
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePorous supramolecular gels produced by reversible self-gelation of ruthenium-based metal–organic polyhedra†
Javier
Troyano
 *abc,
Fuerkaiti
Tayier
ad,
Phitchayapha
Phattharaphuti
ad,
Takuma
Aoyama
e,
Kenji
Urayama
e and
Shuhei
Furukawa
*ad
*abc,
Fuerkaiti
Tayier
ad,
Phitchayapha
Phattharaphuti
ad,
Takuma
Aoyama
e,
Kenji
Urayama
e and
Shuhei
Furukawa
*ad
aInstitute for Integrated Cell-Material Sciences (WPI-iCeMS), Kyoto University, Yoshida, Sakyo-ku, 606-8501 Kyoto, Japan. E-mail: shuhei.furukawa@icems.kyoto-u.ac.jp
bDepartment of Inorganic Chemistry, Autonomous University of Madrid, 28049 Madrid, Spain. E-mail: javier.troyano@uam.es
cInstitute for Advanced Research in Chemical Sciences (IAdChem), Autonomous University of Madrid, 28049 Madrid, Spain
dDepartment of Synthetic Chemistry and Biological Chemistry, Graduate School of Engineering, Kyoto University, Katsura, Nishikyo-ku, Kyoto 615-8510, Japan
eDepartment of Macromolecular Science and Engineering, Kyoto Institute of Technology, Matsugasaki, Sakyo-ku, Kyoto 606-8585, Japan
First published on 15th August 2023
Abstract
Supramolecular gels based on metal–organic polyhedra (MOPs) represent a versatile platform to access processable soft materials with controlled porosity. Herein, we report a self-gelation approach that allows the reversible assembly of a novel Ru-based MOP in the form of colloidal gels. The presence of cationic mixed-valence [Ru2(COO)4]+ paddlewheel units allows for modification of the MOP charge via acid/base treatment, and therefore, its solubility. This feature enables control over supramolecular interactions, making it possible to reversibly force MOP aggregation to form nanoparticles, which further assemble to form a colloidal gel network. The gelation process was thoroughly investigated by time-resolved ζ-potential, pH, and dynamic light scattering measurements. This strategy leads to the evolution of hierarchically porous aerogel from individual MOP molecules without using any additional component. Furthermore, we demonstrate that the simplicity of this method can be exploited for the obtention of MOP-based gels through a one-pot synthetic approach starting from MOP precursors.
Introduction
Supramolecular gels are soft materials, in which molecular units are hierarchically assembled to give a three-dimensional network that spans the volume of a liquid and entraps it therein.1,2 The formation of such gel systems brings several advantages. First, the characteristics of the molecular building units are transferred upwards the macroscale, allowing the production of shaped objects exhibiting predefined properties.2 In addition, the weak and reversible nature of the interactions mediating the assembly favours the emergence of attractive features, such as self-healing3 or stimuli-responsiveness capacities.4 Furthermore, the resulting gel networks can be processed into the corresponding porous aerogels, leading to low-density solids displaying higher surface areas and favoured molecular transport.5,6Among a variety of molecular units, those that contain a defined and accessible cavity in their structure constitute a great platform for accessing functional supramolecular gels.7,8 Here, the resulting properties of the gel shall be determined by virtue of the shape, size, and chemical functionalization of the selected cavities, as well as by the resulting structure derived from their assembly. In this sense, metal–organic polyhedra (MOPs) or cages (MOCs) provide an excellent choice.9,10 MOPs are modular supramolecular porous units constructed from coordination-driven self-assembly of organic ligands and metal ions, in which the geometry, pore size, and external/internal functionalization, among others, can be tuned by design.11–13 In addition, MOPs are intrinsically porous, and therefore, long-range order is not required for accessing permanently porous materials.14
All these aspects have moved researchers to explore the construction of MOP-based supramolecular gels, leading to a steadily growing number of examples.15,16 In most cases, the construction of such gels relies on the same concept: the formation of a polymeric network, in which MOPs act as nodes that are interconnected through organic moieties. For instance, supramolecular MOP-based gels have been synthesised by combining metal ions and polymeric organic precursors designed to act as ligands, leading to the creation of well-defined MOPs and, at the same time, connecting them.17–20 Besides, the polymerization of MOP to form supramolecular gels has also been achieved by covalent21–23 or coordinative24 crosslinking of previously synthesised MOPs. Here, the coordination of organic linkers to available open metal sites of MOPs has been the most explored approach as it permits precise control over assembly through the regulation of coordination bonds, leading to MOP-based materials with controlled porosity.25–27
A much less explored approach consists in the ‘self-gelation’ of MOPs without any linker, where the assembly is only governed by supramolecular interactions. Despite the simplicity of this strategy, to date, the number of cages capable to undergo ‘self-gelation’ is very limited. Previous examples include one Cu(II)-MOP and some Pd(II)-based coordination cages that present certain functional groups, such as –NO2,28 cholesteryl groups,29 metalloligands,30 or photoactive moieties,31 that drive the assembly. However, the obtention of permanently porous aerogels that are purely built from MOPs has yet to been explored. This scarcity can be explained by several factors. First, it remains challenging to predict whether a MOP will form a gel or not due to the delicate balance between intermolecular MOP–MOP and MOP–solvent interactions.29,31 Another obstacle lies in the low stability of many MOPs under potential conditions for gelation (i.e. solvent, pH, temperature), limiting the discovery of novel MOP gelators. In addition to that, once the gel is formed, the MOP must be stable enough to allow solvent molecules removal without structural collapse to demonstrate permanent porosity.
Herein, we present a novel cuboctahedral MOP based on stable mixed-valence diruthenium Ru25+ tetracarboxylates units with external hydroxyl groups at its surface, capable of undergoing reversible self-gelation upon heating. Compared to other carboxylate paddlewheels based on divalent metals [Ru2(COO)4]+ units are positively charged. This feature not only provides a MOP with good solubility in different solvents, which leads to stable solutions due to inter-MOP repulsive interactions, but also enables the destabilization of such solutions by adjusting the MOP charge. By exploiting this charge switch approach, we show that Ru-based MOPs can reversibly assemble into colloidal gels, which can be further converted into permanently porous supramolecular aerogels. The resulting monolithic aerogels present a hierarchical porous structure that features both micro- and meso-porosity, which can be ascribed to the retention of the ‘intrinsic’ porosity of the inner cavity of the MOP, and the creation of additional ‘extrinsic’ porosity between the MOPs. We also present a straightforward one-pot synthesis route to produce Ru-based MOP colloidal gels directly from MOP building blocks.
Results and discussion
Synthesis of soluble OH–RuMOP[BF4]12
The reaction between cationic mixed-valence diruthenium complex [Ru2(OAc)4(THF)2]BF4 and 5-hydroxy-1,3-benzenedicarboxylic acid in DMA at 120 °C in the presence of Na2CO3 yielded brown single crystals of [Ru24(OH-bdc)24(OH)12(H2O)4(DMA)8] (OH–RuMOP(OH)12, where OH-bdc = 5-hydroxy-1,3-benzenedicarboxylate, DMA = N,N-dimethylacetamide) (Fig. 1a and S1†).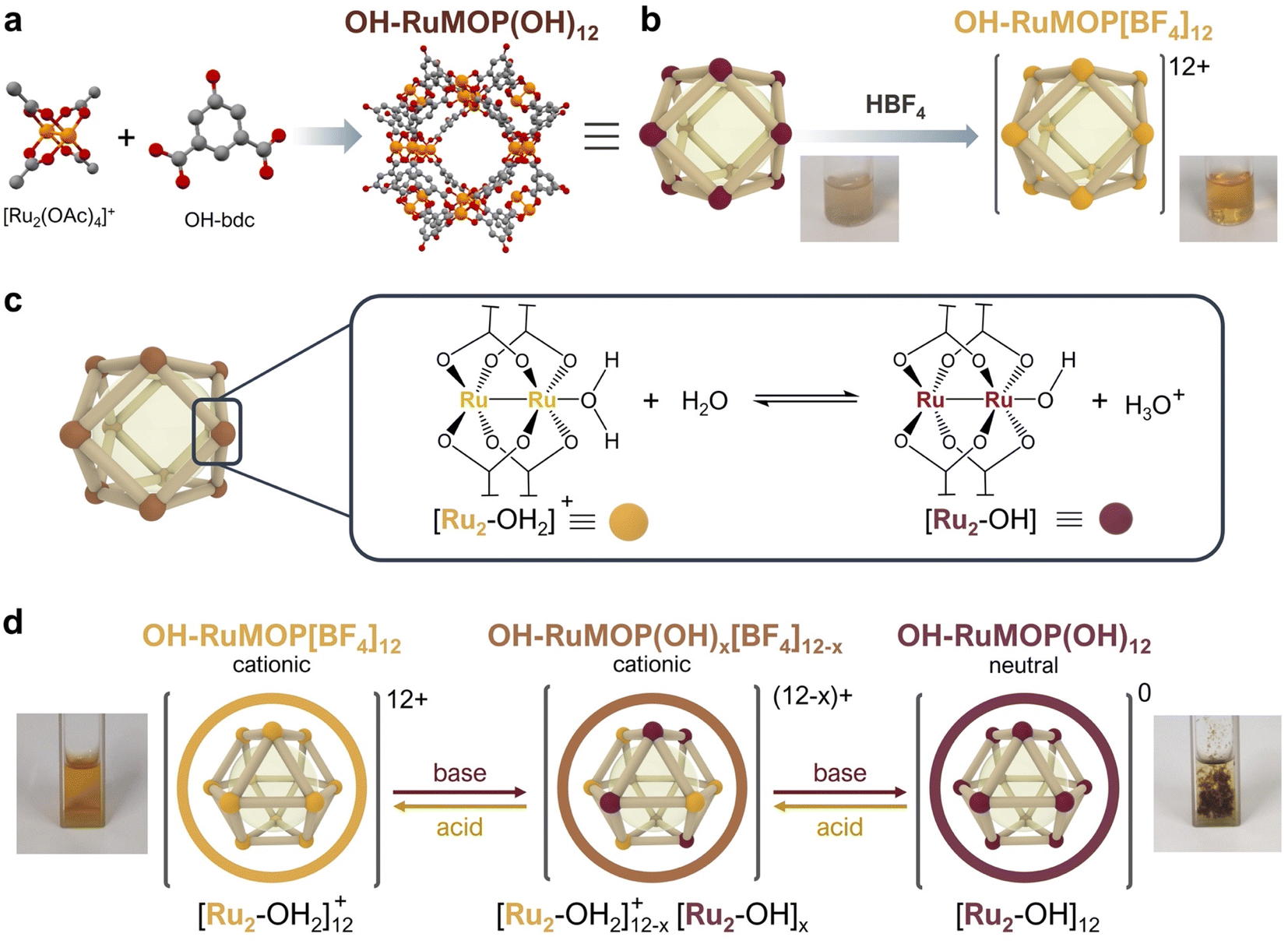 | ||
| Fig. 1 (a) Schematic representation of the synthesis of OH–RuMOP(OH)12 from its building blocks. (b) Transformation of non-soluble OH–RuMOP(OH)12 into soluble OH–RuMOP[BF4]12 due to the removal of coordinated OH− ligands upon acid treatment with HBF4. (c) Acid–base equilibrium between water-coordinated and hydroxo-coordinated Ru2(CO2)4 paddlewheel units in water solutions. (d) Schematic representation of the gradual interconversion between cationic OH–RuMOP[BF4]12 and neutral OH–RuMOP(OH)12 upon acid/base treatment. | ||
Single-crystal X-ray diffraction (SC-XRD) experiments showed that OH–RuMOP(OH)12 crystallizes in the tetragonal space group I4/m (Table S1†). OH–RuMOP(OH)12 displays a cuboctahedral structure, analogous to other M24(OH-bdc)24 MOPs,32–37 in which twelve Ru2 paddlewheel units are connected through twenty-four OH-bdc ligands. The size of the MOP, defined by the average distance between the O atoms in the phenol groups of the opposing dicarboxylate ligands, is 27.3 Å. The pore size, defined by the average distance between the opposing internal Ru atoms, is 16.7 Å. The SC-XRD analysis also showed that eight DMA solvent molecules were located at external axial sites, while the remaining axial positions were refined as oxygen atoms (Fig. S2†). The Ru–Ru bond distances in OH–RuMOP(OH)12 (2.269 and 2.275 Å) correspond to those found in previously reported [Ru2(COO)4]0/+ paddlewheel complexes;38 however, it is in general not possible to distinguish between the oxidation states by the Ru–Ru bond distance.38,39 Fourier-transform infrared (FT-IR) analysis of the crystals confirmed that the mixed oxidation number of Ru25+ unit was maintained during MOP synthesis, as evidenced by the values of symmetric, νsym(CO2), and asymmetric, νasym(CO2), carboxylate stretching vibration modes and their separation (Δν = 67 cm−1) (Fig. S3†).38,40 FT-IR also revealed the disappearance of the strong band at ca. 1050 cm−1 corresponding to BF4− group41 (Fig. S4†). The absence of BF4− in the crystals was further confirmed by energy-dispersive X-ray (EDX) spectroscopy (Fig. S5†). SC-XRD analysis did not allow us to determine the presence of any anion. Thus, we assumed that the required negative charges to compensate positive [Ru2(COO)4]+ units were caused by the coordination of twelve OH− anions. This situation gives rise to a neutral MOP, in which twelve OH− and four H2O indistinguishable ligands occupy four external an all internal Ru2 axial sites.
Due to the lack of solubility of as-synthesised OH–RuMOP(OH)12 crystals in any conventional organic solvents or water, we explored the post-synthetic acid treatment of OH–RuMOP(OH)12 with HBF4 with the aim of generating a soluble salt with poorly coordinating BF4− as a counter anion (Fig. 1b). The addition of HBF4 to a suspension of OH–RuMOP(OH)12 in MeOH resulted in a brown solution, which was further precipitated with diethyl ether to yield a brown-orange amorphous solid, OH–RuMOP[BF4]12 (Fig. S1†). The FT-IR spectrum confirmed the occurrence of BF4− (Fig. S4†), while the EDX analysis revealed a F/Ru atomic ratio of 2.03 (Fig. S6†), which corresponds to the expected [Ru24(OH-bdc)24][BF4]12 composition. After acid treatment, the isolated OH–RuMOP[BF4]12 cage presented good solubility in polar organic solvents, such as DMF, DMA, DMSO, MeOH, EtOH, 2-propanol, acetone and MeCN, but was found to be insoluble in water. We then explored the use of H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMF mixtures by first dissolving the MOP in DMF and then adding water. By following this approach, we achieved the dissolution of OH–RuMOP[BF4]12 in H2O:DMF (9:1, v/v).
:DMF mixtures by first dissolving the MOP in DMF and then adding water. By following this approach, we achieved the dissolution of OH–RuMOP[BF4]12 in H2O:DMF (9:1, v/v).
The resulting solutions of OH–RuMOP[BF4]12 in H2O:DMF (9:1, v/v) were studied through UV-vis, dynamic light scattering (DLS), ζ-potential and pH measurements. UV-vis spectrum (Fig. S7†) showed an absorption band at 430 nm, similarly to that observed in [Rh2(OAc)4]+, which is likely attributed to the π(Ru–Ru) → π*(Ru–Ru) transition.42 DLS measurements confirmed its solubility, showing an average size of ca. 2.8 nm (Fig. S8†), which is in accordance to the size of the MOP determined by the SC-XRD analysis. The ζ-potential measurements showed a value of +26 mV, confirming the positive surface of the MOP (Fig. S9†).
Acid–base behaviour of OH–RuMOP[BF4]12
Analogously to aqueous solutions of [Ru2(OAc)4]BF4 complex, solutions of OH–RuMOP[BF4]12 in H2O:DMF (9:1, v/v) were acidic (pH = 3.0, 0.2 mM). The acidic nature of OH–RuMOP[BF4]12 can be explained by the Lewis acid character of Ru2 units. The coordination of water to Ru implies the weakening of O–H bond, which favours the donation of one proton to surrounding water molecules, lowering the pH (Fig. 1c). Correspondingly, we assumed that the addition of a base would push equilibrium towards the incorporation of more OH− ligands, leading to a neutral, non-soluble OH–RuMOP(OH)12 (Fig. 1d). Thus, we added controlled amounts of triethylamine to a 0.2 mM solution of OH–RuMOP[BF4]12 in H2O:DMF (9:1, v/v) and measured the pH changes. We found that upon addition of excess triethylamine an insoluble dark brown solid was precipitated as OH–RuMOP(OH)12 (Fig. S10†) at pH = 3.6. The FT-IR spectrum (Fig. S11†) indicated the loss of BF4−, which was further corroborated by EDX analysis (Fig. S12†). The precipitation at such relative low pH value allowed us to discard the deprotonation of the external hydroxyl groups of the OH-bdc linker (phenol, pKa = 9.88), supporting the idea that precipitation occurred due to the coordination of OH− ligands.
Self-gelation of OH–RuMOP[BF4]12
The capacity to regulate the acid–base equilibrium of OH–RuMOP[BF4]12, pointed to the possibility to prepare supramolecular gels through controlled aggregation of MOPs. However, the addition of base resulted in non-homogeneous precipitation, which limited its applicability for controlled gel synthesis. Therefore, we explored an indirect method for such purpose. It is known that DMF easily hydrolyses upon heating under acid conditions, leading to a more basic medium.43 Accordingly, we hypothesised that upon heating, acid–base equilibrium would move towards formation of OH–RuMOP(OH)12 as a consequence of the in situ DMF hydrolysis, leading to a more homogeneous aggregation. A 0.2 mM solution of OH–RuMOP[BF4]12 in H2O:DMF (9:1, v/v) was heated at 80 °C for 24 h, resulting in the formation of a dark brown solid after ca. 10 h (Fig. S13†). After washing and drying, this solid was characterised by FT-IR showing the same spectrum that the OH–RuMOP(OH)12 solid obtained by base addition (Fig. S11†). Here, EDX also confirmed the loss of BF4− anion (Fig. S14†). We also monitored the spectral UV-vis changes of solutions of OH–RuMOP[BF4]12 in H2O:DMF (9:1, v/v) upon (i) addition of base (Fig. S15†), and (ii) heating (Fig. S16†). In both cases, we found the appearance of a weak band at ca. 620 nm, together with the main band corresponding to the Ru2 unit (λmax = 436 nm), concomitant with solid precipitation. These results suggested that the same chemical transformation took place during base addition and upon heating, generating neutral OH–RuMOP(OH)12.
Having demonstrated that heating solutions of OH–RuMOP[BF4]12 in H2O–DMF mixture resulted in the precipitation of neutral OH–RuMOP(OH)12, we then attempted to exploit this phenomenon for synthesizing supramolecular MOP-based gels by increasing the MOP concentration. We found that heating a 1 mM solution of OH–RuMOP[BF4]12 in H2O:DMF (9:1, v/v) at 80 °C, resulted in the formation of a gel after few hours (Fig. 2a).
 | ||
| Fig. 2 (a) Photographs showing the evolution of a 1 mM OH–RuMOP[BF4]12 solution in H2O:DMF (9:1, v/v) upon heating at 80 °C. (b) Time-resolved dynamic light scattering, pH, and ζ-potential measurements for 1 mM OH–RuMOP[BF4]12 solutions in H2O:DMF (9:1, v/v) upon heating at 70 °C indicating the different stages of the gelation process. (c) Storage (E′) and loss (E′′) Young's modulus for gels prepared by heating at 80 °C 1 mM H2O:DMF (9:1, v/v) OH–RuMOP[BF4]12 solutions for at different incubation times (ti). (d) Proposed mechanism for the formation of a colloidal gel from OH–RuMOP[BF4]12 solutions through self-gelation. | ||
To gain more insight into the self-gelation dynamics we performed a set of experiments to monitor the changes in ζ-potential, pH, and DLS over time (Fig. 2b). We initially used 1 mM solutions of OH–RuMOP[BF4]12 in H2O:DMF (9:1, v/v), heating at 70 °C (maximum operating temperature for the zeta potential cell). Analyses of the changes in ζ-potential and pH during the first steps of reaction were useful to understand the self-gelation mechanism. Initially, a slight reduction of ζ-potential was recorded, moving from starting +26 mV to +18 mV after ca. 100 min heating. Afterwards, a pronounced ζ-potential decay occurred until it reached a value of zero (t ≈ 125 min), revealing the loss of net charge at the surface of the MOPs due to charge neutralization. After this point, ζ-potential did not show any change up to 240 min, when the gelation happened. Besides, pH did not show any variation up to ca. 110 min. However, at longer periods of time, a very slight increase from the initial state (pH = 2.4) to the gelation point (pH = 2.7) was registered. In situ time-resolved DLS (TR-DLS) measurements were also conducted to gain more information about changes in particle size. During the first 100 min, the particle size was maintained below 3 nm, indicating that MOPs stayed as individual soluble molecules. Then, heating induced their assembly into larger colloidal particles up to ca. 12 nm in size. After that, the formation of a gel network led to the loss of mobility of the particles, making the measurement of particle size unfeasible. TR-DLS was also used to analyse the changes of the time averaged scattering intensity, 〈I〉T, which allowed us to determine the gelation time (tgel). This point corresponds to the time at which random fluctuations of 〈I〉T appear due to the loss of homogeneity upon sol–gel transition (tgel ≈ 240 min).
We also evaluated the role of the H2O–DMF ratio on self-gelation by using OH–RuMOP[BF4]12 solutions with lower water content (H2O:DMF 7:3 and 1:1, v/v). The estimated tgel values derived from 〈I〉T plots clearly indicated that a reduction in the water content led to a significant delay in gelation time from 240 min (H2O:DMF 9:1, v/v) to 320 min (H2O:DMF 7:3, v/v) and to 560 min (H2O:DMF 1:1, v/v) (Fig. S17†). Note here that the same trend was observed when the same gelation experiments were carried out at 80 °C (Fig. S18 and S19†). The elongation of tgel values can be rationalised by comparing the time required for gelation after charge neutralization. For instance, at 70 °C, the H2O:DMF 9:1 v/v mixture required ca. 125 min to achieve charge neutralization, whereas the H2O:DMF 1:1 v/v mixture needed ca. 195 min (Fig. S20†). However, in the former case gelation occurred ca. 125 min after charge neutralization, but in the latter gelation took place ca. 360 min after charge neutralization. The observed trend can be attributed to the lower capacity of water to stabilize neutral OH–RuMOP(OH)12 compared to DMF, making water-rich mixtures to accelerate the gelation process once charge neutralization is achieved. Note that control experiments were also carried out by heating at 80 °C solutions of OH–RuMOP[BF4]12 in pure DMF and different organic solvents (i.e. i-PrOH, MeCN). However, after 72 h heating no precipitation was observed in all the cases (Fig. S21†), confirming that water molecules are required for acid–base reaction and subsequent MOP aggregation.
From the above results the following gelation mechanism can be deduced. As depicted in Fig. 2d, positively charged OH–RuMOP[BF4]12 initially form an stable solution because of the repulsive electrostatic forces. Upon heating, the MOPs steadily incorporate OH− ligands due to the in situ hydrolysis of DMF in acidic medium (stage I). This process ends when charge neutralization is achieved, which corresponds to the reduction of ζ-potential to zero. Beyond this point, the aggregation occurs due to the attractive interactions between neutral MOPs, leading to colloidal clusters, as evidenced by TR-DLS experiments (stage II). The aggregation of such colloidal clusters further leads to a colloidal percolation that spans the full liquid medium and yields a supramolecular gel at tgel (stage III).
Preparation of robust OH–RuMOP-based gels through aging process
The typical gels formed by the assembly of OH–RuMOP[BF4]12 at 80 °C 1 mM H2O:DMF (9:1, v/v) solutions of for 3 h (tgel = 80 min) was found to be not mechanically stable and not self-standing when placed out of the vial (Fig. 2a). We also observed that this weak gel was transformed to the sol state by strongly shaking by hand, followed by the recovery of gel state upon standing at room temperature after a few minutes (Fig. S22 and Movie S1†). This thixotropic response was determined by performing several shearing–resting cycles, confirming the reversible sol–gel transition (Fig. S23†).
The lack of mechanical stability of these gels prompted us to extend the incubation time (ti), defined as the total time under heating, to improve the hardness of the gels. Thus, we prepared a series of gels by heating at 80 °C the solution of 1 mM H2O:DMF (9:1, v/v) OH–RuMOP[BF4]12 for 8, 12, 16, 20 and 24 h. Then, the mechanical properties of the gels were investigated by determining their compression storage (E′) and loss (E′′) moduli as a function of the oscillatory deformation frequency ω (rad s−1), at a fixed strain amplitude (1%). The gel obtained at ti = 8 h was not self-standing, thus the rheological property was measured by shear moduli (G′ and G′′) and converted to E′ and E′′ by the equation of E = 3G. As depicted in Fig. 2c, at ti = 8 h the resulting gel showed a solid-like elastic behaviour (E′ > E′′) at low ω. However, E′ and E′′ become similar as ω increases, originated from the viscous components in the gel. At ti = 12 h, the gel was found to be self-standing and strong enough to directly measure the compression moduli. The gel showed much higher E′ and E′′ values exhibiting a linear viscoelastic response. Further extension of ti resulted in stiffer gels with higher E′ and E′′, being less pronounced as ti increased. At ti = 24 h, the resulting gel, named as OH–RuMOP-gel, showed values of E′ = 3.9 × 103 Pa and E′′ = 0.5 × 103 Pa at 100 rad s−1, comparable to that found for other crosslinked MOP-based gels.28,44 The stiffness increment upon using longer incubation times is most likely attributed to the rearrangement of the colloidal particles at the nano/mesoscopic level, leading to gradual changes in the structure of the gel.
We also evaluated the effect of the solvent mixture on the aging process by preparing gels (ti = 24 h) with different H2O:DMF ratios (7:3 and 1:1, v/v). Rheology measurements of the resulting dark brown transparent gels were carried out, with E′ = 2.5 × 103 and 2.0 × 103 Pa and E′′ = 1.2 × 102 and 0.9 × 102 Pa values at 100 rad s−1 for gels prepared with H2O:DMF 7:3 v/v, and 1:1 v/v, respectively (Fig. S24†). These results evidenced the formation of more robust gels when more water was present in the mixture, which correlates with the previously observed reduction of tgel values at higher H2O:DMF ratio (vide supra). We attribute this effect to the role of water as a v/v solvent, which favours attractive interactions between aggregates, accelerating the formation of the gel and the further enhancement of the gel stiffness.
Aerogel production and characterization
To investigate the structures and the porous property of OH–RuMOP-gel, the wet gel was transformed into the corresponding aerogel, OH–RuMOP-agel, by supercritical CO2 (SC-CO2) drying process (Fig. 3a and the detail synthetic procedure in ESI†). Note here that the obtained OH–RuMOP-gel and OH–RuMOP-agel were not soluble in any of the solvents used for dissolving the pristine MOP. Field-emission scanning electron microscopy (FE-SEM) images of OH–RuMOP-agel showed the formation of colloidal particles with the size of 29 ± 6 nm (Fig. S25†), which are further interconnected to each other to form hierarchical meso- and macroporous colloidal networks (Fig. 3b). This was indicative that colloidal particles increased their size after formation of initial gel at tgel, when particle size was estimated to be ca. 16 nm by DLS (Fig. S18†). The FT-IR analysis of OH–RuMOP-agel showed no changes in carboxylate stretching vibrations, compared to that of the pristine MOP (Fig. 3c), which confirms that the MOP did not undergo neither decomposition nor reduction. The FT-IR analysis also revealed the absence of BF4− anion, further supported by EDX analysis (Fig. S26†). We also checked the reversibility of the assembly process. The aqueous solution of HBF4 was added to a suspension of the aerogel in DMF, resulting in the immediate dissolution of the solid (Fig. 3a). The resulting DMF/HBF4 solution was investigated by DLS measurements (Fig. S27†), revealing an average particle size of 3 nm that corresponds to the size of OH–RuMOP[BF4]12. Further, the integrity of the MOP after the acid-triggered disassembly was checked by the UV-vis absorption spectrum of the resulting solution (Fig. 3d) and the FT-IR measurement of the brown solid obtained after precipitation with diethyl ether (Fig. 3c), confirming the reversibility of the gelation process. | ||
| Fig. 3 (a) Photographs of OH–RuMOP-agel produced by supercritical CO2 (SC-CO2) drying of OH–RuMOP-gel, and the resulting solution obtained after addition of HBF4 to a suspension of the aerogel in DMF. (b) FE-SEM images of OH–RuMOP-agel. The right part image is the magnification of the corresponding yellow square. The scale bars for lower and higher magnification are 1 μm and 100 nm, respectively. (c) FT-IR spectra of OH–RuMOP-agel (magenta), and the solid obtained after dissolving it in HBF4/DMF and precipitating with diethyl ether (blue), compared to OH–RuMOP[BF4]12 (orange). The presence/absence of BF4− at ca. 1050 cm−1 is highlighted. (d) UV-vis absorption spectra of the solution obtained after dissolving OH–RuMOP-agel in HBF4/DMF (blue), compared to OH–RuMOP[BF4]12 (orange). | ||
Porous properties
Despite both OH–RuMOP[BF4]12 and OH–RuMOP-agel were obtained as amorphous solids (Fig. S28†), the intrinsic microporosity of the OH–RuMOP, prompted us to investigate their sorption capabilities.45 It is known that the assembly of MOPs into hierarchical porous aerogels can lead to improved gas sorption capacities, by favouring the gas diffusion through an open mesostructured network.24 In addition, the production of aerogels by only using MOPs without any additional linker, offered a good opportunity to evaluate the effect of aerogel-processing by comparing the sorption behaviour of pure OH–RuMOP[BF4]12 and derived supramolecular aerogel OH–RuMOP-agel.Fig. 4a showed the N2 sorption of OH–RuMOP[BF4]12 at 77 K with a typical type-I isotherm, with high sorption at low relative pressure (≈150 cm3 g−1 at P/P0 = 0.1). The Brunauer–Emmett–Teller surface area (SBET) analysed using BETSI46 was estimated to be 615 m2 g−1 (Fig. S29†), which is the highest value among all reported M24(OH-bdc)24 (M = Cr, Co, Ni, Cu, Rh) cuboctahedral MOPs.32,47–49 The pore size distribution (PSD) of pure OH–RuMOP[BF4]12 was estimated by non-local density functional theory (NLDFT), revealing a micropore of 0.6 nm in size (Fig. 4b), which corresponds to the size of the MOP cavity, as it has been evidenced by previous studies of analogous cuboctahedral MOPs.25,44,50
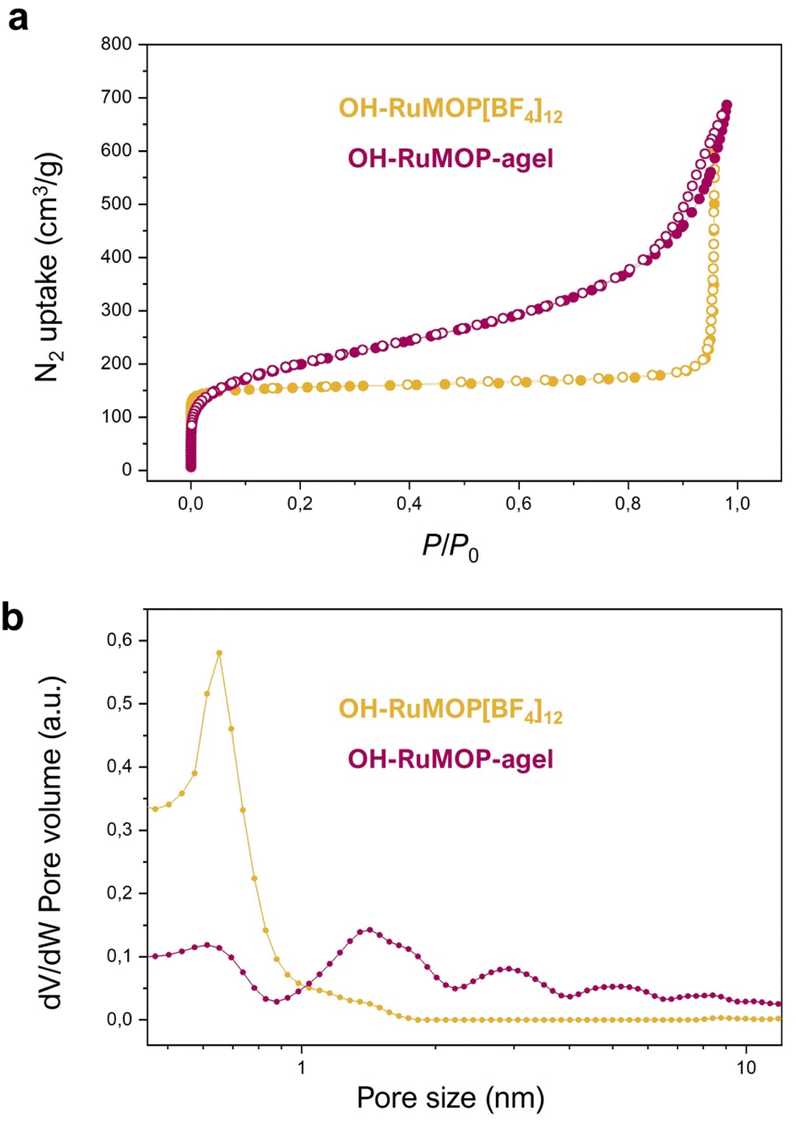 | ||
| Fig. 4 (a) N2 adsorption (filled circle) and desorption (open circle) at 77 K of OH–RuMOP[BF4]12 (orange) and OH–RuMOP-agel (magenta). (b) Pore size distribution (PSD) of OH–RuMOP[BF4]12 (orange) and OH–RuMOP-agel (magenta) estimated from N2 isotherms by NLDFT on a slit pore model. | ||
Besides, OH–RuMOP-agel showed a type-II isotherm with high sorption at the low relative pressure (≈170 cm3 g−1 at P/P0 = 0.1), and a SBET (BETSI) value of 717 m2 g−1 (Fig. S30†), which represents a moderate increment compared to OH–RuMOP[BF4]12. We attribute this improvement to the removal of BF4− anions during gel formation, which can fill and/or block the pores resulting in a reduction of adsorption capacity of the pure MOP.50 The OH–RuMOP-agel displayed the higher adsorption amount at the high-pressure region, thanks to the macro-and mesopores present in the hierarchically structured network. At P/P0 ≈ 0.5, OH–RuMOP[BF4]12 adsorbed 170 cm3 g−1, while aerogel adsorbed 267 cm3 g−1. The PSD analysis for OH–RuMOP-agel (Fig. 4b) showed the presence of a micropore at 0.6 nm, corresponding to the prevalence of the MOP cavity as well as the different pore sizes at both micro- and mesoporous regions, which most likely correspond to the voids generated by the random packing of the MOPs in the solid state.25
One-pot synthesis of OH–RuMOP-based gels
The fact that the formation of OH–RuMOP-gel does not require any additional component motivated us to develop a one-pot synthesis strategy for the preparation of MOP-based gels directly from MOP precursors (Fig. 5a). Conversely to the synthesis of OH–RuMOP(OH)12 crystals, which requires the addition of Na2CO3 as base to the DMA solution, we hypothesised that the basic medium generated upon heating H2O–DMF solvent mixture could result in the primary formation of the MOP and subsequent sol–gel transition. To this end, H2O–DMF 9:1 v/v mixtures containing [Ru2(OAc)4(THF)2]BF4 and OH–H2bdc at different concentrations ([Ru2] = 12 to 72 mM) were heated at 80 °C for 24 h. While fine brown powders were formed at lower concentrations (12–36 mM), the solution with higher concentration (>36 mM) resulted in the formation of gels (Fig. S31†). However, due to the low solubility of OH–H2bdc in water, the gels contained a certain amount of unreacted linker. We found a compromise between concentration and solubility of the linker at [Ru2] = 48 mM, corresponding to a [OH–RuMOP] = 4 mM for theoretical yield. The resulting gel, named as OH–RuMOP-gelOP, revealed higher values of the compression moduli E′ = 9.9 × 103 Pa and E′′ = 9.8 × 102 Pa at 100 rad s−1, compared to that found for OH–RuMOP-gel (Fig. 5c), as expected from the use of higher concentrations.
 | ||
| Fig. 5 (a) Schematic representation of the one-pot production of OH–RuMOP-gelOP and its further transformation into OH–RuMOP-agelOP, showing photographs of the resulting materials. (b) FE-SEM images at different magnifications of OH–RuMOP-agelOP. The right part image is the magnification of the corresponding yellow square. The scale bars for the left and right images are 500 and 100 nm, respectively. (c) Storage (E′) and loss (E′′) Young's modulus for OH–RuMOP-agelOP (purple) compared to OH–RuMOP-agel (magenta). (d) N2 adsorption (filled circle) and desorption (open circle) at 77 K of OH–RuMOP-agelOP (purple) compared to OH–RuMOP-agel (magenta). (e) Pore size distribution (PSD) of OH–RuMOP-agelOP (purple) compared to OH–RuMOP-agel (magenta) estimated from N2 isotherms by NLDFT on a slit pore model. | ||
OH–RuMOP-gelOP was processed analogously as was done for OH–RuMOP-gel, yielding the corresponding aerogel OH–RuMOP-agelOP (Fig. 5b). The FT-IR characterization of OH–RuMOP-agelOP was identical to that exhibited by OH–RuMOP-agel, which confirmed the successful formation of the MOP as well as the lack of BF4− anion in the final structure (Fig. S32†). The FE-SEM images evidenced the formation of an analogous hierarchical three-dimensional colloidal network, built-up by the assembly of the particle with the size of 31 ± 5 nm (Fig. 5b and S33†).
The porous properties of OH–RuMOP-agelOP were also investigated by N2 adsorption measurements at 77 K (Fig. 5d), revealing an almost identical behaviour to that observed in OH–RuMOP-agel. Thus, as expected from its hierarchical porous structure, OH–RuMOP-agelOP showed a type-II N2 isotherm with a steep increase at higher pressures, and a SBET (BETSI) value of 704 m2 g−1 (Fig. S34†).
Conclusions
In summary, a new approach for producing porous supramolecular gels based on MOP through self-gelation is presented. This strategy was demonstrated with a novel cuboctahedral MOP in which mixed-valence diruthenium Ru25+ units provide the possibility to control solubility by adjusting the pH. Thus, it can be post-synthetically treated with acid to yield a soluble cationic MOP, exhibiting permanent porosity. The cationic MOP solutions can be destabilised by base addition, leading to a non-soluble neutral MOP. This phenomenon was exploited to create MOP-based supramolecular colloidal gels, in which charge neutralization drives the aggregation of MOPs into colloidal particles that finally assemble into a three-dimensional gel network. The reversible self-gelation was studied in detail by means of time resolved ζ-potential, pH, and DLS measurements, which revealed the different stages of the process. The rheological properties of the resulting gels at different heating times indicated that aging is required to obtain robust self-standing gels, which were next converted into hierarchically porous aerogels. Notably, the porosity of the resulting aerogel was slightly higher to that found for MOP powder. To the best of our knowledge, this is the first time that gels built-up from only MOPs without any other component present permanent microporosity. Furthermore, an alternative one-pot strategy to produce gels by simply reacting MOP precursors was demonstrated. Remarkably, the resulting (aero)gel exhibited the same porosity found in the materials formed through a stepwise method.We envision that this approach could be applied to other MOPs containing Ru25+ units,39 in which different shapes/external functionalization can lead to the structural control of the resulting porous gels. Further, considering the well-known redox activity of Ru25+ units,51 this strategy paves the way to the production of amorphous soft gels showing redox-responsive or charge transfer capacities.52–55
Author contributions
J. T. and S. F. conceived and designed the project. J. T., F. T. and P. P. performed all synthetic and characterization experiments. J. T., T. A. and K. U. performed rheology measurements. J. T. and S. F. analysed the data and wrote the manuscript. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. T. is grateful to the Japan Society for the Promotion of Science (JSPS) for Postdoctoral Fellowship and the Spanish Ministry of Universities and the European Union through the Funds Next GenerationEU through grant Maria Zambrano-UAM Grant Number CA3/RSUE/2021-00698. This study was supported by JSPS KAKENHI Grant Number 21K18192 (Challenging Research (Pioneering)) for S. F. The authors acknowledge the iCeMS Analysis Centre for access to analytical facilities.References
- E. R. Draper and D. J. Adams, Chem, 2017, 3, 390–410 CAS.
- P. R. A. Chivers and D. K. Smith, Nat. Rev. Mater., 2019, 4, 463–478 CrossRef CAS.
- Z. Wei, J. H. Yang, J. Zhou, F. Xu, M. Zrínyi, P. H. Dussault, Y. Osada and Y. M. Chen, Chem. Soc. Rev., 2014, 43, 8114–8131 RSC.
- S. Panja and D. J. Adams, Chem. Soc. Rev., 2021, 50, 5165–5200 RSC.
- S. P. Patil, V. G. Parale, H.-H. Park and B. Markert, Ceram. Int., 2021, 47, 2981–2998 CrossRef CAS.
- S. Q. Zeng, A. Hunt and R. Greif, J. Non-Cryst. Solids, 1995, 186, 264–270 CrossRef CAS.
- J. A. Foster and J. W. Steed, Angew. Chem., Int. Ed., 2010, 49, 6718–6724 CrossRef CAS PubMed.
- Z. Qi and C. A. Schalley, Acc. Chem. Res., 2014, 47, 2222–2233 CrossRef CAS PubMed.
- I. Jahović, Y.-Q. Zou, S. Adorinni, J. R. Nitschke and S. Marchesan, Matter, 2021, 4, 2123–2140 CrossRef.
- N. Hosono and S. Kitagawa, Acc. Chem. Res., 2018, 51, 2437–2446 CrossRef CAS PubMed.
- A. Khobotov-Bakishev, L. Hernández-López, C. von Baeckmann, J. Albalad, A. Carné-Sánchez and D. Maspoch, Adv. Sci., 2022, 9, 2104753 CrossRef CAS PubMed.
- J. Albalad, L. Hernández-López, A. Carné-Sánchez and D. Maspoch, Chem. Commun., 2022, 58, 2443–2454 RSC.
- S. Lee, H. Jeong, D. Nam, M. S. Lah and W. Choe, Chem. Soc. Rev., 2021, 50, 528–555 RSC.
- A. J. Gosselin, C. A. Rowland and E. D. Bloch, Chem. Rev., 2020, 120, 8987–9014 CrossRef CAS PubMed.
- E.-S. M. El-Sayed and D. Yuan, Chem. Lett., 2020, 49, 28–53 CrossRef CAS.
- E. Sánchez-González, M. Y. Tsang, J. Troyano, G. A. Craig and S. Furukawa, Chem. Soc. Rev., 2022, 51, 4876–4889 RSC.
- N. J. Oldenhuis, K. P. Qin, S. Wang, H.-Z. Ye, E. A. Alt, A. P. Willard, T. Van Voorhis, S. L. Craig and J. A. Johnson, Angew. Chem., Int. Ed., 2020, 59, 2784–2792 CrossRef CAS PubMed.
- Y. Gu, E. A. Alt, H. Wang, X. Li, A. P. Willard and J. A. Johnson, Nature, 2018, 560, 65–69 CrossRef CAS PubMed.
- J. A. Foster, R. M. Parker, A. M. Belenguer, N. Kishi, S. Sutton, C. Abell and J. R. Nitschke, J. Am. Chem. Soc., 2015, 137, 9722–9729 CrossRef CAS PubMed.
- A. V. Zhukhovitskiy, M. Zhong, E. G. Keeler, V. K. Michaelis, J. E. P. Sun, M. J. A. Hore, D. J. Pochan, R. G. Griffin, A. P. Willard and J. A. Johnson, Nat. Chem., 2016, 8, 33–41 CrossRef CAS PubMed.
- D.-H. M. H.-J. Jung and H.-P. Chun, Bull. Korean Chem. Soc., 2011, 32, 2489 CrossRef.
- X.-Y. Xie, F. Wu, X.-Q. Liu and L.-B. Sun, Dalton Trans., 2019, 48, 17153–17157 RSC.
- G. Lal, M. Derakhshandeh, F. Akhtar, D. M. Spasyuk, J.-B. Lin, M. Trifkovic and G. K. H. Shimizu, J. Am. Chem. Soc., 2019, 141, 1045–1053 CrossRef CAS PubMed.
- A. Carné-Sánchez, G. A. Craig, P. Larpent, T. Hirose, M. Higuchi, S. Kitagawa, K. Matsuda, K. Urayama and S. Furukawa, Nat. Commun., 2018, 9, 2506 CrossRef PubMed.
- Z. Wang, C. Villa Santos, A. Legrand, F. Haase, Y. Hara, K. Kanamori, T. Aoyama, K. Urayama, C. M. Doherty, G. J. Smales, B. R. Pauw, Y. J. Colón and S. Furukawa, Chem. Sci., 2021, 12, 12556–12563 RSC.
- Z. Wang, T. Aoyama, E. Sánchez-González, T. Inose, K. Urayama and S. Furukawa, ACS Appl. Mater. Interfaces, 2022, 14, 23660–23668 CrossRef CAS PubMed.
- A. Legrand, G. A. Craig, M. Bonneau, S. Minami, K. Urayama and S. Furukawa, Chem. Sci., 2019, 10, 10833–10842 RSC.
- Y. Qin, L.-L. Chen, W. Pu, P. Liu, S.-X. Liu, Y. Li, X.-L. Liu, Z.-X. Lu, L.-Y. Zheng and Q.-E. Cao, Chem. Commun., 2019, 55, 2206–2209 RSC.
- L. Zeng, Y. Xiao, J. Jiang, H. Fang, Z. Ke, L. Chen and J. Zhang, Inorg. Chem., 2019, 58, 10019–10027 CrossRef CAS PubMed.
- Y. Zhang, Q.-F. Zhou, G.-F. Huo, G.-Q. Yin, X.-L. Zhao, B. Jiang, H. Tan, X. Li and H.-B. Yang, Inorg. Chem., 2018, 57, 3516–3520 CrossRef CAS PubMed.
- S.-C. Wei, M. Pan, Y.-Z. Fan, H. Liu, J. Zhang and C.-Y. Su, Chem.–Eur. J., 2015, 21, 7418–7427 CrossRef CAS PubMed.
- A. Carné-Sánchez, J. Albalad, T. Grancha, I. Imaz, J. Juanhuix, P. Larpent, S. Furukawa and D. Maspoch, J. Am. Chem. Soc., 2019, 141, 4094–4102 CrossRef PubMed.
- H. Kim, M. Oh, D. Kim, J. Park, J. Seong, S. K. Kwak and M. S. Lah, Chem. Commun., 2015, 51, 3678–3681 RSC.
- J. Lee, J. H. Kwak and W. Choe, Nat. Commun., 2017, 8, 14070 CrossRef CAS.
- J.-R. Li, A. A. Yakovenko, W. Lu, D. J. Timmons, W. Zhuang, D. Yuan and H.-C. Zhou, J. Am. Chem. Soc., 2010, 132, 17599–17610 CrossRef CAS PubMed.
- J.-R. Li and H.-C. Zhou, Nat. Chem., 2010, 2, 893–898 CrossRef CAS PubMed.
- Z. Niu, S. Fang, X. Liu, J.-G. Ma, S. Ma and P. Cheng, J. Am. Chem. Soc., 2015, 137, 14873–14876 CrossRef CAS PubMed.
- M. A. S. Aquino, Coord. Chem. Rev., 1998, 170, 141–202 CrossRef CAS.
- M. D. Young, Q. Zhang and H.-C. Zhou, Inorg. Chim. Acta, 2015, 424, 216–220 CrossRef CAS.
- S. Furukawa and S. Kitagawa, Inorg. Chem., 2004, 43, 6464–6472 CrossRef CAS PubMed.
- F. A. Urbanos, M. C. Barral and R. Jiménez-Aparicio, Polyhedron, 1988, 7, 2597–2600 CrossRef CAS.
- M. H. Chisholm, G. Christou, K. Folting, J. C. Huffman, C. A. James, J. A. Samuels, J. L. Wesemann and W. H. Woodruff, Inorg. Chem., 1996, 35, 3643–3658 CrossRef CAS.
- N. K. Noel, M. Congiu, A. J. Ramadan, S. Fearn, D. P. McMeekin, J. B. Patel, M. B. Johnston, B. Wenger and H. J. Snaith, Joule, 2017, 1, 328–343 CrossRef CAS.
- Z. Wang, G. A. Craig, A. Legrand, F. Haase, S. Minami, K. Urayama and S. Furukawa, Chem.–Asian J., 2021, 16, 1092–1100 CrossRef CAS PubMed.
- G. R. Lorzing, A. J. Gosselin, B. A. Trump, A. H. P. York, A. Sturluson, C. A. Rowland, G. P. A. Yap, C. M. Brown, C. M. Simon and E. D. Bloch, J. Am. Chem. Soc., 2019, 141, 12128–12138 CrossRef CAS PubMed.
- J. W. M. Osterrieth, J. Rampersad, D. Madden, N. Rampal, L. Skoric, B. Connolly, M. D. Allendorf, V. Stavila, J. L. Snider, R. Ameloot, J. Marreiros, C. Ania, D. Azevedo, E. Vilarrasa-Garcia, B. F. Santos, X.-H. Bu, Z. Chang, H. Bunzen, N. R. Champness, S. L. Griffin, B. Chen, R.-B. Lin, B. Coasne, S. Cohen, J. C. Moreton, Y. J. Colón, L. Chen, R. Clowes, F.-X. Coudert, Y. Cui, B. Hou, D. M. D'Alessandro, P. W. Doheny, M. Dincă, C. Sun, C. Doonan, M. T. Huxley, J. D. Evans, P. Falcaro, R. Ricco, O. Farha, K. B. Idrees, T. Islamoglu, P. Feng, H. Yang, R. S. Forgan, D. Bara, S. Furukawa, E. Sanchez, J. Gascon, S. Telalović, S. K. Ghosh, S. Mukherjee, M. R. Hill, M. M. Sadiq, P. Horcajada, P. Salcedo-Abraira, K. Kaneko, R. Kukobat, J. Kenvin, S. Keskin, S. Kitagawa, K.-i. Otake, R. P. Lively, S. J. A. DeWitt, P. Llewellyn, B. V. Lotsch, S. T. Emmerling, A. M. Pütz, C. Martí-Gastaldo, N. M. Padial, J. García-Martínez, N. Linares, D. Maspoch, J. A. Suárez del Pino, P. Moghadam, R. Oktavian, R. E. Morris, P. S. Wheatley, J. Navarro, C. Petit, D. Danaci, M. J. Rosseinsky, A. P. Katsoulidis, M. Schröder, X. Han, S. Yang, C. Serre, G. Mouchaham, D. S. Sholl, R. Thyagarajan, D. Siderius, R. Q. Snurr, R. B. Goncalves, S. Telfer, S. J. Lee, V. P. Ting, J. L. Rowlandson, T. Uemura, T. Iiyuka, M. A. van der Veen, D. Rega, V. Van Speybroeck, S. M. J. Rogge, A. Lamaire, K. S. Walton, L. W. Bingel, S. Wuttke, J. Andreo, O. Yaghi, B. Zhang, C. T. Yavuz, T. S. Nguyen, F. Zamora, C. Montoro, H. Zhou, A. Kirchon and D. Fairen-Jimenez, Adv. Mater., 2022, 34, 2201502 CrossRef CAS PubMed.
- G. R. Lorzing, B. A. Trump, C. M. Brown and E. D. Bloch, Chem. Mater., 2017, 29, 8583–8587 CrossRef CAS.
- A. J. Gosselin, C. A. Rowland, K. P. Balto, G. P. A. Yap and E. D. Bloch, Inorg. Chem., 2018, 57, 11847–11850 CrossRef CAS PubMed.
- T.-H. Chen, L. Wang, J. V. Trueblood, V. H. Grassian and S. M. Cohen, J. Am. Chem. Soc., 2016, 138, 9646–9654 CrossRef CAS PubMed.
- J. Troyano, S. Horike and S. Furukawa, J. Am. Chem. Soc., 2022, 144, 19475–19484 CrossRef CAS PubMed.
- E. Van Caemelbecke, T. Phan, W. R. Osterloh and K. M. Kadish, Coord. Chem. Rev., 2021, 434, 213706 CrossRef CAS.
- M. C. Barral, R. González-Prieto, R. Jiménez-Aparicio, J. L. Priego, M. R. Torres and F. A. Urbanos, Inorg. Chim. Acta, 2005, 358, 217–221 CrossRef CAS.
- H. Miyasaka, T. Izawa, N. Takahashi, M. Yamashita and K. R. Dunbar, J. Am. Chem. Soc., 2006, 128, 11358–11359 CrossRef CAS PubMed.
- H. Miyasaka, N. Motokawa, S. Matsunaga, M. Yamashita, K. Sugimoto, T. Mori, N. Toyota and K. R. Dunbar, J. Am. Chem. Soc., 2010, 132, 1532–1544 CrossRef CAS PubMed.
- H. Miyasaka, Acc. Chem. Res., 2013, 46, 248–257 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2263447. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc02888g |
| This journal is © The Royal Society of Chemistry 2023 |