
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA liquid metal-fluoropolymer artificial protective film enables robust lithium metal batteries at sub-zero temperatures†
Hongbao
Li‡
,
Rong
Hua‡
,
Yang
Xu‡
,
Da
Ke
,
Chenyu
Yang
,
Quanwei
Ma
,
Longhai
Zhang
,
Tengfei
Zhou
 * and
Chaofeng
Zhang
*
* and
Chaofeng
Zhang
*
Institutes of Physical Science and Information Technology, Leibniz Joint Research Center of Materials Sciences, Anhui Graphene Engineering Laboratory, Key Laboratory of Structure and Functional Regulation of Hybrid Material (Ministry of Education), Anhui University, Hefei 230601, China. E-mail: tengfeiz@ahu.edu.cn; cfz@ahu.edu.cn
First published on 29th August 2023
Abstract
Batteries that are both high-energy-density and durable at sub-zero temperatures are highly desirable for deep space and subsea exploration and military defense applications. Our design incorporates a casting membrane technology to prepare a gallium indium liquid metal (LM)/fluoropolymer hybrid protective film on a lithium metal anode. The LM not only spontaneously forms a passivation alloy layer with lithium but also reduces the nucleation potential barrier and homogenizes the Li+ flux on the surface of the lithium anode. The fluoropolymer's polar functional groups (–C–F–) effectively induce targeted dispersion of gallium indium seeds, and the unique pit structure on the surface provides oriented sites for lithium plating. By implementing these strategies optimally, the protected lithium metal anode remains in operation at a current density of 20 mA cm−2 with an over-potential of about 50.4 mV after 500 h, and the full cells have a high capacity retention rate of up to 98.5% at a current density of 0.5 C after 100 cycles. Furthermore, the battery shows improved low temperature performance at −30 °C, validating the potential of the protective film to enable battery operation at sub-zero temperatures.
Introduction
The increasing maturity of lithium metal battery (LMB) technology has expanded its usage from microsatellites and high orbit satellites to deep space exploration.1,2 Simultaneously, the demand for rechargeable batteries with a high energy density in harsh environments has become increasingly urgent, particularly for electric vehicles used in high latitudes, upper air, military/defense applications, and space/subsea exploration at sub-zero temperatures.3,4 Unfortunately, traditional carbonate electrolytes used in LMBs currently suffer from decreased ion mobility at low temperatures, which limits their practical use.5–8 During the plating/stripping process, dendrite lithium structures can intensify electrolyte decomposition, ultimately leading to low coulombic efficiency (CE), capacity loss, and volume change, leading to battery failure.9–11 To mitigate this issue, various strategies have been proposed, such as adjusting the electrolyte composition, using functional electrolyte additives, constructing artificial solid-electrolyte interphase (SEI) layers, replacing the liquid electrolyte with a solid/composite electrolyte of a high modulus, constructing lithium alloy layers with low activity, and designing 3D lithium host structures.12–15 Although these strategies have made progress in stabilizing lithium metal anodes, they still face challenges in effectively improving low-temperature performance and preventing lithium dendrite growth.16 Proper structure design is crucial in achieving high-performance lithium metal batteries,17 with the final morphology of lithium deposition determined by the active nucleation process. Therefore, exploring novel lithium metal anodes with high lithium-ion conductivity, low electrical conductivity, and chemical/electrochemical stability is necessary.Metallic gallium and gallium-based liquid metal (LM) alloys have attracted significant attention due to their physicochemical properties,18–20 including their amorphous state, good fluidity, chemical stability, high thermal conductivities, low viscosity (2 mPa s), high surface tension, low melting point (<30 °C), high deformability, and self-healing.21–23 Liquid metals such as GaSn and GaInSnZn are used in anodes for LMBs and possess unique self-healing properties and high lithium affinity,24,25 demonstrating their potential in the field of energy storage. Once the LM is deposited on the bottom, it spontaneously alloys with the lithium metal to form a LixLMy passivation layer of LixLMy, which stabilizes the Li-metal anode due to its virtues as a flexible physical barrier that separates Li from the electrolyte, affords a higher lithium-ion diffusion coefficient than bare Li, accelerates the diffusion kinetics, inhibits the growth of Li dendrites, has good compatibility with the Li-metal anode, exhibits fine Li wettability, and decreases the nucleation barrier of lithium in the plating process.3,23–28 However, the spatial distribution of the liquid metal significantly impacts its usage and area of effectiveness, ultimately affecting the energy density and cycle life of a battery. Previous studies have used gel polymer electrolytes (GPEs) or solid polymer electrolytes (SPEs) with excellent electrochemical properties, flexibility, and low crystallinity to enable safe lithium metal batteries.29–33 These properties facilitate the absorption of liquid electrolytes, providing better ionic conductivity and mechanical stability at low temperatures.34,35 Therefore, combining the advantages of a gel polymer or its analogue and LM would be a good choice to improve the overall performance of LMBs.
In this study, we developed a hybrid surface protective film (SPF) with a pit microstructure obtained using a simple casting membrane technology. On one hand, the introduced liquid metal can form a passivation layer of LixLMy spontaneously upon contact with the lithium metal anode, thereby enhancing the stability of the anode. Furthermore, the inclusion of indium in the gallium–indium alloy leads to a significant reduction in its solidification point when compared to pure gallium metal. Moreover, the poly(vinylidene-co-hexafluoropropylene) (PVDF-HFP) polymers act as the skeleton of the hybrid protective film, featuring polar functional groups (–C–F–) that promote the dissociation of lithium salts, accelerate transport of lithium ions, and induce targeted dispersion of the LM. During the dehydration process, trace amounts of water molecules present in the bis(trifluoromethane)sulfonimide lithium (LiTFSI) salt and fluoropolymer form pits on the polymer membrane. This unique pit structure provides a large area for lithium deposition, enabling the PVDF-HFP-protected Li anode to circulate stably at high current densities. Additionally, the surface structure improves the affinity for the electrolyte and quickly stabilizes the electrolyte/electrode interface. The cells can be cycled in carbonate electrolytes with a low polarization voltage for more than 4500 h at a current density of 0.25 mA cm−2 and for more than 500 h at a higher current density of 20 mA cm−2, while also exhibiting improved low temperature performance.
Experimental section
Synthesis of the LM-SPF and SPF
First, GaIn (LM) was synthesized by stirring gallium and indium with a mass ratio of 75.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :24.5 at 160 °C for 0.5 hours.36 Subsequently, 0.4 g of poly(vinylideneco-hexafluor-opropylene) (PVDF-HFP, MW = 150000, Macklin) and 0.08 g of bis(trifluoromethane)sulfonimide lithium salt (LiTFSI, Aldrich) were added to 3 mL of N-methyl-pyrrolidone (NMP, ≥99.9%, Macklin) and stirred at 60 for 8 hours to obtain a homogeneous solution. After that, the obtained LM was added to the above uniform mixture (volume ratio of 1:30). The ultrasound treatment lasted for 10 minutes and was followed by continuous stirring at room temperature for 12 h to form a homogeneous solution. Subsequently, the solution was applied to a Teflon template and dried at 70 °C overnight to produce the LM-SPF.
:24.5 at 160 °C for 0.5 hours.36 Subsequently, 0.4 g of poly(vinylideneco-hexafluor-opropylene) (PVDF-HFP, MW = 150000, Macklin) and 0.08 g of bis(trifluoromethane)sulfonimide lithium salt (LiTFSI, Aldrich) were added to 3 mL of N-methyl-pyrrolidone (NMP, ≥99.9%, Macklin) and stirred at 60 for 8 hours to obtain a homogeneous solution. After that, the obtained LM was added to the above uniform mixture (volume ratio of 1:30). The ultrasound treatment lasted for 10 minutes and was followed by continuous stirring at room temperature for 12 h to form a homogeneous solution. Subsequently, the solution was applied to a Teflon template and dried at 70 °C overnight to produce the LM-SPF.
Synthesis of the SPF
The SPF synthesis followed a similar procedure to that of the LM-SPF. In this case, a homogeneous solution without the introduction of the LM was directly placed on the Teflon template and dried overnight at 70 °C under vacuum.Synthesis of LM-SPF@Li and SPF@Li
The freestanding LM-SPF was obtained and transferred to an Ar-filled glovebox for storage. The LM side of the membrane was close to the lithium sheet and pressed tightly. Similarly, the synthesis of LM-SPF@Cu and SPF@Cu was based on a similar method to that of LM-SPF@Li and SPF@Li.Structural and morphological characterization
The morphology of the samples was observed using a field-emission scanning electron microscope (FESEM, JEOL-6300F), which was coupled with energy-dispersive X-ray (EDX) spectroscopy. X-ray diffraction (XRD) was investigated on a D2 PHASER with Cu Kα radiation. X-ray photoelectron spectroscopy (XPS, PHi5000) was used to investigate the composition and chemical bonds of the lithium anode interface before and after circulation. Stress/strain experiments were carried out at 10 mm min−1 using a universal test machine (UTM2502) at room temperature. At least three specimens of each polymer were tested. The test samples have a 14 mm gauge length, a 2 mm width, and a thickness of 0.12 mm.Electrochemical measurements
All coin-type cells (CR2032) were assembled in an Ar-filled glove box, in which the content of water and oxygen was controlled below 0.01 ppm. Electrochemical impedance spectroscopy (EIS) spectra were measured using a Princeton multichannel electrochemical workstation; the frequency ranges from 0.1 to 100 MHz with a voltage of 5 mV. The electrochemical stability window was obtained on a NEWARE tester with a voltage range of 2–5 V at sweep rates of 1mV s−1 by assembling Li‖SPF‖SS cells. The ionic conductivity of the SPF, studied by assembly of SS‖SPF‖SS cells, was performed on a PARSTATMC electrochemical workstation (PMC1000). The value of conductivity can be obtained from the following equation:where L, R, and S represent the bulk resistance, thickness, and area of the film, respectively. The electrolyte was 1.0 M LiTFSI in 1,3-dioxolane (DOL)/1,2-dimethoxyethane (DME) (1
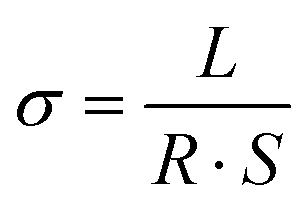 :1 volume ratio), with a 2.0 wt% LiNO3 additive, to investigate the lithium plating/stripping process and to determine the coulombic efficiency (CE) of lithium metal batteries. At a current density of 1.0 mA cm−2, by galvanostatic discharge tests, lithium of different current densities was deposited in Cu or SPF@Cu, and then Li was stripped up to 1 V. The ion transference number (t+) of Li was determined using symmetric cells Li‖SPF‖Li, and then the value was obtained using the following equation:
:1 volume ratio), with a 2.0 wt% LiNO3 additive, to investigate the lithium plating/stripping process and to determine the coulombic efficiency (CE) of lithium metal batteries. At a current density of 1.0 mA cm−2, by galvanostatic discharge tests, lithium of different current densities was deposited in Cu or SPF@Cu, and then Li was stripped up to 1 V. The ion transference number (t+) of Li was determined using symmetric cells Li‖SPF‖Li, and then the value was obtained using the following equation:Here, I0 and R0 represent the initial current and resistance, respectively. Is and Rs severally represent the steady-state current and resistance. V is the constant polarization voltage (10 mV). For the preparation of LFP electrodes, a mixed slurry of LFP, carbon black, and polyvinylidene fluoride with a weight ratio of 8
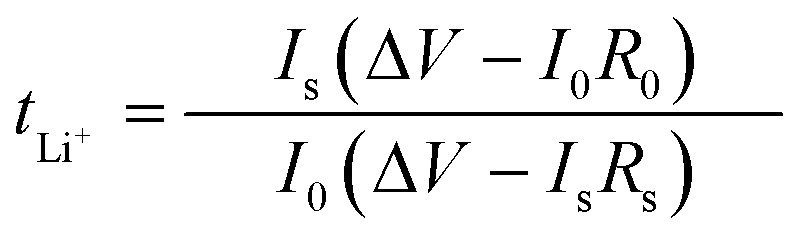 :1:1 in N-methyl-2-pyrrolidinone solvent was prepared. Afterward, it was paired with the SPF@Li anode to assemble a full cell, in a glove box. All of these cells (named LM-SPF@Li‖LFP, SPF@Li, and Li‖LFP) were galvanostatically cycled with a voltage range of 2.5 to 3.9 V.
:1:1 in N-methyl-2-pyrrolidinone solvent was prepared. Afterward, it was paired with the SPF@Li anode to assemble a full cell, in a glove box. All of these cells (named LM-SPF@Li‖LFP, SPF@Li, and Li‖LFP) were galvanostatically cycled with a voltage range of 2.5 to 3.9 V.
Theoretical simulations
COMSOL multiphysics was used to study the distributions of electric and Li ion concentrations near the Li metal anode, regardless of the possible side effects. The whole simulation is a transient model of the plating and stripping process of Li; therefore, the mass conservation (Li+ and electrolyte anions) and Nernst–Planck equation were selected to define it. The LM-SPF was placed on the surface of the Li metal in the model. The coefficients of the chemical equivalent of Li+ in the electrolyte and the initial concentration of Li+ were established at 1.Results and discussion
In our study, we provide a detailed synthesis procedure for a hybrid protection film, which is illustrated in Scheme 1. The LM-SPF is synthesized using a facile and cost-effective method. Initially, a homogeneous mixture of the prepared solutions is applied onto a Teflon mold, and then the solvent is evaporated to obtain the LM-SPF. Optical photos of the suspension before and after the introduction of the LM are shown in Fig. S1.† LiTFSI serves as a lithium-ion conductor and TFSI anion, contributing to the formation of a stable SEI layer. By maintaining humidity at around 30%, a free-standing LM-SPF with a pit shape on one side of the surface can be formed after the drying process. The color of the LM-SPF appears uniform, indicating the uniform dispersion of GaIn. Additionally, the internal illustration demonstrates a high degree of flexibility, as shown in Fig. 1a. When the humidity is lower than 30%, a relatively dense polymer film is obtained (Fig. S2a†). Conversely, when the humidity exceeds 30%, we can prepare a porous composite membrane (Fig. S2b†).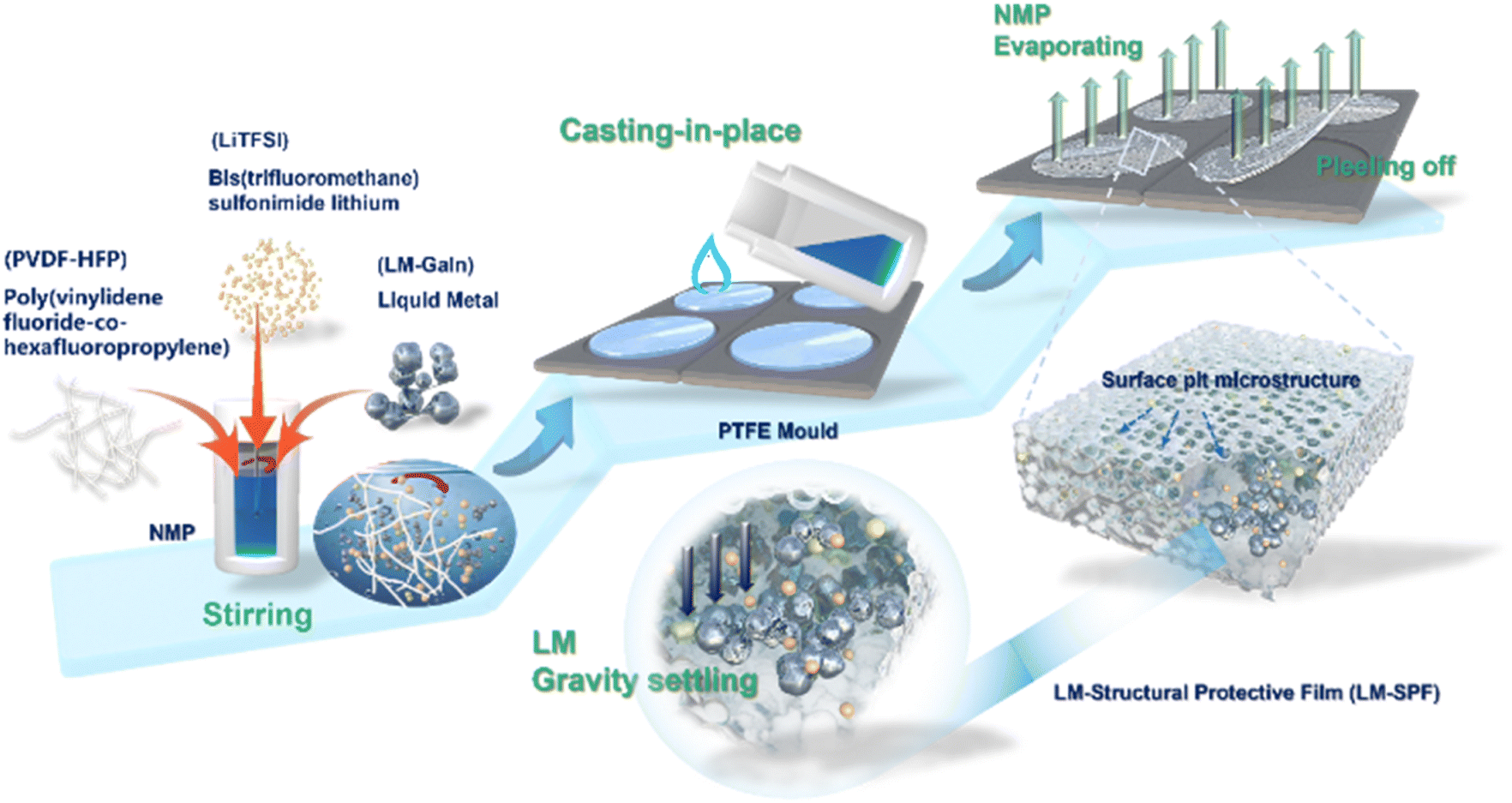 | ||
| Scheme 1 Schematic illustration of the synthesis processes for the LM-SPF. | ||
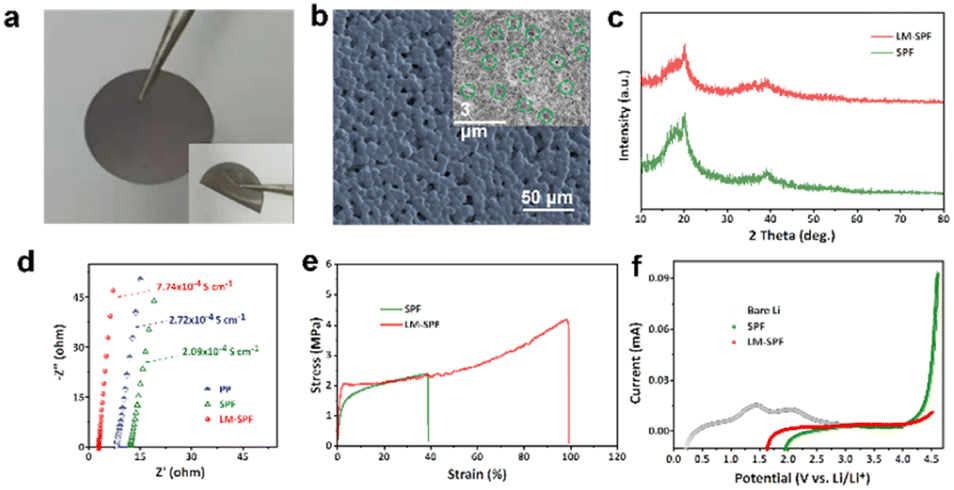 | ||
| Fig. 1 Characterization of the membrane of the LM-SPF. (a) Optical photograph. (b) SEM images of the upper surface. (c) XRD patterns. (d) Nyquist plots of the LM-SPF, the SPF, and PP. (e) Stress–strain curves of the LM-SPF and SPF. (f) LSV curves of the LM-SPF, the SPF and bare Li at room temperature. | ||
Scanning electron microscopy (SEM) was employed to observe the unique morphology of the LM-SPF. Fig. 1b shows that the upper surface of the LM-SPF contains many pits, with diameters ranging from 2.0 to 5.0 μm. Additionally, there are many micropores inside, which are not present on the backside (Fig. S3a†). During the drying process, due to phase separation and water evaporation, a free-standing LM-SPF with a unique hierarchical structure in which all elements are evenly distributed (Fig. S3b and c†) is obtained. We performed XRD to investigate the composition of the composite film. As shown in Fig. 1c, the characteristic peaks at 2θ = 19° and 40° weaken and the overall peak becomes broad. This indicates that the LM particles were successfully polymerized in PVDF-HFP without affecting its crystal structure. Moreover, the increase in the amorphous portion suggests a decrease in the crystallinity of PVDF-HFP, thereby improving the ionic conductivity of the film. As a result, the ionic conductivity of the LM-SPF at room temperature was measured at 7.74 × 10−4 S cm−1, which is higher than that of the SPF (2.09 × 10−4 S cm−1) and PP (2.72 × 10−4 S cm−1) (Fig. 1d). The LM-SPF also exhibited strong tensile strength, which was more than twice that of the SPF (Fig. 1e). Therefore, the stress during extrusion was low, effectively inhibiting dendrite growth. In addition to ionic conductivity and tensility, electrochemical stability was an essential factor in the application of the LM-SPF. The results of linear sweep voltammetry (LSV) revealed that the electrochemical stability of the LM-SPF improved by over 4.7 V when compared to the SPF and bare Li (Fig. 1f). The stable interface compatibility of the film resulted in its great potential to be paired with high-voltage cathodes for high energy density. The Li+ transference number (t+) of the LM-SPF was also higher than that of the SPF (Fig. S4†). Moreover, the wettability of the membrane to the electrolyte is critical to achieve reversible lithium plating/stripping and rapid interfacial ion transfer. The introduction of the LM increased the film's flexibility, resulting in a greater affinity to the electrolyte (Fig. S5†).
We further investigated the electrochemical performance of the composite membrane by evaluating the galvanostatic cycling performance of different symmetric cells at a gradient current density, as shown in Fig. 2. At a current density of 0.25 mA cm−2 for 0.25 mA h cm−2, the LM-SPF@Li symmetric cell exhibited better stability, remaining stable for approximately 4500 h with a low overpotential of 16.8 mV. The SPF@Li and bare Li symmetric cells were short-circuited after 312 h and 525 h with high overpotential, respectively, attributed to uneven nucleation and severe growth of lithium dendrite formation (Fig. 2a). At a current density of 5 mA cm−2, the LM-SPF@Li symmetric cell exhibited a remarkably low overpotential of approximately 39.5 mV and an impressively long cycle life exceeding 3000 hours. In stark contrast, both the SPF@Li and bare Li symmetric cells experienced rapid short-circuiting, as depicted in Fig. 2b. Notably, even at a high current density of 20 mA cm−2, LM-SPF@Li displayed exceptional cycle stability for more than 500 hours, accompanied by a minimal overpotential of 50.4 mV, as presented in Fig. 2c. In contrast, the cycle life of the SPF@Li and bare Li symmetric cells was found to be limited, less than 100 h, due to slow mass transfer, electrode crushing, and significant voltage polarization. The LM-SPF@Li symmetric cells exhibited good rate performance stability within the current density range of 0.2 to 10 mA cm−2 (Fig. 2d), even at a low temperature of −30 °C, with negligible overpotential (Fig. 2e). The unique structure of the composite membrane facilitates uniform lithium deposition, which is evident in the bright surface of the LM-SPF@Li electrode after 100 cycles (Fig. S6a†), compared to the bare Li electrode, which exhibited a high amount of “dead Li” due to the irreversible growth of lithium dendrites (Fig. S6b†). Furthermore, our method exhibited even better long-cycle stability, making it a promising approach for lithium anode protection layers. Fig. S7 and Table S1† demonstrate that our method outperformed other techniques commonly used in lithium anode protection layers.37–48 Our approach has several advantages, including the self-healing ability of the introduced lipophilic “seeds” of the LM and the unique structure of the composite membrane.
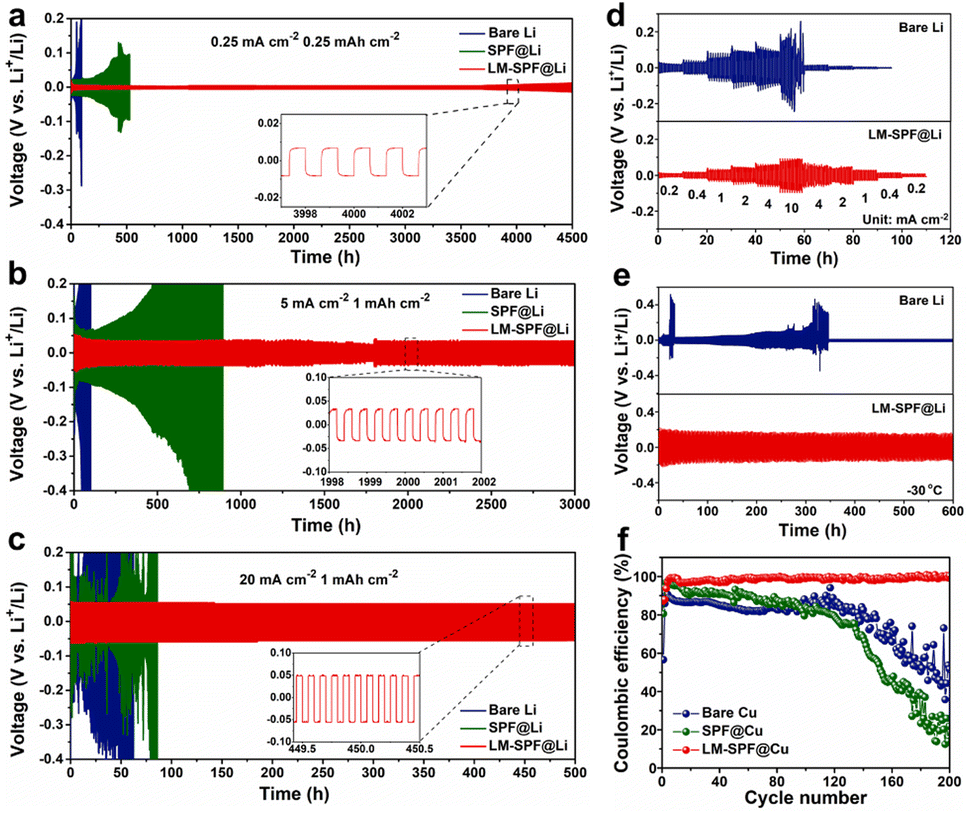 | ||
| Fig. 2 Electrochemical performances of symmetric and half-cells of different electrodes. Electrostatic cycling of the symmetric cells of LM-SPF@Li‖LM-SPF@Li, SPF@Li‖SPF@Li, and Li‖Li, (a) at 0.25 mA cm−2 for 0.25 mA h cm−2, the inset shows a detailed voltage profile from the 3997 to 4003rd cycles, (b) at 5 mA cm−2 for 1 mA h cm−2 with a detailed voltage profile from the 1998 to 2002nd cycles, and (c) at 20 mA cm−2 for 1 mA h cm−2 with a detailed voltage profile from the 449.5 to 450.5th cycles. (d) The rate capability of LM-SPF@Li and bare Li symmetric cells at various current densities. (e) Low temperature applicability of symmetric cells with a current density of 0.5 mA cm−2 for 1 mA h cm−2 at −30 °C. (f) CEs of various electrodes at 1 mA cm−2 for 1 mA h cm−2. | ||
In the present study, the nucleation behavior of lithium on various substrates was examined as it is crucial for achieving uniform lithium nucleation and high-performance lithium metal batteries, and it also helps in assessing the seed effect of lipophilic GaIn (Fig. S8†). For instance, using a bare Cu electrode, the voltage dropped rapidly to around 0.3 V (vs. Li/Li+) with a high nucleation overpotential of approximately 68.5 mV and a substantial voltage hysteresis during the initial Li plating process. In contrast, the nucleation overpotential of the LM-SPF@Cu electrode was impressively low, at around 10.2 mV, which is far less than that of the bare Cu electrode. This significant reduction may be attributed to the regulation of the halophilic seeds of LM nanoparticles and the decrease in local current density. Additionally, to examine the reversibility of Li plating/striping in various half cells, the average coulombic efficiency (CE) was determined by an electrodeposition test (Fig. 2f). The LM-SPF@Cu electrode exhibited a significantly higher and more stable coulombic efficiency (CE) of 99.2% after 200 hours of long cycles at a current density of 1 mA cm−2. In contrast, the bare Cu electrode displayed approximately 70% CE during the initial 40 cycles, followed by instability and rapid decline. On the other hand, the SPF@Cu electrode exhibited an initial high CE ranging from 80 to 90% during the first 80 cycles, which also abruptly decreased. The superior coulombic efficiency of LM-SPF@Cu showcases the high reversibility of plating/stripping on the Li anode, emphasizing the advantageous impact of the specifically structured composite film. This film contributes to the consistent deposition of Li and enhances its electrochemical properties.
This study aimed to investigate how the unique structured composite film affects the Li deposition behavior. To achieve this aim, we deposited varying amounts of Li on different electrodes and conducted SEM observations. The Li plating and stripping behavior of LM-SPF@Cu and bare Cu electrodes is captured in Fig. 3, with Fig. 3a and e outlining the changes in the schematic diagram of the morphology of the electrode's Li plating and stripping behavior. Moreover, to further characterize this process, Li was deposited on Cu foil through the LM-SPF, as confirmed by the cross-sectional SEM image shown in Fig. S9.† During plating, substantial growth of Li dendrites was observed on the bare Cu electrode after plating with 2.0 mA h cm−2, and “tip spots” of uneven Li nucleation were formed (Fig. 3b). When plated further up to 4.0 mA h cm−2, the growth of Li dendrites worsened, and clumps formed instead (Fig. 3c). This not only expedited the growth of lithium dendrites but also the consumption of electrolytes. Subsequently, when the same capacity of 4.0 mA h cm−2 was completely removed, a lot of “dead Li” accumulated on the surface of the bare Cu electrode, attributed to the accumulation of lithium dendrites and the products of electrolyte decomposition, further slowing down the kinetics (Fig. 3d). In sharp contrast, the LM-SPF@Cu electrode filled the pits on the surface of the composite film initially when the deposition area increased from 2 mA h cm−2 to 4 mA h cm−2 (Fig. 3f) and then deposited in the plane area (Fig. 3g). Remarkably, at the same charge density at current 1 V after complete stripping, Li deposited on the LM-SPF@Cu electrode, no “dead Li” formed, and the pits on the film surface were restored (Fig. 3h). This suggests that LM nanoparticles play a pivotal role in the nucleation and deposition processes of Li metal, as well as in the composite film's excellent reversibility.
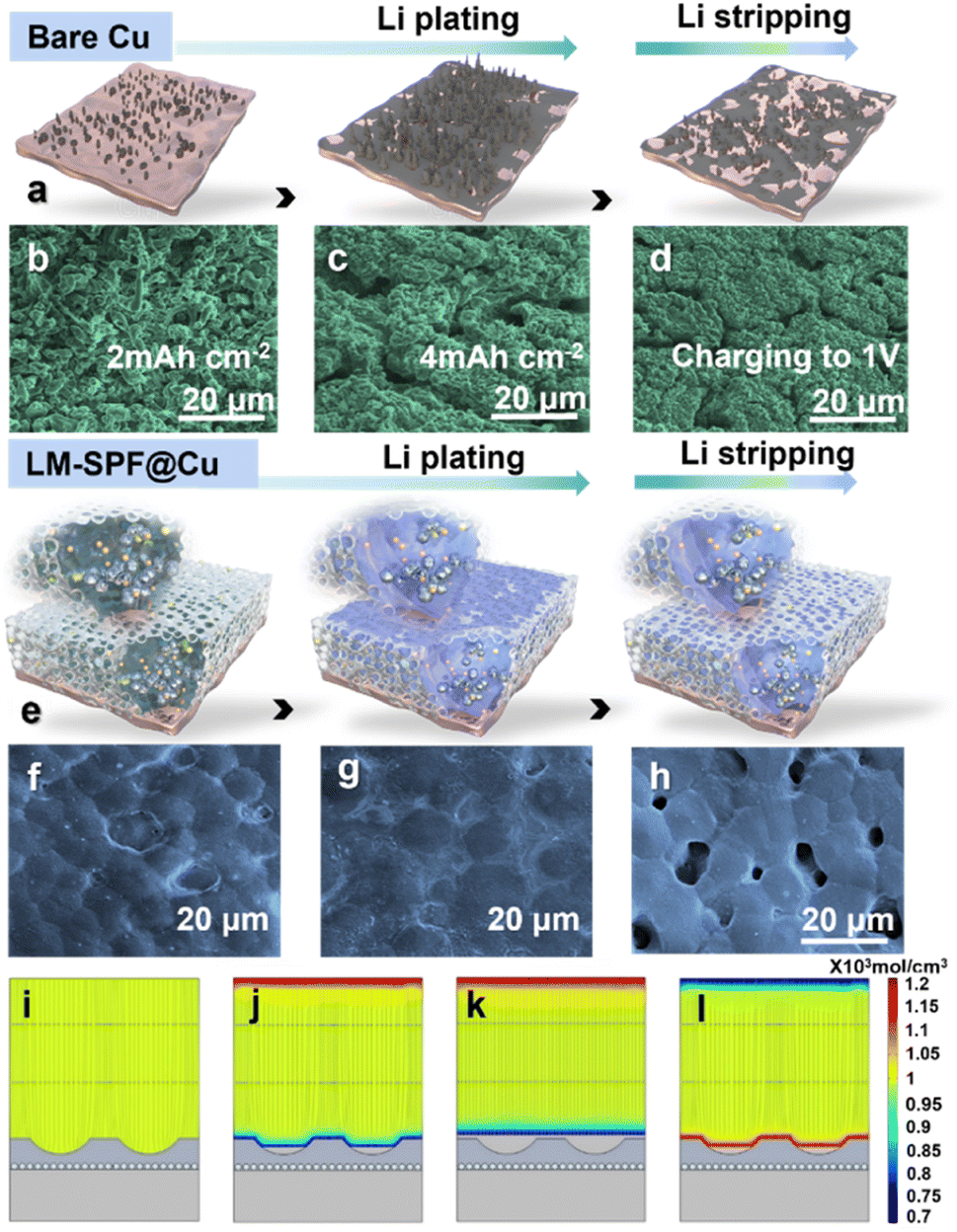 | ||
| Fig. 3 Evolution and simulation of different electrode morphologies during Li plating/stripping. Schematic illustration of different-state Li plating/stripping of (a) bare Cu and (e) LM-SPF@ Cu electrode. SEM images of bare Cu after Li plating at (b) 2 mA h cm−2 and (c) 4 mA h cm−2 and (d) fully stripping at 1 mA cm−2. SEM images of LM-SPF@Cu after Li plating at (f) 2 mA h cm−2 and (g) 4 mA h cm−2 and (h) fully stripping at 1 mA cm−2. COMSOL simulation of LM-SPF@Li in (i) original state, (j) plating at 2 mA h cm−2 and (k) 4 mA h cm−2, and (l) fully stripping density of 1 mA cm−2, with the relatively lower initial CE being attributable to the consumption of lithium due to the formation of solid electrolyte interfaces and the transformation of the LM into LixLMy. In comparison, the CE of the bare Cu electrode was about 70% during the first 40 cycles, and then became unstable and decreased rapidly, whereas the SPF@Cu electrode initially recorded a high CE of roughly 80–90% during the first 80 cycles but also suddenly declined. The superior CE of LM-SPF@Cu demonstrates the high reversibility of plating/stripping on the Li anode and the beneficial effect of the uniquely structured composite film, which provides a robust support for the uniform deposition and superior electrochemical properties of Li. | ||
The COMSOL multiphysics method was utilized to validate the above conclusion by simulating the distribution of the electric ion and lithium ion concentrations. Initially, the concentration and electric field were uniformly distributed on the surface (Fig. 3i). As the amount of lithium deposition increased from 2 to 4 mA h cm−2, lithium ions were preferentially deposited at the bottom of the cavities (Fig. 3j) and then deposited in the plane area (Fig. 3k). This indicated that Li was preferentially deposited in the surface pits rather than the top flat region, indicating that the surface pits could act as additional Li plating hosts, increasing the deposition area of Li, which would reduce the local current density, thereby inhibiting dendrite growth. Stripping at the same current density resulted in the reappearance of the pits (Fig. 3l), demonstrating that the lithiophilicity of the LM could effectively induce an orderly and uniform deposition of lithium, which was consistent with SEM observations (Fig. 3f and h). The unique structure of the LM and polymer network skeleton aided in making the flux uniform and reducing the local current density, which resulted in stable lithium plating behavior and the formation of a dense lithium plating layer that could be reversibly stripped and restored on the LM-SPF@Cu electrode (Fig. 3h). Conversely, bare Cu electrodes lacked a stable SEI, leading to dispersed Li nucleation.
This, in turn, resulted in non-uniform Li deposition on the electrode surface, which ultimately led to dendrite formation and SEI cracks. Subsequently, upon stripping, a significant amount of bulk dead lithium remained on the surface of the Cu electrode (Fig. 3d).
The activation energy (Ea) for Li+ migration inside the film was calculated using the Arrhenius equation σ(T) = Aexp(−Ea/RT),49,50 where temperature-dependent EIS spectra were utilized (Fig. S10a–c†) to investigate the effect of this unique structure composite film on Li deposition behavior. As demonstrated in Fig. 4a, the LM-SPF exhibited a significantly lower activation energy (1.77 eV) than the SPF (2.03 eV) and bare Li (2.16 eV), indicating the easiest ion transfer in the film. As a result, the protective film modification was deemed effective in inhibiting the formation of a thick solvent-derived SEI layer and reducing the accumulation of “dead Li,” which could substantially hinder Li+ transport and elevate the activation energy. Furthermore, to gain insight into the mechanism through which the composite film enhances the lithium anode, electrochemical impedance spectroscopy (EIS) was conducted on LM-SPF@Li and bare Li electrodes before and after 50 cycles, as shown in Fig. 4b. The semicircle observed in the EIS curve represents the electrochemical charge transfer resistance (Rct) of the symmetric cells. Notably, LM-SPF@Li exhibited a significantly lower Rct compared to the bare Li electrodes, which can be attributed to the presence of the multifunctional Li-base alloy layers (LixLMy) within the protective film (Fig. 4c). These layers contribute to the considerable improvement in interfacial kinetics of the Li metal electrode, as demonstrated.1,51–61 In addition, X-ray photoelectron spectroscopy (XPS) spectra were obtained to assess the chemical properties of the electrode surface components in carbonate electrolytes after 10 and 100 cycles. Initially, the LM-SPF@Li electrode primarily consisted of Li2CO3 (55.5 eV, Li 1s; 289.8 eV, C 1s), LiPF6 (137.0 eV, P 2p), and LixPOyFz (136.5 eV, P 2p) (Fig. 4d–f),13,18 indicating the stability of the surface SEI (solid electrolyte interface). As the cycling proceeded, the content of LixLMy increased gradually, as mentioned earlier (Fig. 4c and g), indicating the progressive formation of the protective film. Remarkably, even after 100 cycles, LM-SPF@Li demonstrated excellent stability, with no significant change observed in the composition of the surface ingredients, further confirming the excellent stability of the SEI. In comparison, for the bare Li electrode, it is noteworthy that the content of O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and Li2CO3 significantly increased after 100 cycles, as depicted in Fig. 4h. Moreover, the P 2p spectrum revealed peaks corresponding to LiPF6 and LixPOyFz at 136.7 eV and 133.5 eV respectively (Fig. 4i), indicative of the increased presence of the LixPOyFz by-product as LiPF6 decreased with cycling. Overall, the above analysis of those changes indicates that the bare Li electrode cannot maintain a stable surface SEI during cycling, resulting in continuous electrolyte consumption; nevertheless, the LM-SPF@Li electrode can reach a steady state more quickly.
O and Li2CO3 significantly increased after 100 cycles, as depicted in Fig. 4h. Moreover, the P 2p spectrum revealed peaks corresponding to LiPF6 and LixPOyFz at 136.7 eV and 133.5 eV respectively (Fig. 4i), indicative of the increased presence of the LixPOyFz by-product as LiPF6 decreased with cycling. Overall, the above analysis of those changes indicates that the bare Li electrode cannot maintain a stable surface SEI during cycling, resulting in continuous electrolyte consumption; nevertheless, the LM-SPF@Li electrode can reach a steady state more quickly.
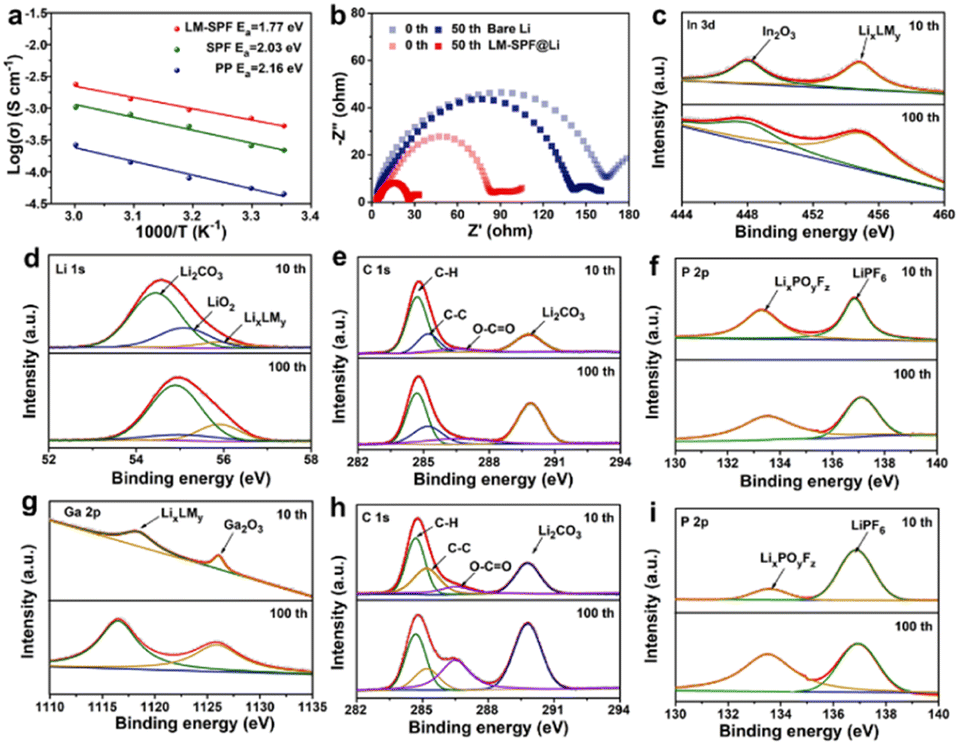 | ||
| Fig. 4 Chemical stability properties of the electrode. (a) Corresponding Arrhenius curves. (b) Nyquist plots of the LM-SPF@Li electrode and the bare Li electrode. High-resolution XPS spectra (c) In 3d, (d) Li 1s, (e) C 1s, (f) P 2p, (g) Ga 2p of LM-SPF@Li after 10 and 100 cycles, respectively; and (h) C 1s, (i) P 2p of the bare Li electrode after 10 and 100 cycles, respectively. | ||
To assess the practical feasibility of LM-SPF@Li for use in LMBs, the electrochemical properties of Li/LFP cells were examined. At various current densities ranging from 0.1 C to 5 C, LM-SPF@Li‖LFP discharged capacities of 163.6, 148.1, 135.7, 130.2, 127.9, and 118.4 mA h g−1 were recorded, indicating a fast Li+ transfer rate and stable Li plating/stripping even at high current densities.61 More impressively, the electrode displayed excellent rate capability, with the capacity returning to its original value when the current density reverted to 0.1 C. Moreover, the cycle stability of LM-SPF@Li was found to be significantly improved, with 98.5% capacity retention after 100 cycles at 0.5 C and with an improved LFP electrode polarization voltage (Fig. 5b). The long-term cycling performance of LM-SPF@Li‖LFP was also tested at a current density of 2 C and recorded a high reversible capacity of 128 mA h g−1 after 1000 cycles with no obvious decay (Fig. 5e). All of these observations point to the significantly enhanced performance of LM-SPF@Li in LMBs, implying its potential application prospects.
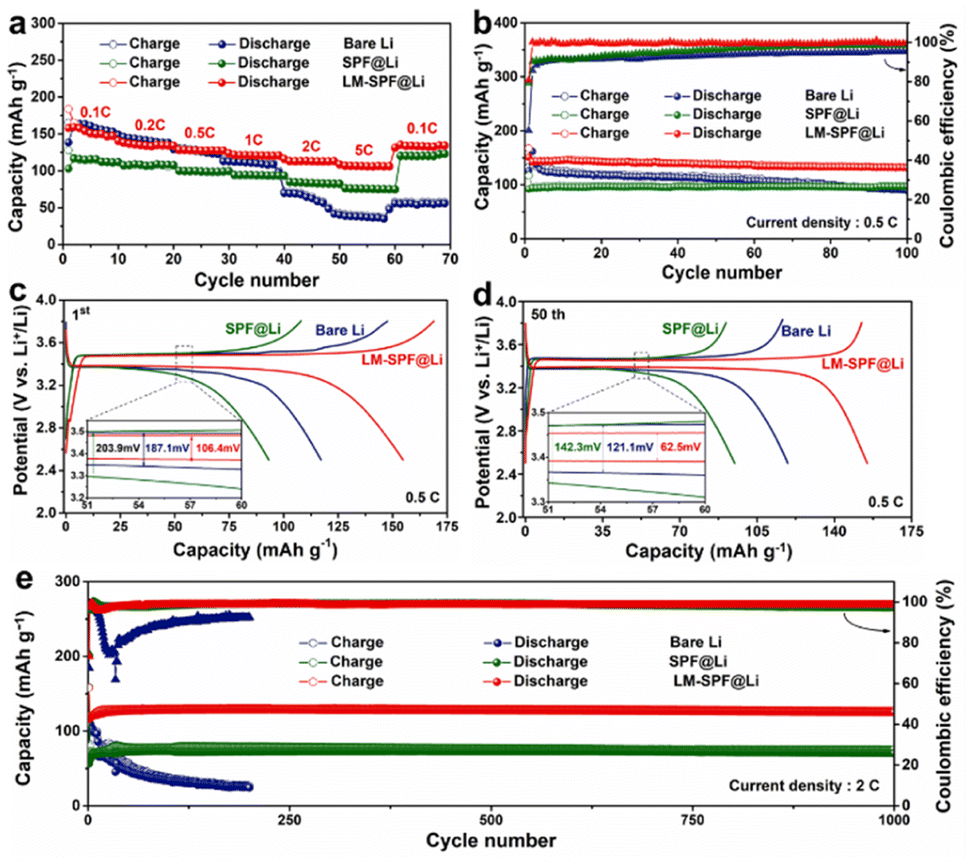 | ||
| Fig. 5 Electrochemical properties of the full cells. (a) The rate capacity ranges from 0.1 C to 3 C (1 C = 170 mA h g−1). (b) Cycle performances of LM-SPF@Li‖LFP, SPF@Li‖LFP, and bare Li‖LFP at a rate of 0.5 C, and the corresponding charge/discharge curves of (c) 1st and (d) 50th cycles. (e) Long-term cycling performance at 2 C. | ||
Conclusions
In this study, we have introduced a new approach for stabilizing the lithium metal anode by employing a unique protective film that features a pit structure on the surface. The film is formed by encapsulating an LM in a polymer network, allowing for a high deposition surface area, excellent mechanical properties, high infiltration of the electrolyte, and high ionic conductivity. As a result, the growth of lithium dendrites is inhibited and a stable electrode/electrolyte interface is formed, leading to good cycling stability of lithium metal anodes. Through extensive testing, we have demonstrated that the symmetric cell can maintain stable cycling for more than 4500 h at a current density of 0.25 mA cm−2 and can also handle a high current density of 20 mA cm−2 with a small overpotential of about 50.4 mV after 500 h. Additionally, the full cell of LM-SPF@Li‖LFP can retain a high capacity rate of up to 98.5% after 100 cycles at a current density of 0.5 C in carbonate electrolyte, and even a capacity of 128 mA h g−1 can be achieved at a current density of 2 C. Overall, this work not only provides a promising solution for improving the electrochemical performance of lithium-metal anodes but also opens up new opportunities for the development of other metal anodes in the energy storage field.Data availability
All the data supporting this article have been included in the main text and the ESI.†Author contributions
H. L. and R. H. designed the experiments, prepared all the materials, conducted the electrochemical measurements, and drafted the initial version of the manuscript. X. Y. provided assistance with additional tests and experiments. D. K. contributed to creating the images. C. Y., Q. M., L. Z., and Z. C. F. participated in analyzing and discussing the characterization data. C. Z. and T. Z. conceived the project and provided guidance and supervision throughout the work. All authors have approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support from the National Natural Science Foundation of China (52172173 and 51872071), the Natural Science Foundation of Anhui Province for Distinguished Young Scholars (2108085J25), the Natural Science Foundation of Anhui Province for Outstanding Young Scholar (2208085Y05), the Anhui Provincial Scientific Reuter Foundation for Returned Scholars (2022LCX030), the Excellent Research and Innovation Team Project of Anhui Province (2022AH010001), the Natural Science Foundation of Anhui Province (2208085QE130), the Distinguished Youths Research Project of Anhui Province (2022AH020013), and the Open Fund of Guangdong Provincial Key Laboratory of Advance Energy Storage Materials (AESM202106). We acknowledge the High-performance Computing Platform of Anhui University for providing computing resources.Notes and references
- B. Han, D. Xu, S. S. Chi, D. He, Z. Zhang, L. Du, M. Gu, C. Wang, H. Meng and K. Xu, Adv. Mater., 2020, 32, 2004793 CrossRef CAS.
- B. K. Park, H. S. Kim, S. A. Han, H. J. Leem, T. Kim, Y. G. Kwon, J. H. Yang, J. Mun, J. Yu and M. S. Park, ACS Appl. Mater. Interfaces, 2023, 15, 6923–6932 CrossRef CAS PubMed.
- Z. L. Zhuang, C. Liu, Y. Y. Yan, P. C. Ma and D. Q. Tan, J. Mater. Chem. A, 2021, 9, 27095 RSC.
- F. Zhao, P. Zhai, Y. Wei, Z. Yang, Q. Chen, J. Zuo, X. Gu and Y. Gong, Adv. Sci., 2022, 9, 2103930 CrossRef CAS.
- Y. Ye, Y. Zhao, T. Zhao, S. Xu, Z. Xu, J. Qian, L. Wang, Y. Xing, L. Wei and Y. Li, Adv. Mater., 2021, 33, 2105029 CrossRef CAS.
- S. Zhang, K. Liu, J. Xie, X. Xu, J. Tu, W. Chen, F. Chen, T. Zhu and X. Zhao, ACS Appl. Mater. Interfaces, 2023, 15, 6594–6602 CrossRef CAS.
- Z. L. Zhuang, F. M. Zhang, Y. N. Zhou, Y. F. Niu, Y. H. Yan and D. Q. Tan, Mater. Today Energy, 2022, 30, 101192 CrossRef CAS.
- L. Luo, S. Xia, X. Zhang, J. Yang and S. Zheng, Adv. Sci., 2022, 9, 2104391 CrossRef CAS.
- F. He, Z. Hu, W. Tang, A. Wang, B. Wen, L. Zhang and J. Luo, Adv. Funct. Mater., 2022, 32, 2201465 CrossRef CAS.
- Y. Zou, C. Chen, Y. Sun, S. Gan, L. Dong, J. Zhao and J. Rong, Chem. Eng. J., 2021, 418, 128616 CrossRef CAS.
- Z. Zhang, L. Tang, C. Chen, H. Yu, H. Bai, L. Wang, M. Qin, Y. Feng and W. Feng, J. Mater. Chem. A, 2021, 9, 875–883 RSC.
- P. Zhai, L. Liu, Y. Wei, J. Zuo, Z. Yang, Q. Chen, F. Zhao, X. Zhang and Y. Gong, Nano Lett., 2021, 21, 7715–7723 CrossRef CAS PubMed.
- D. Yang, J. Li, F. Yang, J. Li, L. He, H. Zhao, L. Wei, Y. Wang, X. Wang and Y. Wei, Nano Lett., 2021, 21, 7063–7069 CrossRef CAS.
- T. Cao, X. Cheng, M. Wang, J. Lu, J. Niu, H. Liu, X. Liu and Y. Zhang, ACS Appl. Mater. Interfaces, 2023, 15, 6666–6675 CrossRef CAS.
- J. Wu, Z. Rao, X. Liu, Y. Shen, C. Fang, L. Yuan, Z. Li, W. Zhang, X. Xie and Y. Huang, Adv. Mater., 2021, 33, 2007428 CrossRef CAS PubMed.
- C. Wei, L. Tan, Y. Tao, Y. An, Y. Tian, H. Jiang, J. Feng and Y. Qian, Energy Storage Mater., 2021, 34, 12–21 CrossRef.
- H. Wang, P. Hu, X. Liu, Y. Shen, L. Yuan, Z. Li and Y. Huang, Adv. Sci., 2021, 8, 2100684 CrossRef CAS PubMed.
- W. Wang, X. Zhu and L. Fu, CCS Chem., 2021, 3, 686–695 CrossRef CAS.
- P. C. Ma, Z. L. Zhuang, J. C. Cao, B. W. Ju and X. M. Xi, ACS Appl. Energy Mater., 2022, 5, 6417–6422 CrossRef CAS.
- M. A. Lee, H. J. Leem, J. Yu and H. S. Kim, ACS Appl. Mater. Interfaces, 2022, 14, 35645–35653 CrossRef CAS.
- X. Guo, Y. Ding, L. Xue, L. Zhang, C. Zhang, J. B. Goodenough and G. Yu, Adv. Funct. Mater., 2018, 28, 1804649 CrossRef.
- F. Li, Q. Qin, Y. Zhou, Y. Wu, W. Xue, S. Gao, J. Shang, Y. Liu and R. W. Li, Adv. Mater. Technol., 2018, 3, 1800131 CrossRef.
- Z. L. Zhuang, Y. T. Tang, B. W. Ju and F. Y. Tu, Sustainable Energy Fuels, 2021, 5, 2433 RSC.
- C. Wei, H. Fei, Y. Tian, Y. An, G. Zeng, J. Feng and Y. Qian, Small, 2019, 15, 1903214 CrossRef CAS PubMed.
- Y. Lou, H. Liu and J. Zhang, Chem. Eng. J., 2020, 399, 125732 CrossRef CAS.
- G. Li, S. Liu, Z. Liu and Y. Zhao, Small, 2021, 17, 2102196 CrossRef CAS.
- B. Feng, X. Jiang, G. Zou, W. Wang, T. Sun, H. Yang, G. Zhao, M. Dong, Y. Xiao and H. Zhu, Adv. Funct. Mater., 2021, 31, 2102359 CrossRef CAS.
- H. Chen, D. Adekoya, L. Hencz, J. Ma, S. Chen, C. Yan, H. Zhao, G. Cui and S. Zhang, Adv. Energy Mater., 2020, 10, 2000049 CrossRef CAS.
- Z. L. Zhuang, B. W. Ju, P. C. Ma, L. Z. Yang and F. Y. Tu, Ionics, 2021, 27, 1069–1079 CrossRef CAS.
- H. Duan, Y.-X. Yin, Y. Shi, P. F. Wang, X. D. Zhang, C. P. Yang, J. L. Shi, R. Wen, Y. G. Guo and L. J. Wan, J. Am. Chem. Soc., 2018, 140, 82–85 CrossRef CAS.
- S. S. Chi, Y. Liu, N. Zhao, X. Guo, C. W. Nan and L. Z. Fan, Energy Storage Mater., 2019, 17, 309–316 CrossRef.
- Y. Zheng, S. Xia, F. Dong, H. Sun, Y. Pang, J. Yang, Y. Huang and S. Zheng, Adv. Funct. Mater., 2021, 31, 2006159 CrossRef CAS.
- H. Wu, H. Jia, C. Wang, J. G. Zhang and W. Xu, Adv. Energy Mater., 2021, 11, 2003092 CrossRef CAS.
- P. Wang, X. Dai, P. Xu, S. Hu, X. Xiong, K. Zou, S. Guo, J. Sun, C. Zhang, Y. Liu, T. Zhou and Y. Chen, eScience, 2023, 3, 100088 CrossRef.
- J. Meng, Y. Zhang, X. Zhou, M. Lei and C. Li, Nat. Commun., 2020, 11, 3716 CrossRef CAS PubMed.
- P. Zhang, Q. Wang, R. Guo, M. Zhang, S. Wang, C. Lu, M. Xue, J. Fan, Z. He and W. Rao, Mater. Horiz., 2019, 6, 1643–1653 RSC.
- J. You, H. Deng, X. Zheng, H. Yan, L. Deng, Y. Zhou, J. Li, M. Chen, Q. Wu and P. Zhang, ACS Appl. Mater. Interfaces, 2022, 14, 5298–5307 CrossRef CAS.
- Y. Sun, M. Amirmaleki, Y. Zhao, C. Zhao, J. Liang, C. Wang, K. R. Adair, J. Li, T. Cui and G. Wang, Adv. Energy Mater., 2020, 10, 2001139 CrossRef CAS.
- R. Xu, X. Q. Zhang, X. B. Cheng, H. J. Peng, C. Z. Zhao, C. Yan and J. Q. Huang, Adv. Funct. Mater., 2018, 28, 1705838 CrossRef.
- S. Chen, T. Liu, J. Ge, J. Hong and Y. Wang, ACS Appl. Energy Mater., 2021, 5, 539–548 CrossRef.
- S. Guo, L. Wang, Y. Jin, N. Piao, Z. Chen, G. Tian, J. Li, C. Zhao and X. He, Nano Convergence, 2020, 7, 1–10 CrossRef PubMed.
- Z. Jiang, L. Jin, Z. Han, W. Hu, Z. Zeng, Y. Sun and J. Xie, Angew. Chem., Int. Ed., 2019, 58, 11374–11378 CrossRef CAS PubMed.
- Z. Zhou, Y. Feng, J. Wang, B. Liang, Y. Li, Z. Song, D. M. Itkis and J. Song, Chem. Eng. J., 2020, 396, 125254 CrossRef CAS.
- Z. Zhang, X. Zhou and Z. Liu, ACS Appl. Energy Mater., 2021, 4, 7288–7297 CrossRef CAS.
- Y. Kim, J. Choi, J. H. Youk, B.-S. Lee and W. R. Yu, RSC Adv., 2022, 12, 346–354 RSC; J. Xiao, P. Zhai, Y. Wei, X. Zhang, W. Yang, S. Cui, C. Jin, W. Liu, X. Wang and H. Jiang, Nano Lett., 2020, 20, 3911–3917 CrossRef CAS PubMed.
- J. Xiao, P. Zhai, Y. Wei, X. Zhang, W. Yang, S. Cui, C. Jin, W. Liu, X. Wang and H. Jiang, Nano Lett., 2020, 20, 3911–3917 CrossRef CAS.
- G. Wang, C. Chen, Y. Chen, X. Kang, C. Yang, F. Wang, Y. Liu and X. Xiong, Angew. Chem., Int. Ed., 2020, 59, 2055–2060 CrossRef CAS PubMed.
- X. Sun, S. Yang, T. Zhang, Y. Shi, L. Dong, G. Ai, D. Li and W. Mao, Nanoscale, 2022, 14, 5033–5043 RSC.
- L. Zou, K. Shi, Z. Xu, Z. Yang and W. Zhang, ACS Omega, 2021, 7, 994–1002 CrossRef PubMed.
- K. Pan, L. Zhang, W. Qian, X. Wu, K. Dong, H. Zhang and S. Zhang, Adv. Mater., 2020, 32, 2000399 CrossRef CAS PubMed.
- B. He, S. Liu, X. Zhao, J. Liu, Q. Ye, S. Liu and W. Liu, ACS Appl. Nano Mater., 2020, 3, 10115–10122 CrossRef CAS.
- H. Jiang, C. Yang, M. Chen, X. Liu, L. Yin, Y. You and J. Lu, Angew. Chem., Int. Ed., 2023, 135, e202300238 CrossRef.
- Q. Zhang, Z. Wang, S. Zhang, T. Zhou, J. Mao and Z. Guo, Electrochem. Energy Rev., 2018, 1, 625–658 CrossRef CAS.
- S. Liu, J. Mao, L. Zhang, W. K. Pang, A. Du and Z. Guo, Adv. Mater., 2021, 33, 2006313 CrossRef CAS.
- S. Liu, J. Vongsvivut, Y. Wang, R. Zhang, F. Yang, S. Zhang, K. Davey, J. Mao and Z. Guo, Angew. Chem., Int. Ed., 2023, 62, e202215600 CrossRef CAS PubMed.
- Y. Lyu, J. Yuwono, P. Wang, Y. Wang, F. Yang, S. Liu, S. Zhang, B. Wang, K. Davey and J. M. Z. Guo, Angew. Chem., Int. Ed., 2023, 62, e202303011 CrossRef CAS.
- J. Peng, Z. Chen, Y. Li, S. Hu, Q. Pan, F. Zheng, H. Wang and Q. Li, Rare Met., 2022, 41, 951–959 CrossRef CAS.
- D. Wang, Y. Chang, Y. Li, S. Zhang and S. Xu, Rare Met., 2021, 40, 3156–3165 CrossRef CAS.
- H. Zhang, C. Wang, H. Luo, J. Chen, M. Kuang and J. Yang, Angew. Chem., Int. Ed., 2023, 62, e202217071 CrossRef CAS PubMed.
- G. Zhu, R. Guo, W. Luo, H. Liu, W. Jiang, S. X. Dou and J. P. Yang, Natl. Sci. Rev., 2021, 8, nwaa152 CrossRef CAS PubMed.
- W. Cao, Q. Li, X. Yu and H. Li, eScience, 2022, 2, 47–78 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc03884j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |