
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal–organic framework-derived CoNx nanoparticles on N-doped carbon for selective N-alkylation of aniline†
Ved
Vyas
,
Priyanka
Maurya
and
Arindam
Indra
 *
*
Department of Chemistry, Indian Institute of Technology (BHU), Varanasi, UP-221005, India. E-mail: arindam.chy@iitbhu.ac.in
First published on 9th October 2023
Abstract
N-alkylation of anilines by alcohols can be used as an efficient strategy to synthesise a wide range of secondary amines. In this respect, a hydrogen borrowing methodology has been explored using precious metal-based catalysts. However, the utilisation of cheap and readily available transition metal based catalysts is required for large-scale applications. In this work, we have reported metal–organic framework-derived CoNx@NC catalysts for the selective N-alkylation of anilines with different types of alcohols. The Co–N coordination in CoNx@NC was found to be extremely important to improve the conversion efficiency and yield of the product. As a result, CoNx@NC produced 99% yield of the desired amines, which is far better than that of Co@C (yield = 65%). In addition, CoNx@NC showed remarkable recyclability for six cycles with a minimum drop in the yield of the desired product.
Introduction
Amines play an important role as intermediates in the chemical industry and life sciences.1 Their wide range of industrial applications include pharmaceuticals, organic dyes, agrochemicals, polymers, lubricants, and so on.2 Additionally, the amine and nitrogen moieties play a key role in drug molecules.3 Due to their high significance, numerous methods have been developed for the selective formation of C–N bonds.1–4In general, secondary amines have been prepared by both oxidative and reductive methods.5,6 In the reductive process, the carbonyl compound reacts with primary amines in the presence of a suitable catalyst and stoichiometric amounts of reducing agents (borohydride/formic acid/ammonium/formate/silanes).7 This process generates a significant amount of waste and some of these are hazardous and difficult to handle.7,8
In the last few decades, oxidative catalytic approaches for C–N bond formation have been followed to enhance the activity and selectivity. One of the efficient catalytic oxidative approaches include N-alkylation of amines.9 This process relies on hydrogen borrowing from the alcohol for the hydrogenation of imine, which is produced by the reaction of alcohol and primary amine.10 As alcohols are abundant and only water is formed as the by-product, the method can be considered as an atom-economic and environmentally friendly approach.10,11
In recent years, N-alkylation reactions have successfully been carried out using homogenous noble metal complexes of Ru,12,13 Ir,2,14,15 Pd,16 Au,17 Pt,18etc.19 These catalysts are highly effective for one-pot reactions. Although homogeneous catalysts show high efficiency, they have the typical drawbacks of limited recovery and reuse.20,21 In addition, the use of expensive metals, ligands, and external additives makes the use of homogeneous catalysts impractical.22 In this respect, heterogeneous noble metal catalysts of Pt,23 Ru,24,25 Pd,26 Rh,27etc. have been used for N-alkylation of primary amines.28 These catalysts also suffer from low selectivity, and the use of cocatalysts, high temperature and pressure, and high metal loading are required to improve the selectivity of N-alkylated products.28
On the other hand, transition metal-based heterogeneous catalysts have been demonstrated with promising catalytic activity for organic reactions.29 Transition metal-based catalysts were found to produce high catalytic activity and selectivity for N-alkylation reactions.30 As supported catalysts increase the surface area and provide more active sites for catalytic reactions, a series of solid-supported 3d transition metal-based catalysts were reported for the N-alkylation of amines.31 For example, silica-supported Co,32 Ni33,34 and Cu35,36 nanoparticles have been explored for the N-alkylation of amines. Other supports like metal oxides,37,38 carbon,39etc. have been demonstrated for the immobilization of nanoparticles. The strong interaction between nanoparticles and the support is also important to stabilize nanoparticles on the support with minimum agglomeration during the catalytic process.40,41
Recently, nitrogen-doped carbon (NC) has been explored as a support material for heterogeneous catalysts due to its beneficial effect on tuning the chemical properties of catalysts.42 NC-supported materials not only have high activity in electrocatalysis, they also exhibit excellent catalytic properties for N-alkylation reactions.29,30 For example, Beller and co-workers developed heterogeneous Co@NC for the N-alkylation of primary amines and ammonia.43 Further, MNx@NC catalysts were found to be highly efficient in catalytic organic transformations like N-alkylation of amines,44 ester formation,45 transfer hydrogenation,46 hydrogenation,47etc. In this respect, the formation of M−N coordination was found to be useful to improve the catalyst–support interaction and hence enhance the stability of the system.48
The use of a metal–organic framework (MOF) having a N-containing bridging ligand as the precursor of MNx@NC (M = transition metal, x > 1) has been explored by us and other groups.49–51 The MOF helps in the uniform distribution of the metal nodes and provides the required coordination and electronic environment for the metal.50 The pyrolysis of the MOF produces uniformly distributed nanoparticles on the surface of N-doped carbon, formed by the pyrolysis of the N-containing ligand.52 The M−N coordination also stabilizes the nanoparticles on the N-doped carbon surface, which minimizes the particle agglomeration during the catalytic process and improves the stability of the catalyst.42 The coordinated N to the metal center offers an environment, similar to the ligand in homogeneous catalysts, and manipulates the electron density on catalytic sites during the reaction.42,50 Further, the adsorption of the substrate and reaction intermediates can also be modulated by the coordinated N. As a result, an improvement in the selectivity and catalytic activity can be achieved for organic transformations.42,53,54
Mostly, Co@NC,49 Co–N–C,55 and Fe–N–C29 nanocatalysts with core–shell structures containing N-doped carbon (NC) shells have been reported for electrochemical energy conversion processes. The use of MOF-derived MNx@NC nanocatalysts for organic transformations has been rarely studied.
Inspired by the above results, we report here the synthesis of a heterogeneous catalyst CoNx@NC for the N-alkylation of amines. For this purpose, we have chosen ZIF-67@C as the precursor and pyrolyzed it to form CoNx@NC. The ZIF-67 provides a unique coordination environment around the Co-center to form CoNx@NC whereas the pyrolysis of the ligand produces N-doped graphene. The CoNx@NC works as a hydrogen-borrowing catalyst for the alkylation of primary amines. The N-doped carbon supported CoNx@NC showed excellent catalytic activity for N-alkylation reactions of amines with alcohols with the best yield of >99% for the desired product. Moreover, the catalyst was found to be stable enough during recycling for six catalytic cycles without having any significant drop in the catalytic activity and selectivity.
Results and discussion
Syntheses and characterization of the catalysts
The catalyst precursor ZIF-67@C was synthesized by the reaction of cobalt(II)nitrate and 2-methylimidazole in the presence of Vulcan-XC-72R carbon as the support (Scheme 1). The powder X-ray diffraction pattern (PXRD) of ZIF-67@C showed the characteristic crystal planes of ZIF-67 (JCPDS: 08-60-513, Fig. S1†). The pyrolysis of ZIF-67@C in an inert atmosphere at different temperatures produced CoNx@NCs (temperature = 700, 800, and 900 °C) (Scheme 1). The PXRD pattern of CoNx@NC nanoparticles showed the face-centre cubic (fcc) crystal of cobalt (JCPDS: No.15-0806) (Fig. S2†).48 In CoNx@NC, the (111) peak of cubic cobalt was shifted towards the right side (2Θ = 0.3°) due to the coordination with nitrogen (JCPDS: 08-60-513).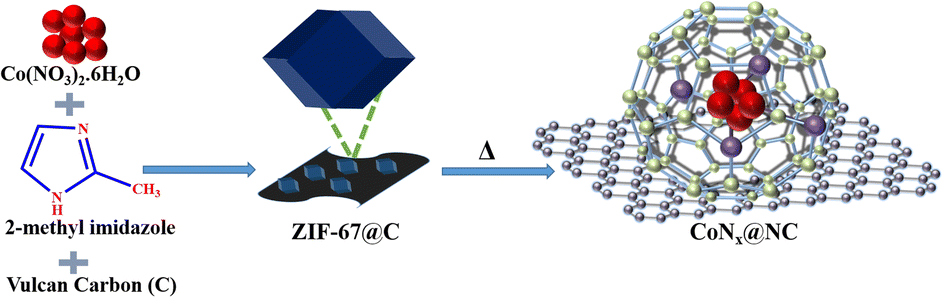 | ||
| Scheme 1 The schematic representation for the synthesis of ZIF-67@C and its pyrolysis to produce CoNx@NC. | ||
Further, the degree of graphitization of N-doped carbon (NC) in CoNx@NC was investigated by Raman spectroscopy. The Raman spectrum showed the characteristic D and G bands of carbon at 1322 cm−1 and 1596 cm−1, respectively (Fig. S3†).56 The ID/IG ratio of 1.59 indicates a defect rich structure of NC. The classical Eg and A1g vibronic modes of metallic Co were detected at 475 cm−1 and 682 cm−1, respectively.
The nature of carbon was further analysed by X-ray photoelectron spectroscopy (XPS, Fig. 1). The C 1s XPS was fitted into five peaks for: sp2 hybridised carbon of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (284.7 eV), sp3 hybridised carbon C–C bond (284.9 eV), CN (285.9 eV), C–N (287.6 eV), and COOH (290.1 eV).57 The ratio of the graphitic carbon to other carbon is determined to be 1.62, which closely matches with the Raman data.
C (284.7 eV), sp3 hybridised carbon C–C bond (284.9 eV), CN (285.9 eV), C–N (287.6 eV), and COOH (290.1 eV).57 The ratio of the graphitic carbon to other carbon is determined to be 1.62, which closely matches with the Raman data.
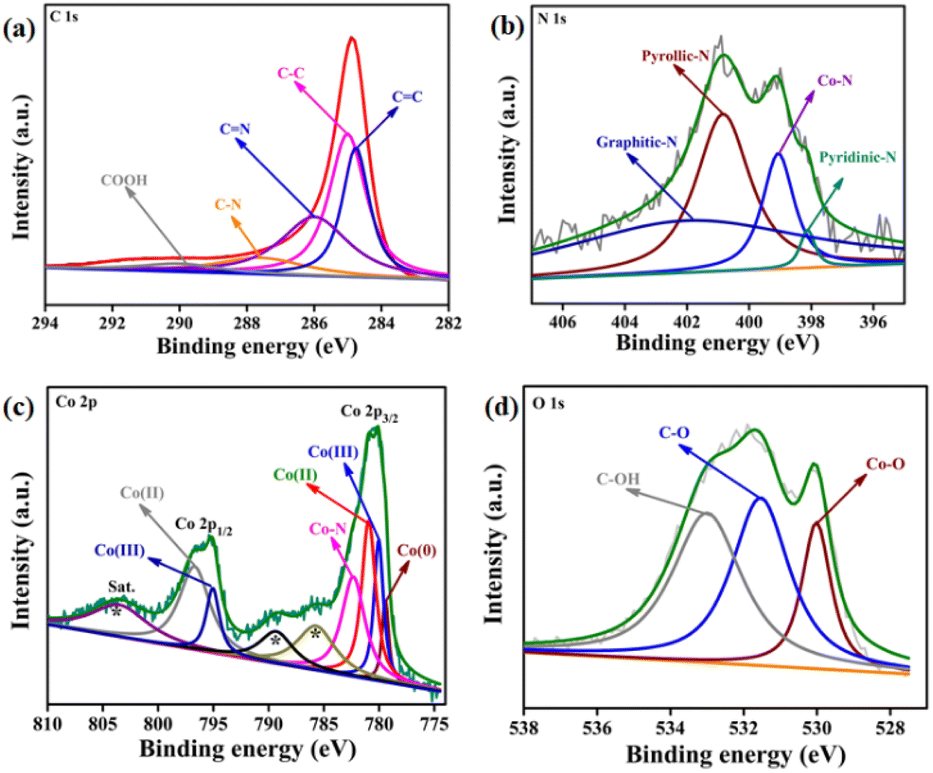 | ||
| Fig. 1 (a) The C 1s spectrum of CoNx@NC was deconvoluted into five peaks for CC, C–C, CN, C–N, and COOH species. (b) The N 1s spectrum was fitted into four peaks-corresponding to pyridinic, Co–N, pyrrolic, and graphitic nitrogen. (c) Co 2p XPS was fitted into peaks for Co(0), Co–N, Co(III), and Co(II) species. The * marked peaks originate as the satellite peaks of Co(II). (d) O 1s spectrum of CoNx@NC. | ||
The N 1s XPS was deconvoluted into four peaks at binding energies 398.1, 399.0, 400.8, and 401.7 eV corresponding to pyridinic, Co–N, pyrrolic, and graphitic nitrogen (Fig. 1b).39 This result confirms the doping of N in the carbon matrix and the formation of the Co–N bond. Therefore, in the CoNx@NC, the Co atoms are coordinated to the pyrolic-N and pyridinic-N and offer active sites during catalytic reactions.
The Co 2p XPS was fitted into two peaks corresponding to Co 2p3/2 (780.1 eV) and Co 2p1/2 (795.3 eV) (Fig. 1c).55 The Co 2p3/2 peak was further deconvoluted into the peaks for Co0 (779.3 eV) and Co–N (782.3 eV), Co(III) (779.9 eV), and Co(II) (780.8 eV) species. The presence of Co(III) and Co(II) species can be explained by the surface oxidation of cobalt nanoparticles. The O 1s XPS was deconvoluted into three peaks at 529.9, 531.5, and 533.0 eV corresponding to Co–O, C–O, and C–OH species (Fig. 1d).58
The scanning electron microscopy (SEM) images showed the rhombic dodecahedron morphology of CoNx@NC where the Co nanoparticles of various sizes were densely embedded on NC (Fig. 2a and S4†). The energy dispersive X-ray (EDX) spectrum of CoNx@NC revealed the presence of Co, N, and C (Fig. S5†) whereas the EDX elemental mapping showed the homogeneous distribution of Co on N-doped carbon (Fig. S6†).
 | ||
| Fig. 2 (a) FE-SEM image of CoNx@NC showing the rhombic dodecahedron morphology of the particles. (b and c) TEM images of CoNx@NC with spherical morphology and the size of the particles was calculated to be 4–12 nm. (d) HR-TEM image showing the inter-planar spacing between the graphene layers and the inset shows the SAED pattern of the CoNx@NC. | ||
Further, the morphological details of the catalyst were confirmed by transmission electron microscopy (TEM, Fig. 2b–d). The TEM images of CoNx@NC demonstrated that the Co nanoparticles with varying sizes (4–12 nm) were distributed in the N-doped carbon frameworks (Fig. S7†). The selected area electron diffraction (SAED) pattern of CoNx@NC showed diffraction rings corresponding to the crystal planes (111), (200), and (220) of fcc cobalt (Fig. 2d-inset). The inter-planar spacing of the N-doped carbon layer was calculated to be 0.36 nm, corresponding to the (002) plane of graphene (Fig. 2d).
Optimization of the catalytic reaction conditions
First, the catalytic performance of CoNx@NC was optimized for the N-alkylation reaction of benzyl alcohol and aniline (Table S1†). The reaction of benzyl alcohol with aniline can produce N-benzylaniline (1) and N-benzylideneaniline (2) as the major products. Therefore, control over the selectivity to produce 1 as the major product is a challenging task. The hydrogen borrowing route can be effectively employed to synthesize N-benzylaniline.Interestingly, CoNx@NC was found to be highly selective for the production of 1 when full conversion was achieved. For the comparison purpose, we used Co@C and found that the coordination of Co with N is extremely important for the improved selectivity and activity. While CoNx@NC can produce 1 with 99% yield after the full conversion (entry 1, Table S1†), Co@C can reach the maximum yield of 65% (87% conversion) under the same reaction conditions (entry 4, Table S1†). The other catalysts had poor conversion and low selectivity for 1 (entries 5–7, Table S1†).
The pyrolysis temperature of the catalysts has also a significant effect on the conversion and selectivity of the products. Comparison of the catalysts prepared at different pyrolysis temperatures (700–900 °C) showed that CoNx@NC (prepared at 800 °C) produced the best activity and selectivity for 1 (entries 1–3, Table S1†).
Further, the effect of different bases on the yield of 1 has been determined. The yield of 1 dropped in the absence of a base, and in the presence of bases, the yield of 1 followed the order: tert-BuOK > KOH > Cs2CO3 > K2CO3 (entries 8–12, Table S1†). The optimized amount of the CoNx@NC catalyst was found to be 10 mg and poorer yield of 1 was obtained with lower (5 mg) or higher (15 mg) amount of catalyst (entries 13–15, Table S1†). Next, we optimized the effect of solvent and the best conversion and selectivity for 1 were recorded in toluene. Other non-polar aprotic solvents also produced high selectivity in the following order: toluene > THF > dioxane > acetonitrile (entries 16–19, Table S1†). In polar protic solvents like methanol and ethanol, we observed the formation of a mixture of products and we did not continue the reaction further.
Scope of different alcohols
With the optimized reaction conditions, the N-alkylation of aniline with different benzyl alcohols was studied (Scheme 2: 3a–3i and Table S2†). In all the cases, secondary amines (3) were found to be the major product (yield = 81–99%). The best yield (99%) was obtained for the reaction of aniline with benzyl alcohol after 18 h of reaction (Scheme 2, 3a and Table S2†). The methoxy-substitution at the para-position of benzyl alcohol slowed down the reaction but the yield of the secondary amine 3b was not affected. However, a decrease in the yield of 3c was observed when 2,4-dimethoxy-benzyl alcohol was reacted with aniline, most probably due to the increasing steric hindrance. On the other hand, the electron withdrawing substituent in the meta- and para-position of benzyl alcohol slightly decreases the yield of the corresponding secondary amines when reacted with aniline (Scheme 2, 3d–3f and Table S2†).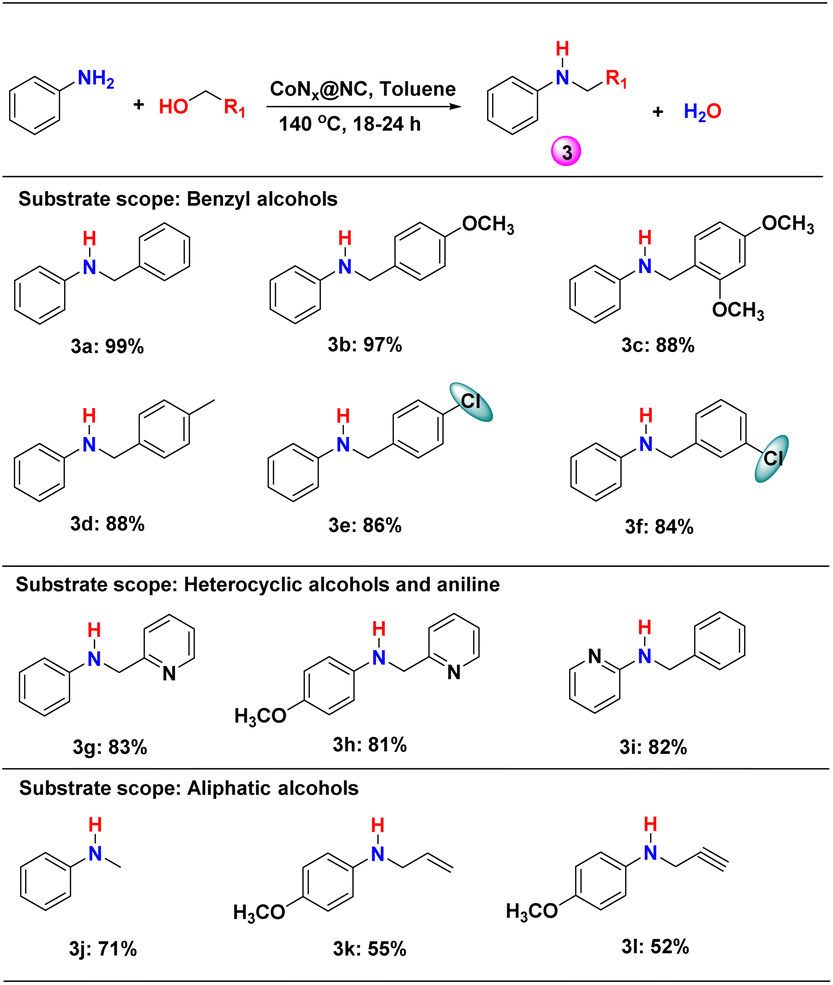 | ||
| Scheme 2 N-alkylation of aniline and substituted-anilines with different benzyl alcohols using the CoNx@NC catalyst. Reaction conditions: 1 mmol substituted benzyl alcohol, 0.5 mmol aniline, 10 mg catalyst, 0.5 mmol tert-BuOK, 2 mL toluene, 140 °C, 24 h. For 3a (18 h), 3e (18 h) and 3f (18 h) optimized at different times. | ||
The use of heterocyclic benzyl alcohol (pyridine-2-yl-methanol) decreased the yield of 3g and 3h (Scheme 2, Table S2†). Even, heterocyclic aniline with benzyl alcohol produced a high yield of secondary heterocyclic amines (Scheme 2, 3i). The aliphatic alcohols were found to produce lower yield (52–71%) for N-alkylation than the benzylic alcohols (Scheme 2, 3j–3l and Table S2†). The yield of the N-alkylated product decreased from methanol to propylene alcohol to propyne alcohol.
Scope of different anilines
Next, we explored the scope of different anilines for N-alkylation reactions with benzyl alcohol. Anilines with different electron donating and electron withdrawing substituents in the phenyl ring produced high yield (>80%) for secondary amines and the effect of the electronic factor on the product yield was not clear (Scheme 3 and Table S2†). However, a series of substituted anilines were selectively converted into secondary amines. The effect of steric crowding is distinct and the substitution in the ortho-position of anilines resulted in low yields (Scheme 3, 4b, 4e, 4g, 4h, 4n and 4q). The lowest yield (72%) among the studied substituted anilines was obtained with 2-nitroaniline due to the introduction of both steric crowding and electron-withdrawing effects (Scheme 3, 4h).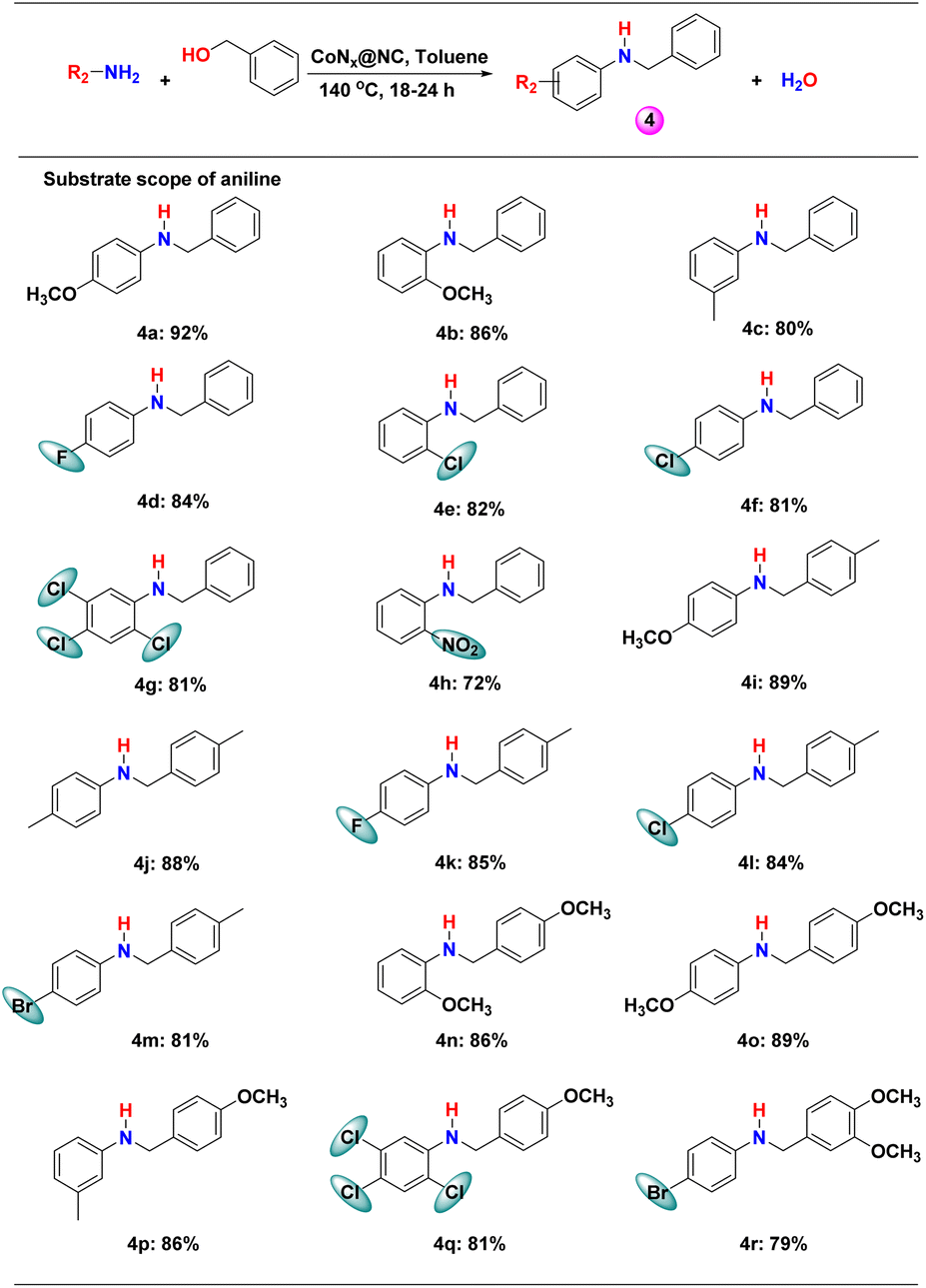 | ||
| Scheme 3 CoNx@NC catalysed N-alkylation reaction of different anilines with benzyl alcohols. Reaction conditions: 1 mmol substituted benzyl alcohol, 0.5 mmol aniline, 10 mg catalyst, 0.5 mmol tert-BuOK, 2 mL toluene, 140 °C, 24 h. For 4d (18 h), 4e (20 h) and 4f (20 h) optimized at different times. | ||
When the steric crowding in both substrates was increased, a significant drop in the yield of the product was observed (Scheme 3, 4q and 4r). Therefore, CoNx@NC is highly efficient in catalysing the N-alkylation of a series of anilines with different alcohols (benzyl alcohols, aliphatic primary alcohols, allylic alcohol, and alkyne alcohol).
Scope of o-phenylenediamine
As N-containing heterocyclic benzimidazole compounds are important components in anticancer, antibacterial, antitumor, anti-HIV and anthelmintic related drugs, we have explored the reaction of o-phenylenediamine with substituted benzyl alcohols to form benzimidazoles.59 The reaction of o-phenylenediamine with benzyl alcohols having an electron donating and electron withdrawing group formed benzimidazoles with high yield (>78%) (5a–c, Scheme 4 and Table S2†).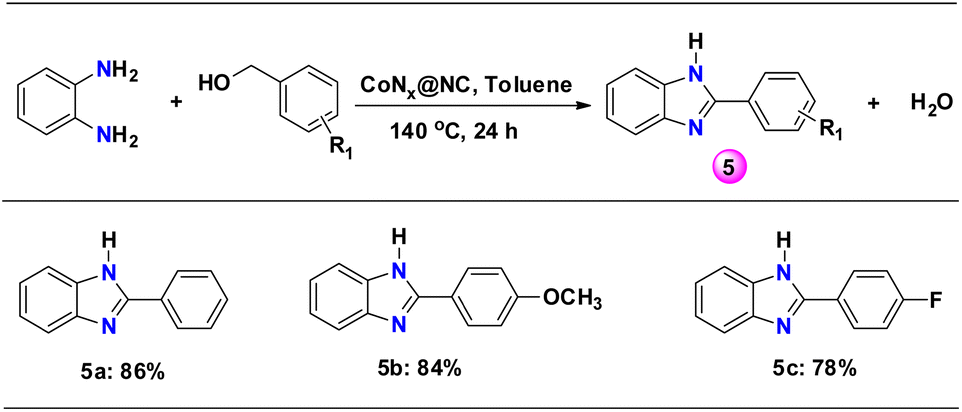 | ||
| Scheme 4 N-alkylation reaction of o-phenylenediamine with substituted benzyl alcohols. Reaction conditions: 1 mmol substituted benzyl alcohol, 0.5 mmol o-phenylenediamine, 10 mg catalyst, 0.5 mmol Cs2CO3, 2 mL toluene, 140 °C, 24 h. | ||
To get an insight of the reaction pathway, the advancement of the reaction with time was monitored. The reaction of benzyl alcohol with aniline produced both N-benzylidene aniline (imine) and N-benzylaniline (secondary amine) (Fig. S8†). The time-dependent profile of the products clearly indicates that a consecutive reaction mechanism is followed for the formation of N-benzylaniline. Initially, the yield of the imine was high and it was converted to N-benzylaniline with time (Fig. S8†).
The hydrogen borrowing reaction mechanism was also established by a series of controlled reactions (Fig. S9†). According to the hydrogen borrowing methodology, the first step is the dehydrogenation of benzyl alcohol by [Co] to form benzaldehyde and H–[Co]–H (Scheme 5). We were able to detect 4-methoxy benzaldehyde as the major product when aniline was not introduced into the reaction mixture (Fig. S9†). Separately, the reaction of 4-methoxy benzaldehyde and 4-methoxyaniline in the presence of CoNx@NC produced imine as the major product by the condensation process (Fig. S9†). The absence of the hydrogen borrowing process in the first step hindered the formation of H–[Co]–H and hence the production of secondary amine by the hydrogenation of imine was restricted (Fig. S9†). These reactions have clearly established that the first step is the dehydrogenation of benzyl alcohol to form benzaldehyde, which undergoes condensation with aniline to form imine, followed by the hydrogenation of imine to form secondary amine. The catalyst CoNx@NC provides active sites for all three processes – dehydrogenation, condensation, and hydrogenation.
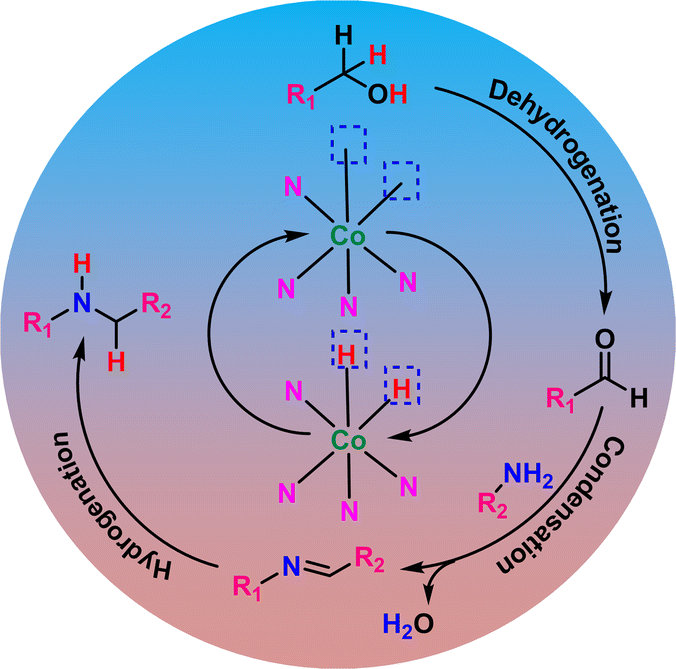 | ||
| Scheme 5 Reaction mechanism for the N-alkylation reaction. | ||
Recycling of the catalyst
We have investigated the recyclability of CoNx@NC for the N-alkylation reaction of aniline and benzyl alcohol. After each reaction, the catalyst was separated from the reaction mixture by centrifugation and used for a new batch of reaction. The catalyst was recycled six times with a minor loss in the activity and yield (Fig. S10†).Conclusions
In conclusion, MOF-derived CoNx@NC catalysts have been developed for the N-alkylation of anilines with alcohols to produce secondary amines. In CoNx@NC, the Co–N coordination, accessed through the pyrolysis of the MOF, was found to be beneficial to modulate the electron density on cobalt, optimize the binding of substrates and reaction intermediates on the catalyst surface and improve the yield of the desired product. The scope of a series of alcohols and anilines was explored and moderate to high yield of secondary amines was observed depending on substrates. Moreover, the catalyst has remarkable stability and versatile substrate scope for the synthesis of secondary amines.Data availability
The data supporting this article have been uploaded as part of the ESI.†Author contributions
VV synthesized and characterized the catalysts, carried out the catalytic reactions, interpreted the data and prepared the manuscript. PM was involved in the characterization of the catalysts. AI designed and conceptualized the project and edited the manuscript.Conflicts of interest
There are no conflicts of interest.Acknowledgements
This work is financially supported by CSIR [01(2977)/19/EMR-II], Govt. of India. VV thanks CSIR for providing the senior research fellowship.Notes and references
- S. Elangovan, J. Neumann, J. B. Sortais, K. Junge, C. Darcel and M. Beller, Nat. Commun., 2016, 7, 1–8 CrossRef PubMed
.
- Y. Wang, F. L. Zhang, Z. J. Liu and Z. J. Yao, Inorg. Chem., 2022, 61, 10310–10320 CrossRef CAS PubMed
- S. Chakraborty, R. Mondal, S. Pal, A. K. Guin, L. Roy and N. D. Paul, J. Org. Chem., 2023, 88, 771–787 CrossRef CAS PubMed
- J. Bariwal and E. Van Der Eycken, Chem. Soc. Rev., 2013, 42, 9283–9303 RSC
- J. I. Ramsden, R. S. Heath, S. R. Derrington, S. L. Montgomery, J. Mangas-Sanchez, K. R. Mulholland and N. J. Turner, J. Am. Chem. Soc., 2019, 141, 1201–1206 CrossRef CAS PubMed
- C. Zhang, Q. Liang, W. Yang, G. Zhang, M. Hu and G. Zhang, Green Chem., 2022, 24, 7368–7375 RSC
- K. Murugesan, T. Senthamarai, V. G. Chandrashekhar, K. Natte, P. C. J. Kamer, M. Beller and R. V. Jagadeesh, Chem. Soc. Rev., 2020, 49, 6273–6328 RSC
- E. Pedrajas, I. Sorribes, K. Junge, M. Beller and R. Llusar, Green Chem., 2017, 19, 3764–3768 RSC
- P. Ruiz-Castillo and S. L. Buchwald, Chem. Rev., 2016, 116, 12564–12649 CrossRef CAS PubMed
- B. G. Reed-Berendt, D. E. Latham, M. B. Dambatta and L. C. Morrill, ACS Cent. Sci., 2021, 7, 570–585 CrossRef CAS PubMed
- T. Irrgang and R. Kempe, Chem. Rev., 2019, 119, 2524–2549 CrossRef CAS PubMed
- M. H. S. A. Hamid, C. L. Allen, G. W. Lamb, A. C. Maxwell, H. C. Maytum, A. J. A. Watson and J. M. J. Williams, J. Am. Chem. Soc., 2009, 131, 1766–1774 CrossRef CAS
- J. J. A. Celaje, X. Zhang, F. Zhang, L. Kam, J. R. Herron and T. J. Williams, ACS Catal., 2017, 7, 1136–1142 CrossRef CAS
- O. Saidi, A. J. Blacker, M. M. Farah, S. P. Marsden and J. M. J. Williams, Chem. Commun., 2010, 46, 1541–1543 RSC
- I. Borthakur, M. Maji, A. Joshi and S. Kundu, J. Org. Chem., 2022, 87, 628–643 CrossRef CAS PubMed
- T. T. Dang, B. Ramalingam, S. P. Shan and A. M. Seayad, ACS Catal., 2013, 3, 2536–2540 CrossRef CAS
- M. Stratakis and H. Garcia, Chem. Rev., 2012, 112, 4469–4506 CrossRef CAS PubMed
- T. Ohshima, Y. Miyamoto, J. Ipposhi, Y. Nakahara, M. Utsunomiya and K. Mashima, J. Am. Chem. Soc., 2009, 131, 14317–14328 CrossRef CAS PubMed
- A. Corma, J. Navas and M. J. Sabater, Chem. Rev., 2018, 118, 1410–1459 CrossRef CAS
- A. Indra, M. Greiner, A. K. Gericke, R. Schlögl, D. Avnir and M. Driess, ChemCatChem, 2014, 6, 1935–1939 CrossRef CAS
- B. Goel, V. Vyas, N. Tripathi, A. Kumar Singh, P. W. Menezes, A. Indra and S. K. Jain, ChemCatChem, 2020, 12, 5743–5749 CrossRef CAS
- S. Rojas-Buzo, P. Concepción, A. Corma, M. Moliner and M. Boronat, ACS Catal., 2021, 11, 8049–8061 CrossRef CAS
- C. Chaudhari, S. M. A. H. Siddiki, K. Kon, A. Tomita, Y. Tai and K. I. Shimizu, Catal. Sci. Technol., 2014, 4, 1064–1069 RSC
- R. Cano, D. J. Ramón and M. Yus, J. Org. Chem., 2011, 76, 5547–5557 CrossRef CAS
- L. Wang, X. Jv, R. Wang, L. Ma, J. Liu, J. Sun, T. Shi, L. Zhao, X. Zhang and B. Wang, ACS Sustain. Chem. Eng., 2022, 10, 8342–8349 CrossRef CAS
- V. Goyal, J. Gahtori, A. Narani, P. Gupta, A. Bordoloi and K. Natte, J. Org. Chem., 2019, 84, 15389–15398 CrossRef CAS PubMed
- P. Tomkins, C. Valgaeren, K. Adriaensen, T. Cuypers and D. E. D. Vos, ChemCatChem, 2018, 10, 3689–3693 CrossRef CAS
- E. Podyacheva, O. I. Afanasyev, D. V. Vasilyev and D. Chusov, ACS Catal., 2022, 12, 7142–7198 CrossRef CAS
- W. Liu, L. Zhang, X. Liu, X. Liu, X. Yang, S. Miao, W. Wang, A. Wang and T. Zhang, J. Am. Chem. Soc., 2017, 139, 10790–10798 CrossRef CAS
- P. Y. Wu, G. P. Lu and C. Cai, Green Chem., 2021, 23, 396–404 RSC
- V. Goyal, N. Sarki, A. Narani, G. Naik, K. Natte and R. V. Jagadeesh, Coord. Chem. Rev., 2023, 474, 214827 CrossRef CAS
- V. G. Chandrashekhar, K. Natte, A. M. Alenad, A. S. Alshammari, C. Kreyenschulte and R. V. Jagadeesh, ChemCatChem, 2022, 14, e202101234 CrossRef CAS
- K. I. Shimizu, N. Imaiida, K. Kon, S. M. A. Hakim Siddiki and A. Satsuma, ACS Catal., 2013, 3, 998–1005 CrossRef CAS
- A. Afanasenko, S. Elangovan, M. C. A. Stuart, G. Bonura, F. Frusteri and K. Barta, Catal. Sci. Technol., 2018, 8, 5498–5505 RSC
- H. Liu, G. K. Chuah and S. Jaenicke, J. Catal., 2015, 329, 262–268 CrossRef CAS
- M. Dixit, M. Mishra, P. A. Joshi and D. O. Shah, Catal. Commun., 2013, 33, 80–83 CrossRef CAS
- K. Natte, H. Neumann, R. V. Jagadeesh and M. Beller, Nat. Commun., 2017, 8, 1–9 CrossRef CAS
- F. Kallmeier, R. Fertig, T. Irrgang and R. Kempe, Angew. Chem., Int. Ed., 2020, 59, 11789–11793 CrossRef CAS PubMed
- C. Li, Y. Meng, S. Yang and H. Li, ChemCatChem, 2021, 13, 5166–5177 CrossRef CAS
- S. Rösler, M. Ertl, T. Irrgang and R. Kempe, Angew. Chem., Int. Ed., 2015, 54, 15046–15050 CrossRef PubMed
- A. Tomer, F. Wyrwalski, C. Przybylski, J. F. Paul, E. Monflier, M. Pera-Titus and A. Ponchel, J. Catal., 2017, 356, 111–124 CrossRef CAS
- A. Indra, T. Song and U. Paik, Adv. Mater., 2018, 30, 1–25 CrossRef
- Z. Ma, B. Zhou, X. Li, R. G. Kadam, M. B. Gawande, M. Petr, R. Zbořil, M. Beller and R. V. Jagadeesh, Chem. Sci., 2022, 13, 111–117 RSC
- F. Kallmeier, R. Fertig, T. Irrgang and R. Kempe, Angew. Chem., Int. Ed., 2020, 59, 11789–11793 CrossRef CAS PubMed
- H. Su, K. X. Zhang, B. Zhang, H. H. Wang, Q. Y. Yu, X. H. Li, M. Antonietti and J. S. Chen, J. Am. Chem. Soc., 2017, 139, 811–818 CrossRef CAS
- Z. An and J. Li, Green Chem., 2022, 24, 1780–1808 RSC
- J. Gao, R. Ma, L. Feng, Y. Liu, R. Jackstell, R. V. Jagadeesh and M. Beller, Angew. Chem., Int. Ed., 2021, 60, 18591–18598 CrossRef CAS PubMed
- X. Liu, L. Xu, G. Xu, W. Jia, Y. Ma and Y. Zhang, ACS Catal., 2016, 6, 7611–7620 CrossRef CAS
- H. Guo, Q. Feng, J. Zhu, J. Xu, Q. Li, S. Liu, K. Xu, C. Zhang and T. Liu, J. Mater. Chem. A, 2019, 7, 3664–3672 RSC
- B. Singh and A. Indra, Mater. Today Energy, 2020, 16, 100404 CrossRef
- J. Li, B. Wang, Y. Qin, Q. Tao and L. Chen, Catal. Sci. Technol., 2019, 9, 3726–3734 RSC
- H. Konnerth, B. M. Matsagar, S. S. Chen, M. H. G. Prechtl, F. Shieh and K. C. Wu, Coord. Chem. Rev., 2020, 416, 213319 CrossRef CAS
- G. Li, H. Yang, H. Zhang, Z. Qi, M. Chen, W. Hu, L. Tian, R. Nie and W. Huang, ACS Catal., 2018, 8, 8396–8405 CrossRef CAS
- R. V. Jagadeesh, K. Murugesan, A. S. Alshammari, H. Neumann, M. M. Pohl, J. Radnik and M. Beller, Science, 2017, 358, 326–332 CrossRef CAS PubMed
- A. K. Singh, S. Ji, B. Singh, C. Das, H. Choi, P. W. Menezes and A. Indra, Mater. Today Chem., 2022, 23, 100668 CrossRef CAS
- Y. Xue, N. N. T. Pham, G. Nam, J. Choi, Y. Y. Ahn, H. Lee, J. Jung, S. G. Lee and J. Lee, Chem. Eng. J., 2021, 408, 127305 CrossRef CAS
- Z. Lin, G. Waller, Y. Liu, M. Liu and C. P. Wong, Adv. Energy Mater., 2012, 2, 884–888 CrossRef CAS
- C. Yuan, J. Li, L. Hou, X. Zhang, L. Shen and X. W. Lou, Adv. Funct. Mater., 2012, 22, 4592–4597 CrossRef CAS
- K. Chakrabarti, M. Maji and S. Kundu, Green Chem., 2019, 21, 1999–2004 RSC
Footnote |
| † Electronic supplementary information (ESI) available: Syntheses and characterization of the catalysts, catalytic reactions and characterization of the products. See DOI: https://doi.org/10.1039/d3sc02515b |
| This journal is © The Royal Society of Chemistry 2023 |