
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Designing complex Pb3SBrxI4−x chalcohalides: tunable emission semiconductors through halide-mixing†
Alison N.
Roth
ab,
Yunhua
Chen
ab,
Anuluxan
Santhiran
 ab,
Jemima
Opare-Addo
ab,
Eunbyeol
Gi
ab,
Emily A.
Smith
ab,
Aaron J.
Rossini
ab and
Javier
Vela
*ab
ab,
Jemima
Opare-Addo
ab,
Eunbyeol
Gi
ab,
Emily A.
Smith
ab,
Aaron J.
Rossini
ab and
Javier
Vela
*ab
aUS DOE Ames National Laboratory, Ames, Iowa 50010, USA
bDepartment of Chemistry, Iowa State University, Ames, Iowa 50011, USA. E-mail: vela@iastate.edu
First published on 16th October 2023
Abstract
Chalcohalides are desirable semiconducting materials due to their enhanced light-absorbing efficiency and stability compared to lead halide perovskites. However, unlike perovskites, tuning the optical properties of chalcohalides by mixing different halide ions into their structure remains to be explored. Here, we present an effective strategy for halide-alloying Pb3SBrxI4−x (1 ≤ x ≤ 3) using a solution-phase approach and study the effect of halide-mixing on structural and optical properties. We employ a combination of X-ray diffraction, electron microscopy, and solid-state NMR spectroscopy to probe the chemical structure of the chalcohalides and determine mixed-halide incorporation. The absorption onsets of the chalcohalides blue-shift to higher energies as bromide replaces iodide within the structure. The photoluminescence maxima of these materials mimics this trend at both the ensemble and single particle fluorescence levels, as observed by solution-phase and single particle fluorescence microscopy, respectively. These materials exhibit superior stability against moisture compared to traditional lead halide perovskites, and IR spectroscopy reveals that the chalcohalide surfaces are terminated by both amine and carboxylate ligands. Electronic structure calculations support the experimental band gap widening and volume reduction with increased bromide incorporation, and provide useful insight into the likely atomic coloring patterns of the different mixed-halide compositions. Ultimately, this study expands the range of tunability that is achievable with chalcohalides, which we anticipate will improve the suitability of these semiconducting materials for light absorbing and emission applications.
Introduction
Lead halide perovskites are well known semiconducting materials for light harvesting and light emitting devices.1 Their impressive optical properties, such as tunable band gaps over the visible and near-IR regions2 and high photoluminescence quantum yields (PLQY)3 are often attainable due to their high defect tolerance.4 However, halide perovskites notoriously suffer from poor thermal and phase stability under irradiation due to the weak ionic interactions of metal–halide bonds, often resulting in halide migration, phase segregation, and PL instability.5,6 This challenge continues to limit full-scale implementation of halide perovskites into their anticipated technologically relevant applications.7,8In light of this challenge, chalcohalides are gaining attention as promising alternative materials to halide perovskites.9–11 In principle, chalcohalides are expected to combine the optimal features of both chalcogenides and halide perovskites: that is, enhanced stability and light absorbing efficiency, respectively.12,13 Multinary chalcohalides have already demonstrated favorable structural and optical properties for photovoltaics.14,15 Other compositions are also being explored for applications including thermoelectrics,16,17 photocatalysis,18 and second harmonic generation.19,20 While solid-state reactions are primarily used to isolate these materials, the solution-phase chemistry of colloidal chalcohalides is rapidly expanding as a way to achieve better control over phase purity, particle size and morphology and, consequently, optoelectronic properties.21–23
An exciting opportunity is to explore the effects of halide-alloying on chalcohalide semiconductors.24 Similar to mixed-halide perovskites (MHPs), introducing various types of halide ions into the structure of chalcohalides could lead to desirable band gap tunability while maintaining the enhanced stability and other characteristics displayed by chalcogenide-based materials. A suitable candidate for exploring this halide-alloying strategy is one of the few existing mixed-halide chalcohalides, P21/m Pb3SBrI3. Recently isolated from a hydrothermal process, Pb3SBrI3 features a pseudo-two-dimensional layered structure with a narrow indirect band gap (Fig. 1).25 The low dimensionality of Pb3SBrI3 makes it unique among other mixed-halide chalcohalide compositions,25,26 as these features may be suggestive of anisotropic optical properties that are worth exploring in more detail.27,28 Furthermore, based on similarities with related quaternary chalcohalides, such as Pb2SbS2I3, we hypothesized that Pb3SBrI3 and other mixed-halide compositions may be accessible using a thiocyanate-based solution-phase synthesis.21–23
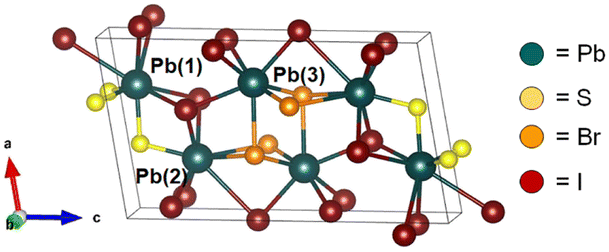 | ||
| Fig. 1 Unit cell of monoclinic (P21/m) Pb3SBrI3.25 | ||
In this work, we explore the structural and optical properties of quaternary mixed-halide chalcohalides with the general composition Pb3SBrxI4−x (1 ≤ x ≤ 3). X-ray diffraction (XRD) in combination with 207Pb solid-state (ss) NMR and electron microscopy (EM) confirm successful halide alloying and reveal that the materials adopt highly anisotropic morphologies. Interestingly, solution-phase spectroscopy and fluorescence microscopy show that the mixed-halide chalcohalides are photoluminescent both as an ensemble and at the single particle level. Their band gap and PL emission energies blue-shift as the content of bromine relative to iodine increases. We used DFT calculations to assess the atomic coloring pattern of the bromine-rich mixed-halide compositions and their corresponding optical properties. Our results suggest that halide-alloying may be a useful strategy for further modifying and enhancing the opto-electronic properties of chalcohalide semiconductors.
Results and discussion
Chalcohalide synthesis and halide mixing
Pb3SBrI3 adopts a monoclinic (P21/m) structure composed of three lead sites with possible coordination environments of [PbS3I5], [PbSBr2I5], and [PbBr3I4], respectfully (Fig. 1). We prepare Pb3SBrI3 from solution using a heat-up approach—additional Experimental details appear in the ESI File† (Scheme 1). Standard starting mixtures consist of 19 mM Pb(SCN)2, 19–57 mM each of PbBr2 and PbI2, 0.25 mL each of oleylamine (oleylNH2) and oleic acid, and 10 mL of 1-octadecene (ODE). Upon heating to temperatures above 150 °C, the reaction mixture initially undergoes a distinct color change from light yellow to dark red, indicating decomposition of the thiocyanate precursor.29,30 Once the temperature reaches 180 °C, the reaction mixture gradually transforms to a vivid yellow-orange color over the course of ≥1 h, indicating the formation of the chalcohalide. Varying the initial concentrations of PbI2 and PbBr2, respectively, from 57 mM and 19 mM, to 38 mM and 19 mM, 19 mM and 19 mM, and 19 mM and 38 mM enables the preparation of Pb3SBrxI4−x (1 ≤ x ≤ 3) phases with nominal halide synthetic loadings of 25%, 33%, 50%, and 66% Br, respectively. These reactions generally occur at 180–200 °C for 60–90 min, resulting in bright yellow-colored suspensions (Table 1, see ESI†).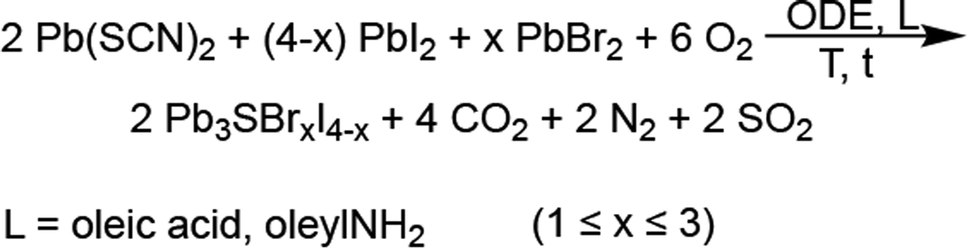 | ||
| Scheme 1 Solution-phase synthesis of quaternary mixed-halide chalcohalides.23,29 | ||
| Precursors | Ligandsa (mL) | T (°C) | t (min) | Productb | Band gapc (eV) | Widthd (Scherrer)e (w, nm) | Lengthd (l, μm) | Aspect ratio (l/w) | |
|---|---|---|---|---|---|---|---|---|---|
| Pb–S (mmol) | Pb![[X with combining low line]](https://www.rsc.org/images/entities/char_0058_0332.gif) 2 (mmol) 2 (mmol) |
||||||||
| a ODE = 1-octadecene (10 mL); oleylNH2 = oleylamine. b From EDS. c From Tauc plots.50 d From SEM. e From XRD. | |||||||||
| Pb(SCN)2 (0.2) | Br (0.2), I (0.6) | OleylNH2 (0.25) | 180 | 90 | Pb2.9SBr1.1I3.0 | 2.22 | 94 ± 22 (70 ± 31) | 1.0 ± 0.4 | 11 |
| Oleic acid (0.25) | |||||||||
| Pb(SCN)2 (0.2) | Br (0.2), I (0.4) | OleylNH2 (0.25) | 180 | 60 | Pb3.0SBr1.2I2.8 | 2.26 | 150 ± 30 (129 ± 56) | 3.1 ± 1.3 | 21 |
| Oleic acid (0.25) | |||||||||
| Pb(SCN)2 (0.2) | Br (0.2), I (0.2) | OleylNH2 (0.25) | 180 | 60 | Pb2.8SBr1.8I1.9 | 2.29 | 170 ± 28 (152 ± 92) | 3.0 ± 0.9 | 18 |
| Oleic acid (0.25) | |||||||||
| Pb(SCN)2 (0.2) | Br (0.4), I (0.2) | OleylNH2 (0.25) | 200 | 60 | Pb3.0SBr2.6I1.5 | 2.33 | 270 ± 80 (245 ± 116) | 4.3 ± 1.5 | 16 |
| Oleic acid (0.25) | |||||||||
Powder X-ray diffraction (XRD) shows that the reaction with a synthetic loading of 25% Br—relative to total halides—results in the known P21/m Pb3SBrI3 phase, although a few reflections appear broadened and/or selectively attenuated (Fig. 2). We hypothesized that this may be caused by size (Scherrer) effects as well as by anisotropic (preferred) particle growth, similar to what is observed for mixed-cation chalcohalide nanorods23,31 and Pb5S2I6 nanoparticles.32 Indeed, scanning electron microscopy (SEM) confirms that the mixed-halide chalcohalides consist of elongated, high-aspect ratio, rod- and needle-like particles (Fig. 2d). The average rod length of the mixed-halides is between 1–4 μm, and the average rod diameter 100–300 nm, in close agreement with their average Scherrer size from XRD (see Table 1). In general, the average rod diameter increases with higher %Br synthetic loading, suggesting a higher reactivity for the bromide precursor.33
 | ||
| Fig. 2 (a) Powder XRD patterns of mixed-halide chalcohalides prepared with different concentrations of PbI2 and PbBr2 (see details in ESI†). (b) Normalized XRD patterns showing the shift in the (200) reflection to higher 2θ with increasing %Br. (c) Position of the (200) and (003) reflections and a lattice parameter as a function of %Br. (d) SEM images showing the rod-like morphology of the mixed-halide chalcohalides: Pb2.9SBr1.1I3.0, Pb3.0SBr1.2I2.8, Pb2.8SBr1.8I1.9, and Pb3.0SBr2.6I1.5. (e) EDS analysis of mixed-halide chalcohalides as a function of %Br. | ||
Powder X-ray diffraction (XRD) shows that higher Br synthetic loadings between 33% and 66% result in crystalline solids with patterns similar to that of Pb3SBrI3. However, the diffraction peaks in these patterns progressively shift to higher 2θ values—corresponding to lower d-spacings—with the largest shift observed for the material with the highest (66%) Br loading (Fig. 2a–c). These XRD features are very characteristic of similar materials such as mixed-halide perovskites (MHPs), where XRD reflections shift to wider (2θ) angles with increasing incorporation of smaller halide ions.34–36 The shift in XRD reflections indicates a steady reduction of lattice parameters and, consequently, a compression of the unit cell volume relative to the original Pb3SBrI3 crystal structure. Thus, the volume compression observed with increasing %Br is consistent with smaller Br− (rcrys = 1.82 Å) replacing larger I− (rcrys = 2.06 Å)37 within the structure, supporting the idea of successful halide-alloying in Pb3SBrxI4−x (1 ≤ x ≤ 3). Interestingly, attempts to incorporate more Br into the chalcohalide were unsuccessful. For example, 75% or higher Br synthetic loadings lead to powder XRD patterns that showed crystalline impurities along with the quaternary material.
Energy dispersive spectroscopy (EDS) confirms mixed-halide phases within the successful alloying regime become richer in Br and poorer in I with increasing Br loading, yielding quaternary phases corresponding to Pb2.9SBr1.1I3.0 (25% Br), Pb3.0SBr1.2I2.8 (33% Br), Pb2.8SBr1.8I1.9 (50% Br), and Pb3.0SBr2.6I1.5 (66% Br), respectively (Fig. 2e). High-resolution transmission electron microscopy (HRTEM) of Pb2.8SBr1.8I1.9 shows that (002) and (001) lattice planes stack along the narrow diameter dimension of the rods while (1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 1) lattice planes stack along the longer rod length (see ESI†). These findings are consistent with the rods growing along the [010] b-direction (rod length) and their diameter being along the [001] c-direction (rod width). In a few cases, high resolution TEM revealed a few small ≤4–5 nm spots—PbI2 or PbS—along the rods. Nonetheless, EM confirms that the quaternary chalcohalides are indeed alloyed with bromine and iodine, in agreement with the XRD results.
1) lattice planes stack along the longer rod length (see ESI†). These findings are consistent with the rods growing along the [010] b-direction (rod length) and their diameter being along the [001] c-direction (rod width). In a few cases, high resolution TEM revealed a few small ≤4–5 nm spots—PbI2 or PbS—along the rods. Nonetheless, EM confirms that the quaternary chalcohalides are indeed alloyed with bromine and iodine, in agreement with the XRD results.
Effect of precursor on phase evolution
As part of our initial efforts aimed at finding suitable syntheses for mixed chalcohalide semiconductors, we screened different sulfur precursors. Under identical conditions to those in the first row of Table 1 (see ESI†), the use of Pb(SCN)2 results in quick formation of chalcohalide; only the desired Pb3SBrI3 quaternary phase is observed after 0.5 h reaction (Fig. 3). In contrast, both thiourea (SC(NH2)2) and elemental sulfur (S8) react much more slowly, achieving only ca. 40–50% quaternary phase purity after 1.5 h reaction. In both cases, unreacted crystalline PbI2 is still present; further, the ternary chalcohalide Pb5S2I6 is also observed. Because the appearance of Pb3SBrI3 coincides with the disappearance of Pb5S2I6, we speculate that the formation of the lower order, ternary phase may precede the formation of the desired, mixed-halide, quaternary phase.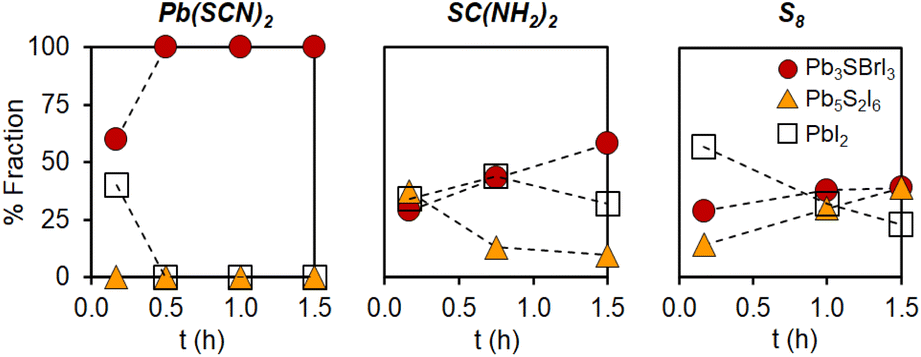 | ||
| Fig. 3 Phase evolution of mixed-halide quaternary chalcohalide Pb3SBrI3vs. ternary and binary impurities over time as a function of sulfur precursor used (under identical conditions to those in the first row of Table 1; see Experimental in the ESI†). | ||
We believe the key to understanding these differences is the existence of preformed Pb–S bonds in Pb(SCN)2.21,23,29 According to the literature, Pb–S bonds (ΔHdiss = 398 kJ mol−1) are much stronger than Pb–X bonds (X = Br or I, ΔHdiss = 248–194 kJ mol−1).38 The stronger, more robust Pb–S bonds thus require a higher activation energy to make (or break) than the Pb-halide bonds (this trend is also evident when the solubility product constants of the corresponding binaries are used as surrogates for a stability measure: Ksp = 3.0 × 10−28 for PbS, 9.8 × 10−9 for PbI2, and 6.6 × 10−6 for PbBr2).38 Because Pb(SCN)2 already contains preformed Pb–S bonds, it is able to more quickly react and deliver [Pb–S] building blocks needed for the effective nucleation and growth of multinary chalcohalide nanocrystals. Thiourea and elemental sulfur lack this advantage, and must overcome a larger kinetic barrier to form Pb–S bonds; therefore, these alternative precursors are inferior in kinetics, phase purity, and yield compared to Pb(SCN)2.
Enhanced stability of chalcohalides
In order to further assess the stability of mixed-halide chalcohalides compared to traditional halide perovskites, we compared them to standard samples of CsPbBr3 and CsPbI3 as reference materials.35 After stirring all individual samples in water for 48 h, the chalcohalides demonstrate impressive stability compared to the halide perovskites. Specifically, powder XRD reveals that Pb2.9SBr1.1I3.0 and Pb3.0SBr2.6I1.5 remain largely intact with ≥85% of the original semiconductor remaining (Fig. 4, see ESI†) after water treatment; in contrast, CsPbI3 and CsPbBr3 easily decompose to PbI2 and PbBr2, respectively. Furthermore, the chalcohalides display good stability over time under standard atmospheric conditions. After 3 weeks under air at room temperature (21 °C), Pb3.0SBr2.6I1.5 remains intact without any evidence of crystalline impurities or decomposition, while Pb2.9SBr1.1I3.0 only slightly decomposes to PbI2 over time. Thus, the more bromine-rich compositions appear to have higher overall stability compared to the more iodine-rich compositions, suggesting that stability increases with the incorporation of the slightly harder, more electronegative bromine ions into the structure. These findings mirror similar trends in the photo- and thermal stability along the CsPbX3 series (X = Cl, Br, or I).39,40 From these analyses, we conclude that mixed-halide chalcohalides possess better ambient and moisture stability compared to halide perovskites, which we anticipate will be help stimulate their incorporation into energy conversion devices.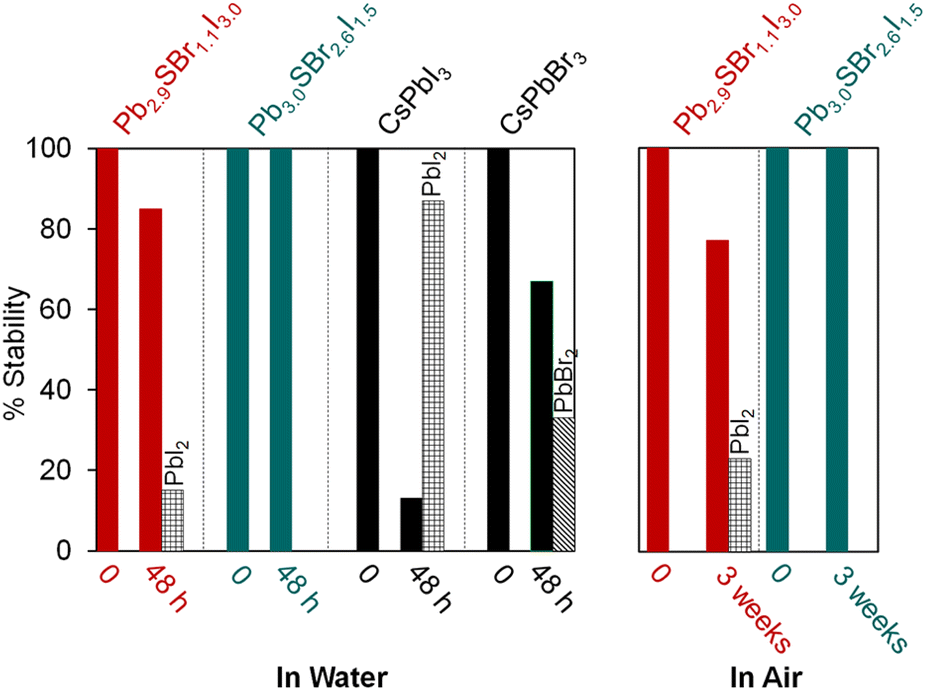 | ||
| Fig. 4 Relative stability of Pb2.9SBr1.1I3.0 and Pb3.0SBr2.6I1.5 over time in water (left) and under air (right). | ||
Surface carboxylates, halides, and hydrogen bonded amines
To determine if any of the ligands used during synthesis coordinates to the chalcohalide particles after purification, we studied their surface chemistry using infrared (IR) spectroscopy (Fig. 5, see ESI†). A combination of v(N–H) (3400–3200 cm−1) and ν(C–N) (1100 cm−1) vibrations indicates the presence of surface-bound amine ligands, which are commonly present on chalcogenide nanocrystals,41,42 as well as halides (see below). Interestingly, the v(N–H) stretch at 3330 cm−1 in Pb2.9SBr1.1I3.0 gradually weakens as additional Br is incorporated into the lattice. This is accompanied by the appearance of a new v(N–H) stretch at ∼3200 cm−1, which gradually increases with Br incorporation and becomes the dominant peak in the most bromine-rich composition, Pb3.0SBr2.6I1.5.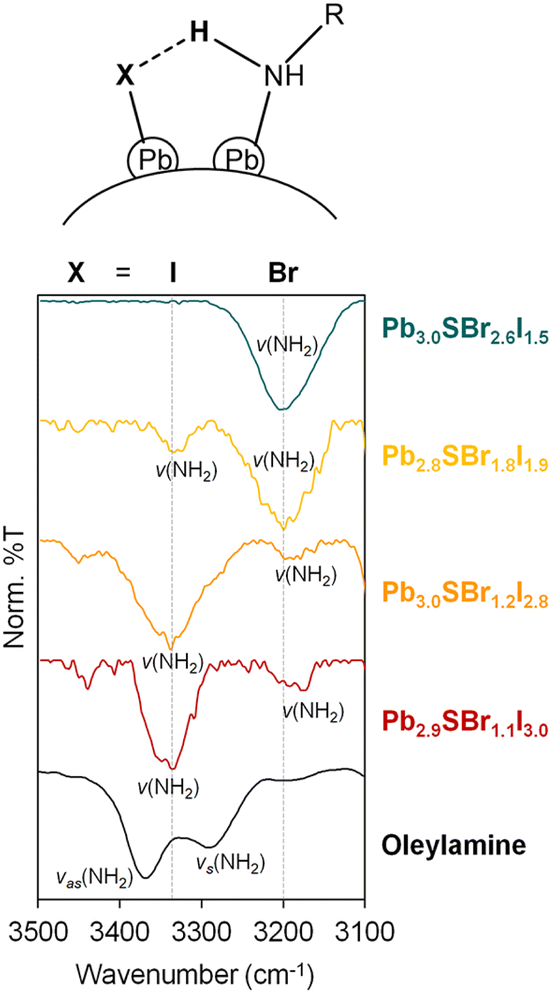 | ||
| Fig. 5 IR spectra of mixed-halide chalcohalides compared to oleylamine in the 3100–3500 cm−1 region. | ||
Free carboxylic acid fails to account for any of these peaks, as the characteristic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O band at ∼1700 cm−1 indicative of a protonated carboxylate (–COOH) is absent from all samples. Rather, we hypothesize that the two v(N–H) peaks correspond to two distinct surface-bound primary amines, each of which is hydrogen bonded to a surface-bound halide. This can be primarily iodide as in Pb2.9SBr1.1I3.0, or bromide as in Pb3.0SBr2.6I1.5, as shown at the top of Fig. 5. Such red-shift (to lower energy) of a hydrogen bonded amine
O band at ∼1700 cm−1 indicative of a protonated carboxylate (–COOH) is absent from all samples. Rather, we hypothesize that the two v(N–H) peaks correspond to two distinct surface-bound primary amines, each of which is hydrogen bonded to a surface-bound halide. This can be primarily iodide as in Pb2.9SBr1.1I3.0, or bromide as in Pb3.0SBr2.6I1.5, as shown at the top of Fig. 5. Such red-shift (to lower energy) of a hydrogen bonded amine 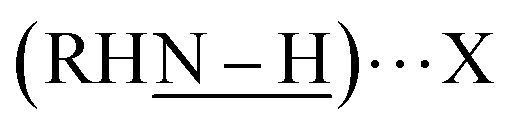 when X goes from I (3330 cm−1) to the more electronegative Br (3200 cm−1) has ample precedent in the literature, for example in methylammonium halides as well as in multiple organometallic compounds.43–47 In addition to halides and hydrogen-bonded amine ligands, we also observe vibrations between 1400–1600 cm−1 that correspond to vas(COO–) and vs(COO–). The small separation between these two bands of less than 100 cm−1 indicates that chelating (η2) carboxylate (oleate) ligands are also bound to the particle surface.48,49
when X goes from I (3330 cm−1) to the more electronegative Br (3200 cm−1) has ample precedent in the literature, for example in methylammonium halides as well as in multiple organometallic compounds.43–47 In addition to halides and hydrogen-bonded amine ligands, we also observe vibrations between 1400–1600 cm−1 that correspond to vas(COO–) and vs(COO–). The small separation between these two bands of less than 100 cm−1 indicates that chelating (η2) carboxylate (oleate) ligands are also bound to the particle surface.48,49
Ensemble and single particle photoluminescence
Diffuse reflectance demonstrates that the chalcohalides absorb strongly within the visible region, consistent with their bright yellow color (Fig. 6). Indirect Tauc plots50—assuming the mixed-halides to be indirect semiconductors based on the electronic structure of P21/m Pb3SBrI3—show that the band gaps gradually widen as more Br is incorporated into the structure. Specifically, the band gap changes from 2.22 eV (Pb2.9SBr1.1I3.0) to 2.26 eV (Pb3.0SBr1.2I2.8), 2.29 eV (Pb2.8SBr1.8I1.9), and 2.33 eV (Pb3.0SBr2.6I1.5), respectively (Table 1). These results are also consistent with the increased band gap energy observed in mixed-halide perovskites (MHPs) with the incorporation of more electronegative—and smaller—halide ions.34,35,51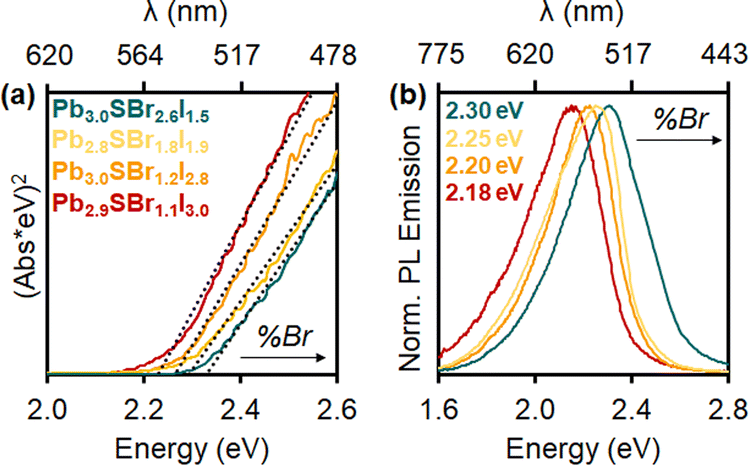 | ||
| Fig. 6 (a) Tauc plots and (b) photoluminescence spectra of Pb2.9SBr1.1I3.0 (red), Pb3.0SBr1.2I2.8 (orange), Pb2.8SBr1.8I1.9 (yellow), and Pb3.0SBr2.6I1.5 (green). | ||
Photoluminescence (PL) measurements reveal that the mixed-halide chalcohalides are emissive in the visible region, with PL emission maxima (PLmax) between 540 and 570 nm (Fig. 6). Furthermore, PLmax values gradually blue-shift with increasing Br-loading and content, in close agreement with the respective band gap energies. Critically, in addition to solution-phase (ensemble) PL measurements, we also successfully measured PL of single mixed-halide chalcohalide particles using fluorescence microscopy. Steady shape-correlated PL emission lacking blinking or OFF periods is observed over time under continuous photoexcitation (see ESI†).23 This finding strongly suggests that the PL originates from the rods themselves rather than from lower order impurities or unreacted precursor(s).
207Pb ssNMR spectroscopy
To probe the chemical speciation of the three mixed-halide chalcohalides, we resorted to solid-state 207Pb NMR spectroscopy (Fig. 7).34,52–54 Our data show that the NMR signals generally shift to more negative chemical shifts with the incorporation of more electronegative Br− ions into the structure, which is consistent with what is observed in binary lead halides,54,55 as well as in mixed-halide perovskites (MHPs).56 Specifically, three observable 207Pb NMR signals for Pb2.9SBr1.1I3.0 resonate at ca. 701.6, −7.9, and −531.7 ppm respectively, with an integral ratio of ∼1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6.5:2.6 (note: the 207Pb NMR spectra are not quantitative because we cannot measure the 207Pb longitudinal (T1) and transverse relaxation time constants
:6.5:2.6 (note: the 207Pb NMR spectra are not quantitative because we cannot measure the 207Pb longitudinal (T1) and transverse relaxation time constants 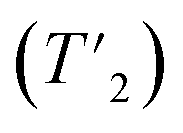 due to low sensitivity). We assign the three distinct signals to the fully occupied sites Pb(1), Pb(2), and Pb(3) (see ESI†). While one could expect other compositions to also show three peaks based on the Pb environments in the chalcohalide crystal structure, the specific chemical shifts of some of these environments are closely positioned, appearing as a single peak. We are able to resolve two distinct Pb chemical shifts in the more bromine-rich compositions. These broadened signals appear at ca. −302.0 and −639.6 ppm with a 1:2.5 integral ratio in Pb2.8SBr1.8I1.9 and at −3.02 and −691.1 ppm with a 1:3.7 integral ratio in Pb3.0SBr2.6I1.5. We conclude that the chalcohalide samples are free of any PbBr2 impurity because of the absence of a peak at ca. −980 ppm.55,57 Further, while it is difficult to discern the presence of PbI2 based on ssNMR chemical shifts alone,58 the XRD patterns of all of the chalcohalides are inconsistent with the presence of any such crystalline binary impurities.
due to low sensitivity). We assign the three distinct signals to the fully occupied sites Pb(1), Pb(2), and Pb(3) (see ESI†). While one could expect other compositions to also show three peaks based on the Pb environments in the chalcohalide crystal structure, the specific chemical shifts of some of these environments are closely positioned, appearing as a single peak. We are able to resolve two distinct Pb chemical shifts in the more bromine-rich compositions. These broadened signals appear at ca. −302.0 and −639.6 ppm with a 1:2.5 integral ratio in Pb2.8SBr1.8I1.9 and at −3.02 and −691.1 ppm with a 1:3.7 integral ratio in Pb3.0SBr2.6I1.5. We conclude that the chalcohalide samples are free of any PbBr2 impurity because of the absence of a peak at ca. −980 ppm.55,57 Further, while it is difficult to discern the presence of PbI2 based on ssNMR chemical shifts alone,58 the XRD patterns of all of the chalcohalides are inconsistent with the presence of any such crystalline binary impurities.
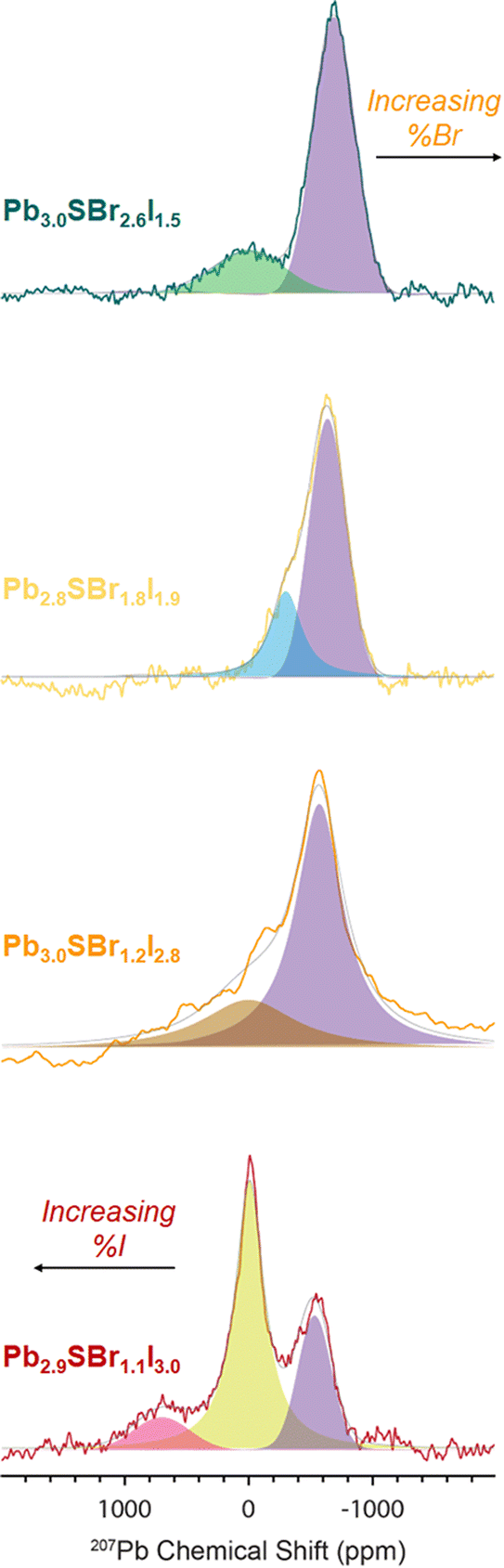 | ||
| Fig. 7 207Pb spin echo ssNMR spectra of Pb2.9SBr1.1I3.0, Pb3.0SBr1.2I2.8, Pb2.8SBr1.8I1.9, and Pb3.0SBr2.6I1.5. | ||
Atom coloring and electronic calculations
To better understand our experimental findings, we evaluated the crystal structure and electronic properties of the different mixed-halide compositions using DFT calculations. After first generating isocompositional coloring models59,60 to represent plausible atomic ordering patterns for Pb3SBrI3, Pb3SBr2I2, and Pb3SBr3I, we fully relaxed their structures using the Vienna Ab initio Simulation Package (VASP).61 We find that the lowest energy structures for the three different mixed-halides all adopt similar atomic coloring patterns. Specifically, the smaller bromine atoms are localized on the Pb(3) site25 toward the center of the unit cell in Pb3SBrI3 and Pb3SBr2I2, which then branch outward onto the Pb(2) site in Pb3SBr3I (Fig. 8). Interestingly, the lowest energy structure for Pb3SBrI3 corresponds to the P21/m structure previously determined from single crystal data.25 Upon closer inspection of the relaxed structures, we observe a steady reduction of the unit cell volume from Pb3SBrI3 to Pb3SBr2I2 and Pb3SB3I (Fig. 8). Even though the calculated volumes are overestimated by ∼9–11% compared to volumes extracted from the refined XRD patterns (see ESI†), the overall trend is fully consistent with the increasing bromine incorporation into the chalcohalide structure.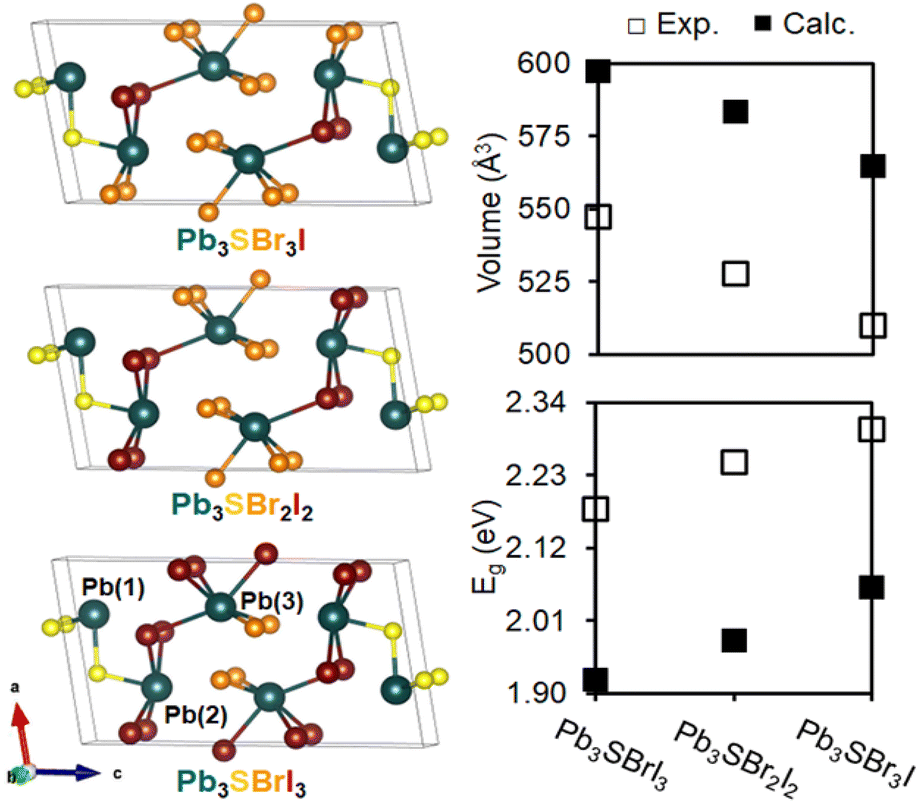 | ||
| Fig. 8 Atomic coloring patterns of the lowest energy unit cells of Pb3SBrI3, Pb3SBr2I2, and Pb3SBr3I. Comparison of the calculated and experimental unit cell volume and band gaps (Eg) of these mixed-halide chalcohalides. | ||
A comparison of density of states (DOS) curves shows that the band gap (Eg) values of the chalcohalides progressively widen from 1.92 eV in Pb3SBrI3 to 1.98 eV and 2.06 eV in Pb3SBr2I2 and Pb3SBr3I, respectively, which agrees well with the experimentally determined values (Fig. 8, see ESI†). The calculated band gaps for the three lowest energy structures underestimate their respective experimental values by ca. 11–13%, well within an acceptable range of underestimation often seen using VASP.21,62 Furthermore, analysis of the partial DOS reveals that the relative Br-p and I-p orbital contributions closely correlate with the relative halide ratio expected in each quaternary chalcohalide. In summary, the results of the electronic structure calculations closely support our experimentally observed trend of band gap widening with increased bromine incorporation into the mixed-halide chalcohalides.
Conclusions
In conclusion, we have demonstrated an effective strategy for synthesizing quaternary chalcohalides with mixed-halide compositions using a solution-phase approach. Electron microscopy reveals that the Pb3SBrxI4−x (1 ≤ x ≤ 3) semiconductors exhibit highly anisotropic growth in the form of rods and needles with high aspect ratios, and their structural features are validated using a combination of XRD and 207Pb ssNMR. Not only do these materials exhibit indirect band gaps that widen with increased Br incorporation, but they also show promising photoluminescence between 2.2–2.3 eV in solution as an ensemble as well as at the single particle level. These materials also demonstrate impressive stability upon exposure to moisture, showing minimal or no degradation when compared to traditional lead halide perovskites. Lastly, relative energy and DOS calculations showcase atomic coloring patterns and electronic structures that agree well with our experimental findings. Overall, this work provides fundamental materials chemistry knowledge that can be applied toward halide-alloying many other complex chalcohalide compositions, which remains a critical goal for tuning the optical properties of next generation semiconductors and devices.Data availability
All necessary information is included in the ESI.†Author contributions
ANR designed the experiments, synthesized, and characterized all materials, performed the electronic structure calculations, and wrote the manuscript. YC and AS performed ssNMR measurements and analyses. JOA performed single particle microscopy experiments. EG performed transmission electron microscopy. All authors contributed to the characterization, data analysis, and discussion. ANR and JV conceived the project, and JV, AJR, and EAS guided and supervised the work. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the US Department of Energy, Office of Science, Basic Energy Sciences, Materials Science and Engineering Division. The Ames National Laboratory is operated for the US Department of Energy by Iowa State University under contract DE-AC02-07CH11358.Notes and references
- J. Ye, M. M. Byranvand, C. O. Martínez, R. L. Z. Hoye, M. Saliba and L. Polavarapu, Angew. Chem., 2021, 133, 21804–21828 CrossRef.
- C. M. Sutter-Fella, Y. Li, M. Amani, J. W. Ager, F. M. Toma, E. Yablonovitch, I. D. Sharp and A. Javey, Nano Lett., 2016, 16, 800–806 CrossRef CAS PubMed.
- C. Bi, S. Wang, W. Wen, J. Yuan, G. Cao and J. Tian, J. Phys. Chem. C, 2018, 122, 5151–5160 CrossRef CAS.
- H. Huang, M. I. Bodnarchuk, S. V. Kershaw, M. V. Kovalenko and A. L. Rogach, ACS Energy Lett., 2017, 2, 2071–2083 CrossRef CAS PubMed.
- P. V. Kamat and M. Kuno, Acc. Chem. Res., 2021, 54, 520–531 CrossRef CAS PubMed.
- A. J. Barker, A. Sadhanala, F. Deschler, M. Gandini, S. P. Senanayak, P. M. Pearce, E. Mosconi, A. J. Pearson, Y. Wu, A. R. S. Kandada, T. Leijtens, F. De Angelis, S. E. Dutton, A. Petrozza and R. H. Friend, ACS Energy Lett., 2017, 2, 1416–1424 CrossRef CAS.
- Z. Ni, H. Jiao, C. Fei, H. Gu, S. Xu, Z. Yu, G. Yang, Y. Deng, Q. Jiang, Y. Liu, Y. Yan and J. Huang, Nat. Energy, 2022, 7, 65–73 CrossRef CAS.
- K. L. Wang, Y. H. Zhou, Y. H. Lou and Z. K. Wang, Chem. Sci., 2021, 12, 11936–11954 RSC.
- U. V. Ghorpade, M. P. Suryawanshi, M. A. Green, T. Wu, X. Hao and K. M. Ryan, Chem. Rev., 2023, 123, 327–378 CrossRef CAS PubMed.
- F. Palazon, Sol. RRL, 2022, 6, 21008291–21008299 Search PubMed.
- D. W. Davies, K. T. Butler, J. M. Skelton, C. Xie, A. R. Oganov and A. Walsh, Chem. Sci., 2018, 9, 1022–1030 RSC.
- Y. T. Huang, S. R. Kavanagh, D. O. Scanlon, A. Walsh and R. L. Z. Hoye, Nanotechnology, 2021, 32, 1320041–13200460 Search PubMed.
- A. M. Ganose, C. N. Savory and D. O. Scanlon, Chem. Commun., 2017, 53, 20–44 RSC.
- R. Nie, B. Kim, S. T. Hong and S. I. Seok, ACS Energy Lett., 2018, 3, 2376–2382 CrossRef CAS.
- C. Yang, Z. Wang, G. He, H. Zhang and C. Liao, ACS Appl. Nano Mater., 2022, 5, 16033–16038 CrossRef CAS.
- P. Govindaraj and K. J. Venugopal, J. Alloys Compd., 2022, 929, 1673471–16734711 CrossRef.
- J. Mark, W. Zhang, K. Maeda, T. Yamamoto, H. Kageyama and T. Mori, J. Mater. Chem. A, 2023, 11, 10213–10221 RSC.
- M. Y. Ran, S. H. Zhou, W. Wei, B. J. Song, Y. F. Shi, X. T. Wu, H. Lin and Q. L. Zhu, Inorg. Chem., 2021, 60, 3431–3438 CrossRef CAS PubMed.
- X. H. Li, Z. H. Shi, M. Yang, W. Liu and S. P. Guo, Angew. Chem., Int. Ed., 2022, 61, e2021158711–e2021158716 Search PubMed.
- J. Li, S. S. Han and S. P. Guo, Eur. J. Inorg. Chem., 2022, e2022004191–e20220041911 Search PubMed.
- S. Toso, Q. A. Akkerman, B. Martín-García, M. Prato, J. Zito, I. Infante, Z. Dang, A. Moliterni, C. Giannini, E. Bladt, I. Lobato, J. Ramade, S. Bals, J. Buha, D. Spirito, E. Mugnaioli, M. Gemmi and L. Manna, J. Am. Chem. Soc., 2020, 142, 10198–10211 CrossRef CAS PubMed.
- M. Imran, L. Peng, A. Pianetti, V. Pinchetti, J. Ramade, J. Zito, F. Di Stasio, J. Buha, S. Toso, J. Song, I. Infante, S. Bals, S. Brovelli and L. Manna, J. Am. Chem. Soc., 2021, 143, 1435–1446 CrossRef CAS PubMed.
- A. N. Roth, J. Opare-Addo, E. Gi, S. Mena, G. Guirado, R. D. Schaller, E. A. Smith and J. Vela, Chem. Mater., 2023, 35, 2165–2172 CrossRef CAS.
- L. Bindia, A. Garavellib, D. Pintob, G. Pratesia and F. Vurro, J. Solid State Chem., 2008, 181, 306–312 CrossRef.
- M. Yan, R. L. Tang, W. Zhou, W. Liu and S. P. Guo, Dalton Trans., 2022, 51, 12921–12927 RSC.
- N. V. Pervukhina, S. A. Magarill, D. Y. Naumov, S. V. Borisov, V. I. Vasil'yev and B. G. Nenashev, J. Struct. Chem., 2006, 47, 312–317 CrossRef CAS.
- L. T. Jiang, M. Z. Li, X. M. Jiang, B. W. Liu and G. C. Guo, Dalton Trans., 2022, 51, 6638–6645 RSC.
- Q. Jing, Z. Yang, S. Pan and D. Xue, Phys. Chem. Chem. Phys., 2015, 17, 21968–21973 RSC.
- Q. A. Akkerman, B. Martín-García, J. Buha, G. Almeida, S. Toso, S. Marras, F. Bonaccorso, U. Petralanda, I. Infante and L. Manna, Chem. Mater., 2019, 31, 8145–8153 CrossRef CAS.
- B. Ptaszyński, E. Skiba and J. Krystek, J. Therm. Anal. Calorim., 2001, 65, 231–239 CrossRef.
- S. M. Islam, C. D. Malliakas, D. Sarma, D. C. Maloney, C. C. Stoumpos, O. Y. Kontsevoi, A. J. Freeman and M. G. Kanatzidis, Chem. Mater., 2016, 28, 7332–7343 CrossRef CAS.
- H. Wang, G. Chen, J. Xu, Y. Xu and Q. Yang, Cryst. Growth Des., 2018, 18, 1987–1994 CrossRef CAS.
- T. P. A. Ruberu, H. R. Albright, B. Callis, B. Ward, J. Cisneros, H. J. Fan and J. Vela, ACS Nano, 2012, 6, 5348–5359 CrossRef CAS PubMed.
- B. A. Rosales, L. Men, S. D. Cady, M. P. Hanrahan, A. J. Rossini and J. Vela, Chem. Mater., 2016, 28, 6848–6859 CrossRef CAS.
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS PubMed.
- A. Sadhanala, S. Ahmad, B. Zhao, N. Giesbrecht, P. M. Pearce, F. Deschler, R. L. Z. Hoye, K. C. Gödel, T. Bein, P. Docampo, S. E. Dutton, M. F. L. De Volder and R. H. Friend, Nano Lett., 2015, 15, 6095–6101 CrossRef CAS PubMed.
- R. D. Shannon, Acta Crystallogr., 1976, A32, 751–767 CrossRef CAS.
- W. M. Haynes, D. R. Lide and T. J. Bruno, CRC Handbook of Chemistry and Physics, CRC Press, Boca Raton, FL, 103rd edn, 2022 Search PubMed.
- B. W. Boote, H. P. Andaraarachchi, B. A. Rosales, R. Blome-Fernández, F. Zhu, M. D. Reichert, K. Santra, J. Li, J. W. Petrich, J. Vela and E. A. Smith, ChemPhysChem, 2019, 20, 2647–2656 CrossRef CAS PubMed.
- M. Liao, B. Shan and M. Li, J. Phys. Chem. Lett., 2019, 10, 1217–1225 CrossRef CAS PubMed.
- A. N. Roth, Y. Chen, M. A. S. Adamson, E. Gi, M. Wagner, A. J. Rossini and J. Vela, ACS Nano, 2022, 16, 12024–12035 CrossRef CAS PubMed.
- A. M. Medina-Gonzalez, B. A. Rosales, U. H. Hamdeh, M. G. Panthani and J. Vela, Chem. Mater., 2020, 32, 6085–6096 CrossRef CAS.
- E. W. Dahl and N. K. Szymczak, Angew. Chem., 2016, 128, 3153–3157 CrossRef.
- A. Cabana and C. Sandordy, Spectrochim. Acta, 1962, 18, 843–861 CrossRef CAS.
- F. Cariati, G. Ciani, L. Menabue, G. C. Pellacani, G. Rassu and A. Sironi, Inorg. Chem., 1983, 22, 1897–1902 CrossRef CAS.
- T. Glaser, C. Müller, M. Sendner, C. Krekeler, O. E. Semonin, T. D. Hull, O. Yaffe, J. S. Owen, W. Kowalsky, A. Pucci and R. Lovrincic, J. Phys. Chem. Lett., 2015, 6, 2913–2918 CrossRef CAS PubMed.
- M. D. Fryzuk, P. A. MacNeil and S. J. Rettig, J. Am. Chem. Soc., 1987, 109, 2803–2812 CrossRef CAS.
- V. Zelenak, Z. Vargova and K. Gyoryova, Spectrochim. Acta, Part A, 2007, 66, 262–272 CrossRef CAS PubMed.
- J. Zhang, H. Zhang, W. Cao, Z. Pang, J. Li, Y. Shu, C. Zhu, X. Kong, L. Wang and X. Peng, J. Am. Chem. Soc., 2019, 141, 15675–15683 CrossRef CAS PubMed.
- B. D. Viezbicke, S. Patel, B. E. Davis and D. P. Birnie, Phys. Status Solidi B, 2015, 252, 1700–1710 CrossRef CAS.
- A. Karmakar, A. Bhattacharya, D. Sarkar, G. M. Bernard, A. Mar and V. K. Michaelis, Chem. Sci., 2021, 12, 3253–3263 RSC.
- J. P. Yesinowski, Top. Curr. Chem., 2011, 306, 229–312 CrossRef PubMed.
- M. P. Hanrahan, L. Men, B. A. Rosales, J. Vela and A. J. Rossini, Chem. Mater., 2018, 30, 7005–7015 CrossRef CAS.
- R. E. Taylor, P. A. Beckmann, S. Bai and C. Dybowski, J. Phys. Chem. C, 2014, 118, 9143–9153 CrossRef CAS.
- A. Glatfelter, C. Dybowski, D. D. Kragten, S. Bai, D. L. Perry and J. Lockard, Spectrochim. Acta, Part A, 2007, 66, 1361–1363 CrossRef PubMed.
- K. Datta, A. Caiazzo, M. A. Hope, J. Li, A. Mishra, M. Cordova, Z. Chen, L. Emsley, M. M. Wienk and R. A. J. Janssen, ACS Energy Lett., 2023, 8, 1662–1670 CrossRef CAS PubMed.
- Y. S. Kye, S. Connolly, B. Herreros and G. S. Harbison, Main Group Met. Chem., 1999, 22, 373–383 CAS.
- R. E. Taylor, P. A. Beckmann, S. Bai and C. Dybowski, J. Phys. Chem. C, 2014, 118, 9143–9153 CrossRef CAS.
- G. J. Miller, Eur. J. Inorg. Chem., 1998, 523–536 CrossRef CAS.
- J. Pham and G. J. Miller, Inorg. Chem., 2018, 57, 4039–4049 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- L. Sun, C. Wang, L. Xu, J. Wang, X. Chen and G. Yi, J. Mater. Chem. C, 2018, 6, 7188–7194 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, XRD, TEM, SEM, EDS, PL movies, unit cell coloring patterns, DOS. See DOI: https://doi.org/10.1039/d3sc02733c |
| This journal is © The Royal Society of Chemistry 2023 |