
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRoom temperature stable E,Z-diphosphenes: their isomerization, coordination, and cycloaddition chemistry†
Jieli
Lin
a,
Shihua
Liu
a,
Jie
Zhang
a,
Hansjörg
Grützmacher
 ab,
Cheng-Yong
Su
a and
Zhongshu
Li
*a
ab,
Cheng-Yong
Su
a and
Zhongshu
Li
*a
aLIFM, IGCME, School of Chemistry, Sun Yat-Sen University, Guangzhou 510006, China. E-mail: lizhsh6@mail.sysu.edu.cn
bDepartment of Chemistry and Applied Biosciences, ETH Zürich, Vladimir-Prelog-Weg 1, Zürich 8093, Switzerland
First published on 11th September 2023
Abstract
E,Z-isomers display distinct physical properties and chemical reactivities. However, investigations on heavy main group elements remain limited. In this work, we present the isolation and X-ray crystallographic characterization of N-heterocyclic vinyl (NHV) substituted diphosphenes as both E- and Z-isomers (L![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–PP–CHL, E,Z-2b; L = N-heterocyclic carbene). E-2b is thermodynamically more stable and undergoes reversible photo-stimulated isomerization to Z-2b. The less stable Z-isomer Z-2b can be thermally reverted to E-2b. Theoretical studies support the view that this E ↔ Z isomerization proceeds via PP bond rotation, reminiscent of the isomerization observed in alkenes. Furthermore, both E,Z-2b coordinate to an AuCl fragment affording the complex [AuCl(η2-Z-2b)] with the diphosphene ligand in Z-conformation, exclusively. In contrast, E,Z-2b undergo [2 + 4] and [2 + 1] cycloadditions with dienes or diazo compounds, respectively, yielding identical cycloaddition products in which the phosphorus bound NHV groups are in trans-position to each other. DFT calculations provide insight into the E/Z-isomerisation and stereoselective formation of Au(I) complexes and cycloaddition products.
CH–PP–CHL, E,Z-2b; L = N-heterocyclic carbene). E-2b is thermodynamically more stable and undergoes reversible photo-stimulated isomerization to Z-2b. The less stable Z-isomer Z-2b can be thermally reverted to E-2b. Theoretical studies support the view that this E ↔ Z isomerization proceeds via PP bond rotation, reminiscent of the isomerization observed in alkenes. Furthermore, both E,Z-2b coordinate to an AuCl fragment affording the complex [AuCl(η2-Z-2b)] with the diphosphene ligand in Z-conformation, exclusively. In contrast, E,Z-2b undergo [2 + 4] and [2 + 1] cycloadditions with dienes or diazo compounds, respectively, yielding identical cycloaddition products in which the phosphorus bound NHV groups are in trans-position to each other. DFT calculations provide insight into the E/Z-isomerisation and stereoselective formation of Au(I) complexes and cycloaddition products.
Introduction
Stable compounds containing a double bond between almost every p-block element have been reported since the independent landmark discoveries of a disilene, Mes2SiSiMes2 (ref. 1), and a diphosphene, Mes*PPMes*2 (A, Fig. 1a), in 1981.3 The species containing heavy main group elements often exhibit unique structures and reactivity patterns compared to their lighter congeners. Some are characterized by “trans-bent” EE double bonds (that is, the molecule R2EER2 is not planar but shows ER2 interplanar angles >0°) and show unique reactivity.3a–e,g–i The kinetic barriers for oligo/polymerization are much lower than for the lighter congeners with E = element from the second period, and the successful isolation of compounds with E from the higher periods requires the utilization of sterically encumbering substituents to introduce kinetic protection. Consequently, cis-folded isomers of R2EER2 or Z-isomers of REER with heavy main group elements are rare due to increased steric repulsion caused by these substituents.3a,b,d,4
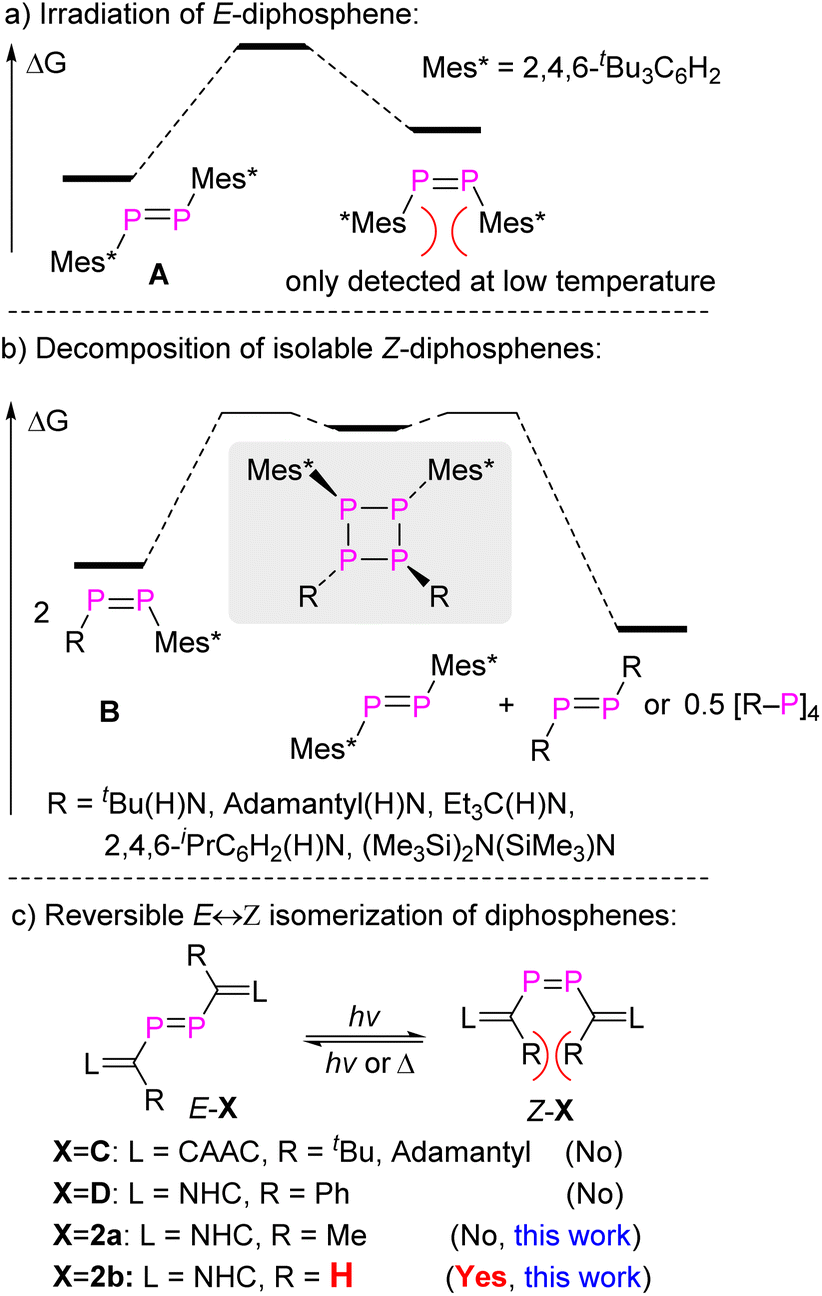 | ||
| Fig. 1 Schematic representation of (a) irradiation of isolable E-diphosphene; (b) decomposition of isolable Z-diphosphenes; and (c) reversible E ↔ Z isomerization of isolable diphosphenes. | ||
Diphosphenes are particularly intriguing; due to the diagonal relationship in the periodic table between carbon and phosphorus they behave to a certain extent like olefins.3b,d,5 Numerous E-diphosphenes were fully characterized, but the synthesis and isolation of Z-diphosphenes remains challenging.4e,6 The first spectroscopic detection of transient Z-diphosphene, Z-A, was found by laser irradiation of E-A at −80 °C. Z-A would revert back to the thermodynamically more stable E-A upon warming the solution to room temperature (RT) (Fig. 1a).6b,c Remarkably, Niecke et al. have isolated and crystallographically characterized a series of Z-diphosphenes (Z-RPPMes*, R = tBu(H)N, adamantyl(H)N, Et3C(H)N, 2,4,6-iPrC6H2(H)N, (Me3Si)2N(SiMe3)N, B, Fig. 1b).7 Among these, one diphosphene RPPMes* with a sterically very demanding amido substituent [R = (Me3Si)2N(SiMe3)N] could be isolated as both the E- and Z-isomers. These compounds displayed E,Z-isomerization in solution at RT.7b To the best of our knowledge, this is the only example where both the E- and Z-isomers of identical molecular composition could be isolated, although these amido-substituted diphosphenes undergo slow metatheses reactions at RT yielding symmetrical diphosphenes Mes*PPMes* and RPPR, likely via cyclotetraphosphanes formed in head-to-head dimerizations as intermediates (Fig. 1b). More recently, both diphosphasilene6a and diphosphene8 substituted Z-diphosphene, [Si]P–PP–P[Si] ([Si] = [PhC(NtBu)2SiN(SiMe3)2]) and LP–PP–PL (L = N-heterocyclic carbene (NHC)), were reported but likewise show limited stability.
Nevertheless, the presence of the RR′N–PP or RP–PP–PR fragments in Z-diphosphenes6a,7 indicates some structural and electronic features for constructing stable Z-diphosphenes: (i) the sterically protecting group points away from the central PP group to impose sufficient kinetic stability while at the same time the steric repulsion between the two substituents in Z-conformation is minimized. (ii) Electron delocalization as provided by co-planar moderately π-electron donating groups across the PP bond should increase the thermodynamic stability of both the E- and Z-isomers. We therefore reasoned that N-heterocyclic vinyl (NHV = LCR) substituted diphosphenes of the type LCR–PP–CRL could likewise be promising candidates for the observation and eventual isolation of persistent Z-diphosphenes. Both the groups of Stephan and Ghadwal independently reported NHV-substituted E-diphosphenes LC(R)–PP–C(R)L, E-C (L = cyclic alkyl amino carbene (CAAC), R = tBu, adamantyl)9 and E-D (L = NHC, R = phenyl)10 (Fig. 1c). In these compounds, the presence of bulky R substituents on the NHV groups prevent the observation of the corresponding Z-isomers. In this study, we have successfully isolated and characterized diphosphene LC(R)–PP–C(R)L 2b in both E- and Z-configurations using the smallest possible substituent R = H. Specifically, E-2b can be converted reversibly to Z-2b by a photo-stimulated process while Z-2b is reverted to E-2b by a thermal process. Notably, only a slight increase in bulkiness in passing from R = H to Me allows only the E-isomer E-2a to be detected (Fig. 1c). The thermodynamic and kinetic parameters for E/Z-isomerisation as well as differences in coordination and cyclo-addition reactions between E- and Z-2b could be studied.
Results and discussion
Synthesis and characterization of 2a and 2b
NHV substituted dichlorophosphines 1a (R = Me) and 1b (R = H) (LC(R)–PCl2, L = SIPr = 1,3-bis-(2,6-diisopropylphenyl)imidazoline-2-ylidine) were prepared as white powders [1a: δ(31P) = 191.9 ppm; 1b: δ(31P) = 183.7 ppm; see the ESI† for further details].10,11 Treatment of 1a with excess Mg powder in tetrahydrofuran (THF) at RT under vigorous stirring overnight in the dark afforded exclusively E-2a (LC(Me)–PP–C(Me)L), which is photolytically and thermally stable. After work-up, E-2a was obtained in pure form as a red powder in 78% yield. Reaction of 1b with Mg powder under the same conditions afforded a mixture of E-2b and Z-2b (LC(H)–PP–C(H)L) in a ratio of 1.0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.3 (Fig. 2a). After evaporation of THF, the solid residue was treated with toluene in order to extract the mixture of E/Z-2b from MgCl2. Heating this solution of E/Z-2b at 110 °C for 20 minutes in the dark led to an enrichment of E-2b (E-2b:Z-2b = 1.0:0.1). In contrast, irradiation of the toluene solution of the mixture of E/Z-2b with UV light (Hg lamp) or LED light (520 nm) at 0 °C for 30 minutes led to the predominant formation of Z-2b (E-2b:Z-2b = 1.0:21.6). Evaporation of solvent toluene from these reaction mixtures after irradiation or heating, and recrystallization of the solid residues from cold pentane at −30 °C yielded a highly air-sensitive red crystalline powder of E-2b or a crystalline orange powder of Z-2b in 62% and 67% yields, respectively.
:0.3 (Fig. 2a). After evaporation of THF, the solid residue was treated with toluene in order to extract the mixture of E/Z-2b from MgCl2. Heating this solution of E/Z-2b at 110 °C for 20 minutes in the dark led to an enrichment of E-2b (E-2b:Z-2b = 1.0:0.1). In contrast, irradiation of the toluene solution of the mixture of E/Z-2b with UV light (Hg lamp) or LED light (520 nm) at 0 °C for 30 minutes led to the predominant formation of Z-2b (E-2b:Z-2b = 1.0:21.6). Evaporation of solvent toluene from these reaction mixtures after irradiation or heating, and recrystallization of the solid residues from cold pentane at −30 °C yielded a highly air-sensitive red crystalline powder of E-2b or a crystalline orange powder of Z-2b in 62% and 67% yields, respectively.
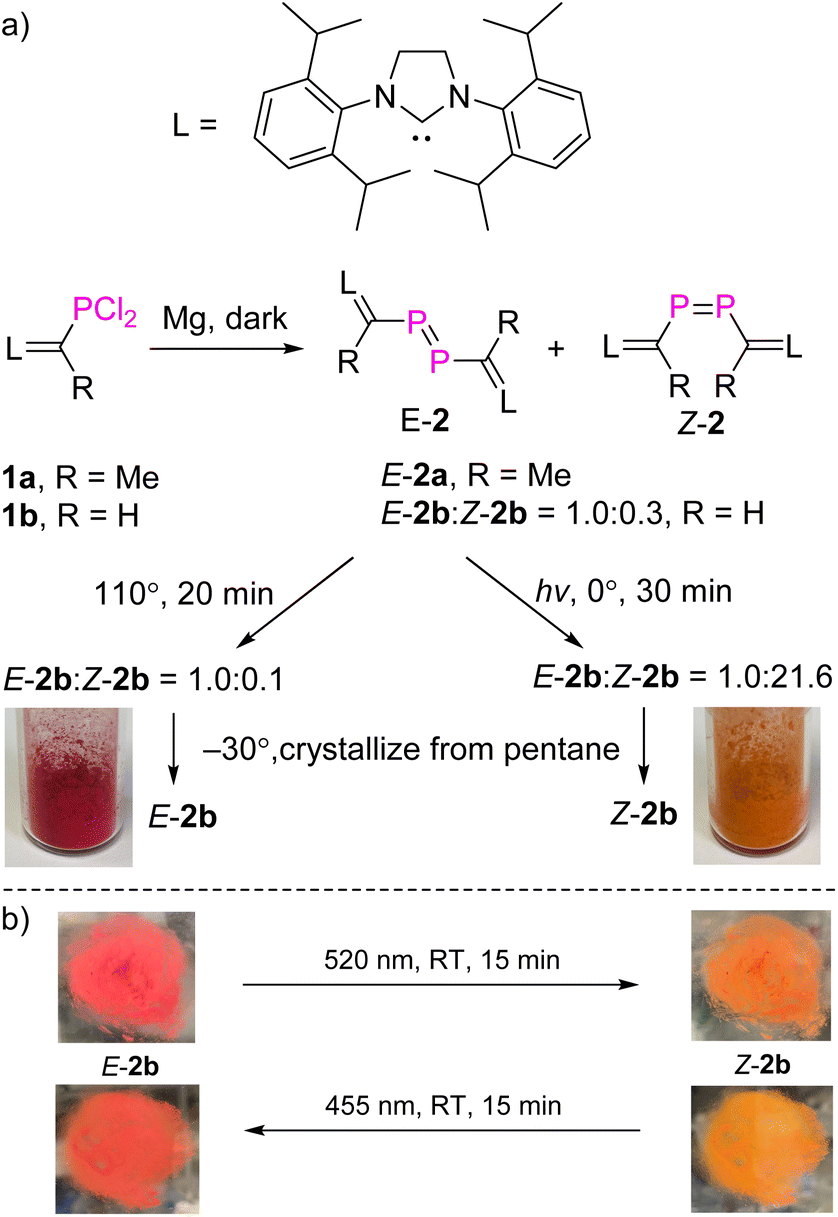 | ||
| Fig. 2 (a) Synthesis of compounds 2a and 2b; (b) the E → Z isomerization of E-2b in the solid state as a thin layer in between two glass plates. | ||
Multi-nuclear NMR spectra, single crystal X-ray diffraction (XRD), and high-resolution mass spectrometry were applied to characterize and confirm the molecular structures of E-2a, E-2b, and Z-2b. Characteristic singlet resonances at δ(31P) = 382.6, 379.6, or 259.5 ppm were observed in the 31P NMR spectra of E-2a, E-2b, and Z-2b, respectively. These 31P NMR shifts at high frequencies are within the range observed for other diphosphenes as well. In agreement with literature reported data, E-configured diphosphenes show resonances by about 100 ppm at higher frequencies compared to those of the Z-isomers.6g,12 In the 1H NMR spectra, singlet resonances at δ(1H) = 1.85 (E-2a PCCH3), δ(1H) = 4.98 (E-2b PCH), and δ(1H) = 4.61 ppm (Z-2b PCH) are attributed to the protons of the R substituents at the NHV moieties, respectively.
The stereo-configurations of 2a and 2b are confirmed unambiguously by XRD methods (Fig. 3). The PP bond lengths in the E-configured isomers E-2a [2.052(7) Å] and E-2b [2.0685(12) Å], and the one in the Z-isomer Z-2b [2.058(6) Å] are remarkably similar and typical of PP double bonds [Σrcov(P–P) = 2.22 Å, Σrcov(PP) = 2.04 Å].13 The P–C and CC bonds [1.765(3)–1.7863(13) Å and 1.362(4)–1.375(2) Å] vary only slightly in 2a and 2b and are typical of π-conjugated delocalized PC or CC bonds [Σrcov(P–C) = 1.86 Å, Σrcov(PC) = 1.69 Å, Σrcov(C–C) = 1.50 Å, Σrcov(CC) = 1.34 Å].13a,b As expected, the C–PP–C fragments are nearly planar in E-2a (∠CPPC = 0.0°) and E-2b (∠CPPC = 0.2°), but show a small deviation from planarity in Z-2b (∠CPPC = 13.3°) indicating a higher steric congestion in the Z-isomer.
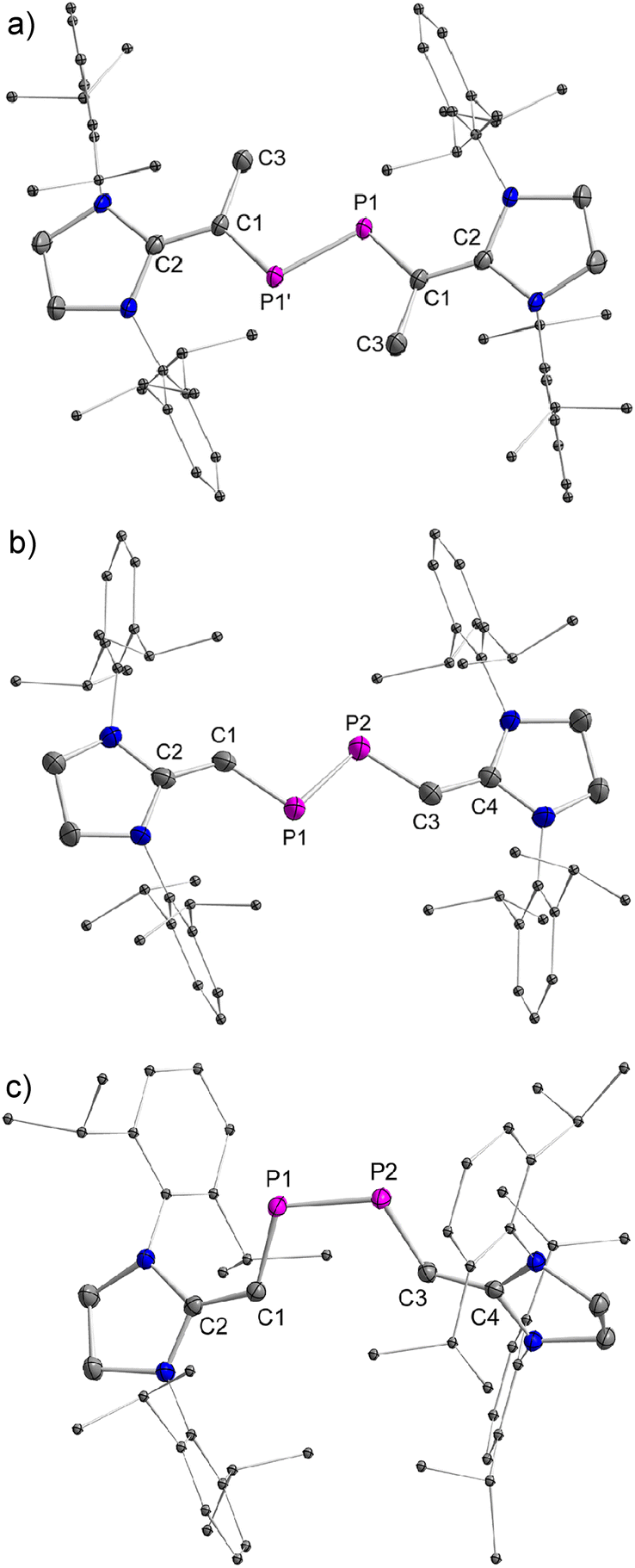 | ||
| Fig. 3 Plots of the molecular structure of E-2a, E-2b and Z-2b (ellipsoids are set to 50% probability; H atoms and solvents are omitted for clarity). Selected distances (Å): (a) E-2a: P1–P1′ 2.0520(7), P1–C1 1.7863(13), C1–C2 1.3689(18), C1–C3 1.5127(19); (b) E-2b: P1–P2 2.0685(12), P1–C1 1.772(3), P2–C3 1.765(3), C1–C2 1.362(4), C3–C4 1.362(4); (c) Z-2b: P1–P2 2.0580(6), P1–C1 1.7848(16), P2–C3 1.7744(16), C1–C2 1.368(2), C3–C4 1.375(2). | ||
Toluene solutions of the E-configured isomers E-2a and E-2b are deep red and show strong absorptions at λmax = 518 (1.8 × 104 M−1 cm−1) and 534 nm (1.8 × 104 M−1 cm−1). The Z-isomer Z-2b exhibits a strong absorption at lower wavenumbers with λmax = 482 nm (2.3 × 104 M−1 cm−1). In combination with TD-DFT calculations (Fig. S73–S78†), these absorptions are mainly attributed to the allowed π–π* transitions (HOMO → LUMO). The larger HOMO–LUMO gap of the Z-isomer is due to a lower HOMO energy (Z-2b: −5.27 eV vs. E-2b: −5.06 eV) and a higher LUMO energy (Z-2b: −0.44 eV vs. E-2b: −0.51 eV). In addition, all compounds show a very weak absorption at shorter wavelengths, namely 416 (2.5 × 103 M−1 cm−1), 401 (3.0 × 103 M−1 cm−1), and 359 (2.3 × 103 M−1 cm−1) nm for E-2a, E-2b, and Z-2b, respectively (Fig. 4a). These are assigned to the forbidden n–π* transitions (HOMO−1 → LUMO). Comparable results have been reported for the structurally similar D (Fig. 1d).10 Note, however, that in aryl substituted diphosphenes such as Mes*PPMes* the ordering of these transitions is inverted and the n–π* is at higher wavenumbers while the π–π* occurs at shorter wavenumbers.14
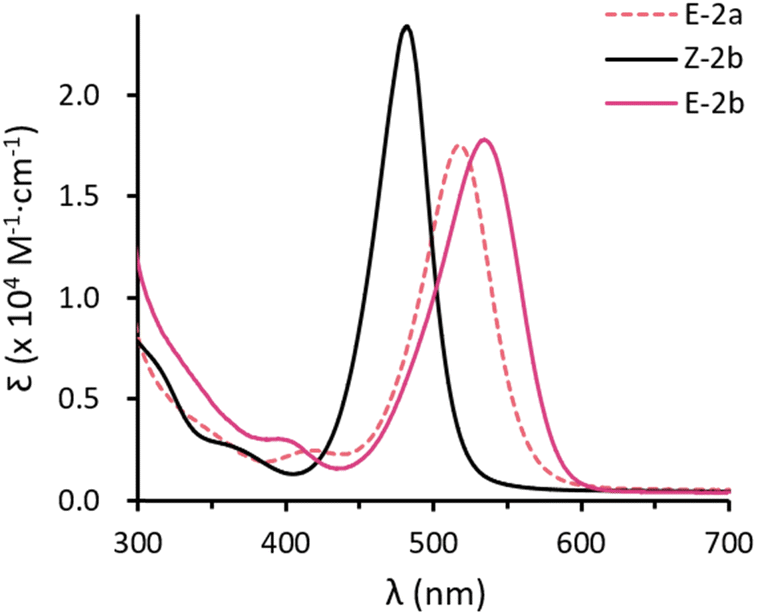 | ||
| Fig. 4 UV-vis absorption spectra for toluene solution of E-2a (dashed red), E-2b (red) and Z-2b (black). | ||
E,Z-isomerization of E-2b and Z-2b
In the solid state and under an inert atmosphere, E,Z-2b are indefinitely stable even at elevated temperature in the dark. Upon irradiation, a gradually reversible E-2b ↔ Z-2b conversion is observed, which is indicated by a colour change of the microcrystalline powder from red to orange or vice versa (Fig. 2b). Similar observations were reported for alkenes or azo species.15 In solution, E-2b is stable for days, whereas Z-2b slowly isomerizes to the thermodynamically more favourable E-2b. For example, approximately 10% of Z-2b converted to E-2b after two days as monitored by 31P NMR spectra. Upon irradiation, E-2b isomerizes to Z-2b at 520 nm and reverts back to E-2b at 455 nm. No significant decomposition is observed for either E- or Z-2b upon irradiation or prolonged heating for hours. Some E-diphosphenes decompose upon irradiation,6b,c,16 but may become stable upon irradiation when armed with extremely bulky substituents.17 In contrast, all reported Z-diphosphenes show limited stability and decompose even at RT in solution.6a–c,7,18 Therefore, Z-2b represents a very rare isolable Z-diphosphene with good thermal and photolytic stability at RT.7,18The kinetics for the E ↔ Z isomerization of 2b was probed by 1H NMR spectroscopy using hexamethylenetetramine as an internal standard. The pseudo-first-order rate constants (k) were calculated based on the consumption of the E-2b or Z-2b for photo- or thermally stimulated E,Z-isomerization. The plots of ln[E-2b] vs. t (Fig. 5a) and ln[Z-2b] vs. t (Fig. S59† and 5b) indicate first order kinetics with k520nm = 4.4 × 10−3 s−1, k455nm = 1.0 × 10−2 s−1, and k348.15K = 5.6 × 10−4 s−1, respectively. The photostationary (PSS) equilibria were established within 300 s or 750 s for the E-2b → Z-2b or E-2b ← Z-2b isomerization process yielding a mixture of E-2b:Z-2b = 0.19 or 4.40 under irradiation at 520 or 455 nm, respectively. The relative molar extinction coefficient of E,Z-2b at 520 or 455 nm was determined to be Z-2b:E-2b = 0.18 or 4.76 (see Fig. S50 and S51† for details). These results fit well to eqn (1) for the PSS ensuring equal quantum yields (Φ) for the photoisomerization reactions in both directions.19
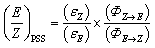 | (1) |
 | ||
| Fig. 5 Plots of (a) ln[E-2b] against reaction time t under irradiation at 520 nm; (b) ln[E-2a] against reaction time t at 348.15 K; (c) ln(k) against 1/T; and (d) ln(Keq) against 1/T at 348.15 K to 368.15 K. | ||
Furthermore, the temperature dependence of the isomerization rate constructed from the kinetic data measured at 5 K intervals from 348.15 K to 368.15 K gives the experimental activation energy for the thermal Z-2b → E-2b conversion as Ea = 23.4 ± 0.9 kcal mol−1. The pre-exponential factor can be expressed at these temperatures by the Arrhenius equation k = 2.72 × 1011 × e−23.4/RT (Fig. 5c). This high pre-exponential factor precludes the population of excited triplet states in the thermal Z-2b → E-2b isomerization process.20 The Eyring plot constructed from these data affords the experimental activation parameters as ΔH‡ = 22.7 ± 0.9 kcal mol−1, ΔS‡ = −8.6 ± 2.4 e.u., and ΔG‡(298.15K) = 25.3 ± 1.6 kcal mol−1 (Fig. S64†). The activation barrier of the thermal isomerization is comparable to those of the previously reported diphosphenes Mes*PPMes* [ΔG‡(273K) = 20.3 kcal mol−1]6b and RPPMes* (ΔG‡(293K) = 25.5 kcal mol−1, R = (Me3Si)2N(Me3Si)N, Fig. 1b).7b For comparison, the activation barriers for stilbene21 or azobenzene22 are about 40 or 23 kcal mol−1, respectively. Moreover, a van't Hoff analysis (ΔH = 1.6 ± 0.1 kcal mol−1, ΔS = 7.6 ± 0.2 e.u.) revealed that the free energy of Z-2b is slightly lower, ΔG(298.15K) = −0.6 ± 0.1 kcal mol−1, than that of E-2b (Fig. 5d). In conclusion, these special factors (small energy difference between E- and Z-2b and relatively high activation barrier for the thermal Z-2b → E-2b isomerization process) allow the isolation of diphosphene 2b in both E- and Z-configurations.
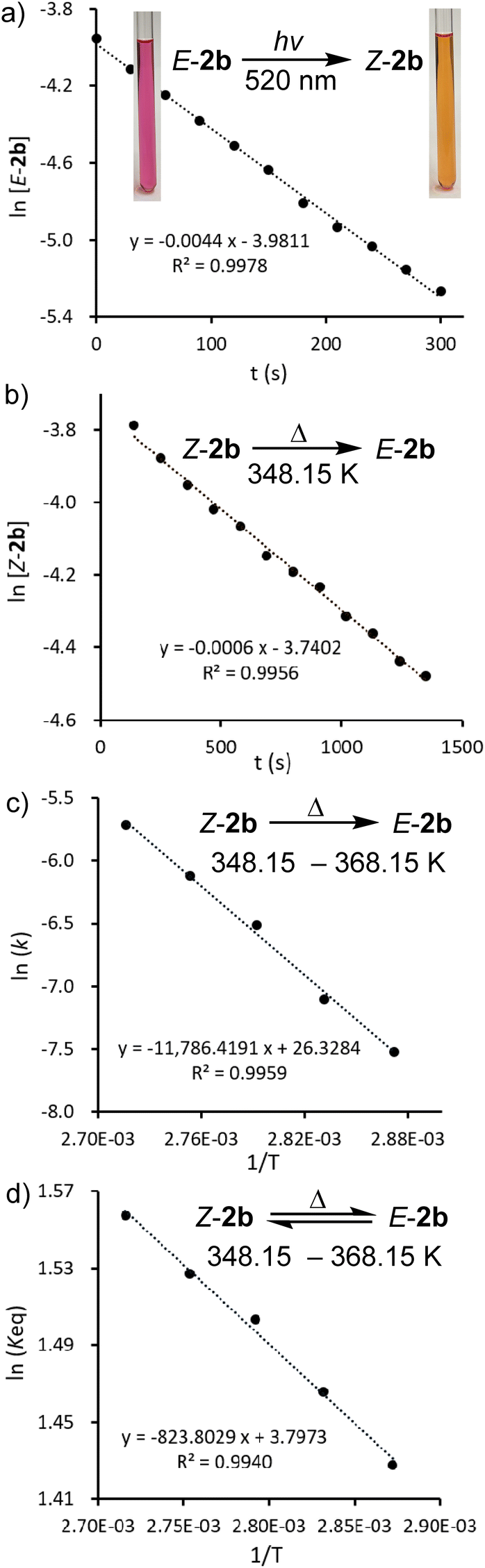
To provide a deeper understanding of the E,Z-isomerization mechanism, density functional theory (DFT) calculations were carried out at the M062X-D3/Def2TZVP-SMD(toluene)//M062X-D3/Def2SVP level of theory.23 Apart from E,Z-2b we included also the methyl-substituted derivates E,Z-2a in this study although an E/Z-isomerism could not be observed experimentally. Possible minimum energy reaction pathways (MERPs) for both the photolytic E-X ↔ Z-X and thermal Z-X → E-X (X = 2a, 2b) are shown in Fig. 6. Upon irradiation, E-2a and E-2b are excited to the triplet states 3T-2a (26.7 kcal mol−1) and 3T-2b (23.6 kcal mol−1).24 The structures of the triplet states 3T-2a and 3T-2b are very similar and notably contain a significantly elongated P–P bond (2.21 Å), which is in the range of a single bond, and C–P–P–C dihedral angles of 106.0°. These structures are very different compared to the singlet ground state structures of E-2a and E-2b (PP: 2.05 Å; C–P–P–C = 180.0°). Subsequently, these excited triplet states relax via a spin flipping process to the E- or Z-isomers in their singlet ground states in exergonic reactions. In case of 2a, the resulting Z-2a will quickly isomerize to the thermodynamically more stable E-2a (−11.2 kcal mol−1) via a relatively small activation barrier TSR-2a (13.9 kcal mol−1). In case of 2b, the E/Z ratio of the resulting mixture depends on the relative molar extinction coefficient of E,Z-2b. At elevated temperatures, Z-2b isomerizes via the activated complexes TSR-2b (22.7 kcal mol−1), which has a singlet electronic configuration, affording the thermodynamically more stable E-isomers E-b (−3.3 kcal mol−1). The calculations are in good agreement with the experimental observations: Z-2a cannot be detected because of its thermodynamic instability versus E-2a (+11.2 kcal mol−1) and low activation barrier for the thermal isomerization (Ea = 13.9 kcal mol−1). On the other hand, Z-2b can be isolated because it is almost isoenergetic with E-2b and Ea for the thermal isomerization is much higher (25.3 ± 1.6 kcal mol−1). Note that the calculated activation barriers predicted at the same level of theory for the Z → E isomerization process of the olefin stilbene [TSR = 43.6 kcal mol−1vs. 40 kcal mol−1 (exp.)]21 is much higher while the one of azobenzene [TSI = 25.5 kcal mol−1vs. 23 kcal mol−1 (exp.)]22 is in the same range (Fig. S79†). For comparison we also investigated possible E/Z-isomerization pathways for the simple divinyl substituted compound CH2CH–PP–CHCH2 and found that the activation barrier EaR = 24.3 kcal mol−1 for a rotation around the P,P-bond is comparable to the one in E-2b (the activation barrier for inversion at one phosphorus center – an alternative process for E/Z-isomerization – is much higher in energy at EaI = 50.1 kcal mol−1) (Fig. S80†). Similar data have been theoretically predicted for the Z → E isomerization of HPPH (Fig. S81†).25
 | ||
| Fig. 6 MERP for the reversible photo- and thermally stimulated E,Z-isomerization of 2a and 2b. | ||
Coordination chemistry of E-2b and Z-2b
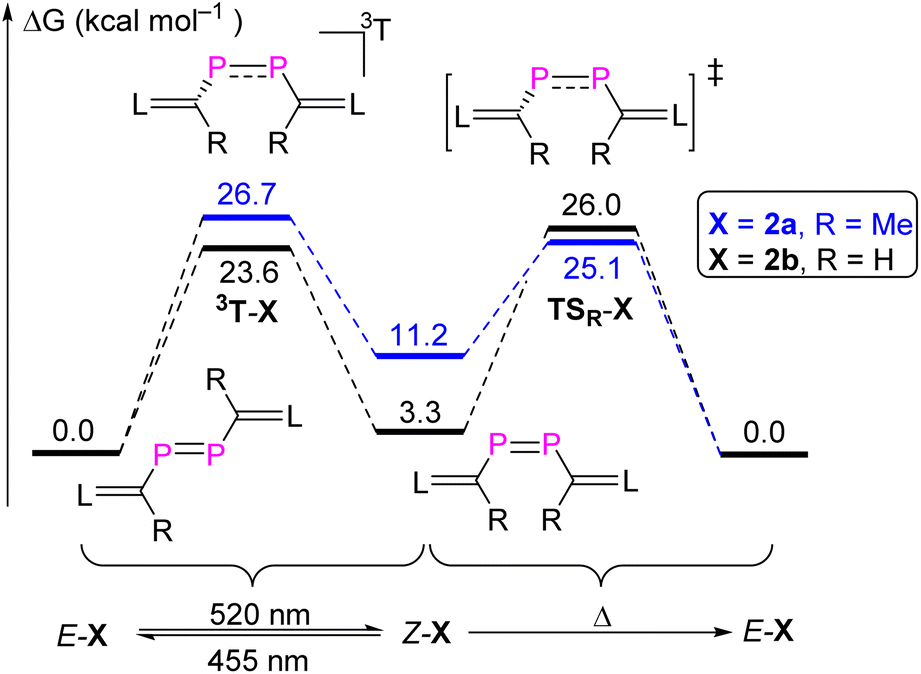
Having the rare opportunity to have stable and pure E- and Z-isomers of a diphosphene of identical molecular composition at hand, we investigated the coordination chemistry versus an AuCl fragment and the cycloaddition chemistry versus dimethylbutadiene (DMBD) and (trimethylsilyl)diazomethane (TDM). Both reactivities are typical of olefins as hydrocarbon counterparts to diphosphenes. Remarkably, treatment of both E-2b and Z-2b with equimolar amounts of [AuCl(SMe2)] in THF at RT afforded selectively the same complex within minutes. After work-up, compound [AuCl(2b)] was obtained as a deep red powder in 92% yield (Fig. 7a). A characteristic singlet resonance at δ(31P) = 153.8 ppm in the 31P NMR spectrum indicates the formation of a symmetric complex. With respect to both E-2b [δ(31P) = 379.6 ppm] and Z-2b [δ(31P) = 259.5 ppm], a significant coordination shift Δδ = δcomplex − δligand of −225.8 ppm or −105.7 ppm respectively, to lower frequencies, is observed. | ||
| Fig. 7 (a) Synthesis of [AuCl(2b)]; (b) plot of the molecular structure of [AuCl(2b)] (thermal ellipsoids are set to 50% probability; H atoms and solvent molecules are omitted for clarity). Selected distances (Å): P1–P2 2.1254(14), P1–C1 1.754(4), P1–Au1 2.4580(11), P2–C3 1.753(4), P2–Au1 2.4280(11), Au1–Cl1 2.3357(12), C1–C2 1.389(6), C3–C4 1.377(5). | ||
The molecular structure of [AuCl(2b)] was determined by XRD methods (Fig. 7b) and shows that 2b is (i) η2-bound to the AuCl moiety and (ii) the substituents adopt a cis-configuration with respect to the P–P vector indicating that, remarkably and unexpectedly, the reaction of E-2b with [AuCl(SMe2)] induces E-2b → Z-2b isomerization. In the complex [AuCl(η2-Z-2b)], the P1–P2 bond length is elongated to 2.1254(12) Å with respect to uncoordinated Z-2b [2.0580(6) Å]. This observation indicates significant electron donation from the d10-valence electron configured Au(I) ion into the π*(PP) anti-bonding orbital. The Au–P1 and Au–P2 bonds are almost identical in length [P1–Au1: 2.4580(11) Å; P2–Au1: 2.4280(11) Å]. The metal center resides in a trigonal planar geometry [sum of bond angles Σ°(Au) = 359.8°]. The dihedral angle between the Au–P1–P2 and C1–P1–P2–C3 planes is 73.9°. Hand in hand with the elongation of the coordinated PP bond goes a reduction of the ∠CPPC torsion angle from 13.3° (Z-2b) to 5.7° in the complex [AuCl(η2-Z-2b)] indicating reduced steric repulsion between two NHV groups. To the best of our knowledge, [AuCl(η2-Z-2b)] represents the first crystallographically characterized mononuclear η2-Z-diphosphene complex and the only η2-bound diphosphene Au(I) complex isolated so far (vide infra).7,18,26
DFT calculations were carried out in order to gain deeper insight into the selective formation of [AuCl(η2-Z-2b)]. Both the full model and a simplified model complex [AuCl(η2-Z-2bH)] where the 2,6-diisopropylphenyl (Dipp) groups were replaced with H atoms were investigated at the BP86-D3BJ/Def2TZVP-SMD(THF)//BP86-D3BJ/Def2SVP level of theory (Fig. 8).23 For the full model, the stability of four different conformers follows the order [AuCl(η2-Z-2b)] (−1.8 kcal mol−1) > [AuCl(η1-E-2b)] (0 kcal mol−1) > [AuCl(η2-E-2b)] (0.1 kcal mol−1) > [AuCl(η1-Z-2b)] (4.4 kcal mol−1). The highest activation barrier of 22.3 kcal mol−1 (TS2-2b) is attributed to the rotation of the PP bond during the [AuCl(η1-E-2b)] → [AuCl(η1-Z-2b)] isomerization process, which is only slightly smaller than the calculated barrier for the E/Z-isomerization of the uncoordinated diphosphene. On the other hand, the conversion from η1- to η2-gold complexes for both E- and Z-isomers proceeds smoothly viaTS1-2b (7.4 kcal mol−1) and TS3-2b (0.6 kcal mol−1). For the simplified model, similar energy barriers are obtained for the intramolecular conversion among these four conformers. However, the stability order differs and follows the order [AuCl(η1-E-2bH)] (0 kcal mol−1) > [AuCl(η1-Z-2bH)] (0.3 kcal mol−1) > [AuCl(η2-Z-2bH)] (0.6 kcal mol−1) > [AuCl(η2-E-2bH)] (2.0 kcal mol−1). The higher stability (although marginal) of the η1-bound complexes agrees with the one previously reported for three Au(I) bisarene-substituted E-diphosphene complexes, in which the PP bond is shortened with respect to the uncoordinated diphosphene.26 Note that diphosphenes in E-configuration are frequently bound in η2-fashion to other transition metal centers.27
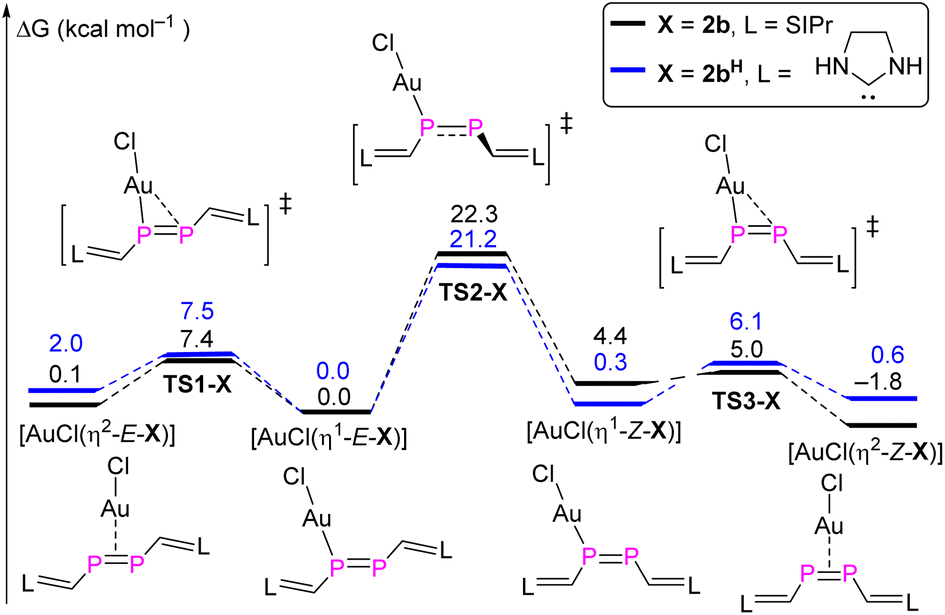 | ||
| Fig. 8 MERP for the intramolecular conversion between η1- and η2-gold complexes with both models 2b and 2bH. | ||
Cycloaddition chemistry of E-2b and Z-2b
Treatment of E-2b with equimolar amounts of DMBD or TDM in toluene at RT resulted in rapid and quantitative conversion to yield the [2 + 4] and [2 + 1] cycloaddition products 3 and 4 within minutes (Fig. 9).6g The 31P NMR spectra of 3 and 4 displayed a singlet at −68.0 ppm, a doublet of a doublet at −161.3 ppm (1JPP = 158.3 Hz, 2JPH = 29.36 Hz) and a doublet at −181.9 ppm (1JPP = 158.3 Hz), respectively. Detailed characterization of three-membered heterocyclic diphosphiranes by multi-nuclear NMR spectroscopic methods has been reported by Jutzi and co-workers.28 Notably, treatment of Z-2b with DMBD or TDM at RT afforded the same products 3 or 4, respectively. However, complete conversion to 3 required more than 8 days or 6 hours to give 4 (Fig. S58 and S63†). The E-isomer E-2b was not detected in the course of these reactions. These findings suggest that Z-2b does not directly react with these substrates but first isomerizes to E-2b, which subsequently is rapidly consumed in the [2 + 4] or [2 + 1] cycloaddition. XRD studies unveiled the molecular structures of 3 and 4 and showed that in both cases the two N-heterocyclic vinyl groups are placed in mutual trans-position to each other (Fig. 10).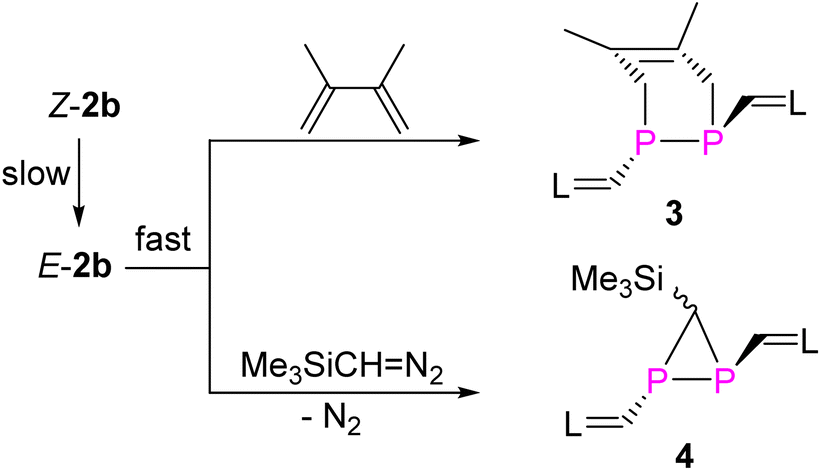 | ||
| Fig. 9 Synthesis of 3 and 4. | ||
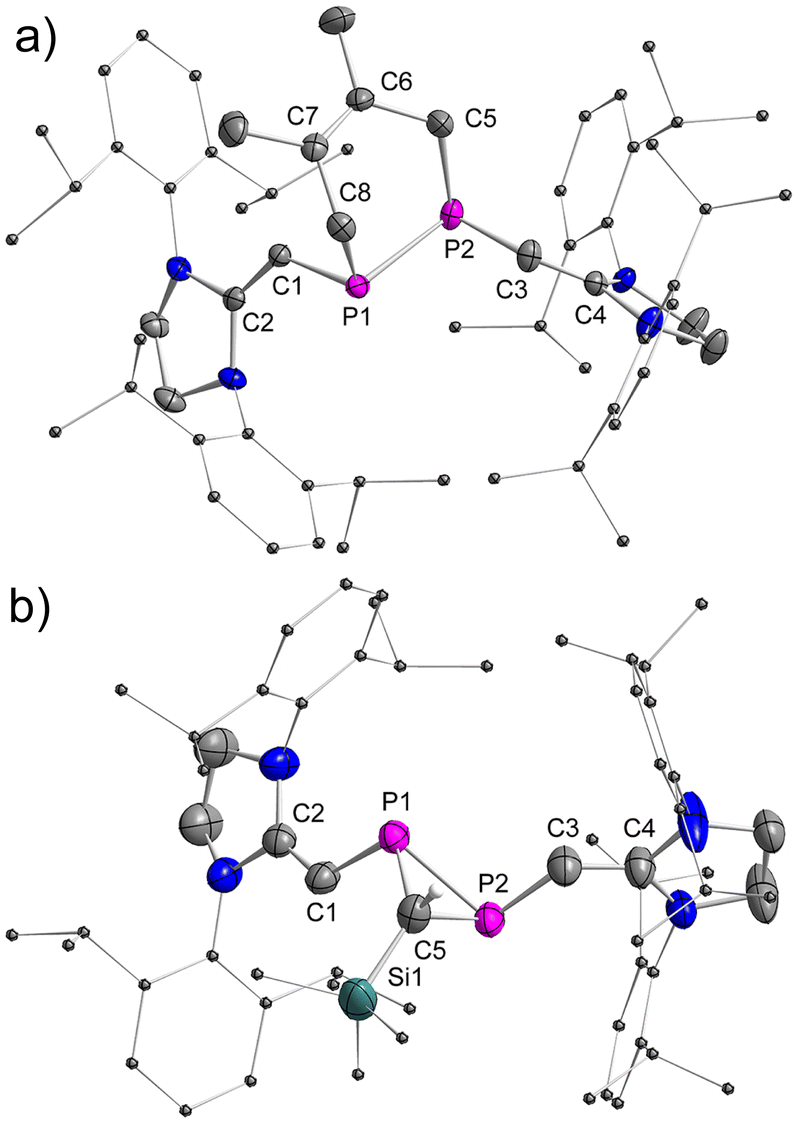 | ||
| Fig. 10 Plot of the molecular structure of 3 and 4 (ellipsoids are set to 50% probability; H atoms except the one on C5 in 4 and solvents are omitted for clarity). Selected distances (Å): (a) 3: P1–P2 2.2389(6), P1–C1 1.8202(16), C1–C2 1.369(2), P2–C3 1.8187(17), C3–C4 1.359(2), P1–C8 1.8999(17), C8–C7 1.512(2), C7–C6 1.352(3), C6–C5 1.505(2), C5–P2 1.9054(19); (b) 4: P1–P2 2.2166(13), P1–C1 1.792(4), C1–C2 1.347(5), P2–C3 1.794(4), C3–C4 1.348(5), P1–C5 1.854(4), P2–C5 1.863(4), C5–Si1 1.854(4). | ||
Again, DFT calculations were performed to provide a better understanding of the reactivity difference in between E-2b and Z-2b. The reaction mechanism for the [2 + 4] cycloaddition reaction between DMBD and model 2b or 2bH was studied at the M062X-D3/Def2TZVP-SMD(toluene)//M062X-D3/Def2SVP level of theory (Fig. 11).23 In both cases, the [2 + 4] cycloaddition reactions proceed in a concerted fashion via one activated complex at the corresponding transition states. In the reaction between DMBD and the simplified E-configured model E-2bH the transition state TS6H (15.2 kcal mol−1) is about 5 kcal mol−1 lower than the one for the reaction with Z-2bH (TS4H = 20.3 kcal mol−1 with respect to Z-2bH). Both products E-3H (−17.0 kcal mol−1) and Z-5H (−12.2 kcal mol−1) are formed in exergonic reactions, which differ by 4.8 kcal mol−1 in favour of the E-isomer. The kinetic and thermodynamic formation of the E-configured isomer becomes even more favourable in the reactions with the sterically more hindered E/Z-2b as diene acceptors. Here TS6 on the MERP in the reaction with E-2b is lower by 9.0 kcal mol−1 than TS4 (on the MERP with Z-2b) and the E-configured product E-3 is more stable by 14.6 kcal mol−1 when compared to Z-3. The latter is formed in an almost thermoneutral reaction (ΔGr = −3.6 kcal mol−1) between Z-2b and DMBD while the formation of the E-isomer E-3 is exergonic by −14.9 kcal mol−1. Not unexpected, with increasing steric encumbrance the transition state energies increase while the product stabilities decrease. But the calculated barriers for the [2 + 4] cycloaddition between E/Z-2b and DMBD are still relatively low and comparable to the barrier for E → Z isomerization to allow a reaction at room temperature, which ultimately leads selectively to the thermodynamically more stable product E-3. Thus, the calculations agree well with the experimental observations.
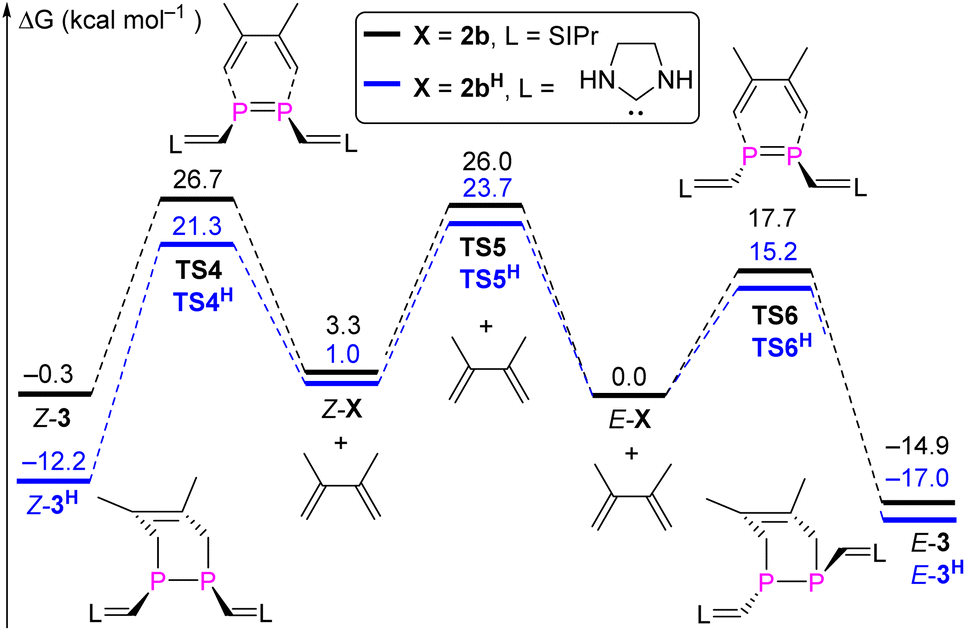 | ||
| Fig. 11 MERP for the formation of 3 and 3H. | ||
Conclusions
In contrast to the E/Z isomerization of olefins or azo compounds, for which this process was discovered more than eight decades ago in 1934 (ref. 21a) and 1937,29 respectively, the direct observation of E,Z-isomerization for compounds with a double bond between two heavy main group elements remains scarce. Specifically, kinetic and thermodynamic data for these isomerizations are lacking as well as comprehensive investigations of their distinct physical properties and chemical reactivities. In this study, we present the successful isolation and unambiguous characterization of the E- and Z-isomers of a diphosphene (E,Z-2b). XRD studies show that both have a short PP double bond of about the same length and confirm their stereo configuration. Kinetic studies reveal the first-order process for both photo- and thermally stimulated E ↔ Z isomerization. In combination, the experimental and theoretical data show that the E ↔ Z isomerization of diphosphene proceeds via rotation around the PP double bond. However, a similar E ↔ Z isomerization has not been reported for the nitrogen analogue of E-2b.30 Both E,Z-2b react with the AuCl fragment to afford exclusively the thermodynamically slightly more favoured complex [AuCl(η2-Z-2b)], which is the first mononuclear metal complex of a diphosphene in Z-conformation. The activation barrier for E → Z-isomerisation of the η1-bound AuCl complexes, which are intermediates on the MERP that give [AuCl(η2-Z-2b)], is slightly smaller (≈4 kcal mol−1) than in the uncoordinated diphosphene. It is presently not clear why in this particular case the Au(I) complex with Z-2b is more stable. The opposite is observed when a mixture of E/Z-2b is employed in a cycloaddition with a diene or diazo compound. Here, the Z-isomer is unreactive and must isomerize slowly to E-2b in order to allow a reaction. Compound E-2b readily engages in a [2 + 4] or [2 + 1] cycloaddition reaction and as a consequence, exclusively the cycloaddition product with an E-arrangement of the substituents with respect to the P–P vector is obtained.
Data availability
The data generated in this study are available in the main text or the ESI.† Crystallographic data has been deposited at the joint Cambridge Crystallographic Data Centre (CCDC 2245822–2245824, 2270969–2270971). Synthesis and characterization of compounds, NMR spectra, crystallographic, and computational details are provided in the ESI.†Author contributions
J. Lin carried out most of the experimental work. S. Liu and J. Zhang assisted with the NMR spectra and X-ray single crystallographic diffraction measurements. Z. Li carried out the computational studies. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22271315, 22171291, 21821003, 21890380), Tip-top Scientific and Technical Innovative Youth Talents of Guangdong Special Support Program (2019TQ05C926), Guangdong Basic and Applied Basic Research Foundation (2021A1515012028, 2023A1515010092), Science and Technology Planning Project of Guangzhou (202102080189), National Key Research and Development Program of China (2021YFA1500401).Notes and references
- R. West, M. J. Fink and J. Michl, Science, 1981, 214, 1343–1344 CrossRef CAS PubMed.
- M. Yoshifuji, I. Shima, N. Inamoto, K. Hirotsu and T. Higuchi, J. Am. Chem. Soc., 1981, 103, 4587–4589 CrossRef CAS.
- (a) M. Kira, Proc. Jpn. Acad., Ser. B, 2012, 88, 167–191 CrossRef CAS PubMed; (b) R. C. Fischer and P. P. Power, Chem. Rev., 2010, 110, 3877–3923 CrossRef CAS; (c) Y. Wang and G. H. Robinson, Chem. Commun., 2009, 5201–5213 RSC; (d) P. P. Power, Chem. Rev., 1999, 99, 3463–3504 CrossRef CAS PubMed; (e) P. P. Power, J. Chem. Soc., Dalton Trans., 1998, 2939–2951 RSC; (f) M. Fischer, M. M. D. Roy, L. L. Wales, M. A. Ellwanger, A. Heilmann and S. Aldridge, J. Am. Chem. Soc., 2022, 144, 8908–8913 CrossRef CAS PubMed; (g) C. Weetman, Chem.–Eur. J., 2021, 27, 1941–1954 CrossRef CAS PubMed; (h) V. Nesterov, N. C. Breit and S. Inoue, Chem.–Eur. J., 2017, 23, 12014–12039 CrossRef CAS PubMed; (i) A. Rammo and D. Scheschkewitz, Chem.–Eur. J., 2018, 24, 6866–6885 CrossRef CAS PubMed; (j) J. Li, Z. Lu and L. L. Liu, J. Am. Chem. Soc., 2022, 144, 23691–23697 CrossRef CAS; (k) E. A. LaPierre, B. O. Patrick and I. Manners, J. Am. Chem. Soc., 2023, 145, 7107–7112 CrossRef CAS PubMed.
- (a) R. Holzner, A. Porzelt, U. S. Karaca, F. Kiefer, P. Frisch, D. Wendel, M. C. Holthausen and S. Inoue, Dalton Trans., 2021, 50, 8785–8793 RSC; (b) M. Kobayashi, N. Hayakawa, T. Matsuo, B. Li, T. Fukunaga, D. Hashizume, H. Fueno, K. Tanaka and K. Tamao, J. Am. Chem. Soc., 2016, 138, 758–761 CrossRef CAS PubMed; (c) D. Wendel, T. Szilvási, C. Jandl, S. Inoue and B. Rieger, J. Am. Chem. Soc., 2017, 139, 9156–9159 CrossRef CAS PubMed; (d) V. Y. Lee and A. Sekiguchi, Organometallic Compounds of Low-Coordinate Si, Ge, Sn and Pb: From Phantom Species to Stable Compounds, Wiley, 2011 Search PubMed; (e) T. Sasamori and N. Tokitoh, Dalton Trans., 2008, 1395–1408 RSC; (f) N. C. Breit, T. Szilvási and S. Inoue, Chem. Commun., 2015, 51, 11272–11275 RSC; (g) L. Zborovsky, A. Kostenko, D. Bravo-Zhivotovskii and Y. Apeloig, Angew. Chem., Int. Ed., 2019, 58, 14524–14528 CrossRef CAS PubMed.
- (a) K. B. Dillon, F. Mathey and J. F. Nixon, Phosphorus: The Carbon Copy: From Organophosphorus to Phospha-organic Chemistry, Wiley, 1998 Search PubMed; (b) G. Rayner-Canham, Found. Chem., 2011, 13, 121–129 CrossRef CAS; (c) F. Mathey, Angew. Chem., Int. Ed., 2003, 42, 1578–1604 CrossRef CAS PubMed; (d) S. Wang, J. D. Sears, C. E. Moore, A. L. Rheingold, M. L. Neidig and J. S. Figueroa, Science, 2022, 375, 1393–1397 CrossRef CAS PubMed; (e) K. B. Dillon, V. C. Gibson and L. J. Sequeira, J. Chem. Soc., Chem. Commun., 1995, 2429–2430 RSC; (f) R. C. Smith, E. Urnezius, K.-C. Lam, A. L. Rheingold and J. D. Protasiewicz, Inorg. Chem., 2002, 41, 5296–5299 CrossRef CAS PubMed.
- (a) S. Khan, R. Michel, S. S. Sen, H. W. Roesky and D. Stalke, Angew. Chem., Int. Ed., 2011, 50, 11786–11789 CrossRef CAS PubMed; (b) A.-M. Caminade, M. Verrier, C. Ades, N. Paillous and M. Koenig, J. Chem. Soc., Chem. Commun., 1984, 875–877 RSC; (c) Y. Masaaki, S. Takahiro and I. Naoki, Chem. Lett., 1988, 17, 1735–1738 CrossRef; (d) L. Weber, F. Ebeler and R. S. Ghadwal, Coord. Chem. Rev., 2022, 461, 214499 CrossRef CAS; (e) N. Tokitoh, J. Organomet. Chem., 2000, 611, 217–227 CrossRef CAS; (f) M. Yoshifuji, J. Chem. Soc., Dalton Trans., 1998, 3343–3350 RSC; (g) L. Weber, Chem. Rev., 1992, 92, 1839–1906 CrossRef CAS; (h) R. S. Ghadwal, Acc. Chem. Res., 2022, 55, 457–470 CrossRef CAS PubMed.
- (a) E. Niecke, B. Kramer and M. Nieger, Angew. Chem., Int. Ed., 1989, 28, 215–217 CrossRef; (b) E. Niecke, O. Altmeyer and M. Nieger, Angew. Chem., Int. Ed., 1991, 30, 1136–1138 CrossRef.
- J. D. Masuda, W. W. Schoeller, B. Donnadieu and G. Bertrand, J. Am. Chem. Soc., 2007, 129, 14180–14181 CrossRef CAS PubMed.
- L. L. Liu, L. L. Cao, J. Zhou and D. W. Stephan, Angew. Chem., Int. Ed., 2019, 58, 273–277 CrossRef CAS PubMed.
- D. Rottschäfer, M. K. Sharma, B. Neumann, H.-G. Stammler, D. M. Andrada and R. S. Ghadwal, Chem.–Eur. J., 2019, 25, 8127–8134 CrossRef.
- (a) I. C. Watson, A. Schumann, H. Yu, E. C. Davy, R. McDonald, M. J. Ferguson, C. Hering-Junghans and E. Rivard, Chem.–Eur. J., 2019, 25, 9678–9690 CrossRef CAS PubMed; (b) C. Hering-Junghans, P. Andreiuk, M. J. Ferguson, R. McDonald and E. Rivard, Angew. Chem., Int. Ed., 2017, 56, 6272–6275 CrossRef CAS PubMed.
- M. Yoshifuji, Eur. J. Inorg. Chem., 2016, 2016, 607–615 CrossRef CAS.
- (a) P. Pyykkö and M. Atsumi, Chem.–Eur. J., 2009, 15, 12770–12779 CrossRef PubMed; (b) F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1–S19 RSC; (c) J. D. Protasiewicz, M. P. Washington, V. B. Gudimetla, J. L. Payton and M. Cather Simpson, Inorg. Chim. Acta, 2010, 364, 39–45 CrossRef CAS.
- (a) A. H. Cowley, A. Decken, N. C. Norman, C. Krüger, F. Lutz, H. Jacobsen and T. Ziegler, J. Am. Chem. Soc., 1997, 119, 3389–3390 CrossRef CAS; (b) H.-L. Peng, J. L. Payton, J. D. Protasiewicz and M. C. Simpson, J. Phys. Chem. A, 2009, 113, 7054–7063 CrossRef CAS PubMed.
- (a) P. Naumov, S. Chizhik, M. K. Panda, N. K. Nath and E. Boldyreva, Chem. Rev., 2015, 115, 12440–12490 CrossRef CAS PubMed; (b) M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS PubMed; (c) H. Koshima, N. Ojima and H. Uchimoto, J. Am. Chem. Soc., 2009, 131, 6890–6891 CrossRef CAS PubMed; (d) A. K. Bartholomew, I. B. Stone, M. L. Steigerwald, T. H. Lambert and X. Roy, J. Am. Chem. Soc., 2022, 144, 16773–16777 CrossRef CAS PubMed.
- (a) M. Yoshifuji, N. Shinohara and K. Toyota, Tetrahedron Lett., 1996, 37, 7815–7818 CrossRef CAS; (b) Y. Masaaki, S. Takahiro and I. Naoki, Bull. Chem. Soc. Jpn., 1989, 62, 2394–2395 CrossRef; (c) V. Cappello, J. Baumgartner, A. Dransfeld, M. Flock and K. Hassler, Eur. J. Inorg. Chem., 2006, 2006, 2393–2405 CrossRef.
- (a) S. Shah, M. C. Simpson, R. C. Smith and J. D. Protasiewicz, J. Am. Chem. Soc., 2001, 123, 6925–6926 CrossRef CAS PubMed; (b) T. Kyoko, F. Yuuichi, S. Shigeru and Y. Masaaki, Chem. Lett., 1997, 26, 855–856 CrossRef; (c) T. Sasamori, N. Takeda and N. Tokitoh, J. Phys. Org. Chem., 2003, 16, 450–462 CrossRef CAS; (d) N. Nagahora, T. Sasamori, N. Takeda and N. Tokitoh, Chem.–Eur. J., 2004, 10, 6146–6151 CrossRef CAS PubMed; (e) K. Tsuji, S. Sasaki and M. Yoshifuji, Heteroat. Chem., 1998, 9, 607–613 CrossRef CAS; (f) E. Urnéžius and J. D. Protasiewicz, Main Group Chem., 1996, 1, 369–372 CrossRef.
- R. Pietschnig and E. Niecke, Organometallics, 1996, 15, 891–893 CrossRef CAS.
- (a) V. B. Gudimetla, A. L. Rheingold, J. L. Payton, H.-L. Peng, M. C. Simpson and J. D. Protasiewicz, Inorg. Chem., 2006, 45, 4895–4901 CrossRef CAS PubMed; (b) T. H. Lowry and K. S. Richardson, Mechanism and Theory in Organic Chemistry, Harper and Row, New York, 1981 Search PubMed.
- (a) J. L. Magee, W. Shand and H. Eyring, J. Am. Chem. Soc., 1941, 63, 677–688 CrossRef CAS; (b) E. R. Talaty and J. C. Fargo, Chem. Commun., 1967, 65–66 RSC; (c) M. Calvin and H. W. Alter, J. Chem. Phys., 1951, 19, 768–770 CrossRef CAS; (d) J.-Å. Andersson, R. Petterson and L. Tegnér, J. Photochem., 1982, 20, 17–32 CrossRef CAS.
- (a) G. B. Kistiakowsky and W. R. Smith, J. Am. Chem. Soc., 1934, 56, 638–642 CrossRef CAS; (b) C. Bastianelli, V. Caia, G. Cum, R. Gallo and V. Mancini, J. Chem. Soc., Perkin Trans. 2, 1991, 679–683 RSC.
- G. S. Hartley, J. Chem. Soc., 1938, 633–642 RSC.
- M. J. Frisch, et al., Gaussian 16, Revision C.01, 2016 Search PubMed.
- (a) H.-L. Peng, J. L. Payton, J. D. Protasiewicz and M. C. Simpson, Dalton Trans., 2012, 41, 13204–13209 RSC; (b) T. Copeland, M. P. Shea, M. C. Milliken, R. C. Smith, J. D. Protasiewicz and M. C. Simpson, Anal. Chim. Acta, 2003, 496, 155–163 CrossRef CAS.
- (a) M. Yoshifuji, T. Hashida, N. Inamoto, K. Hirotsu, T. Horiuchi, T. Higuchi, K. Ito and S. Nagase, Angew. Chem., Int. Ed., 1985, 24, 211–212 CrossRef; (b) S. Sinha Ray, Chem. Phys., 2020, 529, 110555 CrossRef CAS; (c) T. Lu, A. C. Simmonett, F. A. Evangelista, Y. Yamaguchi and H. F. Schaefer III, J. Phys. Chem. A, 2009, 113, 13227–13236 CrossRef CAS PubMed.
- (a) D. V. Partyka, M. P. Washington, T. G. Gray, J. B. Updegraff Iii, J. F. Turner II and J. D. Protasiewicz, J. Am. Chem. Soc., 2009, 131, 10041–10048 CrossRef CAS PubMed; (b) A. Tsurusaki, R. Ura and K. Kamikawa, Organometallics, 2020, 39, 87–92 CrossRef CAS.
- (a) J. Chatt, P. B. Hitchcock, A. Pidcock, C. P. Warrens and K. R. Dixon, J. Chem. Soc., Dalton Trans., 1984, 2237–2244 RSC; (b) R. A. Jones, M. H. Seeberger and B. R. Whittlesey, J. Am. Chem. Soc., 1985, 107, 6424–6426 CrossRef CAS; (c) H. Krautscheid, E. Matern, G. Fritz and J. Pikies, Z. Anorg. Allg. Chem., 2000, 626, 253–257 CrossRef CAS; (d) S. Sabater, D. Schmidt, H. Schmidt, M. W. Kuntze-Fechner, T. Zell, C. J. Isaac, N. A. Rajabi, H. Grieve, W. J. M. Blackaby, J. P. Lowe, S. A. Macgregor, M. F. Mahon, U. Radius and M. K. Whittlesey, Chem.–Eur. J., 2021, 27, 13221–13234 CrossRef CAS PubMed; (e) S. Gómez-Ruiz, S. Zahn, B. Kirchner, W. Böhlmann and E. Hey-Hawkins, Chem.–Eur. J., 2008, 14, 8980–8985 CrossRef PubMed; (f) I. G. Phillips, R. G. Ball and R. G. Cavell, Inorg. Chem., 1992, 31, 1633–1641 CrossRef CAS; (g) M. K. Sharma, D. Rottschäfer, B. Neumann, H.-G. Stammler, S. Danés, D. M. Andrada, M. van Gastel, A. Hinz and R. S. Ghadwal, Chem.–Eur. J., 2021, 27, 5803–5809 CrossRef CAS PubMed; (h) S. Gómez-Ruiz and E. Hey-Hawkins, Dalton Trans., 2007, 5678–5683 RSC; (i) N. Nagahora, T. Sasamori and N. Tokitoh, Organometallics, 2008, 27, 4265–4268 CrossRef CAS; (j) S. Kurz and E. Hey-Hawkins, J. Organomet. Chem., 1993, 462, 203–207 CrossRef CAS; (k) D. Fenske and K. Merzweiler, Angew. Chem., Int. Ed., 1984, 23, 635–637 CrossRef.
- P. Jutzi and S. Opiela, Z. Anorg. Allg. Chem., 1992, 610, 75–82 CrossRef CAS.
- G. S. Hartley, Nature, 1937, 140, 281 CrossRef CAS.
- L. Y. M. Eymann, P. Varava, A. M. Shved, B. F. E. Curchod, Y. Liu, O. M. Planes, A. Sienkiewicz, R. Scopelliti, F. Fadaei Tirani and K. Severin, J. Am. Chem. Soc., 2019, 141, 17112–17116 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of compounds, NMR spectra, and crystallographic and computational details. CCDC 2245822–2245824 and 2270969–2270971. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc04506d |
| This journal is © The Royal Society of Chemistry 2023 |