
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly enantioselective Rh-catalyzed asymmetric reductive dearomatization of multi-nitrogen polycyclic pyrazolo[1,5-a]pyrimidines†
Chaochao
Xie
a,
Guiying
Xiao
a,
Qianling
Guo
a,
Xiaoxue
Wu
a,
Guofu
Zi
 a,
Wanjian
Ding
*a and
Guohua
Hou
*ab
a,
Wanjian
Ding
*a and
Guohua
Hou
*ab
aKey Laboratory of Radiopharmaceuticals, College of Chemistry, Beijing Normal University, No. 19 Xinjiekouwai St., Beijing 100875, China. E-mail: ghhou@bnu.edu.cn; dingwanjian@bnu.edu.cn
bShanghai Key Laboratory for Molecular Engineering of Chiral Drugs, School of Chemistry and Chemical Engineering, Shanghai Jiao Tong University, Shanghai 200240, China
First published on 28th July 2023
Abstract
A highly enantioselective rhodium-catalyzed reductive dearomatization of 7-substituted pyrazolo[1,5-a]pyrimidines has been realized for the first time by two strategies to afford chiral 4,5,6,7-tetrahydropyrazolo[1,5-a]pyrimidines with excellent enantioselectivities of up to 98% ee. This method also provides an efficient approach for the synthesis of the powerful BTK inhibitor, zanubrutinib.
Introduction
Chiral 4,5,6,7-tetrahydropyrazolo[1,5-a]pyrimidine is an important structural unit, which is prevalent in pharmaceuticals and bioactive molecules.1 For example, zanubrutinib containing chiral 4,5,6,7-tetrahydropyrazolo[1,5-a]pyrimidine is a new powerful BTK inhibitor that enables a complete and lasting precise inhibition of the BTK target (Fig. 1).2 The compound A is a potent and selective inhibitor of CDK9, which enables short duration of target engagement for the treatment of hematological malignancies.3 Chiral 4,5,6,7-tetrahydropyrazolo[1,5-a]pyrimidine and its derivatives also play an important role in selective inhibitor and selective KV1.5 blockers such as BMS394136.4 Because of this importance, the development of efficient methods for enantioselective synthesis of chiral 4,5,6,7-tetrahydropyrazolo[1,5-a]pyrimidines has attracted considerable attention from chemists.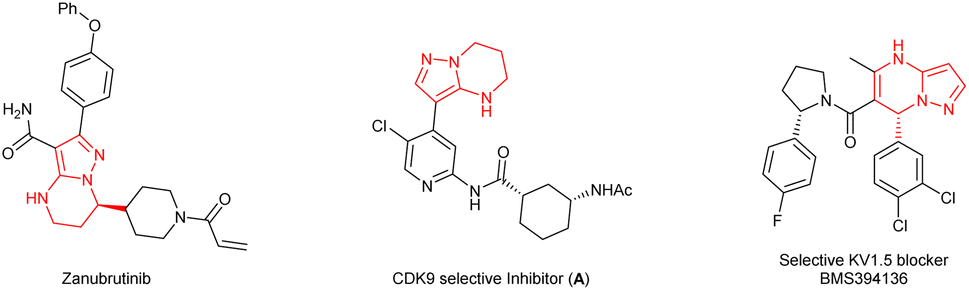 | ||
| Fig. 1 Examples of drug molecules containing polyheteroatom heteroaromatic structures. | ||
Although the chiral 4,5,6,7-tetrahydropyrazolo[1,5-a]pyrimidine skeleton is of significance in natural products and pharmaceuticals, there are few methods for enantioselective synthesis of this class of compounds. The approach to chiral 4,5,6,7-tetrahydropyrazolo[1,5-a]pyrimidines mainly relies on chemical or biocatalytic resolution of racemates.5 Compared with the resolution method, the transition-metal-catalyzed reductive dearomatization of prochiral substituted pyrazolo[1,5-a]pyrimidines obviously is a straightforward method to prepare chiral 4,5,6,7-tetrahydropyrazolo[1,5-a]pyrimidines. To our surprise, the asymmetric reduction of substituted pyrazolo[1,5-a]pyrimidines has not been explored so far. This is perhaps mainly attributed to the following points: (1) the high density of the nitrogen atom which has a strong coordination effect on the poisoning or deactivation of chiral catalysts; (2) the inherent stability of the aromaticity of substrates leading to lower reactivity, and the strict conditions required to break it which will adversely affect the enantioselectivity and chemical selectivity. Generally, low reactivity and poor chemical and stereo selectivity are the main challenges in asymmetric reduction of substituted pyrazolo[1,5-a]pyrimidines. In recent years, great efforts have been devoted to asymmetric reduction of polycyclic N-heterocyclic compounds6 and excellent results have been achieved for pyrrolo[1,2-a]pyrazines and their salts,7,8 pyrrolo/indolo[1,2-a]quinoxalines,9 and imidazo[1,2-a]pyridines10 using a chiral iridium–diphosphine complex and ruthenium–NHC complex as the catalysts by Zhou's and Glorius's group, respectively. In the asymmetric hydrogenation of aromatic N-heterocyclic compounds, catalyst or/and substrate activation is usually required for good reactivity and enantioselectivity.11–14 Although some progress has been made in the hydrogenation of polycyclic nitrogen-containing one or two nitrogen atoms,12–14 there are few attempts on the hydrogenation of the polycyclic N-heterocyclic substrates with more nitrogen atoms, that is, higher nitrogen density. The only example is a chiral ruthenium–NHC complex catalyzed enantioselective hydrogenation of 1,2,3-triazolo-[1,5-a]pyridines with moderate enantioselectivity (up to 66% ee) reported by Glorius and co-workers.10 Therefore, to develop an efficient catalyst for realization of asymmetric reductive dearomatization of polycyclic N-heterocyclic substrates with higher nitrogen density is highly desirable and is of significance for organic and pharmaceutical synthesis. Based on the outstanding work of Zhou and Glorius in the asymmetric reduction of N-heterocyclic arenes15 and our previous great progress in asymmetric hydrogenation of various substrates,16 we hence report an asymmetric reductive dearomatization of substituted pyrazolo[1,5-a]pyrimidines containing three nitrogen atoms by two strategies without any activation of the catalyst and substrate providing chiral 4,5,6,7-tetrahydropyrazolo[1,5-a]pyrimidines with excellent enantioselectivities of up to 98% ee (Scheme 1).
 | ||
| Scheme 1 Rh-catalyzed asymmetric hydrogenation of heteroarenes. | ||
Results and discussion
In our initial study, 7-phenylpyrazolo[1,5-a]pyrimidine-3-carbonitrile 1a was chosen as the model substrate, and hydrogenation was carried out under 80 atm of H2 in THF at 60 °C for 24 h using the complex of [Rh(COD)Cl]2 and (S,S)-f-Binaphane (L1) (Table 1). Disappointingly, the desired product is not obtained under these conditions. Some other bidentate diphosphine ligands (Fig. 2) including (R,R)-QuinoxP* (L2), (R,R)-Me-Duphos (L3), (S)-Binap (L4), (R)-Segphos (L5) and chiral monophosphorus ligand (R)-Monophos (L6) were also evaluated (entries 2–6). Although the enantioselectivities could reach 86% ee using L4 and L5, the conversions were too low (entries 4–5). Exhilaratingly, (S)-DTBM-Segphos (L7) achieved 92% ee with 76% conversion (entry 7). It was found that the solvent had an extremely important influence on the conversion, but had no obvious effect on the enantioselectivity. There was almost no reactivity in MeOH or CHCl3 (entries 9 and 10). Although good to high enantioselectivities were achieved in CH2Cl2, Et2O and toluene, moderate conversions were obtained (entries 8, 11 and 12). When the temperature increased to 80 °C, the conversion was significantly improved (entry 13). At a higher temperature, 90 °C, the reaction could be accomplished without any erosion of the enantioselectivity (entry 14). When we used [Ir(COD)Cl]2 and (S)-DTBM-Segphos complexes as catalysts, the enantioselectivity decreased to 65% ee despite the full conversion (entry 15). More significantly, even on reducing the catalyst loading to 2 mol%, the hydrogenation could still be completed in 48 h with 92% ee maintained (entry 16). When the hydrogen pressure was reduced to 60 atm, the enantioselectivity remained unchanged, but only 85% conversion was achieved (entry 17).|
|
|||||
|---|---|---|---|---|---|
| Entry | Ligand | Solvent | T (°C) | Conv.b | eec |
a Unless otherwise mentioned, all reactions were carried out with a [Rh(COD)Cl]2/diphosphine (monophosphine)/substrate = 2.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5.5:100, 80 atm H2, 60 °C, 24 h.
b Determined by GC analysis.
c Determined by chiral supercritical fluid chromatography (SFC) using a chiral stationary phase.
d 2.5 mol% [Ir(COD)Cl]2, 5.5 mol% (S)-DTBM-Segphos, 80 atm H2, 90 °C, 24 h.
e 1.0 mol% [Rh(COD)Cl]2, 2.2 mol% (S)-DTBM-Segphos, 80 atm H2, 90 °C, 48 h.
f 1.0 mol% [Rh(COD)Cl]2, 2.2 mol% (S)-DTBM-Segphos, 60 atm H2, 90 °C, 48 h. :5.5:100, 80 atm H2, 60 °C, 24 h.
b Determined by GC analysis.
c Determined by chiral supercritical fluid chromatography (SFC) using a chiral stationary phase.
d 2.5 mol% [Ir(COD)Cl]2, 5.5 mol% (S)-DTBM-Segphos, 80 atm H2, 90 °C, 24 h.
e 1.0 mol% [Rh(COD)Cl]2, 2.2 mol% (S)-DTBM-Segphos, 80 atm H2, 90 °C, 48 h.
f 1.0 mol% [Rh(COD)Cl]2, 2.2 mol% (S)-DTBM-Segphos, 60 atm H2, 90 °C, 48 h.
|
|||||
| 1 | L1 | THF | 60 | Trace | — |
| 2 | L2 | THF | 60 | Trace | — |
| 3 | L3 | THF | 60 | Trace | — |
| 4 | L4 | THF | 60 | 18 | 80 |
| 5 | L5 | THF | 60 | 18 | 86 |
| 6 | L6 | THF | 60 | Trace | — |
| 7 | L7 | THF | 60 | 70 | 91 |
| 8 | L7 | CH2Cl2 | 60 | 22 | 92 |
| 9 | L7 | CHCl3 | 60 | Trace | — |
| 10 | L7 | MeOH | 60 | Trace | — |
| 11 | L7 | Et2O | 60 | 54 | 91 |
| 12 | L7 | Toluene | 60 | 42 | 89 |
| 13 | L7 | THF | 80 | 97 | 92 |
| 14 | L7 | THF | 90 | >99 | 92 |
| 15d | L7 | THF | 90 | >99 | 65 |
| 16e | L7 | THF | 90 | >99 | 92 |
| 17f | L7 | THF | 90 | 85 | 91 |
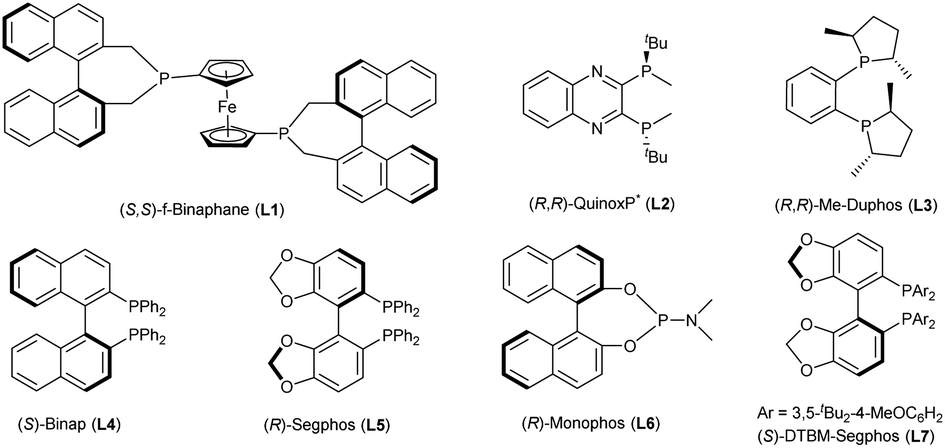 | ||
| Fig. 2 Structures of chiral phosphine ligands screened for 1a. | ||
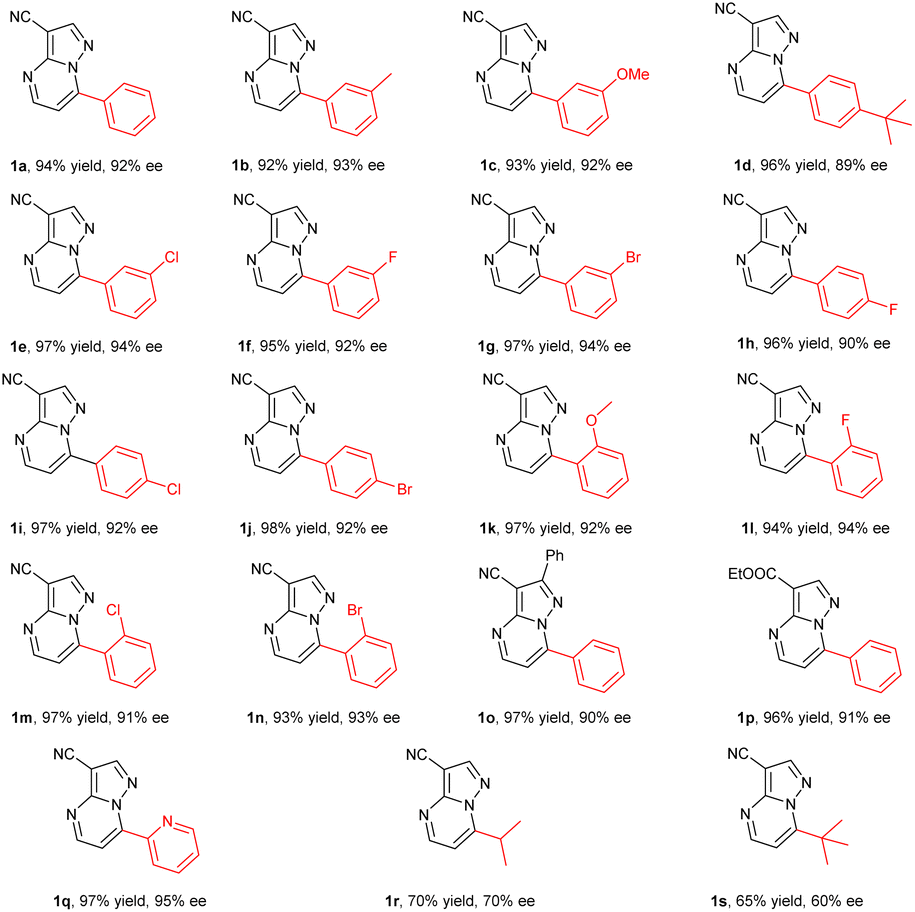
Inspired by the promising results obtained in the hydrogenation of substrate 1a, a variety of pyrazolo[1,5-a]pyrimidines 1b–1s were then prepared and evaluated in the hydrogenation under the optimized conditions. To our delight, all of the substrates could be successfully hydrogenated to afford the corresponding products 2 in high yields with moderate to excellent enantioselectivities (Table 2). In general, the electronic effect of substituents and the position on the phenyl ring had little influence on the reactivity and enantioselectivity. For instance, the substrates 1b, 1c and 1d bearing an electron-donating substituent including methyl, methoxyl or t-butyl groups at the meta- or para-position of the aryl group provided the products in high yields and enantioselectivities of 89–93% ee. The substrates with electron-withdrawing substituents such as F, Cl and Br groups, could be smoothly hydrogenated to produce the desired chiral products with full conversions and 90–94% ee values (1e–1j). The ortho-substituted substrates exhibited comparably high reactivity and enantioselectivity (91–94% ee) regardless of the electron-withdrawing or electron-donating substituents (1k–1n). The substrate 1o with a large conjugated system could still be successfully converted into the corresponding product with good yield and enantioselectivity (97% yield, 90% ee). It was found that replacing the cyano group on the heterocycle with an ester group had no effect on the reactivity or enantioselectivity (1p). Moreover, the pyridinyl substrate 1q bearing four nitrogen atoms could exhibit pretty good reactivity and the corresponding hydrogenation product 2q was obtained in 97% yield with 95% ee. However, replacing the phenyl ring at the 7-position with an alkyl group such as isopropyl or t-butyl led to a decrease in yields and enantioselectivities (1r–1s). This demonstrated that the planarity had a positive effect on the coordination and recognition of the catalyst.
|
|
|---|
|
a All reactions were carried out with a [Rh(COD)Cl]2/(S)-DTBM-Segphos/substrate ratio of 1.0:2.2:100, 80 atm of H2 at 90 °C for 48 h. The conversion was determined by 1H NMR spectroscopy or GC analysis; enantioselectivity was determined by SFC or HPLC analysis using a chiral stationary phase.
|
|
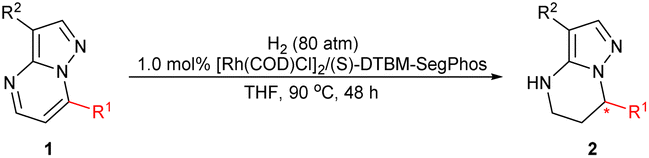
In order to further investigate the substrate scope of the catalyst, we then prepared a variety of 2-substituted substrates 3 and applied them to the hydrogenation under the optimized conditions. Delightedly, all of the substrates examined (3a–3q) could be successfully hydrogenated to produce the corresponding chiral heterocyclic compound with good yields and excellent enantioselectivities, 90–95% ee. As shown by the results in Table 3, the enantioselectivity and reactivity were not affected by the electronic properties or the position of the substituent in the phenyl ring. The substrates 3b–3f with electron-donating substituents (Me and MeO) could be fully hydrogenated to furnish products with pretty good ee values, 90–93% ee. The substrates 3g–3l with an electron-withdrawing substituent including F, Cl, Br or CF3 groups could also achieve a high enantioselectivity of 91–94% ee. In addition, the larger sterically hindered 2-naphthyl substrate 3m also showed similar reactivity and enantioselectivity providing 94% ee. It was noteworthy that the substrates bearing a 2-thiophenyl or 2-furanyl were also successfully hydrogenated with full conversions and excellent enantioselectivities (94–95% ee). Substrate 3p without any substituent at the 2-position exhibited relatively lower reactivity despite the similar enantioselectivity and required more amount of catalyst loading for complete conversion. In comparison, the t-butyl substituted 3q with large steric hindrance could achieve full conversion and 95% ee. Perhaps the substituent at the 2-position could prevent poisoning or deactivation of the chiral catalyst due to steric hindrance from the nitrogen in the pyrazole moiety.
|
|
|---|
|
a All reactions were carried out with a [Rh(COD)Cl]2/(S)-DTBM-Segphos/substrate ratio of 1.1:2.4:100, 80 atm of H2 at 90 °C for 48 h.
b 1.5 mol% [Rh(COD)Cl]2, 3.3 mol (S)-DTBM-Segphos and 80 atm of H2 at 90 °C for 48 h.
c 2.0 mol% [Rh(COD)Cl]2, 4.4 mol (S)-DTBM-Segphos, 80 atm of H2 at 90 °C for 48 h. The conversion was determined by 1H NMR spectroscopy or GC analysis; enantioselectivity was determined by SFC or HPLC analysis using a chiral stationary phase.
|
|
Finally, the absolute configuration of the hydrogenation product 4c was determined and assigned to be the (R) configuration by X-ray crystallographic analysis of the corresponding single-crystal structure (Fig. 3).
 | ||
| Fig. 3 X-ray crystal structure of product (R)-4c. | ||
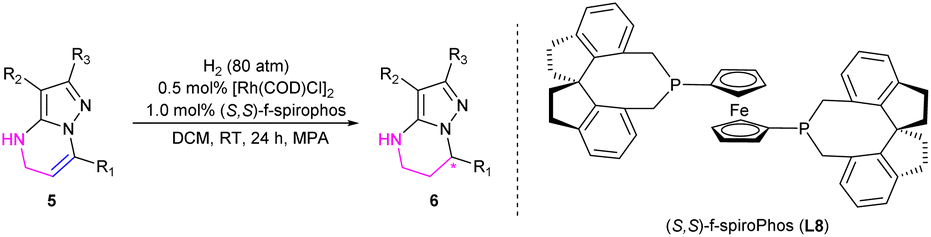
Alternatively, we further developed the strategy of sequential reduction of pyrazolo[1,5-a]pyrimidines for synthesis of chiral 4,5,6,7-tetrahydropyrazolo[1,5-a]pyrimidines. Substrate 1a was first partially reduced by NaBH3CN (sodium cyanoborohydride) to produce intermediate 5a, followed by hydrogenation with a chiral catalyst to finally obtain chiral 4,5,6,7-tetrahydropyrazolo[1,5-a]pyrimidines under mild conditions with this strategy (Scheme 2).
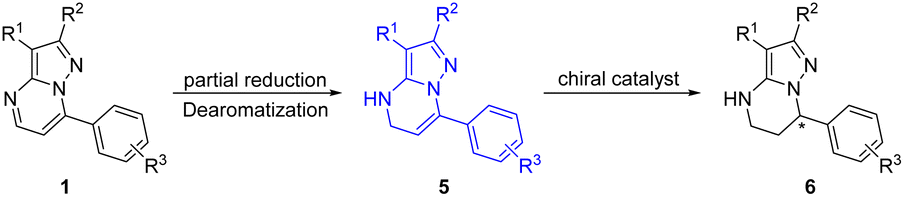 | ||
| Scheme 2 Sequential reduction of pyrazolo[1,5-a]pyrimidines. | ||
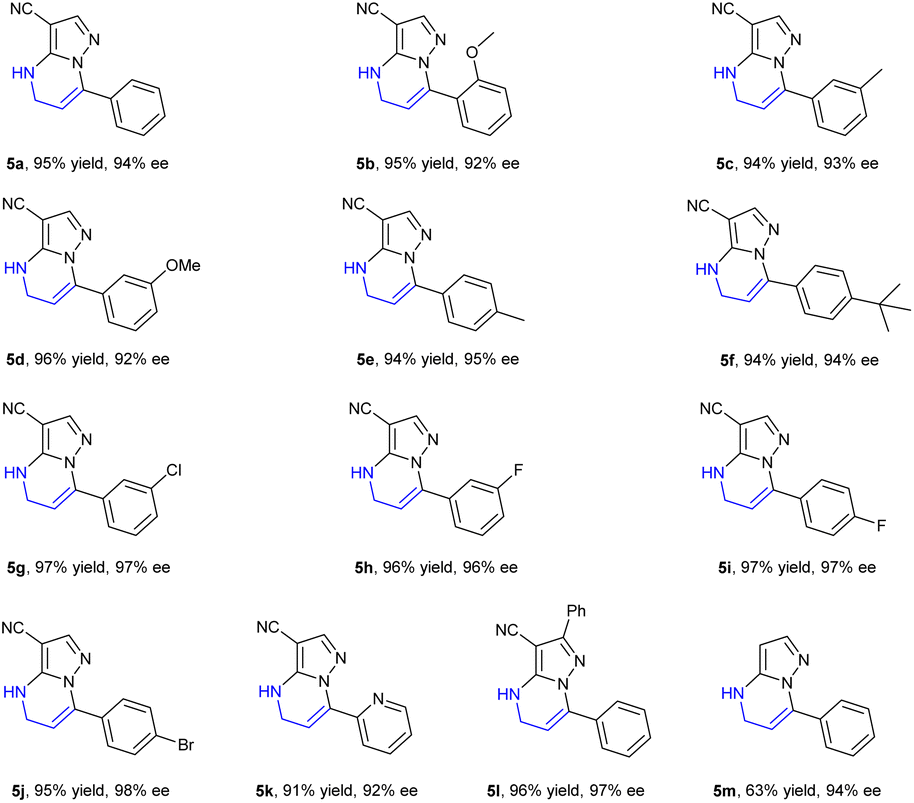
After the optimization of reaction conditions including the screening of chiral ligands, solvents and additives, the substrate 7-phenyl-4,5-dihydropyrazolo[1,5-a]pyrimidine-3-carbonitrile 5a could be completely hydrogenated in CH2Cl2 at room temperature, catalyzed by the complex of [Rh(COD)Cl]2 and (S,S)-f-spiroPhos to produce 6a with a higher enantioselectivity of 94% ee in the presence of N-methyl-p-anisidine (MPA), which could prevent disproportionation.17
Encouraged by the exciting result achieved in the hydrogenation of substrate 5a, a variety of 4,5-dihydropyrazolo[1,5-a]pyrimidines 5b–5m were then prepared and evaluated in the hydrogenation under the optimized conditions. The corresponding chiral products 6a–6m were afforded in high yields with up to 98% ee (Table 4). Generally, the enantioselectivity achieved by the substrates bearing electron-withdrawing substituents on the phenyl ring was higher than those achieved by substrates with electron-donating substituents. The F-, Cl- or Br-substituted substrates 5g–5j achieved higher ee values of up to 98%, while the substrates 5b–5f bearing an electron-donating substituent provided products with 92–95% ee. Notably, the substrates 5k bearing a pyridine heterocycle and 5l with larger conjugated substituents could also be smoothly hydrogenated affording the products 6k and 6l in high yields and enantioselectivities, 92% and 97% ee, respectively. When substrate 5m without a substituent at the 2-position was evaluated, only 63% yield was obtained despite achieving similar enantioselectivity (94% ee), which perhaps demonstrated that the substituents at the 2 or 3 position had a positive effect on avoiding the deactivation of the catalyst.
|
|
|---|
|
a All reactions were carried out with a [Rh(COD)Cl]2/(S,S)-f-spiroPhos/substrate/MPA ratio of 0.5:1.1:100:100, DCM and 80 atm of H2 at rt for 24 h. The conversion was determined by 1H NMR spectroscopy or GC analysis; enantioselectivity was determined by SFC or HPLC analysis using a chiral stationary phase.
|
|
In order to demonstrate the practical utility of this method, we carried out the hydrogenation of substrate 1t under a lower catalyst loading (Scheme 3). The desired product 2t could still be obtained in 96% yield with a high enantioselectivity of 91% ee under 0.3 mol% catalyst loading. More significantly, the product 2t could be used to synthesize the powerful anti-cancer pharmaceutical Zanubrutinib.18 Furthermore, the hydrogenation of 5j was also smoothly accomplished using 0.1 mol% catalyst loading (S/C = 1000) to afford 6j in 92% isolated yield with 94% ee.
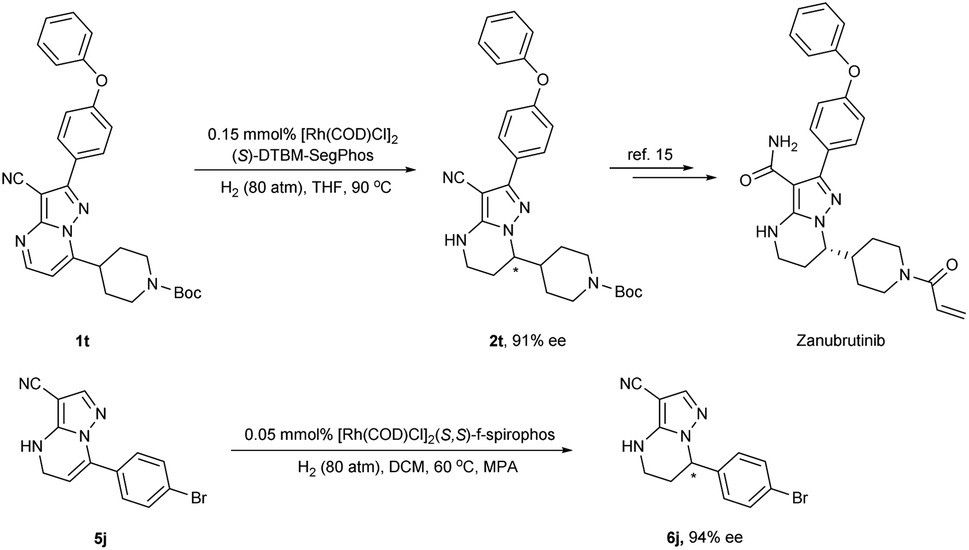 | ||
| Scheme 3 Catalyst loading experiment and application. | ||
To gain insight into the possible catalytic mechanism of this asymmetric hydrogenation, deuterium labelling experiments were conducted (Scheme 4). The asymmetric hydrogenation of substrate 1e was performed under 80 atm of D2 instead of H2 in THF. It was found that the 5,6,7-D incorporated deuteration product 2e was observed. The substrate 5a also underwent deuterium labelling experiments. The results showed that deuterium substitution occurred at positions 5, 6 and 7 of the substrate.
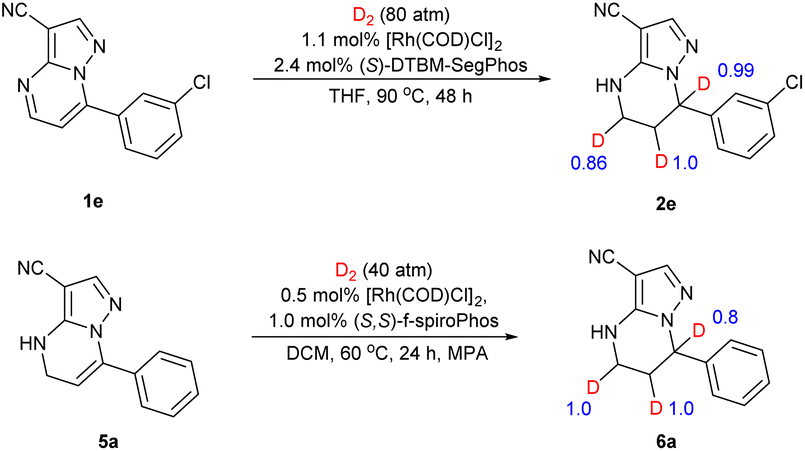 | ||
| Scheme 4 Deuterium labelling experiments. | ||
To probe the origin of the enantioselectivity, density functional theory (DFT) calculations were also performed (Scheme 5). The hydride transfer step which was supposed to be the selectivity determination step in asymmetric hydrogenation19 was paid attention. The DFT calculations revealed that the favored Si-face coordination of the substrate with rhodium (TSa) led to the formation of the major (R) stereoisomer, which is in accordance with the experimental results.
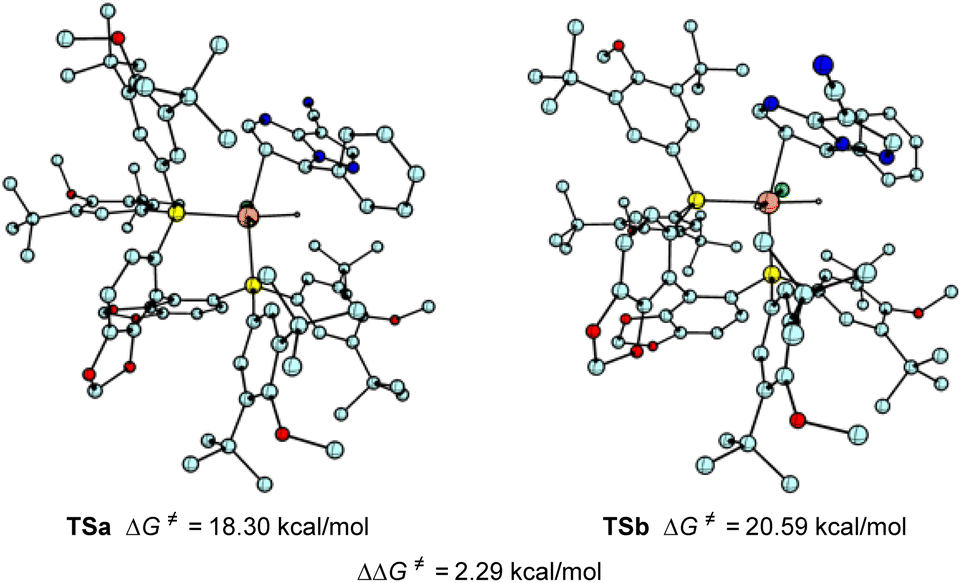 | ||
| Scheme 5 Origin of enantioselectivity. | ||
Based on the results of the deuterium labelling experiments, DFT calculations and the literature,19 we proposed the mechanism of this reaction (Scheme 6). Complex A of rhodium metal and diphosphine ligand first underwent oxidative addition with H2 to form species B. Then substrate 1 coordinated with B to provide species C, followed by hydride transfer to give the species D. The intermediate E was obtained and the complex A was regenerated by reductive elimination of species D. Finally, the intermediate E could be further hydrogenated to produce the final product 2.
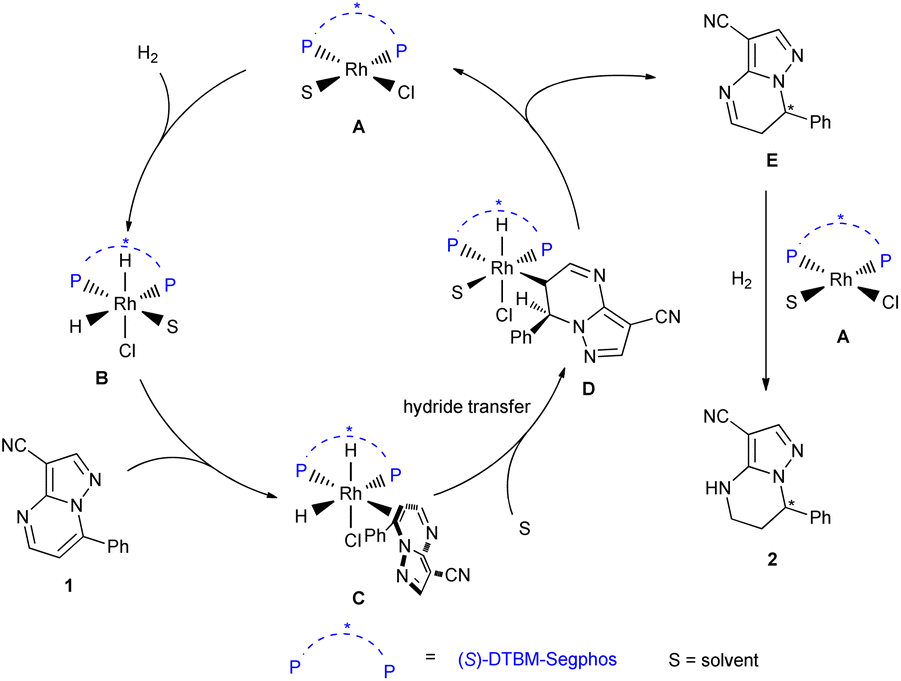 | ||
| Scheme 6 Proposed catalytic cycle. | ||
Conclusions
In conclusion, we realized Rh-catalyzed asymmetric reductive dearomatization of polycyclic pyrazolo[1,5-a]pyrimidines with three nitrogen atoms via two different strategies to afford chiral 4,5,6,7-tetrahydropyrazolo[1,5-a]pyrimidines with excellent enantioselectivities of up to 98% ee. This method also provided an efficient approach for the synthesis of the powerful BTK inhibitor, zanubrutinib.Data availability
General experimental procedures, compound characterization data, analysis of enantioselectivities of products and crystal parameters for the compound 4c are available in the ESI.†Author contributions
G. H. conceived the idea and directed the project. C. X., G. X., Q. G., and X. W. performed the experiments and the analytical characterization. W. D and G. Z. performed the theoretical calculations and modelling. C. X., G. X., and G. H. wrote the manuscript. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (Grant No. 21672024, 21272026, and 21871029) and Beijing Natural Science Foundation (2182025) for generous financial support.Notes and references
- (a) N. A. Lack, P. Axerio-Cilies, P. Tavassoli, F. Q. Han, K. H. Chan, C. Feau, E. LeBlanc, E. T. Guns, R. K. Guy, P. S. Rennie and A. Cherkasov, J. Med. Chem., 2011, 54, 8563–8573 CrossRef CAS PubMed; (b) T. Yakaiah, C. Kurumurthy, B. P. V. Lingaiah, B. Narsaiah, R. Pamanji, L. R. Velatooru, J. Venkateswara Rao, S. Gururaj, T. Parthasarathy and B. Sridhar, Med. Chem. Res., 2012, 21, 4261–4273 CrossRef CAS; (c) T. Asano, H. Yamazaki, C. Kasahara, H. Kubota, T. Kontani, Y. Harayama, K. Ohno, H. Mizuhara, M. Yokomoto, K. Misumi, T. Kinoshita, M. Ohta and M. Takeuchi, J. Med. Chem., 2012, 55, 7772–7785 CrossRef CAS PubMed; (d) E. Alvarez-Ruiz, L. Ballell-Pages, J. Castro-Pichel, L. Encinas, J. Esquivias, F. J. Gamo-Benito, M. C. Garcia-Palancar and M. J. Remuinan-Blanco, WO 2012143522A1, 2012; (e) J. Tassel, M. Thormann, R. Koestler, A. Treml, Z. Pei and M. Yu, WO 2022104206A1, 2022.
- G. Li, X. Liu and X. Chen, Simultaneous development of zanubrutinib in the USA and China, Nat. Rev. Clin. Oncol., 2020, 17, 589–590 CrossRef PubMed.
- B. Barlaam, R. Casella, J. Cidado, C. Cook, C. De Savi, A. Dishington, C. S. Donald, L. Drew, A. D. Ferguson, D. Ferguson, S. Glossop, T. Grebe, C. Gu, S. Hande, J. Hawkins, A. W. Hird, J. Holmes, J. Horstick, Y. Jiang, M. L. Lamb, T. M. McGuire, J. E. Moore, N. O'Connell, A. Pike, K. G. Pike, T. Proia, B. Roberts, M. San Martin, U. Sarkar, W. Shao, D. Stead, N. Sumner, K. Thakur, M. M. Vasbinder, J. G. Varnes, J. Wang, L. Wang, D. Wu, L. Wu, B. Yang and T. Yao, J. Med. Chem., 2020, 63, 15564–15590 CrossRef CAS PubMed.
- J. Lloyd, H. J. Finlay, W. Vacarro, T. Hyunh, A. Kover, R. Bhandaru, L. Yan, K. Atwal, M. L. Conder, T. Jenkins-West, H. Shi, C. Huang, D. Li, H. Sun and P. Levesque, Bioorg. Med. Chem. Lett., 2010, 20, 1436–1439 CrossRef CAS PubMed.
- (a) E. Alvarez-Ruiz, L. Ballell-Pages, J. Castro-Pichel, L. Encinas, J. Esquivias, F. J. Gamo-Benito, M. C. Garcia-Palancar and M. J. Remuinan-Blanco, WO 2012143522A1, 2012; (b) F. Yokokawa, G. Wang, W. L. Chan, S. H. Ang, J. Wong, I. Ma, S. P. S. Rao, U. Manjunatha, S. B. Lakshminarayana, M. Herve, C. Kounde, B. H. Tan, P. Thayalan, S. H. Ng, M. Nanjundappa, S. Ravindran, P. Gee, M. Tan, L. Wei, A. Goh, P.-Y. Chen, K. S. Lee, C. Zhong, T. Wagner, I. Dix, A. K. Chatterjee, K. Pethe, K. Kuhen, R. Glynne, P. Smith, P. Bifani and J. Jiricek, ACS Med. Chem. Lett., 2013, 4, 451–455 CrossRef CAS PubMed.
- (a) A. N. Kim and B. M. Stoltz, ACS Catal., 2020, 10, 13834–13851 CrossRef CAS PubMed; (b) Z.-P. Chen and Y.-G. Zhou, Synthesis, 2016, 48, 1769–1781 CrossRef CAS; (c) R. Gunasekar, R. L. Goodyear, I. P. Silvestri and J. Xiao, Org. Biomol. Chem., 2022, 20, 1794–1827 RSC.
- W.-X. Huang, C.-B. Yu, L. Shi and Y.-G. Zhou, Org. Lett., 2014, 16, 3324–3327 CrossRef CAS PubMed.
- S.-B. Hu, Z.-P. Chen, B. Song, J. Wang and Y.-G. Zhou, Adv. Synth. Catal., 2017, 359, 2762–2767 CrossRef CAS.
- S.-B. Hu, X.-Y. Zhai, H.-Q. Shen and Y.-G. Zhou, Adv. Synth. Catal., 2018, 360, 1334–1339 CrossRef CAS.
- N. Ortega, D.-T. D. Tang, S. Urban, D. Zhao and F. Glorius, Angew. Chem., Int. Ed., 2013, 52, 9500–9503 CrossRef CAS PubMed.
- (a) Z. Yu, W. Jin and Q. Jiang, Angew. Chem., Int. Ed., 2012, 51, 6060–6072 CrossRef CAS PubMed ; For the homogeneous hydrogenation of N-heteroaromatics without activators:; (b) G. E. Dobereiner, A. Nova, N. D. Schley, N. Hazari, S. J. Miller, O. Eisenstein and R. H. Crabtree, J. Am. Chem. Soc., 2011, 133, 7547–7562 CrossRef CAS PubMed; (c) Y. Kita, K. Yamaji, K. Higashida, K. Sathaiah, A. Iimuro and K. Mashima, Chem.–Eur. J., 2015, 21, 1915–1927 CrossRef CAS PubMed.
- Y.-G. Zhou, Acc. Chem. Res., 2007, 40, 1357–1366 CrossRef CAS PubMed.
- M. Rueping, J. Dufour and F. R. Schoepke, Green Chem., 2011, 13, 1084–1105 RSC.
- D.-S. Wang, Q.-A. Chen, S.-M. Lu and Y.-G. Zhou, Chem. Rev., 2012, 112, 2557–2590 CrossRef CAS PubMed.
- (a) K. Wang, Y.-J. Yu, X.-Q. Wang, Y.-Q. Bai, M.-W. Chen and Y.-G. Zhou, J. Org. Chem., 2022, 87, 10398–10407 CrossRef CAS PubMed; (b) G.-S. Feng, Z.-B. Zhao, L. Shi and Y.-G. Zhou, Org. Chem. Front., 2021, 8, 6273–6278 RSC; (c) G.-S. Feng, L. Shi, F.-J. Meng, M.-W. Chen and Y.-G. Zhou, Org. Lett., 2018, 20, 6415–6419 CrossRef CAS PubMed; (d) C.-B. Yu, J. Wang and Y.-G. Zhou, Org. Chem. Front., 2018, 5, 2805–2809 RSC; (e) G.-S. Feng, M.-W. Chen, L. Shi and Y.-G. Zhou, Angew. Chem., Int. Ed., 2018, 57, 5853–5857 CrossRef CAS PubMed; (f) M. P. Wiesenfeldt, D. Moock, D. Paul and F. Glorius, Chem. Sci., 2021, 12, 5611–5615 RSC; (g) W. Zhao, S. Guizzetti, J. A. Schwindeman, D. S. B. Daniels, J. J. Douglas, C. B. Kelly, A. Kosanovich and J. Knight, Org. Process Res. Dev., 2021, 25, 691–702 CrossRef CAS; (h) T. Wagener, A. Heusler, Z. Nairoukh, K. Bergander, C. G. Daniliuc and F. Glorius, ACS Catal., 2020, 10, 12052–12057 CrossRef CAS PubMed; (i) M. Wollenburg, A. Heusler, K. Bergander and F. Glorius, ACS Catal., 2020, 10, 11365–11370 CrossRef CAS PubMed; (j) M. P. Wiesenfeldt, Z. Nairoukh, T. Dalton and F. Glorius, Angew. Chem., Int. Ed., 2019, 58, 10460–10476 CrossRef CAS PubMed.
- (a) C. Wang, F. Xie, Q. Guo, C. Xie, G. Zi, W. Ye, Z. Zhou, G. Hou and Z. Zhang, J. Org. Chem., 2021, 86, 12034–12045 CrossRef CAS PubMed; (b) G. Xiao, S. Xu, C. Xie, G. Zi, W. Ye, Z. Zhou, G. Hou and Z. Zhang, Org. Lett., 2021, 23, 5734–5738 CrossRef CAS PubMed; (c) Q. Yan, G. Xiao, Y. Wang, G. Zi, Z. Zhang and G. Hou, J. Am. Chem. Soc., 2019, 141, 1749–1756 CrossRef CAS PubMed; (d) Y. Zhang, Q. Yan, G. Zi and G. Hou, Org. Lett., 2017, 19, 4215–4218 CrossRef CAS PubMed; (e) Q. Yan, D. Kong, M. Li, G. Hou and G. Zi, J. Am. Chem. Soc., 2015, 137, 10177–10181 CrossRef CAS PubMed.
- T. Nagano, A. Iimuro, R. Schwenk, T. Ohshima, Y. Kita, A. Togni and K. Mashima, Chem.–Eur. J., 2012, 18, 11578–11592 CrossRef CAS PubMed.
- Y. Guo, Y. Liu, N. Hu, D. Yu, C. Zhou, G. Shi, B. Zhang, M. Wei, J. Liu, L. Luo, Z. Tang, H. Song, Y. Guo, X. Liu, D. Su, S. Zhang, X. Song, X. Zhou, Y. Hong, S. Chen, Z. Cheng, S. Young, Q. Wei, H. Wang, Q. Wang, L. Lv, F. Wang, H. Xu, H. Sun, H. Xing, N. Li, W. Zhang, Z. Wang, G. Liu, Z. Sun, D. Zhou, W. Li, L. Liu, L. Wang and Z. Wang, J. Med. Chem., 2019, 62, 7923–7940 CrossRef CAS PubMed.
- (a) I. D. Gridnev, N. Higashi, K. Asakura and T. Imamoto, J. Am. Chem. Soc., 2000, 122, 7183–7194 CrossRef CAS; (b) I. D. Gridnev, M. Yasutake, N. Higashi and T. Imamoto, J. Am. Chem. Soc., 2001, 123, 5268–5276 CrossRef CAS PubMed; (c) N. V. Belkova, L. M. Epstein, O. A. Filippov and E. S. Shubina, Chem. Rev., 2016, 116, 8545–8587 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General experimental procedures, compound characterization data, and analysis of enantioselectivities of products and crystal parameters for the compound 4c. CCDC 2243486. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc02086j |
| This journal is © The Royal Society of Chemistry 2023 |