
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploiting solid-state dynamic nuclear polarization NMR spectroscopy to establish the spatial distribution of polymorphic phases in a solid material†
Samuel F.
Cousin
 a,
Colan E.
Hughes
b,
Fabio
Ziarelli
c,
Stéphane
Viel
ad,
Giulia
Mollica
*a,
Kenneth D. M.
Harris
*b,
Arthur C.
Pinon
*e and
Pierre
Thureau
*a
a,
Colan E.
Hughes
b,
Fabio
Ziarelli
c,
Stéphane
Viel
ad,
Giulia
Mollica
*a,
Kenneth D. M.
Harris
*b,
Arthur C.
Pinon
*e and
Pierre
Thureau
*a
aAix Marseille Univ, CNRS, ICR, Marseille, France. E-mail: giulia.mollica@univ-amu.fr; pierre.thureau@univ-amu.fr
bSchool of Chemistry, Cardiff University, Park Place, Cardiff CF10 3AT, Wales, UK, . E-mail: HarrisKDM@cardiff.ac.uk
cAix Marseille Univ, CNRS, Centrale Marseille, FSCM, Marseille, France
dInstitut Universitaire de France, Paris, France
eSwedish NMR Center, University of Gothenburg, Gothenburg, SE-405 30, Sweden. E-mail: arthur.pinon@gu.se
First published on 23rd June 2023
Abstract
Solid-state DNP NMR can enhance the ability to detect minor amounts of solid phases within heterogenous materials. Here we demonstrate that NMR contrast based on the transport of DNP-enhanced polarization can be exploited in the challenging case of early detection of a small amount of a minor polymorphic phase within a major polymorph, and we show that this approach can yield quantitative information on the spatial distribution of the two polymorphs. We focus on the detection of a minor amount (<4%) of polymorph III of m-aminobenzoic acid within a powder sample of polymorph I at natural isotopic abundance. Based on proposed models of the spatial distribution of the two polymorphs, simulations of 1H spin diffusion allow NMR data to be calculated for each model as a function of particle size and the relative amounts of the polymorphs. A comparison between simulated and experimental NMR data allows the model(s) best representing the spatial distribution of the polymorphs in the system to be established.
Introduction
In the context of organic molecular materials, the phenomenon of polymorphism1–5 arises when a given type of molecule can form two or more solid phases with different crystal structures, and is a critically important concept in understanding fundamental properties of solids and optimizing their applications. In the case of pharmaceutical materials, for example, physicochemical and pharmacokinetic properties such as solubility and bioavailability may differ significantly between different polymorphs of a drug molecule as a consequence of their different crystal structures. For this reason, it is essential to characterize all polymorphic forms that are accessible to the molecule of interest and to establish the relative stabilities of the polymorphs. Importantly, early detection of small amounts of a minor polymorphic phase in a mixture with a major polymorphic phase, of the type that may arise, for example, during solid-state phase transformations between polymorphs, is critical to anticipate the evolution of the molecule of interest towards a polymorph with unfavorable properties.A wide range of diffraction, microscopy and spectroscopy techniques provide key information in polymorphism research, although the ability to detect a small amount of a minor polymorphic phase within a major polymorph remains challenging.
Solid-state NMR spectroscopy is a powerful technique for polymorph characterization as chemical shifts and relaxation times are discriminatingly sensitive to differences in the local environments that exist in different polymorphs.6–16 Unfortunately, the intrinsically low sensitivity of NMR may limit the opportunity to detect a minor polymorphic phase in a mixture with a major polymorphic phase.17–19
Interestingly, recent advances in dynamic nuclear polarization (DNP) NMR,20–27 in which polarization from the electron spin of an exogenous polarizing agent28–32 is transferred to nuclear spins through microwave irradiation, can significantly enhance the ability of NMR to detect small amounts of solid phases in heterogenous materials by enhancing the sensitivity of solid-state NMR measurements by several orders of magnitude. Furthermore, DNP NMR may also generate large polarization gradients, allowing domain sizes between ca. 200 nm and 20 µm to be probed through spin diffusion.33 Transport of DNP-enhanced polarization from the polarizing agent by spin diffusion allows solid-state DNP NMR to be exploited to establish domain sizes in multicomponent blends, for example in biomolecules,34,35 microcrystalline solids,36 pharmaceutical formulations,37 porous materials38 and nanoparticles.39,40
As polymorphs of a given molecule generally have different DNP signal enhancements,41 transport of DNP-enhanced polarization in a solid particle composed of several polymorphs may not be uniform, which represents the basis of a strategy, reported for the first time in this article, to gain detailed insights into the nature of solid materials that comprise mixtures of polymorphic phases.
More specifically, we show that NMR contrast based on distinct transport of DNP-enhanced polarization can be exploited to allow a small amount of a minor polymorphic phase to be detected within a sample of a major polymorphic phase. Furthermore, we show that these experiments can yield quantitative information on the spatial distribution and domain sizes of the two polymorphic phases within particles of the powder sample.
Results and discussion
NMR contrast based on distinct transport of DNP-enhanced polarization
To demonstrate our strategy, we focus on m-aminobenzoic acid (m-ABA; Scheme 1), a system of interest in polymorphism research.42–46 Five polymorphs of m-ABA (denoted Forms I to V) have been reported,42,43 which either contain the zwitterionic tautomer (Forms I, III and IV) or the non-zwitterionic tautomer (Forms II and V) of m-ABA. The crystal structures of Form II (determined42 by single-crystal XRD) and Forms III, IV and V (determined43 from powder XRD data) are known. To date, determination of the crystal structure of Form I has proved elusive, although X-ray photoelectron spectroscopy confirms that Form I contains the zwitterionic tautomer.43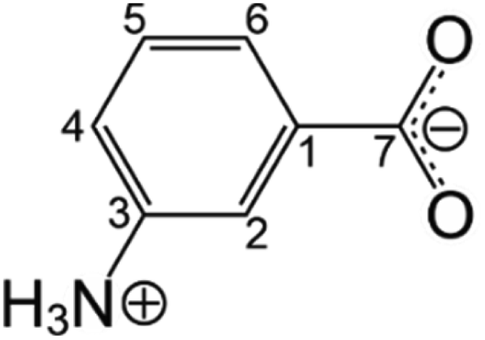 | ||
| Scheme 1 Molecular structure of m-ABA in the zwitterionic tautomer. | ||
Our DNP NMR strategy is demonstrated in studies of freshly prepared samples of Form I of m-ABA at natural isotopic abundance, which show evidence (as discussed below) for the presence of a small amount of Form III. Form I is a meta-stable polymorph that is known43,45 to transform over time to Form III, which is thermodynamically more stable. Direct evidence for the polymorphic transformation of Form I to Form III has been observed by in situ solid-state 13C NMR studies45,46 of the crystallization of m-ABA from methanol.
First, we consider experiments on a freshly prepared powder sample of Form I of m-ABA impregnated with a solution (60 mM) containing the DNP polarizing agent TEKPol in 1,1,2,2-tetrachloroethane.47 Following impregnation, the solution phase containing the polarizing agent is in contact with the surface of the solid particles in the powder sample, but the polarizing agent is not present within the particles.
To assess whether contact between the solution containing the polarizing agent and particles of Form I may induce the polymorphic transformation to Form III, powder XRD data were recorded for a sample of Form I after impregnation with the polarizing solution, both before and after quenching to liquid nitrogen temperature (see ESI; Section 1.1†). The powder XRD data do not show any detectable amount of Form III, indicating that the conditions required for the DNP experiment (i.e., impregnation with the polarizing solution and low-temperature measurements) do not induce any significant extent of formation of Form III within a freshly prepared powder sample of Form I.
Polarization transfer from the polarizing solution to particles in the powder was monitored by 1H–13C CPMAS saturation recovery measurements for different DNP polarization times τ, using the pulse sequence in Fig. 1a. 1H–13C CPMAS NMR spectra recorded under microwave irradiation and using short (τ = 12 s; Fig. 1b) and long (τ = 350 s; Fig. 1c) DNP polarization times both show the spectral features characteristic of Form I.44 However, the difference spectrum (Fig. 1d) obtained by subtracting the spectrum recorded with short polarization time from the spectrum recorded with long polarization time exhibits the spectral features characteristic of Form III (Fig. 1e). Significantly, this result indicates that the freshly prepared powder sample of Form I is actually a blend of polymorphic phases, comprising a major phase (Form I) and a minor phase (Form III). As Form I and Form III both contain the zwitterionic tautomer of m-ABA43 and have similar values of isotropic 13C NMR chemical shifts,44 the presence of a small amount of Form III within a sample of Form I is not readily detected by standard NMR measurements (Fig. 1b and c). We note that the behavior exhibited in Fig. 1b–d (for a sample of Form I impregnated with TEKPol/EtCl4 solution) is also observed for a sample of Form I impregnated with AMUPol/glycerol–D2O solution (see ESI; Section 1.2†).
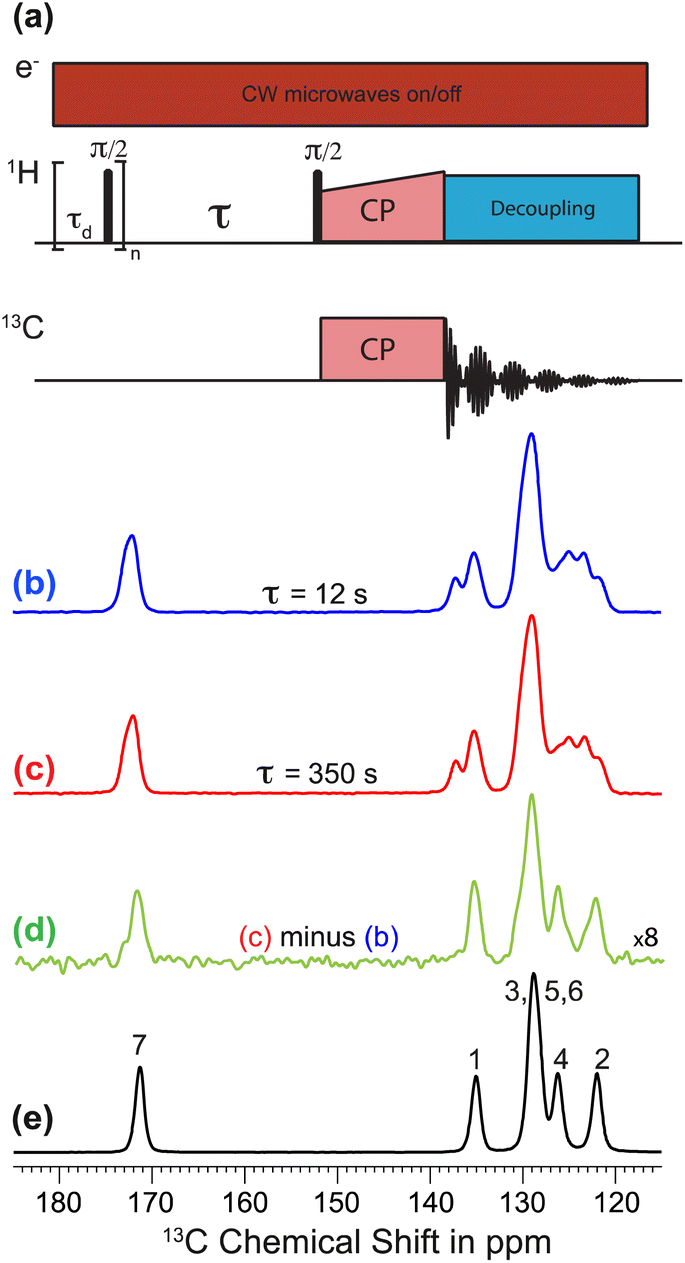 | ||
| Fig. 1 (a) 1H–13C CPMAS saturation-recovery pulse sequence used to record solid-state 13C NMR spectra for different polarization times τ. (b and c) Solid-state 13C NMR spectra recorded at 110 K and under microwave irradiation for a powder sample of Form I of m-ABA impregnated with TEKPol/EtCl4 solution using (b) τ = 12 s and (c) τ = 350 s. (d) Difference spectrum obtained by subtracting the spectrum in (b) from the spectrum in (c), with the intensity scale in the difference spectrum expanded by a factor of 8. (e) Solid-state 13C NMR spectrum recorded at 110 K with τ = 350 s for a powder sample of Form III impregnated with TEKPol/EtCl4 solution. Peak assignments are based on the atom numbering shown in Scheme 1. | ||
Investigation of the spatial distribution and domain sizes of the two polymorphic phases
The distribution of the two polymorphs within the material was explored by examining the build-up of 13C resonances (Fig. 2) in spectra recorded using the 1H–13C CPMAS saturation recovery pulse sequence as a function of the polarization time τ.33 It should be noted that, although solid-state NMR has been exploited to identify the presence of structurally distinct domains within inhomogeneous materials,48–51 it has not been used hitherto to explore the spatial distribution of polymorphic phases of a given molecule within a solid material.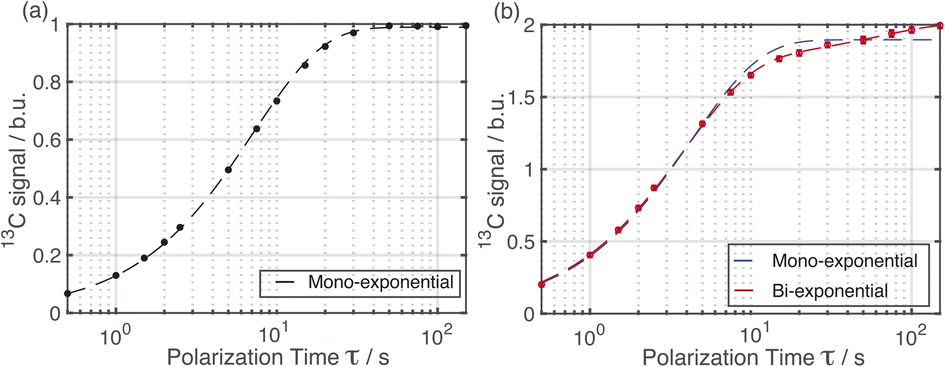 | ||
| Fig. 2 Experimental 13C NMR signal intensities for a powder sample of Form I impregnated with a solution of AMUPol in glycerol/D2O (60/40, v/v). Data were recorded using 1H–13C CPMAS saturation recovery (a) without and (b) with microwave irradiation applied. The signals are normalized to Boltzmann units (b.u.); one Boltzmann unit represents the polarization level reached without DNP or depolarization effects. Fits to the experimental data are shown by dashed lines. The experimental data in (a) are described by a mono-exponential build-up curve with build-up time TB,OFF = 7.2 s. The experimental data in (b) are described by a bi-exponential build-up curve with build-up times TB,ON(1) = 3.8 s and TB,ON(2) = 60 s. | ||
For these measurements, a freshly prepared powder sample of Form I was impregnated with a solution (12 mM) containing the polarizing agent AMUPol in glycerol–D2O (60/40, v/v). In this case, AMUPol was preferred to TEKPol as the DNP properties of AMUPol (e.g. depolarization) are understood in detail,52 allowing a more accurate description of the polarization source in the numerical simulations discussed below. We note that the experimental build-up curves for a powder sample of Form I impregnated with a TEKPol/EtCl4 solution (see ESI; Section 1.3†) are essentially identical to those shown in Fig. 2.
Importantly, the build-up curves derived from solid-state DNP NMR data recorded with and without microwave irradiation are clearly different. Specifically, the build-up curve is mono-exponential with no microwave irradiation applied (Fig. 2a) and bi-exponential with microwave irradiation applied (Fig. 2b). The bi-exponential build-up curve observed with microwave irradiation confirms that the contrast between the two polymorphic phases arises from non-uniform DNP signal enhancements within the sample. We note that the build-up curves shown in Fig. 2 were obtained by monitoring all the 13C NMR resonances between 119 and 138 ppm as a function of the polarizing time τ. Importantly, the build-up curve for the single resonance at 137 ppm (see ESI; Fig. S7†), which is a 13C NMR signal for pure Form I, is mono-exponential with a build-up time of 4 s, which is comparable to the short component of the bi-exponential build-up curve shown in Fig. 2b. Significantly, the same experiments on a freshly prepared powder sample of Form I that had not been impregnated with a solution of AMUPol gave a mono-exponential build-up curve in both cases.
To gain more detailed structural insights from the solid-state DNP NMR data, we have carried out numerical simulations based on five models that represent different spatial distributions of the two polymorphic phases within particles in the powder sample (for more details, see ESI; Section 2†). Our models have been chosen to represent the spatial distribution of the polymorphic phases that would arise from plausible scenarios for the production of Form III by nucleation and growth within particles of Form I, while retaining a level of simplicity (based on justifiable simplifying assumptions) that is required to allow the computational analysis to remain tractable and practicable.
For each model, transport of polarization from the solution containing the polarizing agent to particles in the powder was modelled using simulations of 1H spin diffusion33 based on classical diffusion processes following Fick's second law. This approach allows build-up curves to be computed for the specific spatial distribution of the two polymorphs characteristic of each model, and as a function of the variables that define each model, specifically the particle size and the relative amounts of the two polymorphic phases. By comparison between experimental and simulated build-up curves, the model(s) giving best agreement with the experimental data may be deduced, thus establishing the spatial distribution of the polymorphic phases within the material.
Each model represents a different spatial distribution (shown schematically in Fig. 3) of the two polymorphic phases within particles of the powder sample, and corresponds to a plausible scenario for the production of Form III by nucleation and growth within particles of Form I (which is meta-stable with respect to transformation to Form III). For each model, it is assumed that the material comprises spherical particles of uniform size (defined by radius R) that are initially pure Form I.
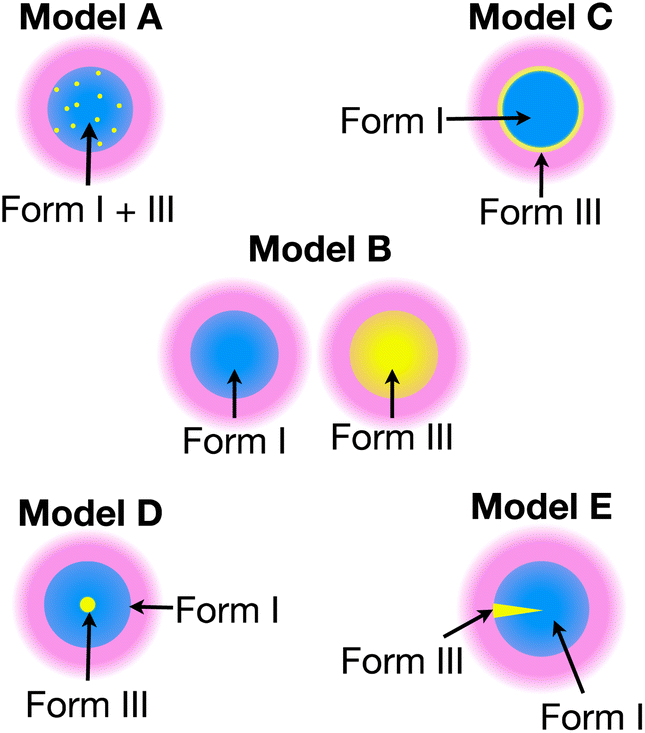 | ||
| Fig. 3 The distribution of the two polymorphs within particles in a powder sample of m-ABA in Models A–E. Each model assumes spherical particles of uniform size (defined by radius R), with regions containing Form I shown in blue and regions containing Form III shown in yellow. The solution (pink) containing the polarizing agent is located on the surface of the particles. | ||
In assessing the quality of agreement between the simulated solid-state DNP NMR data for each model and the experimental data, our numerical simulations involved variation of: (i) the radius R of the spherical particles, and (ii) the relative amounts of Form I and Form III.
Numerical simulations for Models A–E
where D is the spin diffusion coefficient and T1 is the longitudinal relaxation time. We choose a reference spin diffusion coefficient Dref, using the value Dref = 500 nm2 s−1 determined for polystyrene,53 and the spin diffusion coefficient for the material of interest is determined as:
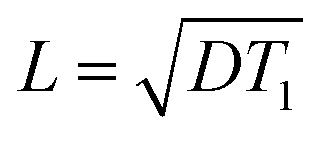
where C is the 1H concentration in the material of interest and Cref = 80 mol dm–3 for the reference material polystyrene. For Form I and Form III of m-ABA, C = 58 mol dm–3. For the polarizing solution containing AMUPol in glycerol–D2O (60/40 v/v), C = 65 mol dm–3. We note that glycerol (with natural isotopic abundances) was used within the solvent mixture in order to increase the polarizing power of the polarizing solution.54–56
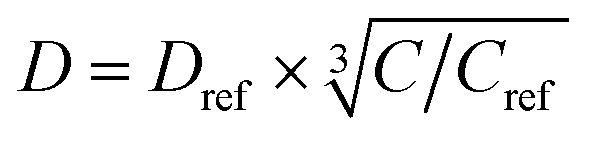
From the equations above, the spin diffusion length is determined to be L = 309 nm for Form III and L = 47 nm for Form I. In a homogeneous distribution in which the domains of Form III are smaller than 10 nm, any polarization gradient between Form I and Form III would be instantly compensated by 1H spin-diffusion, leading to a mono-exponential build-up curve.
Simulated build-up curves for Model E are shown together with the experimental build-up curves in Fig. 4a and b, and experimental and simulated build-up curves for the polarizing solution are shown in Fig. 4c and d. Clearly, excellent agreement is observed between experimental and simulated data for all four curves. In fact, the simulations give mono-exponential behavior for the polarizing solution and for the solid phase not subjected to microwave irradiation, whereas the simulations give bi-exponential behavior under conditions of microwave irradiation. The best agreement between experimental and simulated data for Model E is obtained for particles of radius R = 4.6 µm containing 96% of Form I and 4% of Form III (see ESI; Section 2.3†).
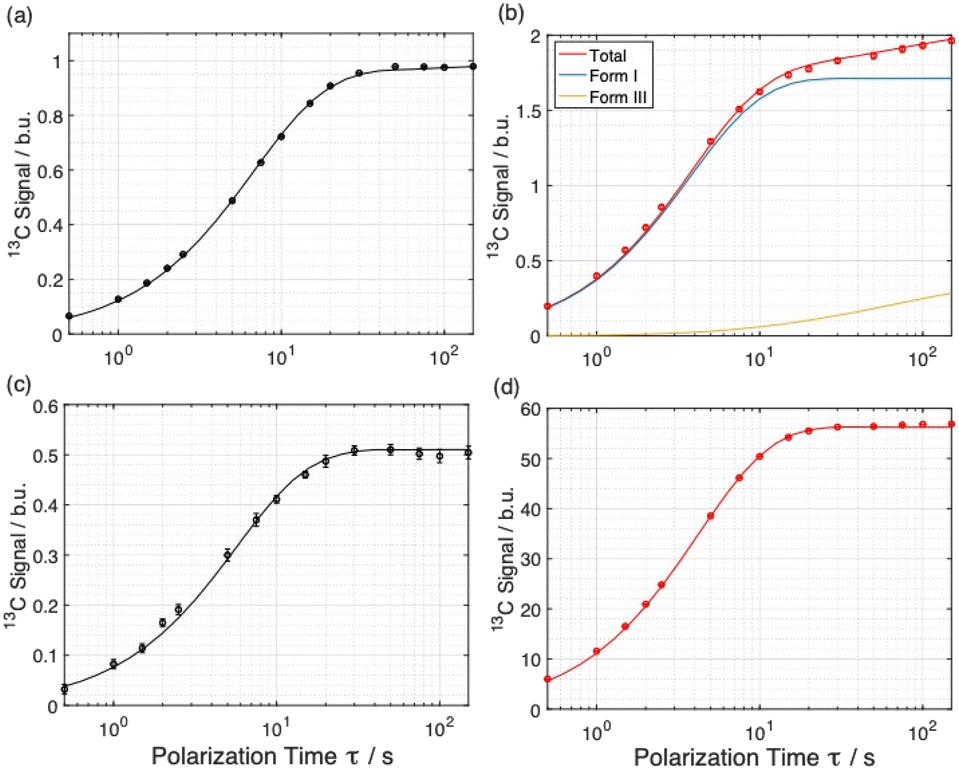 | ||
| Fig. 4 Experimental build-up curves (data points with error bars) for 13C resonances for: (a and b) a powder sample of Form I of m-ABA impregnated with AMUPol/glycerol–D2O solution, and (c and d) the AMUPol/glycerol–D2O solution. Experimental data were recorded using 1H–13C CPMAS saturation recovery as a function of polarization time τ, either (b and d) with or (a and c) without microwave irradiation. Solid lines shown the best-fit simulated build-up curves for Model E obtained with R = 4.6 µm and a composition with 96% of Form I and 4% of Form III. The best-fit simulated build-up curves for the individual components of Form I (blue solid line) and Form III (yellow solid line) are also shown in (b). Signals are expressed in Boltzmann units (b.u.). Note that the error bars are larger than the data points only in (c). | ||
In summary, only the simulated build-up curves for Model B and Model E show good agreement with the experimental build-up curves. In Model B, the two polymorphs are present in different particles, whereas in Model E, the two polymorphs are present as distinct domains within the same particle. Clearly, in order to distinguish whether Model B or Model E is a more accurate representation of the actual distribution of Form III within particles of Form I of m-ABA, information derived from complementary experimental techniques (particularly electron microscopy or Raman microspectrometry) may yield valuable insights.
Concluding remarks
Overall, our results demonstrate the ability of solid-state DNP NMR experiments to detect the presence of a minor polymorphic phase within a major polymorph in a powder sample at natural isotopic abundance, and to establish contrast between the two polymorphic phases. Furthermore, quantitative details of the spatial distribution of the two polymorphic phases within the particles can be obtained through numerical simulations of 1H spin diffusion.Our methodology has been illustrated in a challenging system, for which the NMR signals for the two polymorphs overlap significantly, and for which the crystal structure of the major polymorph (Form I of m-ABA) is unknown. In this regard, it is relevant to highlight the advantages of the methodology reported here compared to the use of powder XRD to characterize the mixture of polymorphic phases. Firstly, we emphasize that powder XRD could not provide insights into the spatial distribution of the different polymorphs within the mixture of polymorphic phases, in contrast to the DNP NMR approach described in this paper; in this regard, we note that Raman microspectrometry is a valuable technique for studying the spatial distributions of different phases (including polymorphic phases) in heterogenous solid materials57–64 on length-scales comparable to the size of the polymorphic domains studied by DNP NMR in the present work. Secondly, with regard to detecting the existence of a small amount (e.g., 5%) of a minor polymorphic phase together with the major polymorphic phase, powder XRD should be able to confirm this fact qualitatively, provided the quality of the powder XRD data is sufficiently high (e.g., with better signal/noise ratio than the powder XRD data in Fig. S1†).
However, reliable quantification of the relative amount of the minor polymorphic phase by analysis of powder XRD data is not necessarily straightforward. The only rigorous way to quantify the relative amounts of the two polymorphic phases by powder XRD would be to carry out a two-phase Rietveld refinement (with the relative amounts of the two phases determined from the refined value of the relative scale factor), but a two-phase Rietveld refinement requires that the structures of both polymorphic phases are already known. In the present case, this approach would not be possible for a mixture of polymorphs involving Form I of m-ABA as the structure of this material is not known. The reason that Rietveld refinement is required in this context is that it allows the effects of “preferred orientation” in the powder sample to be taken quantitatively into account. Simpler approaches (e.g., based on assessing the relative intensities of specific peaks characteristic of the two polymorphs in the powder XRD data) do not provide a reliable or rigorous basis for quantifying the relative amounts of the two polymorphs as this type of approach does not take into account the fact that the two polymorphs in the powder sample may be affected by a different extent of preferred orientation.
Clearly, the method presented here has the potential to reveal the early onset of transformations between polymorphic forms and will thus be a highly valuable addition to existing powder XRD and electron microscopy methods for the investigation of polymorph transformation in powder samples of organic materials.
Methods
Sample preparation and characterization
Solid-state DNP NMR experiments
1H NMR saturation recovery experiments were carried out to determine the different time constants of the (exponential) recovery of the polarization. A train of 90° pulses (comprising 50 pulses separated by 1 ms) was used to saturate the 1H magnetization, with recovery of the 1H magnetization allowed during the polarization delay τ. The magnetization was then transferred to the 13C spin nuclei for detection. The intensities of the 13C NMR signals of m-ABA were measured in the region of the spectrum between 119 and 138 ppm, which was chosen because this region contains the same number of 13C NMR resonances for the two polymorphic forms and the minimum amount of noise.
It is essential to clearly distinguish between the intrinsic build-up time, the effective build-up time in the presence of microwave irradiation, and the effective build-up time in the absence of microwave irradiation. In this regard, T1 denotes the 1H relaxation time measured from a saturation recovery experiment on a pure dry powder sample. The effective 1H build-up time for a powder sample impregnated with the solution containing the polarizing agent is denoted TB,ON (measured in the presence of microwave irradiation) or TB,OFF (measured in the absence of microwave irradiation).
Theoretical build-up curves were simulated using the following parameters measured experimentally: T1,ON(Form I) = 3 s, T1,OFF(Form I) = 6.5 s, T1,ON(Form III) = 214 s, T1,OFF(Form III) = 213 s. An enhancement of 115, a depolarization factor of 0.5, TB,ON(solution) = 4.35 s and TB,OFF(solution) = 5.79 s were measured for the solution containing AMUPol (12 mM) dissolved in glycerol–D2O (60/40 v/v).
The 13C NMR resonances in the high-resolution solid-state 13C NMR spectrum of Form I of m-ABA (see Fig. 1e) were assigned from a 13C–13C DQ dipolar correlation NMR spectrum recorded with a DQ excitation time of 0.4 ms.65
Data availability
ESI† contains additional figures discussed in the text. The code and raw data used to carry out the numerical simulations presented in the manuscript is available at https://zenodo.org/record/8019697.Author contributions
The specific components of the research were carried out as follows: preparation of the sample of m-ABA (C. E. H., A. C. P., S. F. C.); measurement of powder XRD data (C. E. H.); analysis of powder XRD data (C. E. H., K. D. M. H.); measurement of solid-state DNP NMR data (A. C. P., S. F. C., F. Z., S. V., G. M., P. T.); analysis of solid-state DNP NMR data (A. C. P., S. F. C., F. Z., S. V., G. M., K. D. M. H., P. T.); numerical simulations of 1H spin diffusion (A. C. P., S. F. C.). Preparation of the manuscript was coordinated by G. M., K. D. M. H., A. C. P., P. T. with contributions from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Basile Heresanu (CINaM-CNRS/AMU laboratory) and Andrew Williams (Cardiff University) for recording the powder XRD data in Fig. S2.† This project received funding from ERC under the European Union Horizon 2020 research and innovation programme (Grant Agreement No. 758498) and from CNRS under the International Emerging Actions (Grant Agreement No. 00211). We are grateful to Cardiff University for financial support.References
- J. Bernstein, Polymorphism in Molecular Crystals, Oxford University Press, Oxford, 2002 Search PubMed.
- B. Rodríguez-Spong, C. P. Price, A. Jayasankar, A. J. Matzger and N. R. Rodríguez-Hornedo, Adv. Drug Delivery Rev., 2004, 56, 241–274 CrossRef PubMed.
- S. L. Price, Acc. Chem. Res., 2009, 42, 117–126 CrossRef CAS PubMed.
- A. Y. Lee, D. Erdemir and A. S. Myerson, Annu. Rev. Chem. Biomol. Eng., 2011, 2, 259–280 CrossRef CAS PubMed.
- D.-K. Bučar, R. W. Lancaster and J. Bernstein, Angew. Chem., Int. Ed., 2015, 54, 6972–6993 CrossRef PubMed.
- K. D. M. Harris and J. M. Thomas, J. Solid State Chem., 1991, 94, 197–205 CrossRef CAS.
- B. E. Padden, M. T. Zell, Z. Dong, S. A. Schroeder, D. J. W. Grant and E. J. Munson, Anal. Chem., 1999, 71, 3325–3331 CrossRef CAS.
- N. Zumbulyadis, B. Antalek, W. Windig, R. P. Scaringe, A. M. Lanzafame, T. Blanton and M. Helber, J. Am. Chem. Soc., 1999, 121, 11554–11557 CrossRef CAS.
- R. K. Harris, Analyst, 2006, 131, 351–373 RSC.
- J. M. Griffin, D. R. Martin and S. P. Brown, Angew. Chem., Int. Ed., 2007, 46, 8036–8038 CrossRef CAS PubMed.
- R. K. Harris, P. Hodgkinson, C. J. Pickard, J. R. Yates and V. Zorin, Magn. Reson. Chem., 2007, 45, S174–S186 CrossRef CAS PubMed.
- C. Bonhomme, C. Gervais, F. Babonneau, C. Coelho, F. Pourpoint, T. Azaïs, S. E. Ashbrook, J. M. Griffin, J. R. Yates, F. Mauri and C. J. Pickard, Chem. Rev., 2012, 112, 5733–5779 CrossRef CAS PubMed.
- D. Stueber and S. Jehle, J. Pharm. Sci., 2017, 106, 1828–1838 CrossRef CAS PubMed.
- F. M. Paruzzo, A. Hofstetter, F. Musil, S. De, M. Ceriotti and L. Emsley, Nat. Commun., 2018, 9, 4501 CrossRef PubMed.
- P. Hodgkinson, Prog. Nucl. Magn. Reson. Spectrosc., 2020, 118–119, 10–53 CrossRef CAS PubMed.
- C. J. H. Smalley, H. E. Hoskyns, C. E. Hughes, D. N. Johnstone, T. Willhammar, M. T. Young, C. J. Pickard, A. J. Logsdail, P. A. Midgley and K. D. M. Harris, Chem. Sci., 2022, 13, 5277–5288 RSC.
- K. Maruyoshi, D. Iuga, A. E. Watts, C. E. Hughes, K. D. M. Harris and S. P. Brown, J. Pharm. Sci., 2017, 106, 3372–3377 CrossRef CAS PubMed.
- Y. T. A. Wong, R. L. E. G. Aspers, M. Uusi-Penttilä and A. P. M. Kentgens, Anal. Chem., 2022, 94, 16667–16674 CrossRef CAS.
- Y. Du, P. Phyo, M. Li, B. Sorman, M. McNevin, W. Xu, Y. Liu and Y. Su, Anal. Chem., 2022, 94, 15341–15349 CrossRef CAS PubMed.
- Q. Z. Ni, E. Daviso, T. V. Can, E. Markhasin, S. K. Jawla, T. M. Swager, R. J. Temkin, J. Herzfeld and R. G. Griffin, Acc. Chem. Res., 2013, 46, 1933–1941 CrossRef CAS PubMed.
- A. J. Rossini, A. Zagdoun, M. Lelli, A. Lesage, C. Copéret and L. Emsley, Acc. Chem. Res., 2013, 46, 1942–1951 CrossRef CAS PubMed.
- A. J. Rossini, J. Phys. Chem. Lett., 2018, 9, 5150–5159 CrossRef CAS PubMed.
- R. W. Hooper, B. A. Klein and V. K. Michaelis, Chem. Mater., 2020, 32, 4425–4430 CrossRef CAS.
- I. B. Moroz and M. Leskes, Annu. Rev. Mater. Res., 2022, 52, 25–55 CrossRef.
- T. Biedenbänder, V. Aladin, S. Saeidpour and B. Corzilius, Chem. Rev., 2022, 122, 9738–9794 CrossRef PubMed.
- W. Y. Chow, G. De Paëpe and S. Hediger, Chem. Rev., 2022, 122, 9795–9847 CrossRef CAS PubMed.
- D. Xiao, S. Xu, N. J. Brownbill, S. Paul, L.-H. Chen, S. Pawsey, F. Aussenac, B.-L. Su, X. Han, X. Bao, Z. Liu and F. Blanc, Chem. Sci., 2018, 9, 8184–8193 RSC.
- C. Sauvée, M. Rosay, G. Casano, F. Aussenac, R. T. Weber, O. Ouari and P. Tordo, Angew. Chem., Int. Ed., 2013, 52, 10858–10861 CrossRef PubMed.
- G. Mathies, M. A. Caporini, V. K. Michaelis, Y. Liu, K.-N. Hu, D. Mance, J. L. Zweier, M. Rosay, M. Baldus and R. G. Griffin, Angew. Chem., Int. Ed., 2015, 54, 11770–11774 CrossRef CAS PubMed.
- D. J. Kubicki, G. Casano, M. Schwarzwälder, S. Abel, C. Sauvée, K. Ganesan, M. Yulikov, A. J. Rossini, G. Jeschke, C. Copéret, A. Lesage, P. Tordo, O. Ouari and L. Emsley, Chem. Sci., 2016, 7, 550–558 RSC.
- R. Yao, D. Beriashvili, W. Zhang, S. Li, A. Safeer, A. Gurinov, A. Rockenbauer, Y. Yang, Y. Song, M. Baldus and Y. Liu, Chem. Sci., 2022, 13, 14157–14164 RSC.
- T. Halbritter, R. Harrabi, S. Paul, J. van Tol, D. Lee, S. Hediger, S. T. Sigurdsson, F. Mentink-Vigier and G. De Paëpe, Chem. Sci., 2023, 14, 3852–3864 RSC.
- A. C. Pinon, J. Schlagnitweit, P. Berruyer, A. J. Rossini, M. Lelli, E. Socie, M. Tang, T. Pham, A. Lesage, S. Schantz and L. Emsley, J. Phys. Chem. C, 2017, 121, 15993–16005 CrossRef CAS.
- P. C. A. van der Wel, J. R. Lewandowski and R. G. Griffin, J. Am. Chem. Soc., 2007, 129, 5117–5130 CrossRef CAS PubMed.
- A. Jantschke, E. Koers, D. Mance, M. Weingarth, E. Brunner and M. Baldus, Angew. Chem., Int. Ed., 2015, 54, 15069–15073 CrossRef CAS PubMed.
- A. J. Rossini, A. Zagdoun, F. Hegner, M. Schwarzwälder, D. Gajan, C. Copéret, A. Lesage and L. Emsley, J. Am. Chem. Soc., 2012, 134, 16899–16908 CrossRef CAS PubMed.
- A. J. Rossini, C. M. Widdifield, A. Zagdoun, M. Lelli, M. Schwarzwälder, C. Copéret, A. Lesage and L. Emsley, J. Am. Chem. Soc., 2014, 136, 2324–2334 CrossRef CAS PubMed.
- O. Lafon, A. S. L. Thankamony, T. Kobayashi, D. Carnevale, V. Vitzthum, I. I. Slowing, K. Kandel, H. Vezin, J.-P. Amoureux, G. Bodenhausen and M. Pruski, J. Phys. Chem. C, 2013, 117, 1375–1382 CrossRef CAS.
- J. Viger-Gravel, A. Schantz, A. C. Pinon, A. J. Rossini, S. Schantz and L. Emsley, J. Phys. Chem. B, 2018, 122, 2073–2081 CrossRef CAS PubMed.
- A. C. Pinon, U. Skantze, J. Viger-Gravel, S. Schantz and L. Emsley, J. Phys. Chem. A, 2018, 122, 8802–8807 CrossRef CAS PubMed.
- A. C. Pinon, A. J. Rossini, C. M. Widdifield, D. Gajan and L. Emsley, Mol. Pharm., 2015, 12, 4146–4153 CrossRef CAS PubMed.
- A. Théorêt, Spectrochim. Acta, Part A, 1971, 27, 11–18 CrossRef.
- P. A. Williams, C. E. Hughes, G. K. Lim, B. M. Kariuki and K. D. M. Harris, Cryst. Growth Des., 2012, 12, 3104–3113 CrossRef CAS.
- C. E. Hughes, P. A. Williams and K. D. M. Harris, Angew. Chem., Int. Ed., 2014, 53, 8939–8943 CrossRef CAS PubMed.
- K. D. M. Harris, C. E. Hughes and P. A. Williams, Solid State Nucl. Magn. Reson., 2015, 65, 107–113 CrossRef CAS PubMed.
- K. D. M. Harris, C. E. Hughes, P. A. Williams and G. R. Edwards-Gau, Acta Crystallogr., Sect. C: Struct. Chem., 2017, 73, 137–148 CrossRef CAS.
- A. Zagdoun, G. Casano, O. Ouari, M. Schwarzwälder, A. J. Rossini, F. Aussenac, M. Yulikov, G. Jeschke, C. Copéret, A. Lesage, P. Tordo and L. Emsley, J. Am. Chem. Soc., 2013, 135, 12790–12797 CrossRef CAS.
- S. Schantz and N. Ljungqvist, Macromolecules, 1993, 26, 6517–6524 CrossRef CAS.
- T. N. Pham, S. A. Watson, A. J. Edwards, M. Chavda, J. S. Clawson, M. Strohmeier and F. G. Vogt, Mol. Pharm., 2010, 7, 1667–1691 CrossRef CAS.
- P. Duan, M. S. Lamm, F. Yang, W. Xu, D. Skomski, Y. Su and K. Schmidt-Rohr, Mol. Pharm., 2020, 17, 3567–3580 CrossRef CAS.
- D. Stueber and Z. E. X. Dance, Anal. Chem., 2020, 92, 11095–11102 CrossRef CAS PubMed.
- S. Hediger, D. Lee, F. Mentink-Vigier, G. De Paëpe, MAS-DNP Enhancements: Hyperpolarization, Depolarization, and Absolute Sensitivity, eMagRes, 2018, 7, 105–116 Search PubMed.
- T. Wenckebach, Essentials of Dynamic Nuclear Polarization, Spindrift Publications, The Netherlands, 2016 Search PubMed.
- P. Thureau, M. Juramy, F. Ziarelli, S. Viel and G. Mollica, Solid State Nucl. Magn. Reson., 2019, 99, 15–19 CrossRef CAS PubMed.
- K. Kundu, F. Mentink-Vigier, A. Feintuch and S. Vega, eMagRes, 2019, 8, 295–338 CAS.
- N. A. Prisco, A. C. Pinon, L. Emsley and B. F. Chmelka, Phys. Chem. Chem. Phys., 2021, 23, 1006–1020 RSC.
- T. Wu and L. Yu, Pharm. Res., 2006, 23, 2350–2355 CrossRef CAS PubMed.
- M. J. Henson and L. Zhang, Appl. Spectrosc., 2006, 60, 1247–1255 CrossRef CAS PubMed.
- A. Brillante, I. Bilotti, R. G. Della Valle, E. Venuti and A. Girlando, CrystEngComm, 2008, 10, 937–946 RSC.
- K. Greco and R. Bogner, Mol. Pharm., 2010, 7, 1406–1418 CrossRef CAS PubMed.
- J. Marti-Rujas, A. Desmedt, K. D. M. Harris and F. Guillaume, J. Am. Chem. Soc., 2004, 126, 11124–11125 CrossRef CAS.
- B. A. Palmer, A. Le Comte, K. D. M. Harris and F. Guillaume, J. Am. Chem. Soc., 2013, 135, 14512–14515 CrossRef CAS PubMed.
- E. Widjaja, P. Kanaujia, G. Lau, W. K. Ng, M. Garland, C. Saal, A. Hanefeld, M. Fischbach, M. Maio and R. B. H. Tan, Eur. J. Pharm. Sci., 2011, 42, 45–54 CrossRef CAS PubMed.
- J. R. Petriglieri, E. Salvioli-Mariani, L. Mantovani, M. Tribaudino, P. P. Lottici, C. Laporte-Magoni and D. Bersani, J. Raman Spectrosc., 2015, 46, 953–958 CrossRef CAS.
- M. Dekhil, G. Mollica, T. T. Bonniot, F. Ziarelli, P. Thureau and S. Viel, Chem. Commun., 2016, 52, 8565–8568 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc02063k |
| This journal is © The Royal Society of Chemistry 2023 |