assfeld, S. Haacke and P. C. Gros, An Iron-Based Photosensitizer with Extended Excited-State Lifetime: Photophysical and Photovoltaic Properties, Eur. J. Inorg. Chem., 2015, 2015, 2469–2477 CrossRef CAS.
- T. C. B. Harlang, Y. Z. Liu, O. Gordivska, L. A. Fredin, C. S. Ponseca, P. Huang, P. Chabera, K. S. Kjaer, H. Mateos, J. Uhlig, R. Lomoth, R. Wallenberg, S. Styring, P. Persson, V. Sundström and K. Wärnmark, Iron sensitizer converts light to electrons with 92% yield, Nat. Chem., 2015, 7, 883–889 CrossRef CAS PubMed.
- L. Lindh, O. Gordivska, S. Persson, H. Michaels, H. Fan, P. Chábera, N. W. Rosemann, A. K. Gupta, I. Benesperi, J. Uhlig, O. Prakash, E. Sheibani, K. S. Kjaer, G. Boschloo, A. Yartsev, M. Freitag, R. Lomoth, P. Persson and K. Wärnmark, Dye-sensitized solar cells based on Fe N-heterocyclic carbene photosensitizers with improved rod-like push-pull functionality, Chem. Sci., 2021, 12, 16035–16053 RSC.
- A. Reddy-Marri, E. Marchini, V. D. Cabanes, R. Argazzi, M. Pastore, S. Caramori and P. C. Gros, Panchromatic light harvesting and record power conversion efficiency for carboxylic/cyanoacrylic Fe(II) NHC co-sensitized FeSSCs, Chem. Sci., 2023, 14, 4288–4301 RSC.
- A. Reddy-Marri, E. Marchini, V. D. Cabanes, R. Argazzi, M. Pastore, S. Caramori and P. C. Gros, Record power conversion efficiencies for iron(ii)-NHC-sensitized DSSCs from rational molecular engineering and electrolyte optimization, J. Mater. Chem. A, 2021, 9, 3540–3554 RSC.
- A. Reddy-Marri, E. Marchini, V. D. Cabanes, R. Argazzi, M. Pastore, S. Caramori, C. A. Bignozzi and P. C. Gros, A Series of Iron(II)-NHC Sensitizers with Remarkable Power Conversion Efficiency in Photoelectrochemical Cells, Chem. - Eur. J., 2021, 27, 16260–16269 CrossRef PubMed.
- A. Ilic, J. Schwarz, C. Johnson, L. H. M. de Groot, S. Kaufhold, R. Lomoth and K. Wärnmark, Photoredox catalysis via consecutive (2)LMCT- and (3)MLCT-excitation of an Fe(III/II)-N-heterocyclic carbene complex, Chem. Sci., 2022, 13, 9165–9175 RSC.
- P. Zimmer, P. Müller, L. Burkhardt, R. Schepper, A. Neuba, J. Steube, F. Dietrich, U. Flörke, S. Mangold, M. Gerhards and M. Bauer, N-Heterocyclic Carbene Complexes of Iron as Photosensitizers for Light-Induced Water Reduction, Eur. J. Inorg. Chem., 2017, 1504–1509 CrossRef CAS.
- P. Chabera, Y. Liu, O. Prakash, E. Thyrhaug, A. E. Nahhas, A. Honarfar, S. Essen, L. A. Fredin, T. C. Harlang, K. S. Kjaer, K. Handrup, F. Ericson, H. Tatsuno, K. Morgan, J. Schnadt, L. Häggstrom, T. Ericsson, A. Sobkowiak, S. Lidin, P. Huang, S. Styring, J. Uhlig, J. Bendix, R. Lomoth, V. Sundström, P. Persson and K. Wärnmark, A low-spin Fe(III) complex with 100-ps ligand-to-metal charge transfer photoluminescence, Nature, 2017, 543, 695–699 CrossRef CAS PubMed.
- O. Prakash, L. Lindh, N. Kaul, N. W. Rosemann, I. B. Losada, C. Johnson, P. Chabera, A. Ilic, J. Schwarz, A. K. Gupta, J. Uhlig, T. Ericsson, L. Häggström, P. Huang, J. Bendix, D. Strand, A. Yartsev, R. Lomoth, P. Persson and K. Wärnmark, Photophysical Integrity of the Iron(III) Scorpionate Framework in Iron(III)-NHC Complexes with Long-Lived 2LMCT Excited States, Inorg. Chem., 2022, 61, 17515–17526 CrossRef CAS PubMed.
- T. K. Hollis and C. E. Webster, Unsymmetrical salts, ccc-nhc pincer metal complexes, and methods of making the same, International Pat., 2018/175659, World Intellectual Property Organization, 2018.
- W. J. Lin, A. R. Naziruddin, Y. H. Chen, B. J. Sun, A. H. Chang, W. J. Wang and W. S. Hwang, Photofunctional platinum complexes featuring N-heterocyclic carbene-based pincer ligands, Chem. - Asian J., 2015, 10, 728–739 CrossRef CAS.
- E. Ahn, S. Ko, S. Kim, H. Kim, S. Shin, E. Lee, J. Lee, H. Lee, M. Jeon and J. Han, Organometallic compound, organic light-emitting device including the same and apparatus including the organometallic compound, US Pat., 2021/0167301, 2021 Search PubMed.
- K. Majima, M. Watanabe, F. Okuda, M. Haga and L. Yan, Transition metal complex compound and organic electroluminescent element, Japan Pat. 2008-266163, 2008.
- J. Steube, A. Kruse, O. S. Bokareva, T. Reuter, S. Demeshko, R. Schoch, M. A. Argüello Cordero, A. Krishna, S. Hohloch, F. Meyer, K. Heinze, O. Kühn, S. Lochbrunner and M. Bauer, Janus-type emission from a cyclometalated iron(III) complex, Nat. Chem., 2023, 15, 468–474 CrossRef CAS PubMed.
- P. Chabera, K. S. Kjaer, O. Prakash, A. Honarfar, Y. Z. Liu, L. A. Fredin, T. C. B. Harlang, S. Lidin, J. Uhlig, V. Sundström, R. Lomoth, P. Persson and K. Wärnmark, Fe-II Hexa N-Heterocyclic Carbene Complex with a 528 ps Metal-to-Ligand Charge-Transfer Excited-State Lifetime, J. Phys. Chem. Lett., 2018, 9, 459–463 CrossRef CAS PubMed.
- W. Gawelda, A. Cannizzo, V. T. Pham, F. van Mourik, C. Bressler and M. Chergui, Ultrafast nonadiabatic dynamics of [Fe(II)(bpy)(3)](2+) in solution, J. Am. Chem. Soc., 2007, 129, 8199–8206 CrossRef CAS PubMed.
- C. Bressler, C. Milne, V. T. Pham, A. Elnahhas, R. M. van der Veen, W. Gawelda, S. Johnson, P. Beaud, D. Grolimund, M. Kaiser, C. N. Borca, G. Ingold, R. Abela and M. Chergui, Femtosecond XANES study of the light-induced spin crossover dynamics in an iron(II) complex, Science, 2009, 323, 489–492 CrossRef CAS PubMed.
- W. Leis, M. A. Argüello Cordero, S. Lochbrunner, H. Schubert and A. Berkefeld, A Photoreactive Iron(II) Complex Luminophore, J. Am. Chem. Soc., 2022, 144, 1169–1173 CrossRef CAS PubMed.
- C. B. Larsen, J. D. Braun, I. B. Lozada, K. Kunnus, E. Biasin, C. Kolodziej, C. Burda, A. A. Cordones, K. J. Gaffney and D. E. Herbert, Reduction of Electron Repulsion in Highly Covalent Fe-Amido Complexes Counteracts the Impact of a Weak Ligand Field on Excited-State Ordering, J. Am. Chem. Soc., 2021, 143, 20645–20656 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2254082 and 2254083. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc02806b |
| ‡ Shared authorship. |
| This journal is © The Royal Society of Chemistry 2023 |