
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA Pd-based plasmonic photocatalyst for nitrogen fixation through an antenna–reactor mechanism†
Yuanyuan
Yang‡
a,
Henglei
Jia‡
 *a,
Sihua
Su
c,
Yidi
Zhang
a,
Mengxuan
Zhao
a,
Jingzhao
Li
a,
Qifeng
Ruan
*c and
Chun-yang
Zhang
*ab
*a,
Sihua
Su
c,
Yidi
Zhang
a,
Mengxuan
Zhao
a,
Jingzhao
Li
a,
Qifeng
Ruan
*c and
Chun-yang
Zhang
*ab
aCollege of Chemistry, Chemical Engineering and Materials Science, Shandong Normal University, Jinan 250014, China. E-mail: hljia@sdnu.edu.cn; cyzhang@sdnu.edu.cn
bSchool of Chemistry and Chemical Engineering, Southeast University, Nanjing 211189, China. E-mail: zhangcy@seu.edu.cn
cMinistry of Industry and Information Technology Key Lab of Micro-Nano Optoelectronic Information Systems, Guangdong Provincial Key Laboratory of Semiconductor Optoelectronic Materials and Intelligent Photonic Systems, Harbin Institute of Technology, Shenzhen 518055, China. E-mail: ruanqifeng@hit.edu.cn
First published on 5th September 2023
Abstract
Plasmonic metal nanocrystals (e.g., Au, Ag, and Cu) hold great promise for driving photocatalytic reactions, but little is known about the plasmonic properties of Pd nanocrystals. Herein, we constructed a plasmonic Pd/Ru antenna–reactor photocatalyst through the controllable growth of a Ru nanoarray ‘reactor’ on a Pd nano-octahedron ‘antenna’ and demonstrated a plasmonic Pd-driven N2 photofixation process. The plasmonic properties of Pd nano-octahedrons were verified using finite-difference time-domain (FDTD) simulations and refractive index sensitivity tests in water–glycerol mixtures. Notably, the constructed plasmonic antenna–reactor nanostructures exhibited superior photocatalytic activities during N2 photofixation, with a maximum ammonia production rate of 117.5 ± 15.0 μmol g−1 h−1 under visible and near-infrared (NIR) light illumination. The mechanism can be attributed to the ability of the plasmonic Pd nanoantennas to harvest light to generate abundant hot electrons and the Ru nanoreactors to provide active sites for adsorption and activation of N2. This work paves the way for the development of Pd-based plasmonic photocatalysts for efficient N2 photofixation and sheds new light on the optimal design and construction of antenna–reactor nanostructures.
Introduction
Artificial ammonia (NH3) synthesis from the reduction of nitrogen (N2) is crucial for agricultural and industrial production, because NH3 is an important building block for fertilizer and living forms.1–3 NH3 has attracted great interest as a hydrogen storage material because of its high hydrogen capacity (17.6 wt%).4 Although there are abundant N2 resources (approximately 78%) in the atmosphere, it is difficult to efficiently utilize N2 because a quantity of energy is required to overcome the large bond energy (approximately 941 kJ mol−1) of a stable N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N triple bond.5,6 At present, the industrial production of NH3 mainly adopts the classical Haber–Bosch process under high temperature and high pressure in the presence of highly pure N2 and H2, which is not only costly and requires the consumption of energy,7,8 but also leads to the emission of the greenhouse gas CO2 for acquiring H2 from the steam reformation of natural gas.9 Alternatively, solar-driven chemical reactions represent a green and sustainable strategy to address the energy issues.10–12 Effective NH3 synthesis could occur if a green and sustainable N2 photofixation process could be developed with renewable solar energy and H2O as the energy and hydrogen source, respectively.13–16 Among the various metal photocatalysts, ruthenium (Ru) with the appropriate N2 adsorption energy is a satisfactory candidate for N2 fixation, but its weak light-harvesting capability and lack of hot electrons to split the NN triple bond limit its applications in photocatalysis.17–19 Therefore, the design and construction of highly efficient Ru-based photocatalysts are promising but quite challenging.
N triple bond.5,6 At present, the industrial production of NH3 mainly adopts the classical Haber–Bosch process under high temperature and high pressure in the presence of highly pure N2 and H2, which is not only costly and requires the consumption of energy,7,8 but also leads to the emission of the greenhouse gas CO2 for acquiring H2 from the steam reformation of natural gas.9 Alternatively, solar-driven chemical reactions represent a green and sustainable strategy to address the energy issues.10–12 Effective NH3 synthesis could occur if a green and sustainable N2 photofixation process could be developed with renewable solar energy and H2O as the energy and hydrogen source, respectively.13–16 Among the various metal photocatalysts, ruthenium (Ru) with the appropriate N2 adsorption energy is a satisfactory candidate for N2 fixation, but its weak light-harvesting capability and lack of hot electrons to split the NN triple bond limit its applications in photocatalysis.17–19 Therefore, the design and construction of highly efficient Ru-based photocatalysts are promising but quite challenging.
Plasmonic metal nanocrystals (NCs) with localized surface plasmon resonance (LSPR) properties can generate highly energetic hot electrons to reduce the adsorbed molecules and effectively promote the conversion from light energy to chemical energy.20,21 Although substantial endeavours have been devoted to the exploration of Au, Ag, Cu, Al, and Fe as plasmonic metal photocatalysts, the LSPR properties of Pd NCs have rarely been reported.22–25 Interestingly, Pd not only provides sufficient active sites for molecular adsorption and activation, but it also harvests light as the plasmonic core to generate hot electrons through the manipulation of the spatial architecture.26–28 Consequently, the combination of a plasmonic Pd NC ‘antenna’ with a catalytically active Ru ‘reactor’ to fabricate antenna–reactor nanostructures offers the potential for driving N2 photofixation.29–33 Specifically, Pd nano-octahedrons with large cross-sections for light harvesting across a broad spectral range are good candidates as plasmonic nanoantennas.34 Moreover, there has been great interest in Ru nanoarrays due to their larger surface area and a greater amount of active sites as compared to dense nanostructures.35 Despite extensive efforts being devoted to the investigation of Au-, Ag-, and Cu-based antenna–reactor nanostructures in previous works, the fabrication of Pd-based antenna–reactor nanostructures remains a great challenge, but is extremely desirable.
In this work, we demonstrate a facile seed-mediated strategy for the controllable growth of Ru nanoarrays with different densities on a plasmonic Pd nano-octahedron core by precisely manipulating the growth kinetics. Theoretical and experimental studies were conducted to investigate the plasmonic properties of Pd nanoantennas. The plasmonic Pd/Ru antenna–reactor nanostructures exhibited excellent photocatalytic N2 photofixation performance. In this nanostructure, plasmonic Pd nanoantennas harvest light to generate sufficient hot electrons, while the Ru nanoreactor provides active sites to adsorb/activate N2 molecules and subsequently reduce it with hot electrons from the Pd antenna. In addition, the photocatalytic performance of the Pd/Ru antenna–reactor nanostructures during N2 fixation was dependent upon the thickness of the Ru shells, with a maximum NH3 production rate of 117.5 ± 15.0 μmol g−1 h−1. The plasmon-driven N2 fixation mechanism on the Pd/Ru antenna–reactor photocatalyst was systematically investigated as well.
Results and discussion
Fig. 1 illustrates the synthesis process for the preparation of Pd octahedron/Ru array nanostructures. Pd nano-octahedrons were prepared by employing Pd nanocubes as the seed.36 For the overgrowth of Ru nanoarrays on Pd nano-octahedrons, ruthenium(III) acetylacetonate (Ru(acac)3) was reduced by benzyl alcohol, with polyvinylpyrrolidone (PVP) as the surfactant. Because there is a lattice mismatch between Pd and Ru,37 the deposition of Ru atoms on Pd nano-octahedrons prefers to occur in 3D island growth mode (Volmer–Weber mode), and Ru nanoarrays are subsequently formed on the Pd nano-octahedron surface.38,39 By changing the amount of the Ru precursor, the density and diameter of the Ru nanoarrays can be readily adjusted.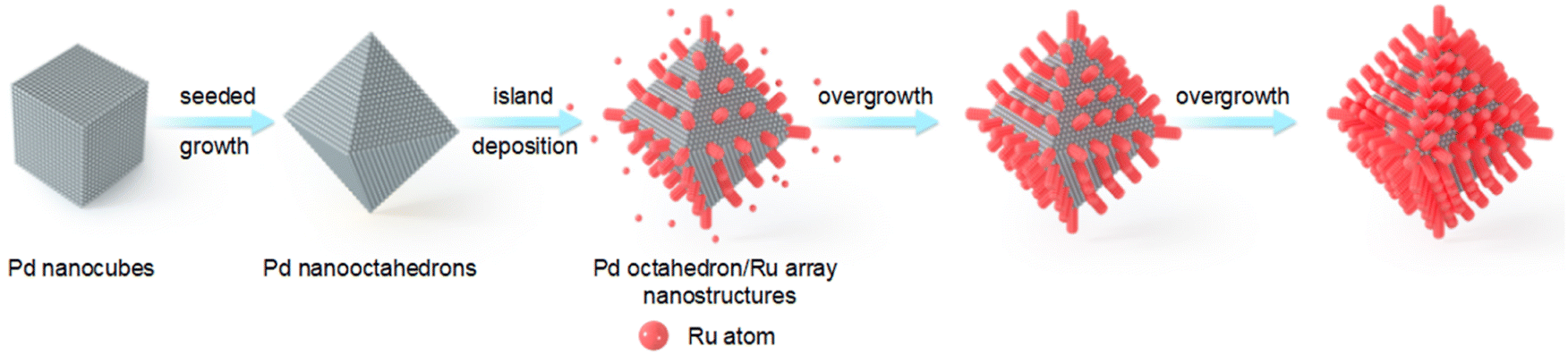 | ||
| Fig. 1 Schematic illustrating the synthesis process of the Pd octahedron/Ru array nanostructures. | ||
Pd nano-octahedrons with an average edge length of 43.4 ± 4.7 nm were employed as the seeds for the overgrowth of Ru nanoarrays (Fig. 2a). The reasons for their utilization as the seeds are as follows: (1) the Pd nano-octahedrons possess an obvious plasmon peak that redshifts to the visible light region after the overgrowth of Ru, and therefore, they are a satisfactory candidate as a visible-light photocatalyst; (2) the finite-difference time-domain (FDTD) simulation results suggest that the extinction values are dominantly contributed by light absorption when the plasmon peak of Pd nano-octahedrons is less than 400 nm. As shown in the transmission electron microscopy (TEM) image, Ru nanoarrays with a thickness of 5.9 ± 0.74 nm have been successfully grown on the surface of Pd nano-octahedrons (Fig. 2b). The spatial architecture of Pd nano-octahedrons (Fig. 2c) and Pd/Ru bimetallic NCs (Fig. 2d) was further verified using scanning electron microscopy (SEM) images. The SEM results confirmed that Pd NCs possess an octahedral structure, and their size and morphology distributions are notably homogeneous. After the overgrowth of Ru nanoarrays, the octahedral morphology remained unchanged. The plasmonic properties of Pd NCs were revealed with extinction spectra, and the presence of an extinction peak at approximately 400 nm was attributed to the LSPR of Pd nano-octahedrons (Fig. 2e).34 Because the LSPR wavelengths of plasmonic NCs are highly sensitive to the surrounding medium, an increase in the refractive index of the surrounding medium can induce a redshift of the plasmon resonance.40 The plasmon peak exhibited a redshift from 403 nm to 502 nm after the Ru nanoarray overgrowth, which was caused by the larger refractive index of Ru as compared to water. To further examine the structure and composition of the Pd octahedron/Ru array nanostructures, high-angle annular dark-field scanning transmission electron microscopy (HAADF STEM) and corresponding energy-dispersive X-ray (EDX) element mapping were carried out (Fig. 2f). The EDX elemental mapping results clearly demonstrate that elemental Pd with an octahedral shape is located inside, while elemental Ru is coated outside. This is consistent with the observation from the elemental profile results (Fig. S1†). The representative high-resolution TEM (HRTEM) image of a single Pd octahedron/Ru array nanostructure suggests that Ru nanoarrays perpendicularly grow on the surface of Pd nano-octahedrons with an array interval of 1.41 ± 0.3 nm (Fig. 2g). As observed in the HRTEM image (Fig. 2h), lattice fringes of 2.239 Å were assigned to face-centered cubic (fcc) Pd {111} facets, while lattice fringes of 2.204 Å corresponded to the fcc Ru {111} facets. The lattice mismatch between Pd and Ru led to the 3D island epitaxial growth of Ru nanoarrays.41 A Ru nanoarray shell with a spatially separated structure is more advantageous for charge separation than a dense shell, implying that it is an ideal photocatalyst for N2 fixation.42,43
 | ||
| Fig. 2 Pd octahedron/Ru array nanostructures. TEM images of (a) Pd nano-octahedrons and (b) Pd octahedron/Ru array nanostructures. SEM images of (c) Pd nano-octahedrons and (d) Pd octahedron/Ru array nanostructures. (e) Extinction spectra of Pd nano-octahedrons (blue) and Pd octahedron/Ru array nanostructures (orange). (f) HAADF-STEM image (top left) and the corresponding EDX maps (top right and bottom) of a single Pd octahedron/Ru array nanostructure. (g) HRTEM image of a single Pd octahedron/Ru array nanostructure. (h) HRTEM image at the Pd–Ru interface. | ||
The crystalline structure and chemical constituents of the Pd octahedron/Ru array nanostructures were further investigated with powder X-ray diffraction (PXRD) and X-ray photoelectron spectroscopy (XPS). There are only two groups of diffraction patterns that correspond to standard Pd and Ru peaks in the XRD spectrum (Fig. S2†), indicating that Pd and Ru exist in the metallic phase in Pd octahedron/Ru array nanostructures.44 In addition, the representative XPS survey spectrum of the Pd octahedron/Ru array nanostructures is in accordance with the PXRD results. The high-resolution Pd 3d XPS spectra display two peaks after in situ Ar+ ion sputtering for 40 s (Fig. S3b†), which were ascribed to the Pd 3d3/2 (340.5 eV) and Pd 3d5/2 (335.3 eV) of metallic Pd.45 The Ru 3d XPS spectra were fitted with three peaks, including two Ru0 peaks (Ru0 3d3/2 at 285.4 eV and Ru0 3d5/2 at 280.2 eV) and one Ru4+ peak (Fig. S3c†). The Ru4+ XPS peak originated from the inevitable occurrence of surface oxidation.46 The Ru4+/Ru0 ratio was calculated to be 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :11, which confirms that elemental Ru exists primarily in the Ru0 state. Moreover, the presence of Ru 3p1/2 and Ru 3p2/3 peaks further identifies the metallic Ru0 state (Fig. S3d†).47
:11, which confirms that elemental Ru exists primarily in the Ru0 state. Moreover, the presence of Ru 3p1/2 and Ru 3p2/3 peaks further identifies the metallic Ru0 state (Fig. S3d†).47
To gain a more complete understanding of the LSPR properties of Pd nano-octahedrons, theoretical calculations and experimental measurements were conducted. First, we calculated the extinction, scattering, and absorption cross sections of Pd nano-octahedrons with edge lengths of 30–80 nm using FDTD simulation (Fig. 3a–f). Fig. 3g shows the dependence of the extinction spectra on the edge length of the Pd nano-octahedrons. The simulated extinction cross-section increases and the longitudinal plasmon peak redshifts with increasing edge length of the Pd nano-octahedrons. A single LSPR peak can be clearly observed when the edge length is larger than approximately 40 nm, which is predominantly contributed by absorption rather than scattering (Fig. 3a–f).48 Despite some discrepancies between the calculated and measured plasmon peaks, the simulation results revealed the plasmonic properties of the Pd nano-octahedrons. The difference was attributed to the nonuniform morphology and wide size distribution of the prepared Pd nano-octahedrons.49 Next, the plasmonic properties of the Pd nano-octahedrons were further examined using a refractive index sensitivity test. Because the LSPR peaks are highly sensitive to the surrounding dielectric environment, the change in the refractive index of the surrounding environment will cause a shift in plasmon resonance peaks.50 In this work, Pd nano-octahedrons were redispersed into a series of water–glycerol mixture solutions of varying volume ratios to detect the refractive index sensitivity.51Fig. 3h displays the refractive index-dependence of the plasmon peaks in different mixture solutions. As the volume ratio of glycerol and the refractive index of the mixtures increased, the plasmon resonance peaks redshifted. Moreover, there is a linear dependence of the plasmon shift on the refractive index of the mixture (Fig. 3i). The slope of this line is 230 nm RIU−1, which indicates the excellent refractive index sensitivity of the Pd octahedron nanoparticles.52 Notably, the plasmon peak of the Pd nano-octahedrons in Fig. 3h is not consistent with the calculation result. The discrepancy may be ascribed to the difference in the size and morphology distribution between the theoretical calculation and the experimental results. The Pd nano-octahedrons used for the FDTD simulations were uniform in shape and size, but the yield of the obtained Pd nano-octahedrons was not 100% and the morphology was not perfectly octahedral, which led to a deviation between the theoretical and experimental results. To further confirm the LSPR properties of Pd nano-octahedrons, we performed surface photovoltage (SPV) spectroscopy. Under plasmon resonance excitation, the local electric field around the Pd nano-octahedrons was largely enhanced, inducing a surface voltage enhancement in the SPV spectrum. As shown in Fig. S4,† the SPV signal faithfully followed the surface plasmon resonance of the Pd nano-octahedrons, confirming the LSPR properties of the Pd nano-octahedrons. Above all, the theoretical and experimental results substantiated the plasmonic property of the Pd nano-octahedrons, suggesting that a Pd nano-octahedron sample is a satisfactory candidate as a nanoantenna for plasmon-driven photocatalysis. Pd nanocubes that were seeds for the preparation of Pd nano-octahedrons were also employed to fabricate Pd cube/Ru nanostructures (Fig. S5†). There were no plasmon resonance peaks in the visible and near-infrared (NIR) regions for Pd nanocubes or Pd cube/Ru nanostructures (Fig. S5c and d†), which may be caused by the small size (13.2 ± 1.2 nm) of the Pd nanocubes. Therefore, Pd nano-octahedrons were selected as plasmonic antennas for visible-/NIR-light photocatalysis.
 | ||
| Fig. 3 Plasmonic properties of Pd nano-octahedrons. (a–f) Simulated extinction (blue), absorption (green), and scattering (red) spectra of Pd nano-octahedrons with an edge length of (a) 80 nm, (b) 70 nm, (c) 60 nm, (d) 50 nm, (e) 40 nm, and (f) 30 nm. (g) Comparison of the simulated extinction spectra of Pd nano-octahedrons with different edge lengths. (h) Normalized extinction spectra of Pd nano-octahedrons dispersed in water–glycerol mixtures of varying compositions. The arrow indicates the direction of increasing volume percentage of glycerol. (i) Dependence of the plasmon shift on the refractive index of the liquid mixture. | ||
Precisely adjusting the thickness of the Ru nanoarray shell is important for its photocatalytic performance. The Pd octahedron/Ru array nanostructures with different shell thicknesses were obtained by increasing the concentration of the Ru(acac)3 precursor from 0.25 mg mL−1 to 2.5 mg mL−1 (Fig. 4 and S6†). The shell thickness displayed a linear dependence on the Ru(acac)3 concentration, with the minimum and maximum values being 3.4 ± 0.4 nm and 9.8 ± 0.9 nm, respectively (Fig. 4i). Ru nanoarrays with sparse spatial distribution were observed when the Ru(acac)3 concentration was less than 0.75 mg mL−1 (Fig. S6a and b†). In addition, the gap distance in the Ru nanoarrays can be controlled in the range of 0.9–2.5 nm by simply adjusting the amount of Ru precursor (Fig. S6†). When the concentration of Ru precursor is equal to or greater than 2.5 mg mL−1, small Ru nanoparticles are generated due to self-nucleation, which may be caused by the greater deposition rate rather than the diffusion rate (Fig. 4g).53
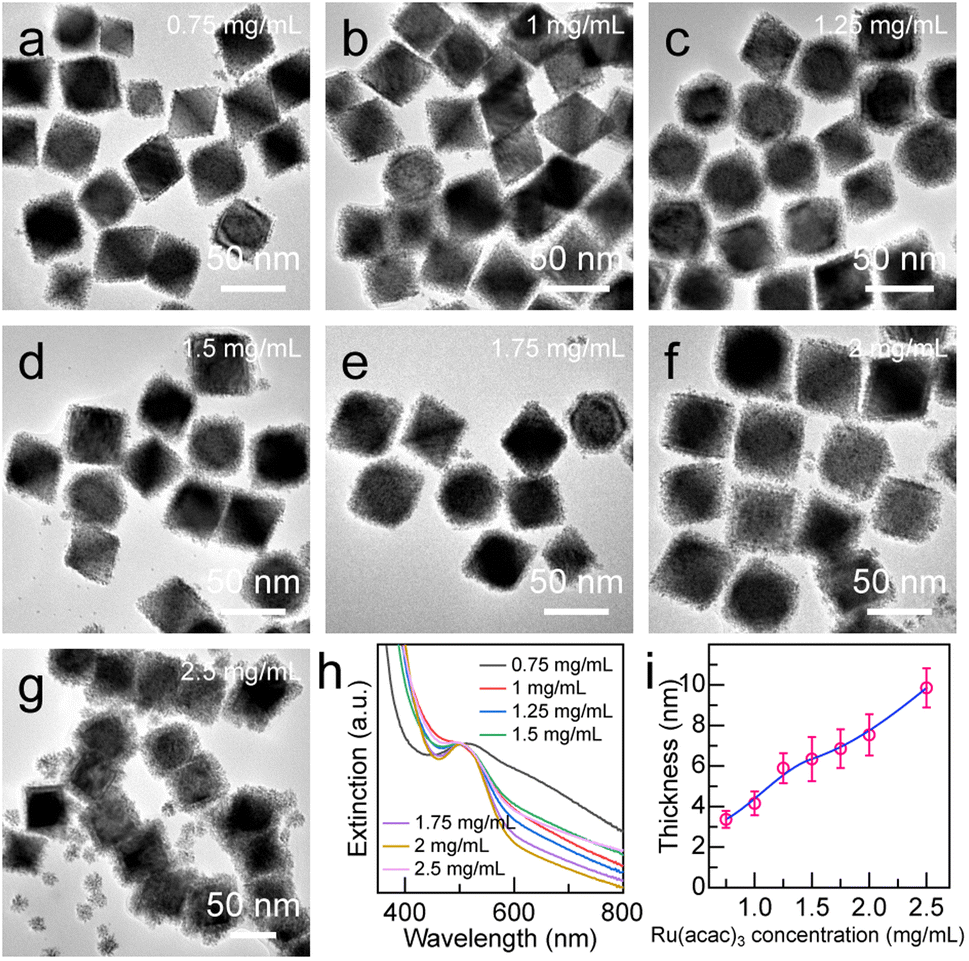 | ||
| Fig. 4 Effect of precursor concentration on Ru shell thickness. (a–g) Representative TEM images of the Pd octahedron/Ru array nanostructures produced at different Ru(acac)3 concentrations. (h) Extinction spectra of the Pd octahedron/Ru array nanostructures obtained when different Ru(acac)3 concentrations were employed for Ru shell growth. (i) Ru shell thickness as a function of Ru(acac)3 concentration. | ||
A few control experiments were conducted to investigate the growth behaviour, including precursor injection rate (Fig. S7†), reaction temperature (Fig. S8†), PVP molecular weight (Fig. S9†), amount of added PVP (Fig. S10†), and Ru precursor species (Fig. S11†). First, the precursor injection rate can affect the growth behaviour.54,55 When the injection rate of the Ru(acac)3 precursor was 2 mL h−1, a large amount of self-nucleated Ru nanoparticles was formed due to the rapid reduction of Ru precursor by benzyl alcohol (Fig. S7a†). When the injection rate of the Ru(acac)3 precursor was greater than 8 mL h−1, the Ru nanoparticles tended to grow on Pd seeds, and Pd octahedron/Ru array nanostructures were obtained (Fig. S7b and c†). Thus, a relatively moderate precursor injection rate is of great importance for the formation of Ru array nanostructures. Next, we investigated the effect of reaction temperature on Ru deposition. A layer of ultrathin Ru shells was deposited on the side edges of Pd nano-octahedrons when the reaction temperature was changed from 180 °C to 140 °C due to the lower reduction rate of the Ru precursor (Fig. S8b†). However, no Ru nanoarrays were observed once the reaction temperature was decreased to 100 °C due to the poor reduction capability of benzyl alcohol at this temperature (Fig. S8a†).56 Third, we investigated the effect of the molecular weight and the amount of PVP molecules added on the growth behaviour. A relatively large molecular weight and an appropriate addition of PVP are of great importance to the overgrowth of Ru nanoarrays on Pd nano-octahedrons (Fig. S9 and S10†). Fourth, when the Ru(acac)3 precursor was replaced with RuCl3, the products of the Pd octahedron/Ru array nanostructures were obtained, but the plasmon peak vanished (Fig. S11†). Finally, hollow Ru nanoshells were obtained by etching the Pd core from the Pd octahedron/Ru array nanostructures (Fig. S12†), indicating the excellent stability of Ru shells.57
The Pd octahedron/Ru array nanostructures with plasmonic Pd antennas for harvesting light and catalytically active Ru reactors for N2 adsorption/activation are satisfactory candidates as plasmonic antenna–reactor catalysts for N2 photofixation. To explore the photocatalytic performance of the Pd octahedron/Ru array nanostructures, N2 reduction experiments were conducted under visible and NIR light illumination (λ > 400 nm). In this study, methanol was selected as the hole scavenger. To investigate the dependence of N2 photofixation activity on Ru array thickness, six types of Pd octahedron/Ru array nanostructures with different array thicknesses (3.4 ± 0.4 nm (Fig. 4a), 4.2 ± 0.6 nm (Fig. 4b), 5.9 ± 0.7 nm (Fig. 4c), 6.4 ± 1.1 nm (Fig. 4d), 6.9 ± 1.3 nm (Fig. 4e), and 7.5 ± 1.0 nm (Fig. 4f)) were synthesized and employed as photocatalysts, named Pd/Ru3.4, Pd/Ru4.2, Pd/Ru5.9, Pd/Ru6.4, Pd/Ru6.9, and Pd/Ru7.5. For comparison, Pd nano-octahedrons (Fig. 2a), Ru NCs (Fig. S13†), Pd cube/Ru nanostructures (Fig. S5b†), and the mixture of Pd nano-octahedrons with Ru NCs were prepared as the photocatalysts. Notably, although most of the surfactants were removed by centrifugation, some residual PVP molecules remained on the catalyst surface to maintain the stability of the colloidal nanoparticles, which may affect the catalytic activity. Although the surfactant molecules can block the active sites and decrease the catalytic activity, the presence of some molecules on catalysts can enhance the catalytic activity and selectivity through the electron donation effect or geometric changes.58–60 In this work, PVP was employed as the surfactant for all the catalysts. Its influence on catalytic activity should be similar and was not taken into account. The catalyst amount and the Pd/Ru molar ratio in the Pd/Ru catalyst were determined by inductively coupled plasma-optical emission spectroscopy (ICP-OES) (Fig. S14 and Table S1†). The amount of NH3 production was quantitatively detected by the indophenol blue method.61 The linear calibration relationship between the standard NH4+ concentrations and the corresponding absorbance peak values is displayed in Fig. S15.†
As shown in Fig. 5a, the individual Pd nano-octahedrons or Ru NCs exhibit weak N2 photofixation activity due to the lack of catalysts for N2 adsorption or the weak light-harvesting capability. The photocatalytic N2 fixation activity on the Pd cube/Ru nanostructures was also weak because there was an absence of plasmonic properties for the Pd nanocubes. In contrast, the NH3 production rate for the Pd/Ru5.9 sample was 3.6-fold higher than that of the mixture sample after 2 h of illumination, indicating that the close coupling between the plasmonic antenna and active reactor is crucial for the antenna–reactor photocatalyst. Pd nano-octahedrons or Ru NCs independently contribute to the photocatalytic performance of the mixture sample. The N2 photofixation activity exhibited a volcano-shaped dependence on the array thickness of the Pd octahedron/Ru array nanostructures (Fig. 5b), with a maximum NH3 production rate of 117.5 ± 15.0 μmol g−1 h−1. A thin Ru shell leads to weak photocatalytic activity due to insufficient Ru reactors on the Pd nanoantennas. Contrarily, a Ru shell that is too thick with high-density Ru nanoarrays can cover the active sites on the plasmonic Pd nanoantennas, which hinders the transfer of hot holes and the entrance of hole scavengers, eventually resulting in charge carrier recombination and a decrease in photocatalytic performance. It was found that an appropriate Ru shell thickness of 5.9 ± 0.7 nm exhibits the most optimal photocatalytic N2 fixation activity. Notably, although the indophenol blue method has been widely applied for the quantitative detection of ammonia concentration, interference may occur due to many factors such as pH value, organic compounds, and impurities. The results will be more convincing if a cross-check of the ammonia concentration can be performed using two different quantitative methods.62–64 To confirm the N2 fixation activity, the generated ammonia concentrations were analyzed using an ion chromatography method (Fig. S16†). The NH3 production rates determined by the ion chromatography method were consistent with those obtained by the indophenol blue method. In addition, we confirmed the N2 fixation rates with a quantitative isotopically labelled 1H nuclear magnetic resonance (NMR) method (Fig. S17†). The N2 photofixation experiment was conducted for 6 h with 3 mg of catalyst, and ammonia concentrations were determined by an indophenol blue method and an NMR method. The values of the produced NH3 determined by the indophenol blue method were consistent with those obtained by the NMR method. Therefore, ion chromatography and NMR methods confirmed the N2 fixation activity of the catalysts.
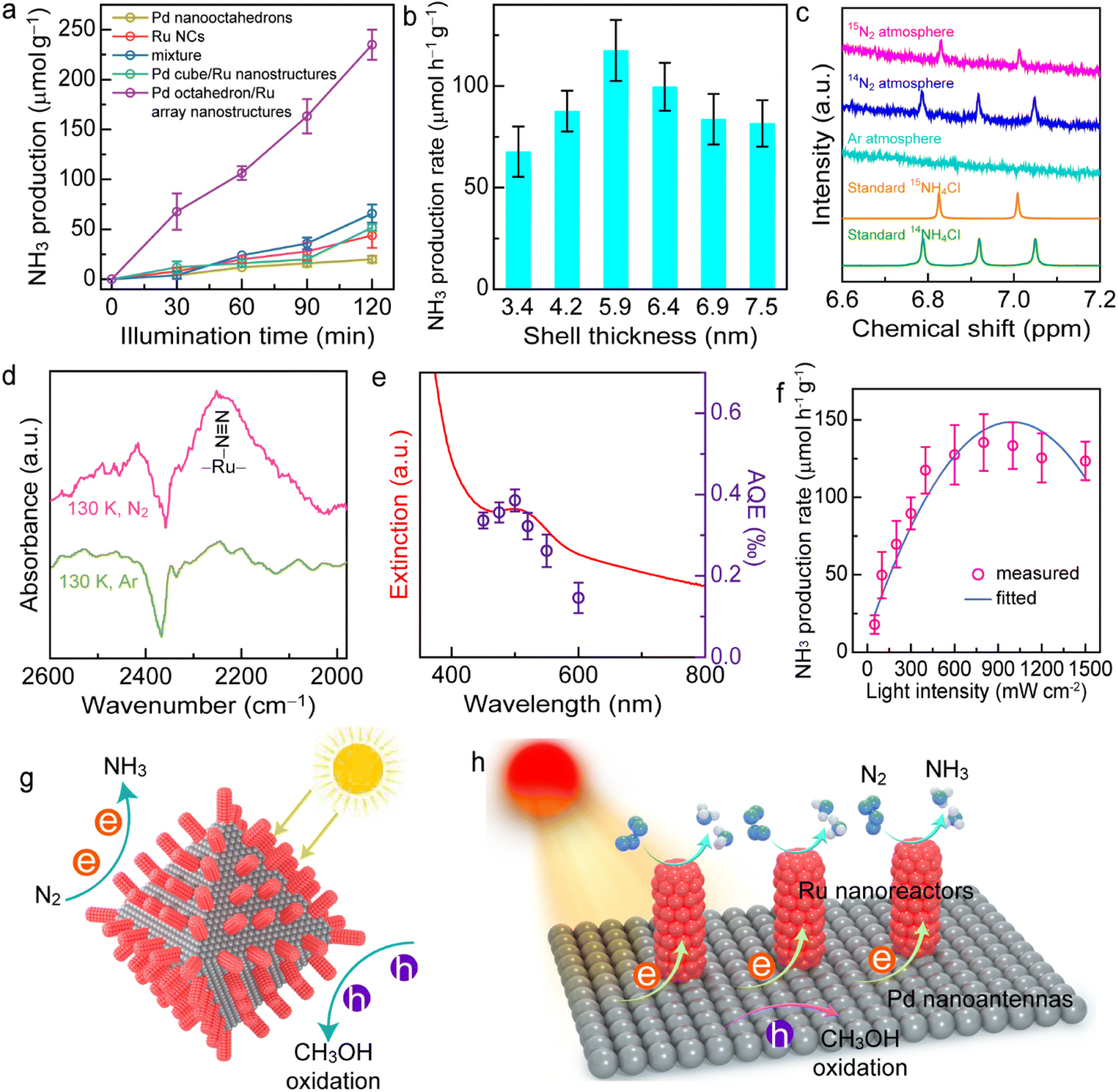 | ||
| Fig. 5 Plasmon-driven N2 photofixation. (a) NH3 production amounts as functions of illumination time using different photocatalysts. (b) Dependence of the N2 photofixation activity upon the Ru shell thickness. (c) 1H NMR spectra of the reaction solutions under Ar, 14N2, and 15N2 atmospheres. (d) LT-FTIR spectra of the Pd/Ru5.9 sample under N2 and Ar atmospheres. (e) Extinction spectrum (left axis) and wavelength-dependent apparent quantum efficiency of NH3 evolution (right axis) of the Pd/Ru5.9 sample. (f) Dependence of the photocatalytic NH3 production rate upon the light intensity. (g) Schematics illustrating N2 photofixation on Pd octahedron/Ru array nanostructures. (h) Mechanism of N2 photofixation on the Pd octahedron/Ru array nanostructure. e, hot electron; h, hot hole. | ||
To investigate the origin of produced NH3, a series of control experiments under different conditions was carried out (Fig. S18†). No NH3 can be detected in the absence of catalysts, light irradiation, or N2 atmosphere, which suggests that NH3 production occurred from photocatalytic N2 fixation. The photocatalytic activity of the Pd octahedron/Ru array nanostructures decreased to 29.7 μmol g−1 h−1 in the absence of a sacrificial agent, revealing that the addition of hole scavengers increased the charge separation efficiency.61 To investigate the effect of hole sacrificial reagents, we employed four types of sacrificial reagents: ethanol, triethanolamine (TEOA), triethylamine (TEA), and Na2SO3. Notably, the presence of TEA in the reaction system led to the aggregation of catalyst. Among the different types of hole scavengers, CH3OH exhibited the strongest N2 fixation performance (Fig. S19†). Thus, CH3OH was selected as the hole sacrificial reagent in this work. To further confirm the nitrogen source for NH3 production, we performed photocatalytic N2 reduction experiments under Ar, 14N2, and 15N2 atmospheres and analyzed the reaction solution using 1H NMR spectroscopy (Fig. 5c and S20†). As displayed in Fig. 5c, it is difficult to observe 14NH4+ under an Ar atmosphere, suggesting that the reduction of possible 14N2 in our system exerted no apparent influence on the NMR peaks. The double peaks associated with 15NH4+ were clearly observed in the 1H NMR spectrum under an 15N2 atmosphere, while three peaks attributed to 14NH4+ were detected under an 14N2 atmosphere, which suggests that the nitrogen source for NH3 production was from N2. In addition, the byproduct N2H4 was not detected in the typical photocatalytic reaction using a spectrophotometric method, suggesting the high selectivity of photocatalytic N2 fixation (Fig. S21†).65 H2 or CO2 may also be generated through either the reduction of water by hot electrons or the oxidation of methanol by hot holes. To investigate whether there is H2 or CO2 evolution in this reaction process, we conducted N2 photofixation experiments in a gastight glass reactor. No H2 was detected by gas chromatography after a 2 h reaction (Fig. S22a†), suggesting that H2 evolution is unfavourable under an N2 atmosphere. However, CO2 was detected in this photocatalytic system, and the amount of CO2 increased over time (Fig. S22b†). The evolution of CO2 originated from the oxidation half-reaction driven by hot holes, which further confirmed that NH3 was from the reduction half-reaction driven by hot electrons. Because an off-line gas chromatograph was employed for the detection of gaseous products, the air may affect this detection. Thus, a CO2 signal was detected at 0 h. To determine the level of recyclability of the Pd octahedron/Ru array nanostructures, the photocatalytic reaction was conducted in three successive cycles. Although there was a 10.6% decrease in the photocatalytic activity after three cycles, the catalyst maintained satisfactory recyclability (Fig. S23†). The decrease in the photocatalytic activity may be caused by the loss of catalyst during centrifugation. Moreover, the stability of the Pd octahedron/Ru array nanostructures was examined by performing TEM imaging, and XRD and XPS measurement of the catalyst after 2 h of N2 photofixation (Fig. S24†). The morphology, crystalline nature, and chemical states of the catalyst remained unchanged, confirming the excellent stability of the photocatalysts.
The excellent photocatalytic performance of the Pd octahedron/Ru array nanostructures was mainly contributed by the antenna–reactor mechanism. First, we investigated the active sites for N2 adsorption using theoretical and experimental studies. Density functional theory (DFT) calculations showed that the adsorption energy of N2 on Ru (−0.534 eV) was more negative than that on Pd (0.145 eV) (Fig. S25†), which revealed that the activation sites for N2 fixation were on the Ru nanoarrays. To gain a deeper insight into the N2 adsorption, we conducted N2 temperature-programmed desorption (TPD) (Fig. S26†) and low-temperature Fourier-transform infrared (LT-FTIR) (Fig. 5d) spectroscopy measurements. Two peaks appeared in the N2 TPD spectrum, with one peak located at 334 °C that was attributed to physisorption and the other peak at 498 °C that originated from N2 chemisorption, revealing the presence of active sites on the Pd octahedron/Ru array nanostructures for N2 adsorption. In addition, a vibration peak located at 2252 cm−1 in the LT-FTIR spectra was detected under an N2 atmosphere, suggesting that N2 molecules adsorb on the Ru catalyst via an end-on configuration.18 Next, we examined the plasmon-driven N2 photofixation process. To reveal the contribution of the photothermal effect upon the N2 fixation, we carried out N2 reduction experiments in water baths set at different temperatures in the dark (Fig. S27†). The results showed that the effect of photothermal heating on the photocatalytic activity was small, although it can hardly be ruled out. To further verify the plasmon-driven N2 fixation process, we calculated the apparent quantum efficiency (AQE) by performing N2 fixation experiments under different levels of monochromatic light illumination, and the action spectrum was therefore acquired (Fig. 5e). The trend of AQE values was in accordance with the absorption spectrum, which definitely confirmed that the photocatalytic N2 fixation was driven by the plasmon resonance of the Pd nano-octahedrons.61 In addition, there was a nearly linear dependence of the NH3 production rate on the Pd octahedron/Ru array nanostructures when the light intensity was less than 800 mW cm−2 (Fig. 5f). Further increase of the light intensity caused a decrease in photocatalytic activity, with the maximum NH3 production rate at 800 mW cm−2. The linear relationship between activity and light intensity suggested that the N2 photofixation was driven by plasmonic hot electrons. On the basis of the above results, we proposed a possible mechanism for N2 photofixation on Pd octahedron/Ru array nanostructures (Fig. 5g and h). In the nanostructures, Ru reactors afford active sites to adsorb and activate N2 molecules, while Pd antennas interact with light and generate hot electrons for N2 reduction. The hot holes are consumed using hole scavengers (CH3OH) to close the photocatalytic cycle. The Ru array nanostructures also facilitated charge separation and promoted the photocatalytic activity.
Conclusions
We constructed a novel Pd-based antenna–reactor plasmonic photocatalyst through the overgrowth of Ru nanoarrays on Pd nano-octahedrons. The plasmonic properties of the Pd nanoantennas were systematically investigated using theoretical and experimental studies. The obtained Pd octahedron/Ru array nanostructures exhibited an excellent photocatalytic N2 fixation performance. The mechanism may be attributed to the fact that Ru reactors adsorb and activate N2 molecules and that Pd octahedron antennas capture light and effectively generate energetic hot electrons for N2 reduction. This work provides a new strategy for the construction of plasmonic Pd-based antenna–reactor nanostructures for photocatalysis and opens a new avenue to explore potential plasmonic metal photocatalysts.Data availability
The data that support the findings of this study are available in the main text and the ESI.†Author contributions
Y. Y. Yang and H. L. Jia contributed equally to this work. H. L. Jia and C.-y. Zhang conceived and supervised the project, and wrote the paper. Y. Y. Yang performed the experiments. S. H. Su and Q. F. Ruan carried out the FDTD simulations and contributed to the data interpretation. Y. D. Zhang, M. X. Zhao, and J. Z. Li assisted with the photocatalytic experiments and participated in discussions. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 21735003), the Natural Science Foundation of Shandong Province (No. ZR2020MB040), and the Natural Science Foundation of Guangdong Province (No. 2023A1515012912).Notes and references
- B. H. R. Suryanto, K. Matuszek, J. Choi, R. Y. Hodgetts, H.-L. Du, J. M. Bakker, C. S. M. Kang, P. V. Cherepanov, A. N. Simonov and D. R. MacFarlane, Science, 2021, 372, 1187–1191 CrossRef CAS PubMed.
- Y. Ashida, K. Arashiba, K. Nakajima and Y. Nishibayashi, Nature, 2019, 568, 536–540 CrossRef CAS PubMed.
- L. C. Seefeldt, Z.-Y. Yang, D. A. Lukoyanov, D. F. Harris, D. R. Dean, S. Raugei and B. H. Hoffman, Chem. Rev., 2020, 120, 5082–5106 CrossRef CAS PubMed.
- J. P. Guo and P. Chen, Chem, 2017, 3, 709–712 CAS.
- J. H. Yang, Y. Z. Guo, W. Z. Lu, R. B. Jiang and J. F. Wang, Adv. Mater., 2018, 30, 1802227 CrossRef PubMed.
- J. Y. Li, R. M. Chen, J. L. Wang, Y. Zhou, G. D. Yang and F. Dong, Nat. Commun., 2022, 13, 1098 CrossRef CAS PubMed.
- S. S. Shang, W. Xiong, C. Yang, B. Johannessen, R. G. Liu, H.-Y. Hsu, Q. F. Gu, M. K. H. Leung and J. Shang, ACS Nano, 2021, 15, 9670–9678 CrossRef CAS PubMed.
- W. S. Huang, L.-Y. Peng, J. Y. Zhang, C. R. Liu, G. Y. Song, J.-H. Su, W.-H. Fang, G. L. Cui and S. W. Hu, J. Am. Chem. Soc., 2023, 145, 811–821 CrossRef CAS PubMed.
- S.-L. Meng, X.-B. Li, C.-H. Tung and L.-Z. Wu, Chem, 2021, 7, 1431–1450 CAS.
- H. W. Lin, S. Q. Luo, H. B. Zhang and J. H. Ye, Joule, 2022, 6, 294–314 CrossRef CAS.
- W. H. Huang, C. Y. Su, C. Zhu, T. T. Bo, S. W. Zuo, W. Zhou, Y. F. Ren, Y. N. Zhang, J. Zhang, M. Rueping and H. B. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202304634 CrossRef CAS PubMed.
- W. H. Huang, T. T. Bo, S. W. Zuo, Y. Z. Wang, J. M. Chen, S. Ould-Chikh, Y. Li, W. Zhou, J. Zhang and H. B. Zhang, SusMat, 2022, 2, 466–475 CrossRef CAS.
- H. Hirakawa, M. Hashimoto, Y. Shiraishi and T. Hirai, J. Am. Chem. Soc., 2017, 139, 10929–10936 CrossRef CAS PubMed.
- N. Zhang, A. Jalil, D. X. Wu, S. M. Chen, Y. F. Liu, C. Gao, W. Ye, Z. M. Qi, H. X. Ju, C. M. Wang, X. J. Wu, L. Song, J. F. Zhu and Y. J. Xiong, J. Am. Chem. Soc., 2018, 140, 9434–9443 CrossRef CAS PubMed.
- H. L. Jia, A. X. Du, H. Zhang, J. H. Yang, R. B. Jiang, J. F. Wang and C.-y. Zhang, J. Am. Chem. Soc., 2019, 141, 5083–5086 CrossRef CAS PubMed.
- H. L. Jia, M. X. Zhao, A. X. Du, Y. R. Dou and C.-y. Zhang, Chem. Sci., 2022, 13, 13060–13067 RSC.
- M. Kitano, Y. Inoue, Y. Yamazaki, F. Hayashi, S. Kanbara, S. Matsuishi, T. Yokoyama, S.-W. Kim, M. Hara and H. Hosono, Nat. Chem., 2012, 4, 934–940 CrossRef CAS PubMed.
- C. Y. Hu, X. Chen, J. B. Jin, Y. Han, S. M. Chen, H. X. Ju, J. Cai, Y. R. Qiu, C. Gao, C. M. Wang, Z. M. Qi, R. Long, L. Song, Z. Liu and Y. J. Xiong, J. Am. Chem. Soc., 2019, 141, 7807–7814 CrossRef CAS PubMed.
- H. L. Jia, Y. Y. Yang, Y. R. Dou, F. Li, M. X. Zhao and C.-y. Zhang, Chem. Commun., 2022, 58, 1013–1016 RSC.
- S. Linic, P. Christopher and D. B. Ingram, Nat. Mater., 2011, 10, 911–921 CrossRef CAS PubMed.
- S. S. Li, H. Huang, L. Shao and J. F. Wang, ACS Nano, 2021, 15, 10759–10768 CrossRef CAS PubMed.
- H. L. Jia, F. Li, T. H. Chow, X. Y. Liu, H. Zhang, Y. Lu, J. F. Wang and C.-y. Zhang, Nano Lett., 2022, 22, 7268–7274 CrossRef CAS PubMed.
- R. C. Elias and S. Linic, J. Am. Chem. Soc., 2022, 144, 19990–19998 CrossRef CAS PubMed.
- L. T. Quynh, C.-W. Cheng, C.-T. Huang, S. S. Raja, R. Mishra, M.-J. Yu, Y.-J. Lu and S. Gwo, ACS Nano, 2022, 16, 5975–5983 CrossRef CAS PubMed.
- H. B. Zhang, T. Wang, J. J. Wang, H. M. Liu, T. D. Dao, M. Li, G. G. Liu, X. G. Meng, K. Chang, L. Shi, T. Nagao and J. H. Ye, Adv. Mater., 2016, 28, 3703–3710 CrossRef CAS PubMed.
- J. Guo, Y. Zhang, L. Shi, Y. F. Zhu, M. F. Mideksa, K. Hou, W. S. Zhao, D. W. Wang, M. T. Zhao, X. F. Zhang, J. W. Lv, J. Q. Zhang, X. L. Wang and Z. Y. Tang, J. Am. Chem. Soc., 2017, 139, 17964–17972 CrossRef CAS PubMed.
- C. Y. Hu, K. Q. Lin, X. L. Wang, S. J. Liu, J. Yi, Y. Tian, B. H. Wu, G. X. Chen, H. Y. Yang, Y. Dai, H. Li and N. F. Zheng, J. Am. Chem. Soc., 2014, 136, 12856–12859 CrossRef CAS PubMed.
- F. A. A. Nugroho, P. Bai, I. Darmadi, G. W. Castellanos, J. Fritzsche, C. Langhammer, J. G. Rivas and A. Baldi, Nat. Commun., 2022, 13, 5737 CrossRef CAS PubMed.
- D. F. Swearer, R. K. Leary, R. Newell, S. Yazdi, H. Robatjazi, Y. Zhang, D. Renard, P. Nordlander, P. A. Midgley, N. J. Halas and E. Ringe, ACS Nano, 2017, 11, 10281–10288 CrossRef CAS PubMed.
- Y. G. Yuan, L. N. Zhou, H. Robatjazi, J. W. L. Bao, J. Y. Zhou, A. Bayles, L. Yuan, M. H. Lou, M. H. Lou, S. Khatiwada, E. A. Carter, P. Nordlander and N. J. Halas, Science, 2022, 378, 889–893 CrossRef CAS PubMed.
- L. Yuan, J. Y. Zhou, M. Zhang, X. L. Wen, J. M. P. Martirez, H. Robatjazi, L. N. Zhou, E. A. Carter, P. Nordlander and N. J. Halas, ACS Nano, 2022, 16, 17365–17375 CrossRef CAS PubMed.
- K. Sytwu, M. Vadai, F. Hayee, D. K. Angell, A. Dai, J. Dixon and J. A. Dionne, Science, 2021, 371, 280–283 CrossRef CAS PubMed.
- H. L. Jia, F. Li, Y. Y. Yang, M. X. Zhao, J. Z. Li and C.-y. Zhang, Chem. Sci., 2023, 14, 5656–5664 RSC.
- R. Long, Z. L. Rao, K. K. Mao, Y. Li, C. Zhang, Q. L. Liu, C. M. Wang, Z.-Y. Li, X. J. Wu and Y. J. Xiong, Angew. Chem., Int. Ed., 2015, 54, 2425–2430 CrossRef CAS PubMed.
- L. Q. Tian, Q. Xin, C. Zhao, G. C. Xie, M. Z. Akram, W. R. Wang, R. P. Ma, X. R. Jia, B. D. Guo and J. R. Gong, Small, 2021, 17, 2006530 CrossRef CAS PubMed.
- M. S. Jin, H. Zhang, Z. X. Xie and Y. N. Xia, Energy Environ. Sci., 2012, 5, 6352–6357 RSC.
- Y. C. Yao, D. S. He, Y. Lin, X. Q. Feng, X. Wang, P. Q. Yin, X. Hong, G. Zhou, Y. Wu and Y. D. Li, Angew. Chem., Int. Ed., 2016, 55, 5501–5505 CrossRef CAS PubMed.
- J. Liu and J. T. Zhang, Chem. Rev., 2020, 120, 2123–2170 CrossRef CAS PubMed.
- H. L. Jia, Y. Y. Yang, T. H. Chow, H. Zhang, X. Y. Liu, J. F. Wang and C.-y. Zhang, Adv. Funct. Mater., 2021, 31, 2101255 CrossRef CAS.
- H. J. Chen, L. Shao, Q. Li and J. F. Wang, Chem. Soc. Rev., 2013, 42, 2679–2724 RSC.
- A. Janssen, Z. H. Lyu, M. Figueras-Valls, H.-Y. Chao, Y. F. Shi, V. Pawlik, M. F. Chi, M. Mavrikakis and Y. N. Xia, Nano Lett., 2022, 22, 3591–3597 CrossRef CAS PubMed.
- Z. B. Luo, C. C. Li, S. S. Liu, T. Wang and J. L. Gong, Chem. Sci., 2017, 8, 91–100 RSC.
- C. W. Liu, Z. L. Qiu, W. L. Meng, J. W. Chen, J. J. Qi, C. Dong and M. T. Wang, Nano Energy, 2015, 12, 59–68 CrossRef CAS.
- K. Kusada, H. Kobayashi, R. Ikeda, Y. Kubota, M. Takata, S. Toh, T. Yamamoto, S. Matsumura, N. Sumi, K. Sato, K. Nagaoka and H. Kitagawa, J. Am. Chem. Soc., 2014, 136, 1864–1871 CrossRef CAS PubMed.
- Z. C. Zhang, Y. Liu, B. Chen, Y. Gong, L. Gu, Z. X. Fan, N. L. Yang, Z. C. Lai, Y. Chen, J. Wang, Y. Huang, M. Sindoro, W. X. Niu, B. Li, Y. Zong, Y. H. Yang, X. Huang, F. W. Huo, W. Huang and H. Zhang, Adv. Mater., 2016, 28, 10282–10286 CrossRef CAS PubMed.
- H. Liu, W. W. Zheng, Y. Zhao and Y. Z. Zhou, Anal. Chem., 2021, 93, 4944–4951 CrossRef CAS PubMed.
- T. L. Jiang, K. Li, S. Park, K. Zheng, Y. H. Meng, Y. Yuan, Z. C. Liu, Z. X. Zhu, X. H. Zheng, S. Liu and W. Chen, Nano Lett., 2022, 22, 1741–1749 CrossRef CAS PubMed.
- H. L. Jia, C. H. Fang, X.-M. Zhu, Q. F. Ruan, Y.-X. J. Wang and J. F. Wang, Langmuir, 2015, 31, 7418–7426 CrossRef CAS PubMed.
- W. H. Ni, X. S. Kou, Z. Yang and J. F. Wang, ACS Nano, 2008, 2, 677–686 CrossRef CAS PubMed.
- J. P. Zheng, X. Z. Cheng, H. Zhang, X. P. Bai, R. Q. Ai, L. Shao and J. F. Wang, Chem. Rev., 2021, 121, 13342–13453 CrossRef CAS PubMed.
- H. J. Chen, L. Shao, K. C. Woo, T. Ming, H.-Q. Lin and J. F. Wang, J. Phys. Chem. C, 2009, 113, 17691–17697 CrossRef CAS.
- R. Bukasov and J. S. Shumaker-Parry, Nano Lett., 2007, 7, 1113–1118 CrossRef CAS PubMed.
- W. C. Wang, X. Li, T. O. He, Y. M. Liu and M. S. Jin, Nano Lett., 2019, 19, 1743–1748 CrossRef CAS PubMed.
- S. F. Xie, S.-I. Choi, N. Lu, L. T. Roling, J. A. Herron, L. Zhang, J. Park, J. G. Wang, M. J. Kim, Z. X. Xie, M. Mavrikakis and Y. N. Xia, Nano Lett., 2014, 14, 3570–3576 CrossRef CAS PubMed.
- Y. N. Xia, K. D. Gilroy, H.-C. Peng and X. H. Xia, Angew. Chem., Int. Ed., 2016, 55, 2–38 CrossRef.
- S. F. Xie, H.-C. Peng, N. Lu, J. G. Wang, M. J. Kim, Z. X. Xie and Y. N. Xia, J. Am. Chem. Soc., 2013, 135, 16658–16667 CrossRef CAS PubMed.
- H. H. Ye, Q. X. Wang, M. Catalano, N. Lu, J. Vermeylen, M. J. Kim, Y. Z. Liu, Y. G. Sun and X. H. Xia, Nano Lett., 2016, 16, 2812–2817 CrossRef CAS PubMed.
- G. X. Chen, C. F. Xu, X. Q. Huang, J. Y. Ye, L. Gu, G. Li, Z. C. Tang, B. H. Wu, H. Y. Yang, Z. P. Zhao, Z. Y. Zhou, G. Fu and N. F. Zheng, Nat. Mater., 2016, 15, 564–569 CrossRef CAS PubMed.
- Z. Cao, D. Kim, D. C. Hong, Y. Yu, J. Xu, S. Lin, X. D. Wen, E. M. Nichols, K. Jeong, J. A. Reimer, P. D. Yang and C. J. Chang, J. Am. Chem. Soc., 2016, 138, 8120–8125 CrossRef CAS PubMed.
- J. H. Hu, H. Mi, N. Wang, H. Y. Zhu, W. Y. Guo, S. R. Zhang, F. Shi, Z. B. Lei, Z.-H. Liu and R. B. Jiang, Nanoscale, 2018, 10, 5607–5616 RSC.
- J. H. Yang, Y. Z. Guo, R. B. Jiang, F. Qin, H. Zhang, W. Z. Lu, J. F. Wang and J. C. Yu, J. Am. Chem. Soc., 2018, 140, 8497–8508 CrossRef CAS PubMed.
- S. Z. Andersen, V. Čolić, S. Yang, J. A. Schwalbe, A. C. Nielander, J. M. McEnaney, K. Enemark−Rasmussen, J. G. Baker, A. R. Singh, B. A. Rohr, M. J. Statt, S. J. Blair, S. Mezzavilla, J. Kibsgaard, P. C. K. Vesborg, M. Cargnello, S. F. Bent, T. F. Jaramillo, I. E. L. Stephens, J. K. Nørskov and I. Chorkendorff, Nature, 2019, 570, 504–508 CrossRef CAS PubMed.
- C. Tang and S.-Z. Qiao, Chem. Soc. Rev., 2019, 48, 3166–3180 RSC.
- Y. X. Zhao, F. Wu, Y. X. Miao, C. Zhou, N. Xu, R. Shi, L.-Z. Wu, J. W. Tang and T. R. Zhang, Angew. Chem., Int. Ed., 2021, 60, 21728–21731 CrossRef CAS PubMed.
- H. Li, J. Shang, Z. H. Ai and L. Z. Zhang, J. Am. Chem. Soc., 2015, 137, 6393–6399 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc02862c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |