
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRhodium(I)-catalyzed cascade C(sp2)–H bond alkylation – amidation of anilines: phosphorus as traceless directing group†
Marie
Peng
a,
Denis
Ari
a,
Thierry
Roisnel
 a,
Henri
Doucet
a and
Jean-François
Soulé
*b
a,
Henri
Doucet
a and
Jean-François
Soulé
*b
aUniv. Rennes, CNRS UMR6226, Rennes F-3500, France
bChimie ParisTech, PSL University, CNRS, Institute of Chemistry for Life and Health Sciences, 75005 Paris, France. E-mail: jean-francois.soule@chimieparistech.psl.eu
First published on 1st August 2023
Abstract
We introduce a versatile Rh(I)-catalyzed cascade reaction, combining C(sp2)–H bond functionalization and amidation between N-arylphosphanamines and acrylates. This innovative approach enables the rapid synthesis of dihydroquinolinone scaffolds, a common heterocycle found in various pharmaceuticals. Notably, the presence of the phosphorus atom facilitates the aniline ortho-C(sp2)–H bond activation prior to N–P bond hydrolysis, streamlining one-pot intramolecular amidation. Moreover, we demonstrate the applicability of this reaction by synthesizing an antipsychotic drug. Detailed mechanistic investigations revealed the involvement of a Rh–H intermediate, with substrate inhibition through catalyst saturation.
Introduction
Benzo-fused N-heterocycles are valuable bulling blocks in molecular sciences, including medicinal and materials chemistry. Remarkably, several drug leads feature dihydroquinolinone scaffolds (Fig. 1A).1 For instance, carteolol is a non-selective beta blocker used to treat glaucoma; cilostazol is a medication used to help the symptoms of intermittent claudication in peripheral vascular disease, and vesnarinone is a cardiotonic agent. Dihydroquinolinones were formerly prepared from aniline derivatives via amidation reaction followed by intramolecular Friedel–Crafts reaction in the presence of an excess amount of Lewis acid, typically AlCl3 (Fig. 1B, left).2 Due to their importance, several strategies considering better process sustainability have been developed, ranging from transition metal-catalyzed cyclization reactions3 and radical cyclizations.4 Since these approaches require multi-step synthesis of the precursors and specific starting materials, they are often limited to preparing a few particular examples. On the other hand, a more direct approach for synthesizing dihydroquinolinones through the concomitant formation of C–C and C–N bonds, so-called C(sp2)–H bond annulation remains unexplored (Fig. 1B, right). This approach is highly desirable as mere anilines and acrylates are the starting materials. However, this C(sp2)–H bond annulation for the formation of dihydroquinolinones has proven to be incredibly challenging given the inherent nucleophilic characters of anilines which are prone to react in 1,4-addition reaction affording trisubstituted anilines (Fig. 1C(1)).5 To swap the selectivity toward C(sp2)–H bond functionalization at the ortho-position of the N-atom, a directing group (DG) has to be installed.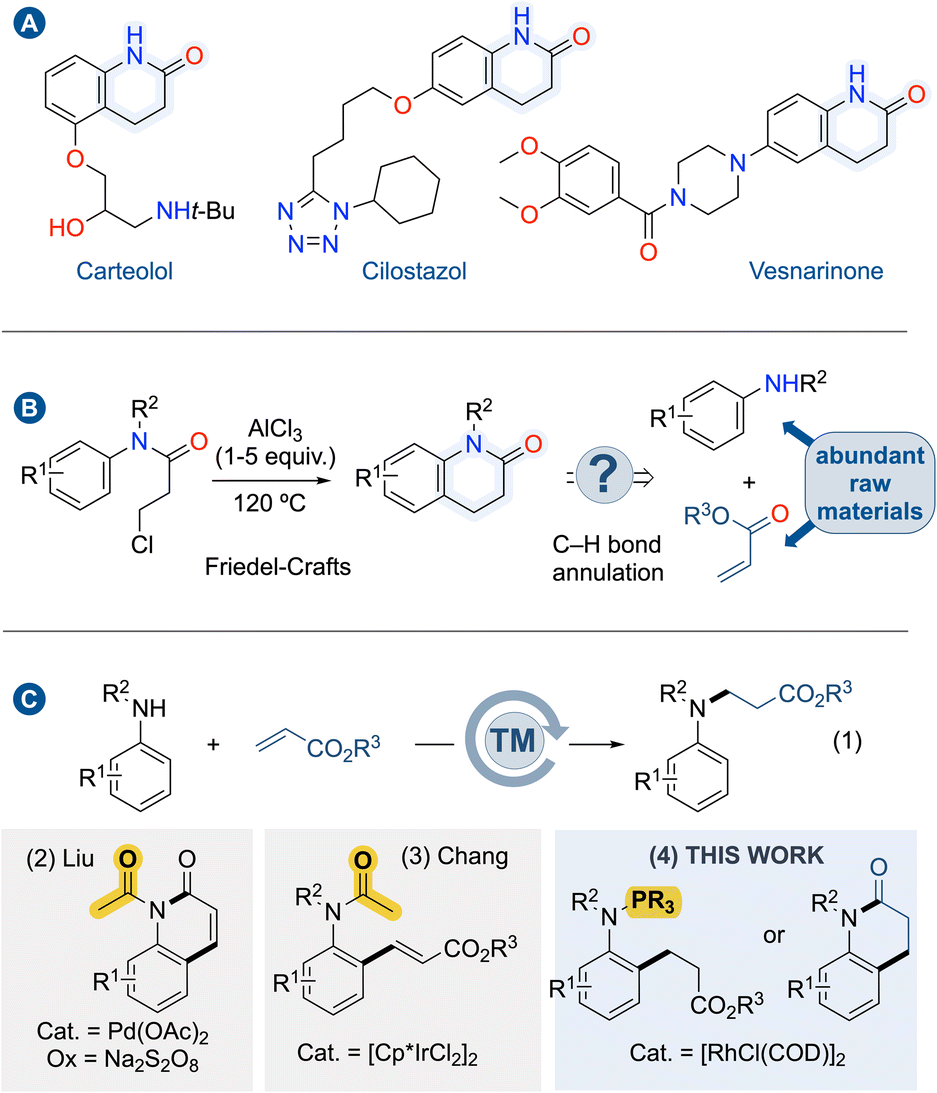 | ||
| Fig. 1 Dihydroquinolinones & their preparation: (A) selected examples of dihydroquinolinones-containing drug leads. (B) Traditional synthesis of dihydroquinolinones vs. C(sp2)–H bond annulation. (C) Key role of the directing group for anilines acrylates couplings: amination, alkenylation vs. alkylation. | ||
The second issue to face is the selectivity between alkenylation versus alkylation. For instance, in 2015, Liu and co-workers demonstrated that the installation of a transient acetamido as a directing group on aniline partners allows the synthesis of 2-quinolinones through a cascade reaction of Pd-catalyzed C(sp2)–H alkenylation – cyclization reaction (Fig. 1C(2)).6 Therefore, a subsequent hydrogenation step of the double bond is required to deliver dihydroquinolinones.7 The alkenylation versus alkylation selectivity generally depends on the choice of the directing group. Indeed, Chang and co-workers demonstrated that the acetamido group affords alkenylation, while pyridine, pyrazole, and pyrimidine lead to alkylation products using [Cp*IrCl2] catalyst (Fig. 1C(3)).8 Nevertheless, utilizing Rh(III)-catalysis, the non-removable directing group of pyridine enables alkenylation even within the annulation process.9 Alternatively, allylic alcohols can be used in Rh(III)-catalyzed ortho-directed C–H bond alkylations of anilines using pyrimidine as non-removable directing group.10 Following the discovery that trivalent phosphorus [P(III)] acts as directing group for the C(sp2)–H bond functionalization of phosphines11 or indoles,12 and given the fact that P(III) favors alkylation products with acrylates using Rh(I) catalysts,13 we hypothesized that if N-arylphosphanamines could be alkylated at ortho-position with acrylates, such intermediate would rapidly undergo intramolecular amidation reaction driven by N–P bond hydrolysis provided that suitable conditions are found (Fig. 1C(4)). Herein, we describe the reaction development with mechanistic aspects showcasing that trivalent phosphorus acts as a traceless directing group in Rh(I)-catalyzed cascade reaction of C(sp2)–H bond activation/alkylation – amidation of anilines with acrylate derivatives for the one-pot synthesis of dihydroquinolinones.
Results and discussion
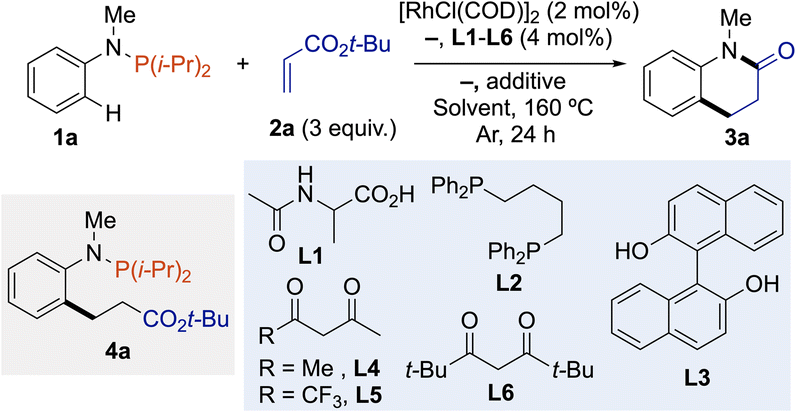
To demonstrate the feasibility of the one-pot cascade of the C(sp2)–H bond alkylation – amidation reaction, we carried out optimization with phosphanamine 1a and tert-butylacrylate (2a) as the annulative agent. Using our previously published method for the C(sp2)–H bond alkylation of phosphines as the starting point,13a we quickly recognized that the most important aspect of reaction optimization was identifying the optimal solvent (and additive) to promote C(sp2)–H bond alkylation followed by the one-pot removal of P(i-Pr)2 directing group to trigger amidation reaction and deliver the dihydroquinolin-2-one scaffold. Indeed, in toluene or 1,4-dioxane, desired product 3a was obtained among intermediate 4a, which could be converted into 3a by acidic hydrolysis (Table 1, entries 1 and 2). In contrast, the one-pot process occurred in 1,2-dichloroethane or DMF, affording dihydroquinolin-2-one 3a in 24% and 40% yield, respectively, without additional treatment (Table 1, entries 3 and 4). The addition of base to favor C(sp2)–H bond cleavage,14 or Lewis acid to promote the amination reaction (see ESI†) have both a negative effect on the reaction outcomes (Table 1, entries 5 and 6). Next, we examined the effect of ligands with the aim of increasing the reactivity. Protected amino-acid such as Ac-Ala (L1) slightly improved the reaction to afford 3a in 54% yield, while diphosphine L2 failed to affect the title reaction (Table 1, entries 7 and 8). Diols, such as BINOL (L3), previously employed by Shi in Rh(I)-catalyzed phosphine-directed C(sp2)–H functionalization,12b did not gave better results (Table 1, entry 9). A set of bidentate acac-type ligands L4–L6 was tested and 2,2,6,6-tetramethylheptane-3,5-dione (L6) was the most efficient, providing 3a in 58% yield (Table 1, entries 10–12). However, the yield in 3a varied when different qualities of solvents were employed. Nevertheless, the reaction proceeded to completion, resulting in the observation of a mixture containing both 3a and 4a, demonstrating the superiority of DMF. The 3a/4a selectivity varaition may be due to the amount of water necessary to hydrolyze the N-arylphosphanamine into aniline derivatives. In the presence of molecular sieves, the reaction was much less effective and intermediate 4a was obtained as the major product in 51%; while in the presence of a small amount of water, reproducible results were obtained, and 10 equivalents was the best stoichiometry affording 3a in 81% isolated yield (Table 1, entries 13–17). Control experiment without L6 but with 10 equivalents of water gave a lower yield of 35% in 1a, revealing the importance of L6 in the catalytic conditions (Table 1, entry 18). It is worth mentioning that reactions performed with methyl or ethyl acrylate gave 3a in similar yields (Table 1, entries 19 and 20).|
|
||||
|---|---|---|---|---|
| Entry | L | Additive | Solvent | 3a (%) |
| a Determined by GC-analysis using n-dodecane as internal standard, isolated yield is shown in parentheses. b Yield obtained after HCl hydrolysis. c Yield of 4a. d Methyl acrylate instead of tert-butyl acrylate. e Ethyl acrylate instead of tert-butyl acrylate. | ||||
| 1 | — | — | Toluene | 42b |
| 2 | — | — | 1,4-Dioxane | 12b |
| 3 | — | — | ClCH2CH2Cl | 24 |
| 4 | — | — | DMF | 40 |
| 5 | — | NaOAc (0.3 equiv.) | DMF | 30 |
| 6 | — | AlMe3 (0.2 equiv.) | DMF | 20 |
| 7 | L1 | — | DMF | 54 |
| 8 | L2 | — | DMF | 36 |
| 9 | L3 | — | DMF | 46 |
| 10 | L4 | — | DMF | 45 |
| 11 | L5 | — | DMF | 22 |
| 12 | L6 | — | DMF | 58 |
| 13 | L6 | MS 4 Å (50 mg) | DMF | 21 (51)c |
| 14 | L6 | H2O (2 equiv.) | DMF | 62 |
| 15 | L6 | H2O (6 equiv.) | DMF | 75 |
| 16 | L6 | H2O (10 equiv.) | DMF | 90 (81) |
| 17 | L6 | H2O (20 equiv.) | DMF | 54 |
| 18 | — | H2O (10 equiv.) | DMF | 35 |
| 19d | L6 | H2O (10 equiv.) | DMF | 85 |
| 20e | L6 | H2O (10 equiv.) | DMF | 84 |
With the aforementioned optimal reaction conditions established, the substrate scopes of this reaction were then examined (Scheme 1). First, different substituents on the aromatic ring of the aniline moiety were examined. Electron-donating (MeO, Me) and electron-withdrawing (OCF3, F, Cl) groups at the para-position of the N-atom were tolerated providing dihydroquinolinones 3b–f in good to high yields. However, the conditions were incompatible with Br substituent on the N-arylphosphanamine partner. When meta-substituted phosphanamines were employed, the C(sp2)–H bond alkylation regioselectivity occurred at the less hindered position, whatever the substituent, as exemplified by products 3h–k containing F, Cl, Me, or MeO groups. From N-(3,5-difluorophenyl)-1,1-diisopropyl-N-methylphosphanamine, dihydroquinolinone 3l was isolated in 72% yield. Reaction with ortho-fluoro-N-arylphosphanamine 1m gave 3m in a moderate yield of 42%. Other alkyl groups on the N-atom such as Et or n-Bu were also tolerated affording 3n–p in 75–83% yields.15 Beyond N-alkyl phosphanamines, 1,2,3,4-tetrahydroquinoline was also subjected to this C(sp2)–H bond annulation to afford tricyclic amide 3q in 70% yield. α-Substituted acrylates such as dimethyl 2-methylenesuccinate (2b) and methyl methacrylate (2c) were also successfully coupled with 1a to produce 3-substituted N-methyl-dihydroquinolinones 3r and 3s in 57% and 60% yield, respectively.
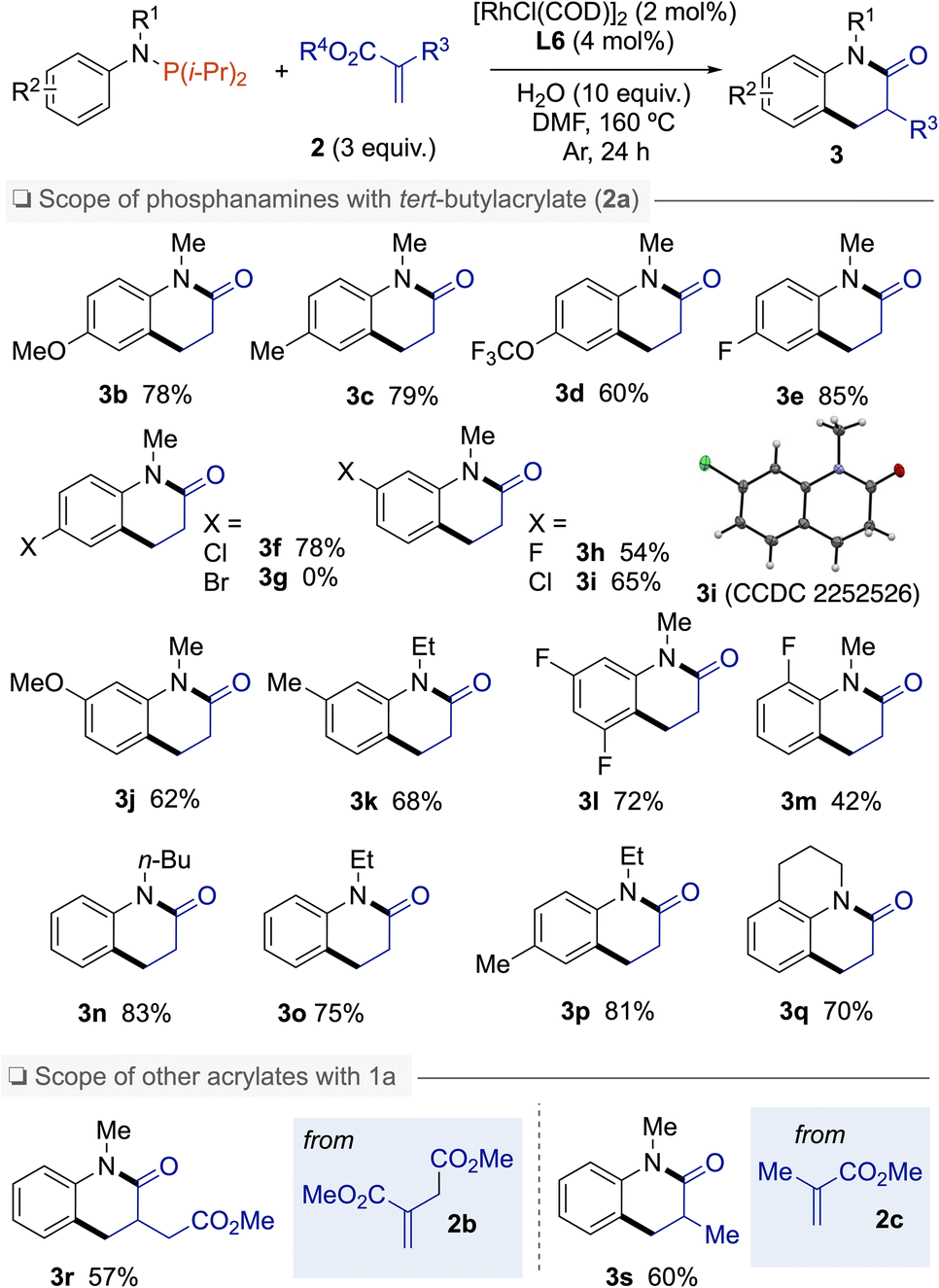 | ||
| Scheme 1 Scope of the Rh(I)-catalyzed C–H bond activation with migrations of nitroarenes. | ||
Satisfied by these results, we then moved to extend this trivalent phosphorus traceless directing group strategy to ortho-C(sp2)–H bond functionalization of anilines with styrene derivatives (Scheme 2). Accordingly, ortho-alkylated N-methyl aniline 5a was obtained in 60% yield from 1a and styrene (2d) using the same catalytic system, namely 2 mol% [RhCl(COD)]2 associated with 4 mol% of L6 in the presence of 10 equivalents of water in DMF. Noteworthy, the C–P bond was not fully hydrolyzed when the reaction was performed without water. Electron-rich 1-(tert-butyl)-4-vinylbenzene displayed a higher reactivity than styrene, as exemplified by forming 5b and 5c in 65% and 73% yield, respectively. This reactivity trend was also confirmed with 1-methoxy-4-vinylbenzene and 2,4-dimethyl-1-vinylbenzene, with which 1a was coupled to give the ortho-alkylated N-methyl anilines 5d and 5e in 75% and 69% yield, respectively. Conversely, the reaction was more sluggish with styrenes holding an electron-withdrawing group such as F and Cl, as the resulting products 5f and 5g were isolated in only 32% and 42% yields.
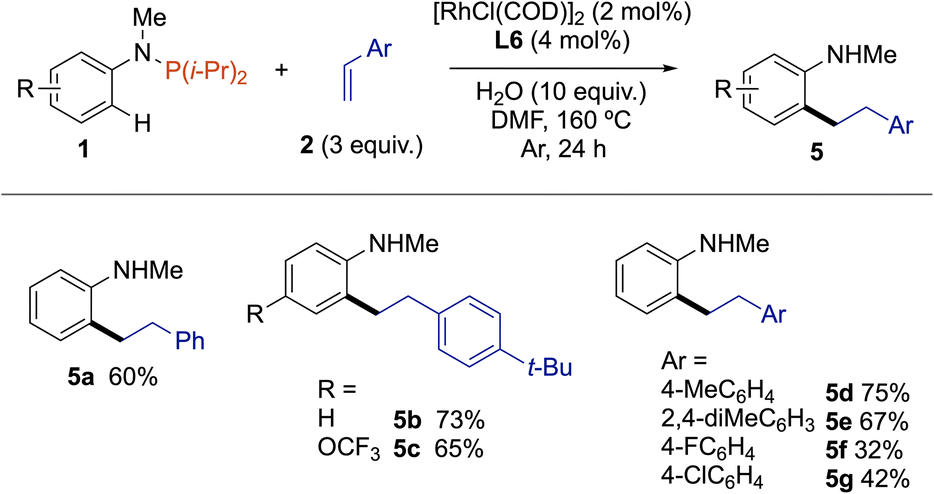 | ||
| Scheme 2 Scope of C–H bond alkylation of N-arylphosphanamines with styrenes. | ||
Having established a method to construct dihydroquinolinones with good functional group compatibility, we investigated the feasibility of applying it to prepare aripiprazole N-methylated analog 8. Aripiprazole treats schizophrenia, bipolar disorder, major depressive and tic disorders.16 In 2020, the aripiprazole market was dominated by two brand companies (Otsuka & Bristol-Myers Squibb) and is expected to grow at a CAGR of 4.60% in the forecast period of 2022–2029. The majority of previous routes toward aripiprazole (or its N-alkyl congeners) involved multi-step synthesis for the construction of the dihydroquinolinone scaffold17 (e.g., intramolecular Friedel–Craft reaction followed by Pd-catalyzed hydrogenation of double bond,18 or through a Beckmann rearrangement from indanone derivative). Owing to the increasing demand for aripiprazole and its N-alkyl analogs, and the lack of synthetic routes for the construction of dihydroquinolinones from simple starting materials prompted us to develop an efficient route to aripiprazole N-methylated analog 8, employing our Rh(I)-catalyzed cascade reaction of C(sp2)–H bond alkylation – amidation of anilines (Scheme 3). A gram-scale reaction (5 mmol) performed from 1,1-diisopropyl-N-(3-methoxyphenyl)-N-methylphosphanamine with 2a gave 0.54 g of 3j (57% yield). Then, classical deprotection of phenol group by BBr3, followed by Williamson reaction using 2 equivalents of 1,4-dibromobutane in the presence of K2CO3 afforded 7 in good yield. Final coupling with 1-(2,3-dichlorophenyl)piperazine in the presence of KI as a relay-nucleofuge and triethylamine as base led to the formation of desired aripiprazole N-methylated analog 8 in 70% yield.
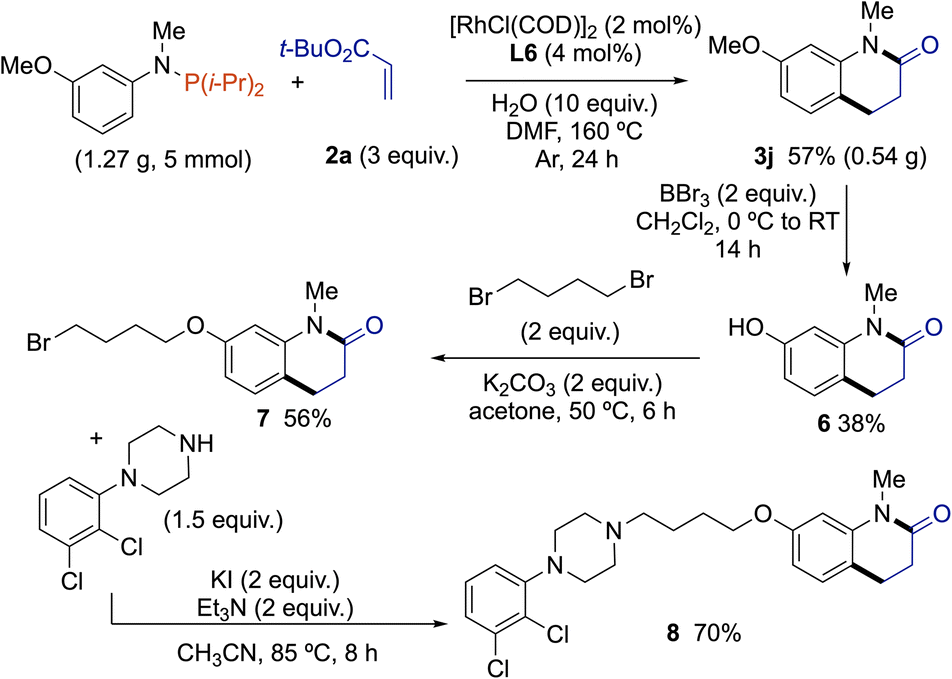 | ||
| Scheme 3 Gram-scale reaction and synthesis of aripiprazole N-methylated analog. | ||
Intrigued by the exact role and the becoming of the phosphorus group along this reaction, we started a mechanistic investigation by carrying out a set of control experiments. No reaction occurred from P,P-diisopropyl-N-methyl-N-phenylphosphinic amide, indicating that trivalent phosphorus is not oxidized at least before the C(sp2)–H bond cleavage. The use of classical acetamide as a directing group or reaction from N-methylaniline also failed to deliver cyclized amide 3a (Fig. 2A). These results indicate that trivalent phosphorus is the active directing group. A standard reaction between phosphanamine 1a and 2a in the presence of 1 equivalent of 3-fluoro-N-methylaniline revealed that there is no phosphorus-directing group scrambling (Fig. 2B). A careful analysis of the reaction outcomes by GC-MS and 31P NMR has allowed us to identify the formation hydroxydiisopropylphosphane (9a) as the side product,19 which may explain that -P(i-Pr)2 acts as a traceless directing group rather than a transient directing group (Fig. 2C).20 To completely rule out the intermediacy of the NH amide substrate, we conducted a control experiment in the presence of one equivalent of 9a, which did not yield the product 3a (Fig. 2D). To determine the role of L6, we decided to prepare the well-defined Rh(L6)(COD) complex, but it was completely inactive for this cascade reaction (Fig. 2E). This result may suggest that L6 has an indirect role, possibly in a catalyst regeneration process, by facilitating the decomplexation of 9a through an unknown mechanism. To get more information on the structure of rhodium intermediates, we then conducted a stoichiometric reaction between [RhCl(COD)]2 and 1a with in situ following by NMR spectroscopies (Fig. 2F). At room temperature, a mixture of two distinct phosphorus containing Rh complexes was observed within two hours. It is generally accepted that treatment of complexes [{Rh-(COD)(μ2-Cl)}2] with phosphine ligands in the absence of silver salts smoothly affords neutral μ2-bridged dinuclear rhodium complexes of the type [{Rh(phosphine)2(μ2-Cl)}2].21 The signal at 87.1 ppm (d, 1JRh,P = 156.7 Hz) is attributed to [{Rh(1a)2(μ2-Cl)}2]; while after isolation, we can attribute the signal at 30.4 ppm (d, 1JRh,P = 140.5 Hz) to [{Rh(9a)2(μ2-Cl)}2]. This complex might be formed through the in situ hydrolysis of P–N bond by water. After 24 h, the proportion of [{Rh(9a)2(μ2-Cl)}2] increases, indicating that it is the resting state catalyst. Although multiple doublet signals are also observed by 31P NMR data, a characteristic Rh–H signal appears as a doublet of doublets at −21.66 ppm (1JRh,H = 34.1 Hz, 2JP,H = 27.6 Hz) in 1H NMR, proving that a C–H bond activation mechanism rather than a CMD mechanism (Fig. 2F). To gain further insight into the reaction mechanism, deuterated labelling experiments were carried out in the standard reaction conditions (Fig. 2G). A standard reaction performed using 10 equivalents of deuterated water gave 57% deuterium incorporation at ortho-position of the NMeP(i-Pr)2 group. This deuterium labelling experiment suggests that the C(sp2)–H bond cleavage is reversible. Moreover, 69% and 20% deuterium incorporations were detected at the α and β-positions of the ester group of 3d. In addition, the deuterium incorporation in the NMe group may result from the formation of amide rhodacycle through C(sp3)–H bond activation. The control experiment involving 3d under deuterium labelling conditions does not show any deuterium incorporation in 3d. The observed H/D isotopic exchanges likely occur due to the formation of Rh–D species, similar to a process described by Martin and Milstein in the C–H bond activation of benzylphosphine by Rh(I) complexes.22 Furthermore, the incorporation of deuterium at the α-position of the ester group should result from an insertion/β-H elimination process.
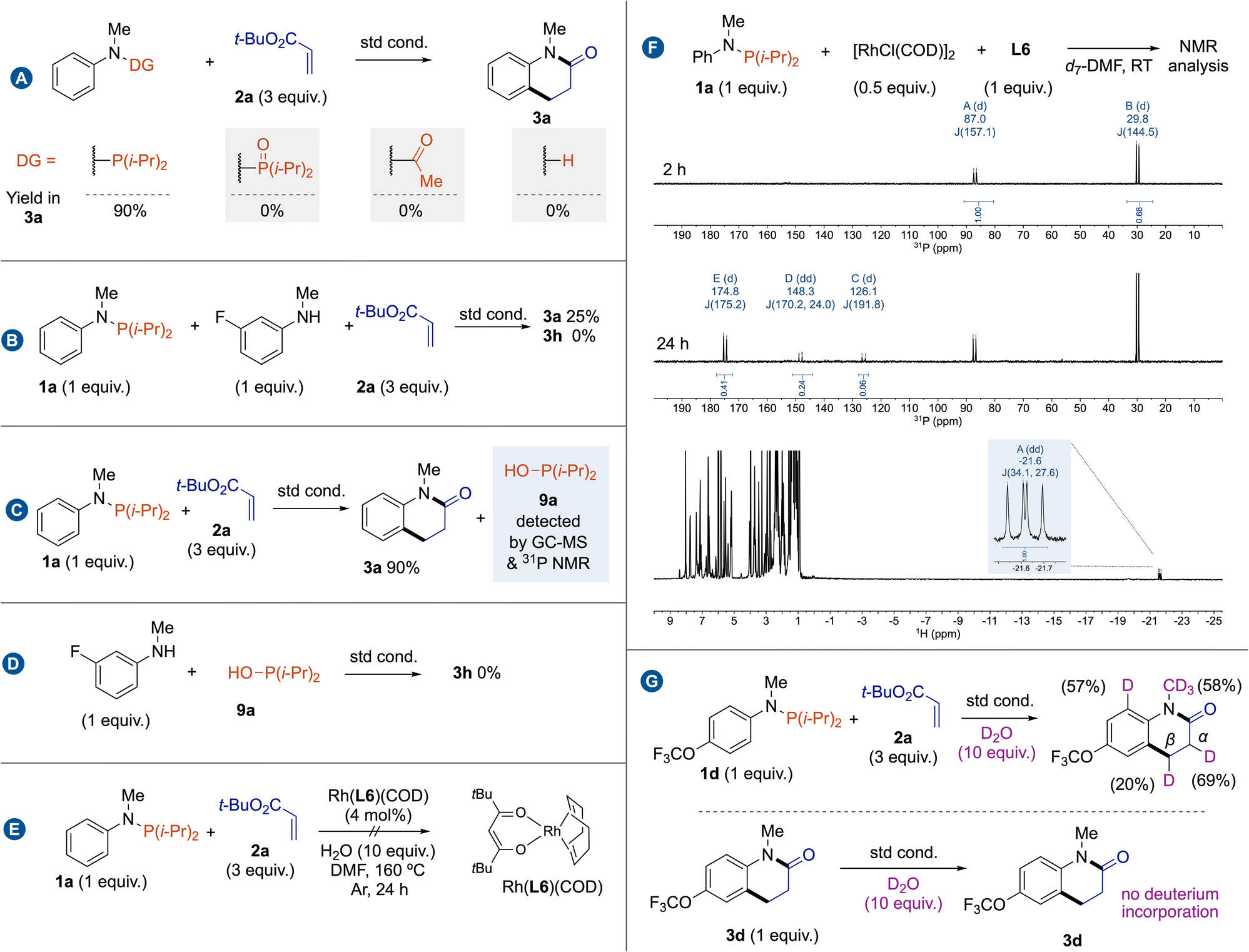 | ||
| Fig. 2 Mechanistic studies: (A) control experiments using different directing groups, (B) parallel control experiment, (C) detection of residual phosphorus, (D) control experiment of the reactivity of NH amide substrate in the presence of 9a, (E) reactivity of RhL6(COD) complex, (F) Stoichiometric reactions (G) deuterium labelling experiments. | ||
Then, we conducted a kinetic study to obtain a better picture of the reaction pathway (Fig. 3). First, we plotted the kinetic profile of the reaction by following the consumption of 1a, the production and consumption of 4a by 31P NMR, while the production of 3a was followed by 1H NMR (Fig. 3A). The Gaussian-shape kinetic profile of 4a indicates that k1 > k2 meaning that 4a is a latent intermediate of the reaction. However, we have never observed hydrolyzed aniline 10a during the reaction, possibly due to a spontaneous cyclization affording 3a. Some de facto approximations were formulated. If [10a] ≈ 0 during the reaction, then k3 ≫ k2; steps 2 and 3 can be considered as a single operation, and the rate determining step (RDS) is the hydrolysis of N–P(i-Pr)2 bond. Our attention next turned toward determining the factors influencing the kinetic efficiency of Rh(I)-catalyzed cascade C(sp2)–H bond alkylation – amidation of anilines. Reaction progress kinetic analysis revealed that catalyst deactivation was not caused by substrates or product formation, including 9a.17 Then, we measured the initial reaction rate for the model reaction as a function of the concentration of water, catalyst, tert-butylacrylate (2a) and phosphanamine 1a in DMF at 160 °C. Interestingly, we observed a zero-order dependence on water for the consumption of 1a, and a first-order dependence for the production of 3a (Fig. 3B). These observations indicate that the water is required for the second step, namely the hydrolysis of N–P(i-Pr)2 bond, and its presence (up to 10 equivalents) has a negligible influence on the Rh(I) catalytic activity.23 In contrast, first-order dependence on Rh/L6 catalytic system was observed for the consumption of 1a (Fig. 3C). The first-order dependence of the reaction rate on acrylate concentration was obtained from the linear plot of initial rate (Δ[1a]/Δt) vs. initial acrylate concentration ([2a]) (Fig. 3D). Saturation kinetics were observed with respect to phosphanamine concentration ([1a]) when similar experiments were carried out by measuring the initial rate over a range of 1a concentrations (Fig. 3E). This rate inhibition by phosphanamine 1a might have been anticipated due the strong ability of P(III)-ligands to coordinate Rh delivering a fully-coordinate stable complexes as resting state catalysts.
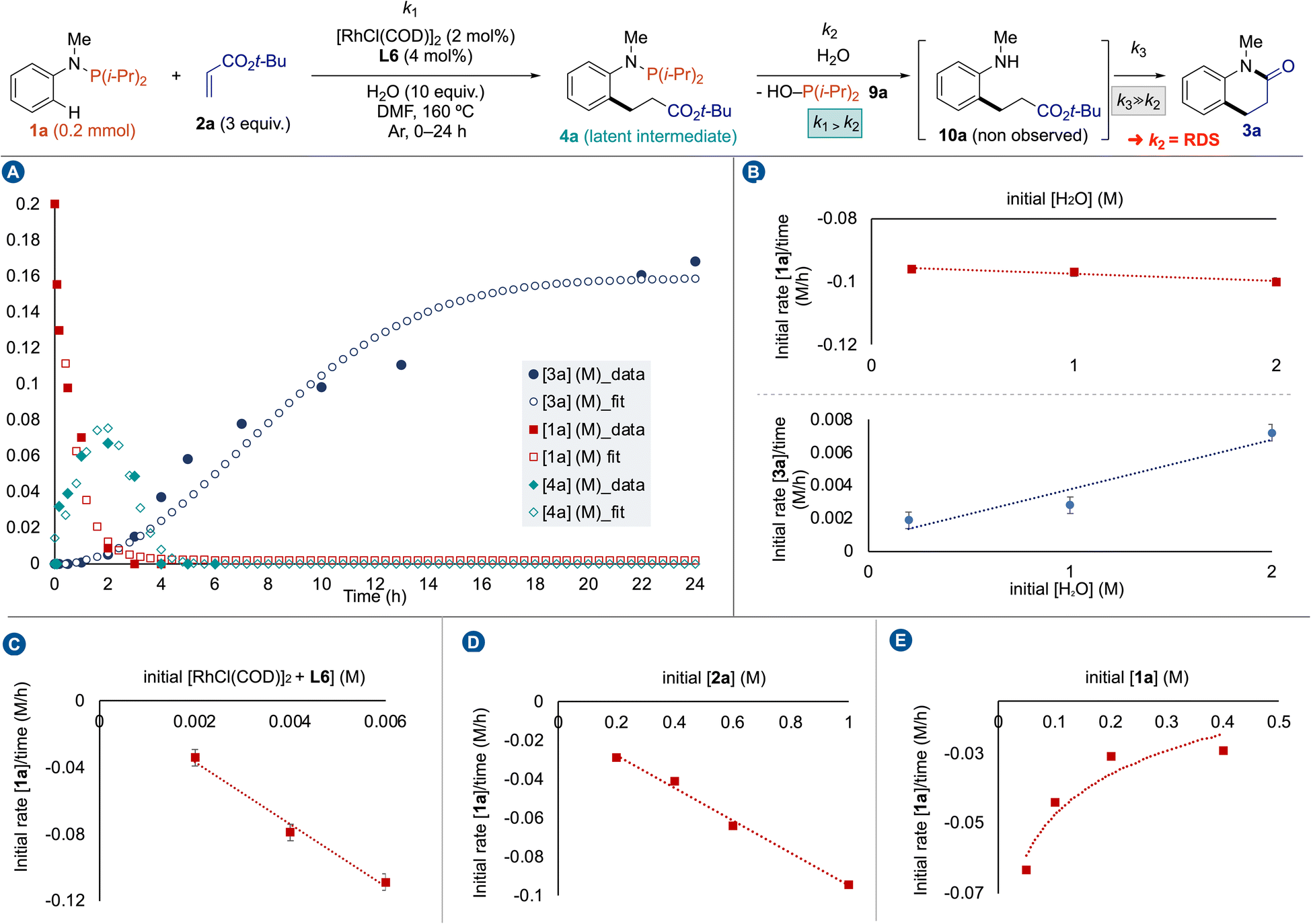 | ||
| Fig. 3 Kinetic study: (A) reaction profile, (B) determination of H2O order, (C) determination of catalyst order, (D) determination of 2a order, (E) determination of 1a order. | ||
Based on our mechanistic and kinetic investigations and the DFT calculation by Bai, Lan on Rh(I)-catalyzed C7-alkylation of indole N–Pt-Bu2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 24 – which demonstrated that μ2-bridged dinuclear rhodium complexes preferentially act as active catalysts because of the endothermic monomerization process – we propose the catalytic cycle depicted in Fig. 4. [RhCl(COD)]2 complex reacts with phosphanamine 1a to form the active catalytic species I. Then, oxidative addition occurs to deliver the Rh(III)–H hydride II. Acrylate 2a may followed by 1,2-insertion to yield III. Then, reductive elimination affords IV. In the presence of water, the coordinated functionalized phosphanamine is prompted to hydrolysis to give the desired product 3a along with Rh complex V surrounded by hydroxydiisopropylphosphane (9a). The next catalytic cycle starts by a ligand exchange between 9a and 1a to give I. However, V or I may react with phosphine derivatives 9a or 1a in an inhibitory manner to form nonproductive coordination complexes [{Rh(9a)2(μ2-Cl)}2] and [{Rh(1a)2(μ2-Cl)}2]. At this stage, the exact role of L6 remains unclear but it may help the decomplexation of one phosphine to regenerate the active catalysts.
24 – which demonstrated that μ2-bridged dinuclear rhodium complexes preferentially act as active catalysts because of the endothermic monomerization process – we propose the catalytic cycle depicted in Fig. 4. [RhCl(COD)]2 complex reacts with phosphanamine 1a to form the active catalytic species I. Then, oxidative addition occurs to deliver the Rh(III)–H hydride II. Acrylate 2a may followed by 1,2-insertion to yield III. Then, reductive elimination affords IV. In the presence of water, the coordinated functionalized phosphanamine is prompted to hydrolysis to give the desired product 3a along with Rh complex V surrounded by hydroxydiisopropylphosphane (9a). The next catalytic cycle starts by a ligand exchange between 9a and 1a to give I. However, V or I may react with phosphine derivatives 9a or 1a in an inhibitory manner to form nonproductive coordination complexes [{Rh(9a)2(μ2-Cl)}2] and [{Rh(1a)2(μ2-Cl)}2]. At this stage, the exact role of L6 remains unclear but it may help the decomplexation of one phosphine to regenerate the active catalysts.
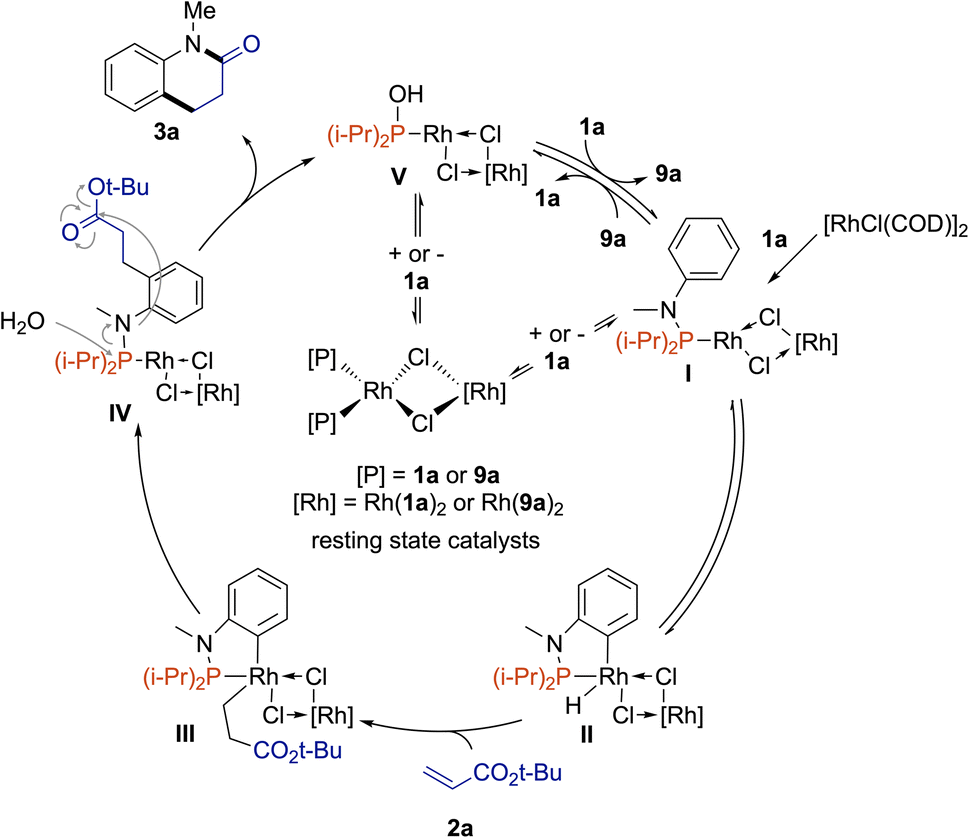 | ||
| Fig. 4 Proposed mechanism. | ||
Conclusions
We have developed a novel method for the construction of functionalized dihydroquinolinone scaffolds based on a cascade reaction of Rh(I)-catalyzed C(sp2)–H bond functionalization of phosphanamines with acrylates followed by N–P hydrolysis and intramolecular amidation. The reaction features good substrate scope with respect to both coupling partners. A mechanistic investigation established a viable catalytic cycle that was consistent with the kinetic data obtained and revealed that P(III)-directed C(sp2)–H bond functionalization with Rh(I) occurred through oxidative addition leading to Rh–H species and an inhibitory effect by the trivalent phosphorus species. The formal C(sp2)–H bond alkylation of anilines was also extended to styrene derivatives to afford the corresponding ortho-alkylated anilines. Finally, the utility of this protocol was demonstrated by the synthesis of aripiprazole N-methylated analog from easily accessible and cheap N-methylaniline. These findings provide a general framework for improving and expanding the scope of Rh(I)-catalyzed C(sp2)–H bond activation in synthetic organic chemistry of N-heterocycles, and the collected mechanistic insights should benefit in the future development of Rh(I)-catalyzed P(III)-directed C–H bond activation.Data availability
General information, detailed experimental procedures, kinetics, characterization data for all new compounds, and NMR spectra are in the ESI.†Author contributions
M. P. co-conceived, performed the laboratory experiments (optimization, substrate scope, application and mechanistic studies) and analyzed the experimental data. D. A. performed the laboratory experiments for substate scope. T. R. did the X-ray analysis. H.-D. took part to the discussions and proofread the manuscript. J.-F. S. conceived, directed the investigations, analyzed the experimental data, and prepared the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the CNRS, UR1, Agence Nationale de la Recherche (Grant No. ANR-20-CE07-0019-01) for providing financial support, and Rennes 1 University for PhD grant to M. Peng. We thank Umicore AG & Co. KG for their generous donation of rhodium(III) chloride hydrate.Notes and references
- (a) J. Fischer and C. R. Ganellin, Analogue-based Drug Discovery, John Wiley & Sons, Weinheim, 2006 CrossRef; (b) R. Sharma, R. K. Yadav, R. Sharma, N. K. Sahu, M. Jain and S. Chaudhary, Curr. Top. Med. Chem., 2021, 21, 1538–1571 CrossRef CAS PubMed.
- (a) M. Cherest and X. Lusinchi, Tetrahedron Lett., 1989, 30, 715–718 CrossRef CAS; (b) M. C. Elliott and S. V. Wordingham, Synlett, 2004, 898–900 CrossRef CAS; (c) K. Li, L. N. Foresee and J. A. Tunge, J. Org. Chem., 2005, 70, 2881–2883 CrossRef CAS PubMed.
- (a) M. Wasa and J.-Q. Yu, J. Am. Chem. Soc., 2008, 130, 14058–14059 CrossRef CAS PubMed; (b) L. Zhang, L. Sonaglia, J. Stacey and M. Lautens, Org. Lett., 2013, 15, 2128–2131 CrossRef CAS PubMed; (c) J.-X. Yan, H. Li, X.-W. Liu, J.-L. Shi, X. Wang and Z.-J. Shi, Angew. Chem., Int. Ed., 2014, 53, 4945–4949 CrossRef CAS PubMed; (d) B. Li, Y. Park and S. Chang, J. Am. Chem. Soc., 2014, 136, 1125–1131 CrossRef CAS PubMed; (e) M. Guan, Y. Pang, J. Zhang and Y. Zhao, Chem. Commun., 2016, 52, 7043–7046 RSC; (f) Z. Kuang, B. Li and Q. Song, Chem. Commun., 2018, 54, 34–37 RSC; (g) H.-Z. Xiao, W.-S. Wang, Y.-S. Sun, H. Luo, B.-W. Li, X.-D. Wang, W.-L. Lin and F.-X. Luo, Org. Lett., 2019, 21, 1668–1671 CrossRef CAS PubMed.
- (a) T. E. Hurst, R. M. Gorman, P. Drouhin, A. Perry and R. J. K. Taylor, Chem.–Eur. J., 2014, 20, 14063–14073 CrossRef CAS PubMed; (b) Q.-F. Bai, C. Jin, J.-Y. He and G. Feng, Org. Lett., 2018, 20, 2172–2175 CrossRef CAS PubMed; (c) H. Cheng, T.-L. Lam, Y. Liu, Z. Tang and C.-M. Che, Angew. Chem., Int. Ed., 2021, 60, 1383–1389 CrossRef CAS PubMed; (d) Z. Liu, S. Zhong, X. Ji, G.-J. Deng and H. Huang, Org. Lett., 2022, 24, 349–353 CrossRef CAS PubMed.
- (a) T.-P. Loh and L.-L. Wei, Synlett, 1998, 1, 975–976 CrossRef; (b) C. Munro-Leighton, E. D. Blue and T. B. Gunnoe, J. Am. Chem. Soc., 2006, 128, 1446–1447 CrossRef CAS PubMed; (c) K. De, J. Legros, B. Crousse and D. Bonnet-Delpon, J. Org. Chem., 2009, 74, 6260–6265 CrossRef CAS PubMed.
- J. Wu, S. Xiang, J. Zeng, M. Leow and X.-W. Liu, Org. Lett., 2015, 17, 222–225 CrossRef CAS PubMed.
- X. Liu and K. K. Hii, J. Org. Chem., 2011, 76, 8022–8026 CrossRef CAS PubMed.
- J. Kim, S.-W. Park, M.-H. Baik and S. Chang, J. Am. Chem. Soc., 2015, 137, 13448–13451 CrossRef CAS PubMed.
- J. Chen, G. Song, C.-L. Pan and X. Li, Org. Lett., 2010, 12, 5426–5429 CrossRef CAS PubMed.
- (a) J. Qi, L. Huang, Z. Wang and H. Jiang, Org. Biomol. Chem., 2013, 11, 8009–8013 RSC; (b) S. M. Khake and N. Chatani, ACS Catal., 2022, 12, 4394–4401 CrossRef CAS.
- (a) Q. Shelby, N. Kataoka, G. Mann and J. Hartwig, J. Am. Chem. Soc., 2000, 122, 10718–10719 CrossRef CAS; (b) X. Qiu, M. Wang, Y. Zhao and Z. Shi, Angew. Chem., Int. Ed., 2017, 56, 7233–7237 CrossRef CAS PubMed; (c) X. Luo, J. Yuan, C.-D. Yue, Z.-Y. Zhang, J. Chen, G.-A. Yu and C.-M. Che, Org. Lett., 2018, 20, 1810–1814 CrossRef CAS PubMed; (d) X. Qiu, H. Deng, Y. Zhao and Z. Shi, Sci. Adv., 2018, 4, eaau6468 CrossRef CAS PubMed; (e) K. Fukuda, N. Iwasawa and J. Takaya, Angew. Chem., Int. Ed., 2019, 58, 2850–2853 CrossRef CAS PubMed; (f) J.-W. Li, L.-N. Wang, M. Li, P.-T. Tang, X.-P. Luo, M. Kurmoo, Y.-J. Liu and M.-H. Zeng, Org. Lett., 2019, 21, 2885–2889 CrossRef CAS PubMed; (g) D. Wang, Y. Zhao, C. Yuan, J. Wen, Y. Zhao and Z. Shi, Angew. Chem., Int. Ed., 2019, 58, 12529–12533 CrossRef CAS PubMed; (h) J. Wen, D. Wang, J. Qian, D. Wang, C. Zhu, Y. Zhao and Z. Shi, Angew. Chem., Int. Ed., 2019, 58, 2078–2082 CrossRef CAS PubMed; (i) S. E. Wright, S. Richardson-Solorzano, T. N. Stewart, C. D. Miller, K. C. Morris, C. J. A. Daley and T. B. Clark, Angew. Chem., Int. Ed., 2019, 58, 2834–2838 CrossRef CAS PubMed; (j) B. Dong, J. Qian, M. Li, Z.-J. Wang, M. Wang, D. Wang, C. Yuan, Y. Han, Y. Zhao and Z. Shi, Sci. Adv., 2020, 6, eabd1378 CrossRef CAS PubMed; (k) Y. Homma, K. Fukuda, N. Iwasawa and J. Takaya, Chem. Commun., 2020, 56, 10710–10713 RSC; (l) G. Li, J. An, C. Jia, B. Yan, L. Zhong, J. Wang and S. Yang, Org. Lett., 2020, 22, 9450–9455 CrossRef CAS PubMed; (m) Z. Shi, M. Wang, H. Luo and D. Wang, Synlett, 2020, 33, 351–356 Search PubMed; (n) J. Wen, B. Dong, J. Zhu, Y. Zhao and Z. Shi, Angew. Chem., Int. Ed., 2020, 59, 10909–10912 CrossRef CAS PubMed; (o) Z. Zhang, M. Cordier, P. H. Dixneuf and J.-F. Soulé, Org. Lett., 2020, 22, 5936–5940 CrossRef CAS PubMed; (p) Z.-Y. Zhang, X. Zhang, J. Yuan, C.-D. Yue, S. Meng, J. Chen, G.-A. Yu and C.-M. Che, Chem.–Eur. J., 2020, 26, 5037–5050 CrossRef CAS PubMed; (q) T. Komuro, J. Asagami, H. Higashi, K. Sato, H. Hashimoto and H. Tobita, Organometallics, 2021, 40, 3113–3123 CrossRef CAS; (r) M. Li, J.-Y. Tao, L.-N. Wang, J.-W. Li, Y.-J. Liu and M.-H. Zeng, J. Org. Chem., 2021, 86, 11915–11925 CrossRef CAS PubMed; (s) L.-N. Wang, P.-T. Tang, M. Li, J.-W. Li, Y.-J. Liu and M.-H. Zeng, Adv. Synth. Catal., 2021, 363, 2843–2849 CrossRef CAS; (t) H.-B. Xu, Y.-J. Chen, X.-Y. Chai, J.-H. Yang, Y.-J. Xu and L. Dong, Org. Lett., 2021, 23, 2052–2056 CrossRef CAS PubMed; (u) N.-J. Zhang, W.-T. Ma, J.-W. Li, Y.-J. Liu and M.-H. Zeng, Asian J. Org. Chem., 2021, 10, 1113–1116 CrossRef CAS; (v) Y. Fu, C.-H. Chen, M.-G. Huang, J.-Y. Tao, X. Peng, H.-B. Xu, Y.-J. Liu and M.-H. Zeng, ACS Catal., 2022, 12, 5036–5047 CrossRef CAS; (w) W.-T. Ma, M.-G. Huang, Y. Fu, Z.-H. Wang, J.-Y. Tao, J.-W. Li, Y.-J. Liu and M.-H. Zeng, Chem. Commun., 2022, 58, 7152–7155 RSC; (x) D. Wang, M. Li, C. Shuang, Y. Liang, Y. Zhao, M. Wang and Z. Shi, Nat. Commun., 2022, 13, 2934 CrossRef CAS PubMed.
- (a) X. Qiu, P. Wang, D. Wang, M. Wang, Y. Yuan and Z. Shi, Angew. Chem., Int. Ed., 2019, 58, 1504–1508 CrossRef CAS PubMed; (b) A. J. Borah and Z. Shi, J. Am. Chem. Soc., 2018, 140, 6062–6066 CrossRef CAS PubMed.
- (a) Z. Zhang, T. Roisnel, P. H. Dixneuf and J.-F. Soulé, Angew. Chem., Int. Ed., 2019, 58, 14110–14114 CrossRef CAS PubMed; (b) J.-W. Li, L.-N. Wang, M. Li, P.-T. Tang, N.-J. Zhang, T. Li, X.-P. Luo, M. Kurmoo, Y.-J. Liu and M.-H. Zeng, Org. Lett., 2020, 22, 1331–1335 CrossRef CAS PubMed; (c) D. Wang, B. Dong, Y. Wang, J. Qian, J. Zhu, Y. Zhao and Z. Shi, Nat. Commun., 2019, 10, 3539 CrossRef PubMed.
- (a) D. L. Davies, S. M. A. Donald and S. A. Macgregor, J. Am. Chem. Soc., 2005, 127, 13754–13755 CrossRef CAS PubMed; (b) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624–655 CrossRef CAS PubMed.
- Reaction of 1,1-diisopropyl-N-phenylphosphanamine with 2a resulted in N-addition reaction.
- (a) M. A. Grady, T. L. Gasperoni and P. Kirkpatrick, Nat. Rev. Drug Discovery, 2003, 2, 427–428 CrossRef CAS PubMed; (b) V. Ozdemir, J. Fourie and F. Ozdener, Curr. Opin. Investig. Drugs, 2002, 3, 113–120 CAS.
- P. Kowalski, J. Jaskowska and Z. Majka, Mini-Rev. Org. Chem., 2012, 9, 374–380 CrossRef CAS.
- S. J. Bonacorsi Jr, S. C. Waller and J. Kent Rinehart, J. Label. Compd. Radiopharm., 2006, 49, 1–9 CrossRef.
- See ESI for more details.†.
- (a) R. B. Bedford, S. J. Coles, M. B. Hursthouse and M. E. Limmert, Angew. Chem., Int. Ed., 2003, 42, 112–114 CrossRef CAS PubMed; (b) R. B. Bedford, M. Betham, A. J. M. Caffyn, J. P. H. Charmant, L. C. Lewis-Alleyne, P. D. Long, D. Polo-Cerón and S. Prashar, Chem. Commun., 2008, 990–992 RSC.
- K. Wang, G. P. Rosini, S. P. Nolan and A. S. Goldman, J. Am. Chem. Soc., 1995, 117, 5082–5088 CrossRef CAS.
- B. Rybtchinski, R. Cohen, Y. Ben-David, J. M. L. Martin and D. Milstein, J. Am. Chem. Soc., 2003, 125, 11041–11050 CrossRef CAS PubMed.
- We have also demonstrated that 1a is stable in the presence of 2 mol% [RhCl(COD)]2 associated with 4 mol% of L6, and 10 equivalents of water in DMF over several hours, suggesting that N–P bond hydrolysis majority occurs on the complexed phosphanamines.
- D. Heng, H. Chen, X. He, S. Liu, L. Zhu, K. Zhong, T. Zhang, R. Bai and Y. Lan, ACS Catal., 2021, 11, 3975–3987 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2252526. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc02992a |
| This journal is © The Royal Society of Chemistry 2023 |