
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Noncovalent interaction guided selectivity of haloaromatic isomers in a flexible porous coordination polymer†
Rohan
Jena
 a,
Subhajit
Laha
a,
Nimish
Dwarkanath
a,
Arpan
Hazra
a,
Ritesh
Haldar
*b,
Sundaram
Balasubramanian
*a and
Tapas Kumar
Maji
*a
a,
Subhajit
Laha
a,
Nimish
Dwarkanath
a,
Arpan
Hazra
a,
Ritesh
Haldar
*b,
Sundaram
Balasubramanian
*a and
Tapas Kumar
Maji
*a
aChemistry and Physics of Materials Unit (CPMU), School of Advanced Materials (SAMat) Jawaharlal Nehru Centre for Advanced Scientific Research (JNCASR) Jakkur, Bangalore-560064, India. E-mail: tmaji@jncasr.ac.in
bTata Institute of Fundamental Research Hyderabad Gopanpally, Hyderabad 500046, Telangana, India
First published on 7th October 2023
Abstract
Porous, supramolecular structures exhibit preferential encapsulation of guest molecules, primarily by means of differences in the order of (noncovalent) interactions. The encapsulation preferences can be for geometry (dimension and shape) and the chemical nature of the guest. While geometry-based sorting is relatively straightforward using advanced porous materials, designing a “chemical nature” specific host is not. To introduce “chemical specificity”, the host must retain an accessible and complementary recognition site. In the case of a supramolecular, porous coordination polymer (PCP) [Zn(o-phen)(ndc)] (o-phen: 1,10-phenanthroline, ndc: 2,6-naphthalenedicarboxylate) host, equipped with an adaptable recognition pocket, we have discovered that the preferential encapsulation of a haloaromatic isomer is not only for dimension and shape, but also for the “chemical nature” of the guest. This selectivity, i.e., preference for the dimension, shape and chemical nature, is not guided by any complementary recognition site, which is commonly required for “chemical specificity”. Insights from crystal structures and computational studies unveil that the differences in the different types of noncovalent host–guest interaction strengths, acting in a concerted fashion, yield the unique selectivity.
Introduction
Dimension, shape, and chemical nature – based on any of these parameters, a mixture of chemicals can be separated using porous materials.1–4 To perform selective separations, new porous materials have been developed over the last two decades.5–8 These are made by connecting specific geometry nodes via dynamic crosslinking bonds.9 As a result, empty spaces or voids can be designed with desirable dimensions and chemical functionalities. By judiciously combining metal ions and complementary organic linkers, porous coordination polymers (PCPs)10 and metal–organic cages11 can be obtained; using organic linkers alone, covalent organic frameworks (COFs),12 conjugated microporous polymers (CMPs),13 and organic cages14 can also be designed. With the advantages of easy structural tailorability, it is evident that pores of specific shapes and sizes can be made for desired chemical separations.15,16 This scheme works in a straightforward manner, but in the case of a mixture of molecules with very similar physical and chemical properties, selectivity decreases drastically. This is because of the fact that molecules are discriminated on the basis of dimension, shape, or chemical nature, not all three at once. Molecular recognition by a combination of all three is non-trivial to design and realize.One way to introduce a more advanced recognition system in these porous materials is by allowing the pores to adjust to the guest molecules. The adjustment will depend on the dimension, shape as well as chemical nature of the guests. Among PCPs, a sub-category, i.e., flexible or soft PCPs exists.17 This class of PCPs is known to adapt its pore structure to the environment.18,19 These structures have flexible metal-nodes or geometrically flexible organic linkers. These features allow the structure to undergo contraction, expansion, or some specific distortion20–29 in response to changes in temperature and pressure, or upon filling of guest molecules in the voids.30,31 These structural responses are neither easy to predict nor to tune. Many previous reports on flexible PCPs illustrate structural changes, such as breathing, net movement in the entangled PCPs and hinge-like motion of 3D nets.19,32–34 In this work, we have realized selectivity by exploiting noncovalent interactions, acting in a concerted way, in a flexible PCP (Scheme 1). Noncovalent interactions, such as hydrogen bonding, aromatic π–π, and C–H–π interactions are much weaker compared to coordination or covalent bonds, and hence allow geometric flexibility, and can also be chemical-nature specific. We explore this possibility in a flexible porous coordination polymer containing a supramolecular pocket with tunable void space.35,36
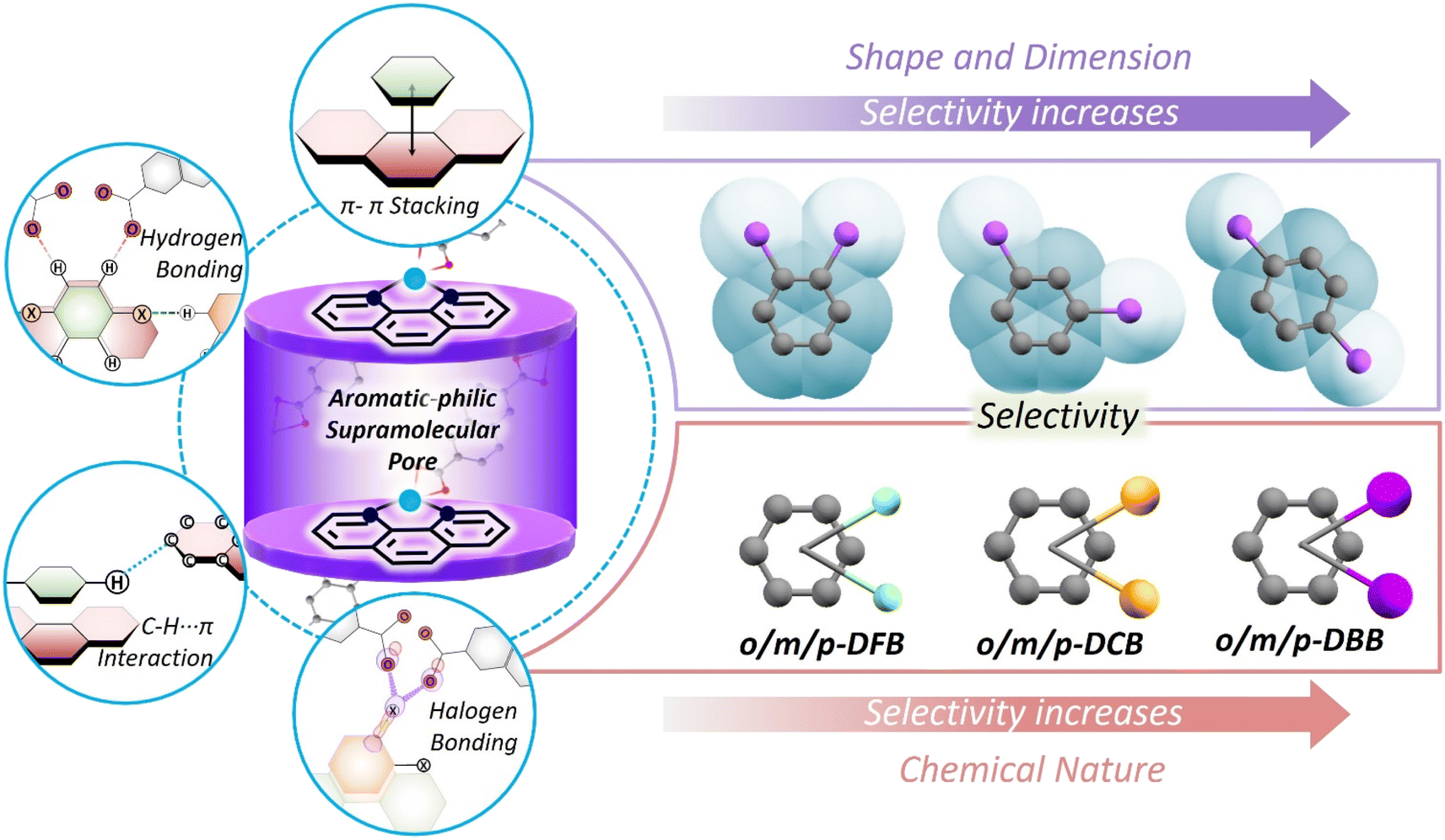 | ||
| Scheme 1 A schematic representation of selective encapsulation of haloaromatic isomers in a flexible porous coordination polymer; left panels show the supramolecular aromatic-philic pores created between the o-phen units of two adjacent 1D-chains of the PCP and the supramolecular interactions affecting the packing of various guest molecules in the nanopore. Right panels show the geometrical and chemical nature driven selectivity for the guest molecules. | ||
To test our hypothesis, we chose a library of positional haloaromatic isomers containing planar benzene rings. Among these selected isomers, dihalobenzenes are widely used as organic solvents, raw materials and intermediates for pharmaceuticals, pesticides and dye industries.37–43 The recognition and separation of these are of high value and remain a grand challenge.44,45 The dihalobenzene isomers have (i) different geometries due to the difference in positions of halogen substitution and (ii) different halogens (fluorine: F, chlorine: Cl, and bromine: Br) having different polarizabilities and sizes.46,47 Considering these differences, we have tested the separation capabilities in a Zn-based PCP {[Zn(o-phen)(ndc)]·(DMF)}, having aromatic-philic supramolecular pockets (o-phen: 1,10-phenanthroline, ndc: 2,6-naphthalenedicarboxylate, DMF: dimethylformamide).48 The preference for aromatic planar molecules is due to the unique pore-environment created by two sandwiching o-phen. Such an arrangement offers optimum space to host planar guest molecules like benzene, toluene, xylene, and substituted anilines.48,49 The combination of π⋯π, C–H⋯π, and hydrogen bonding interactions in this PCP yields unique molecular preferences. By performing in situ crystallization experiments on isomer-mixtures and single crystal structure determination, it was revealed that the host PCP prefers para-isomers (1,4-substituted) by geometric consideration and Br-substituted guests by chemical preference. As a result, ∼100% selective encapsulation was observed for the para-dibromo isomer from the mixture of para/ortho dibromo isomers, while for the para-difluoro- and dichloro-isomers, the host showed lower selectivities. The cooperativity between geometric and chemical preference improves the selectivity factor, and in the following section, we unveil the key parameters responsible, through structural insights of guest-encapsulated crystal structures and first-principles computational studies.
Results and discussion
A large number of flexible PCPs are available;50 we have selected one that can specifically host substituted benzene molecules. The selected PCP is constructed by linking Zn(II)-o-phen node with an NDC linker. This 1D structure, as illustrated in Fig. S1,† self-assembles into a 3D porous structure held together by noncovalent interactions (reported elsewhere,48 see Fig. S1†). This creates void spaces between a pair of o-phen of neighboring chains, and hosts solvent molecules (DMF or aromatic molecules) as shown in Fig. 1a. In earlier work, it has been observed that the PCP is porous in nature with intrinsic guest induced flexibility. The removal of the guest leads to the shrinkage of the pores, which reverts back to it's original porous structure on adsorption of the guest, confirmed by in situ adsorption-PXRD measurements.48 In the presence of DMF, the o-phen⋯o-phen distance is ∼8.5 Å, while for toluene, it reduces to about 7.5 Å.48 Hence the pore size is adjusted in accordance with the chemical nature of the guests. Similar changes were observed earlier, in the case of xylene isomers, i.e., structural adjustments induced by the geometry of the guest.49 The above features of the PCP prompted us to study its selectivities for haloaromatic isomers.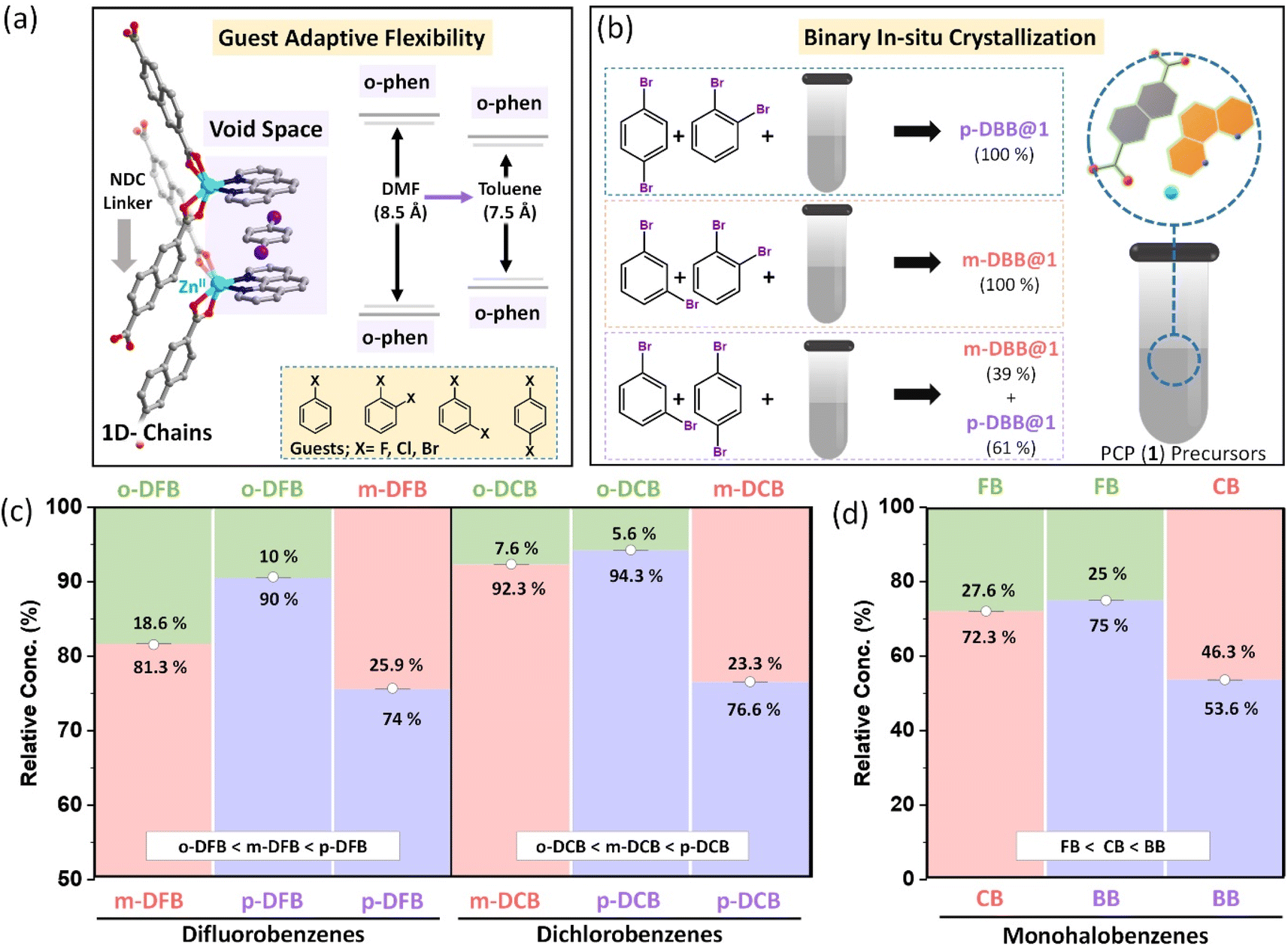 | ||
| Fig. 1 (a) Illustration of a nanospace created between the o-phen of adjacent 1D chains of PCPs, (b) experimental procedure for in situ crystallization of binary aliquot mixtures of dibromobenzenes (DBB), and the right panel shows reaction vessel containing PCP precursors i.e., 2,6-NDC, o-phen, and z Zn(NO3)2⋅6H2O in DMF,bar diagram showing relative amounts of isomers encapsulated after binary in situ crystallization of positional isomers of (c) dihalobenzene and (d) monohalobenzenes. [note: the components of each binary mixture for difluorobenzenes (DFB), dichlorobenzenes (DCB) and monohalobenzenes are mentioned at the top and bottom of the respective plots along with their relative concentrations]. | ||
Two classes of halo-substituted benzene isomers are considered for separation using the selected PCP: ternary or binary mixtures of (i) monohalobenzenes, namely, fluorobenzene (FB), chlorobenzene (CB), and bromobenzene (BB) and (ii) dihalobenzenes with halogen substitutions in ortho, meta and para configurations. For the latter class, only mixtures containing positional isomers of a given dihalobenzene, namely, difluorobenzene (DFB), dichlorobenzene (DCB), and dibromobenzene (DBB) are used. The PCP is assessed for a more challenging task of separating isomers of a given dihalobenzene having similar physicochemical properties rather than those with different halogen substitutions whose boiling points differ substantially. To probe the selectivities we have devised an in situ multicomponent crystallization-based experiment, as illustrated in Fig. 1b. In this method, the PCP precursors and the mono-/di-halobenzene isomer mixtures are mixed together to crystallize in the first set of experiments, and for each of the three dihalobenzenes, we tested ortho/meta, ortho/para, and meta/para mixtures (see ESI section S3 and Table S1, S2† with nomenclature guest@1). After the in situ crystallization experiments, the crystals were washed thoroughly and digested for characterization using 1H-NMR experiments (see ESI section S4 and Fig. S3–S7†). The relative amounts of the isomers estimated by the 1H-NMR experiments are shown in Fig. 1b–d (see Fig. S3–S7†). For all three dihalobenzenes, a common preference of ortho < meta < para is observed. However, there are two noticeable differences in the case of DBB compared to DFB or DCB: (i) the ortho/meta and ortho/para selectivity factors are nearly ∼99 (see ESI Table S3†) for DBB and (ii) the meta/para selectivity factor is lower for DBB (Fig. 1b). Similar experiments were carried out using the ternary mixtures, i.e., with mixtures of ortho, meta, and para isomers of each dihalobenzene (See Fig. S6†). The selectivity trend is ortho < meta < para, consistent with the binary mixture experiments (Fig. S3–S6†). It is worth noting that ortho isomers of DCB and DBB are exclusively rejected in their respective series. These experiments demonstrate that the PCP shows (i) a geometric preference for the para isomers and (ii) relative concentrations as well as selectivity factors, in general, improve with larger halogen size. To confirm this chemical preference, we have carried out in situ multi-component crystallization experiments for the mixture of fluorobenzene (FB), chlorobenzene (CB), and bromobenzene (BB) (see Fig. S7(a–c)†). The preference order is FB < CB < BB (Fig. 1d). This observation is in accordance with the results observed for binary and ternary dihalobenzene isomer mixtures. Such a concurrent geometry and chemical preference is unique. Additionally, the PCP is capable of selectively adsorbing halobenzene isomers, which has been demonstrated from the liquid-phase batch reactions using activated PCPs (see section S5, Fig. S14–S18 and Table S4†). The relative concentrations and selectivity values precisely match with the binary/ternary in situ crystallization results. Furthermore, at a relatively lower concentration of p-DBB in the mixtures of p-DBB and o-DBB, p-DBB is preferentially adsorbed by the PCP, as can be seen from Fig. S18.†
To understand the unique selectivity properties of the PCP, it is important to gain insights into the host–guest noncovalent interactions. To this end, we have carried out (i) crystal structure determination of the individual isomers encapsulated in the supramolecular pores using SCXRD experiments, (ii) periodic-density functional theory (DFT) calculations of the determined crystal structures to quantify the binding energies and study the extent of noncovalent interactions, and (iii) estimation of the enthalpy of guest binding (ΔH) using differential scanning calorimetry (DSC) experiments.
We could synthesize and characterize the individual monohalobenzene and dihalobenzene isomer-encapsulated PCP single crystals (guest@1) (CCDC no. 2266788–2266798). In all cases, the guest molecules are sandwiched between the o-phen rings, as illustrated in Fig. 2 (see ESI section S6†). This spatial geometry allows for facile π–π stacking to stabilize the guest@host crystal structures. In Fig. 2a–c, the three isomers of DFB encapsulated in the supramolecular pocket are shown with details of the orientation and π–π stacking. For o-DFB@1, the C–F bond is oriented inwards with a twist angle of 40° (angle between the a-axis and the C–F bond) and the guest is not sandwiched symmetrically by the two o-phen rings with centroid-to-centroid distances of 3.986 and 4.397 Å. This is because the aromatic planes of the guest and o-phen are not parallel (Fig. 2a). However, for both m-DFB@1 and p-DFB@1, the two centroid-to-centroid distances are identical and are shorter than that for o-DFB@1, yielding distances of 3.921 and 3.970 Å, respectively. This clearly indicates that the π–π stacking interactions are weaker for the ortho isomer and hence it is least preferred (in accordance with the observed selectivity). The spatial geometries of the ortho isomers of DCB and DBB are slightly different compared to the case of DFB, as shown in Fig. 2d and g. The C–Cl/C–Br bonds are projected outward with twist angles of 20° (Cl) and 30° (Br) and the aromatic planes of the guest and o-phen are nearly parallel. However, for the meta and para isomers, spatial positions are very similar to those of any of the halogen substitutions, except for small differences in the centroid-to-centroid distance (see Fig. 2). For the monohalobenzenes, no distinct difference in the guest spatial geometry was observed (Fig. S19†). These insights provide a qualitative explanation of the guest encapsulation and spatial geometry. The origin of preferential encapsulation of guests however, remains unclear.
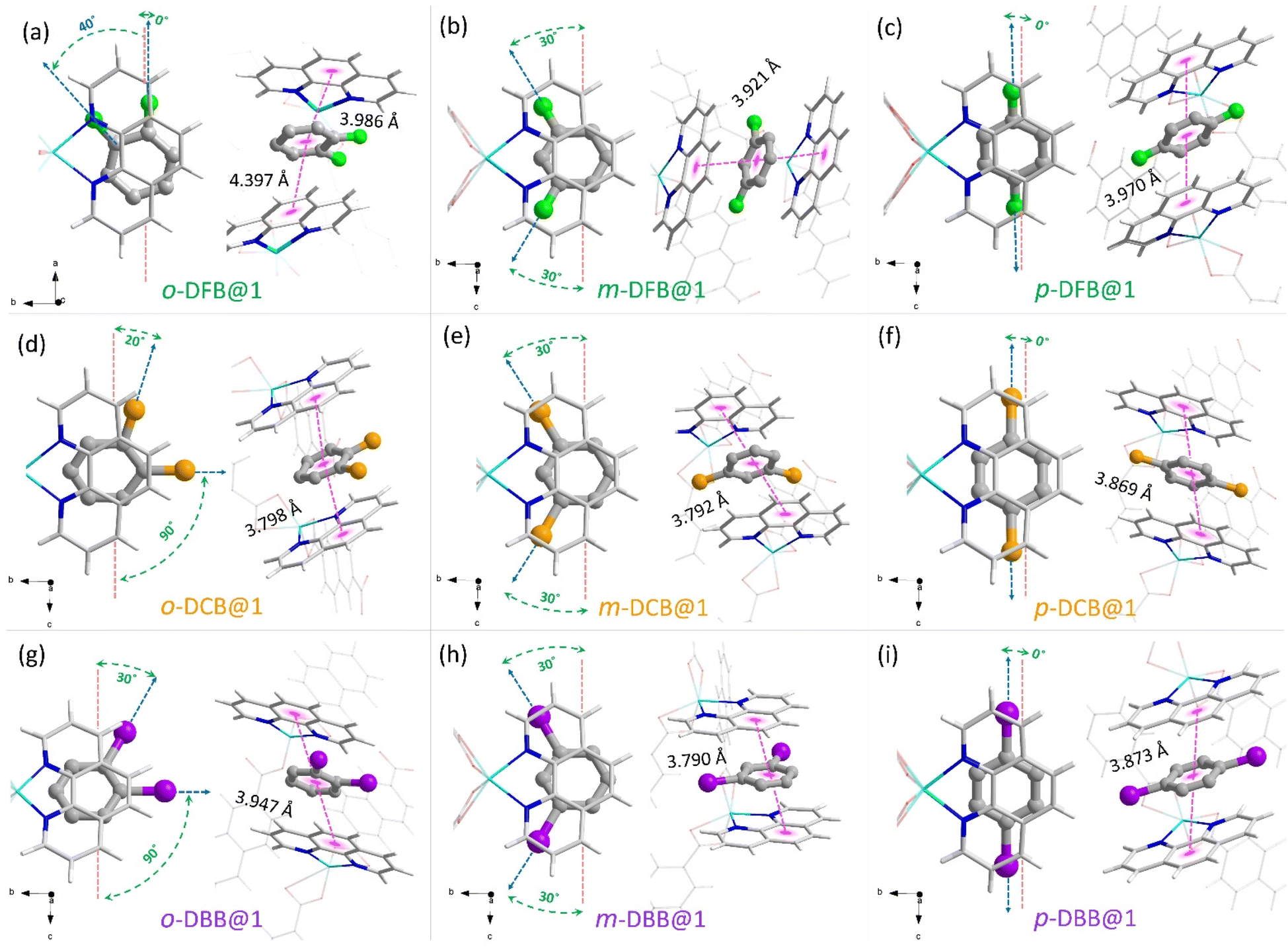 | ||
| Fig. 2 Guest molecule encapsulation in a “dynamic clip” in their respective guest@1 crystal structures determined using SCXRD experiments. Top and side views of the guest encapsulated cavities are shown on the left and on the right sides of each panel, along with their respective centroid-to-centroid distances from the sandwiching o-phen rings. (a–c) Difluorobenzenes, i.e., o-DFB@1, m-DFB@1 and p-DFB@1, (d–f) dichlorobenzenes, i.e., o-DCB@1, m-DCB@1 and p-DCB@1, (g–i) dibromobenzenes, i.e., o-DBB@1, m-DBB@1 and p-DBB@1. Color scheme—framework: hydrogen (white), carbon (silver), nitrogen (blue), oxygen (red), and zinc (cyan). Guests: carbon (gray), fluorine (green), chlorine (orange), and bromine (purple). Hydrogen atoms of the guest molecules are not shown for clarity. The guest molecules are represented by a thicker “ball-and-stick” representation. | ||
To rationalize the observed order of guest preference, a quantification of the host–guest interaction strengths and specific host-guest interactions are required. To this end, we have employed periodic-density functional theory (DFT) calculations using the Gaussian plane wave51 method implemented in Quickstep52,53 of the CP2K-7.1 package53,54 (see the ESI for details). First, periodic-DFT-based cell optimizations (CO) of supercells of all the experimentally determined crystal structures of guest@1 were performed. Later, the optimized structures were used to generate electron density difference maps from single point energy calculations (see section S8†) as a visual aid to identify the regions of PCP which interact with the guest molecules (Fig. 3–5 and S26†). Among DFB isomers, ortho-DFB exhibits four possible interactions – one hydrogen (HB) bonding and three C–H⋯π interactions (see Fig. 3a). For each of the meta and para isomers, eight interactions are seen, as illustrated in Fig. 3b and c. Although the types and number of interactions are the same for the meta and para isomers, p-DFB and m-DFB show prominent gain in electron density around hydrogen atoms in their respective crystal structures (Fig. 3e and f). The calculated binding energies (ESI section S8, Table S17†) are −124.90, −124.70 and −108.60 kJ mol−1 for para, meta, and ortho DFB, respectively, and this order is in accordance with the experimental observation (see 1H NMR results and the selectivity order, Fig. 1c).
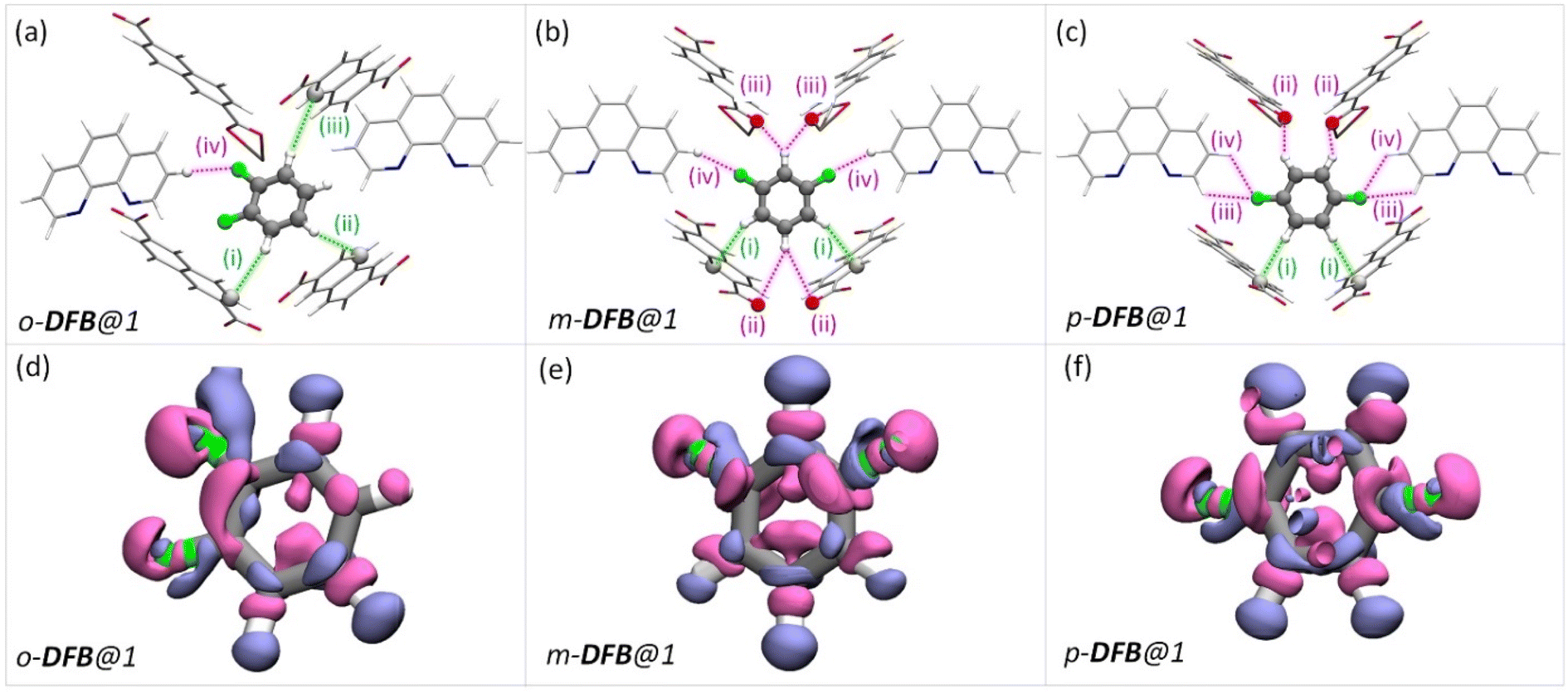 | ||
| Fig. 3 (a–c) The guest encapsulated cavities of (a) o-DFB@1, (b) m-DFB@1, and (c) p-DFB@1, respectively. o-phen rings sandwiching the guest molecules are omitted for clarity. The colored dashed-lines represent the type of supramolecular interactions (green and magenta lines represent C–H⋯π and hydrogen bonding interactions, respectively). Details of atom names, bond lengths, and angles associated with each interaction are shown in Table S18.† (d–f) Electron density differences in the vicinity of the guest in a cavity of (a) o-DFB@1, (b) m-DFB@1, and (c) p-DFB@1, respectively. Violet and magenta surfaces represent electron density gain and loss of magnitude 5.0 × 10−4 a.u., respectively. The guest encapsulated cavities shown here were obtained from the guest@1 super cells optimized using periodic-DFT. The color scheme for the atoms is identical to that in Fig. 2. | ||
In the case of DCB, the ortho isomer exhibits two types of interactions: two of its hydrogen atoms have close contact with the framework, one forming a C–H⋯π and the other forming a HB with NDC and carboxylate oxygen of the framework, respectively. In the case of meta and para isomers, the interactions are more in number, as shown in Fig. 4. However, a noticeable difference in the case of o-DCB is the presence of a halogen bonding interaction with the PCP. When a halogen atom, ‘X’, is bonded to another group ‘R’, X suffers a depletion of electron density near its polar region with respect to the R–X bond, called a “σ-hole”. A halogen bond emerges when an electron-rich group ‘A’ (A = lone pair, π-electrons, etc.) interacts with the electron-depleted region of X. Such a setting demands ∠R−X⋯A = 180°.55 This geometry-based criterion for halogen bonding identification is a necessary condition but not a sufficient one, especially for the case of guests confined within MOF/PCP pores.56 The electronic structure, however, definitively reveals the presence of such interactions. Hence, to elucidate this specific interaction (blue-dashed line in Fig. 4a), we have calculated maximally localized Wannier functions (MLWFs), and their corresponding centers – maximally localised Wannier function centers (MLWFCs),56–58 MLWFs (violet surfaces) and MLWFCs (yellow spheres) of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O1 (Fig. 4g–h) show that the π-electrons of the double bond face the C–Cl axis.
O1 (Fig. 4g–h) show that the π-electrons of the double bond face the C–Cl axis.
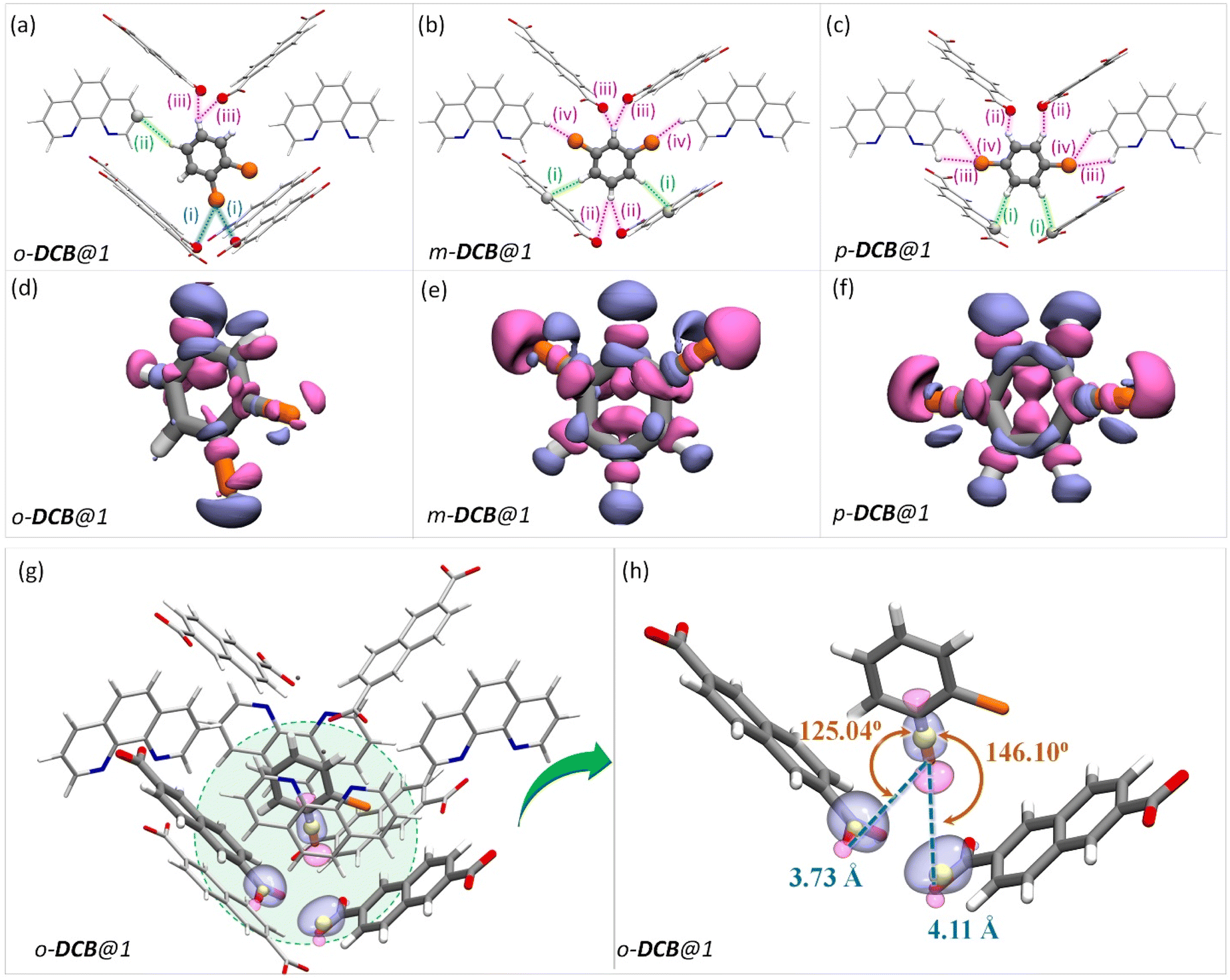 | ||
| Fig. 4 The guest encapsulated cavities of (a) o-DCB@1, (b) m-DCB@1, and (c) p-DCB@1, respectively. o-phen rings sandwiching the guest molecules are omitted for clarity. Details of the atom names, bond lengths and angles associated with each interaction are shown in Table S19.† (d–f) Electron density differences in the vicinity of the guest in a cavity of (a) o-DCB@1, (b) m-DCB@1, and (c) p-DCB@1, respectively. The coloring scheme used in this figure is identical to that in Fig. 2. (g and h) Reproduction of Fig. 4(a) with maximally localised Wannier functions (MLWFs; violet and magenta surfaces) and maximally localised Wannier function centres (MLWFCs; yellow spheres) for the C–Cl σ-bond (of o-DCB) and electron-pair in π-bonds (PB) of the framework CO1 bonds. Violet and magenta surfaces represent MLWFs of opposite phases with an isovalue of 5.0 × 10−2 a.u., with an exception of the σ-bond of C–Cl which is shown for an isovalue of 1.5 × 10−1 a.u. ∠C–Cl⋯PB are 125.04° and 146.10°, confirming that the π-bond electrons indeed interact with the σ-hole of the chlorine atom. All the guest encapsulated cavities shown in this figure were extracted from the respective guest@1 supercells of periodic DFT calculations, post cell optimization. | ||
Such an orientation indicates the interactions between the C–Cl σ-hole and the electron-rich π-clouds of the CO1 bond. This observation explains the difference in the spatial orientation of o-DCB and o-DFB. However, this additional halogen bonding is not substantial enough to affect the selectivity trend noted earlier. Furthermore, among the stacked-benzene dimers, the “slip” geometry is more favored than the “face-on” geometry, and the energy difference is about 4 kJ mol−1.59,60 Hence, p-DCB, the only DCB isomer having a slip w. r. t. the o-phen rings is more stabilized in the cavity than the other two isomers. Furthermore, binding energy calculations corroborate the selectivity preference of ortho < meta < para, with binding energies of −134.40, −142.70, and −144.00 kJ mol−1, in the same order (see Table S17 and Fig. S27†). For the DBB isomers, the nature of interactions is very similar to that of DCB. The o-DBB isomer, again, shows halogen bonding interactions; however the total possible interactions are still fewer compared to those of meta or para isomers. This is illustrated in Fig. 5. A noticeable difference is the change in the halogen bonding angle; for Br the halogen bond angle C–O–Br/Cl is larger, close to the ideal 180° (see Fig. S25†). This indicates stronger halogen bonding for Br, and this is in accordance with the polarizability trend of the halogens.55o-DBB also has a small region of electron density in the vicinity of the other halogen atom (Fig. 5d) alluding to another halogen bonding interaction not present for o-DCB in its crystal structure. Irrespective of this, the selectivity trend remains identical to that of DFB or DCB with binding energies of −138.50, −150.70, and −151.90 kJ mol−1, for o-, m-, and p-DBB, respectively (ESI, Table S17, Fig. S27†).
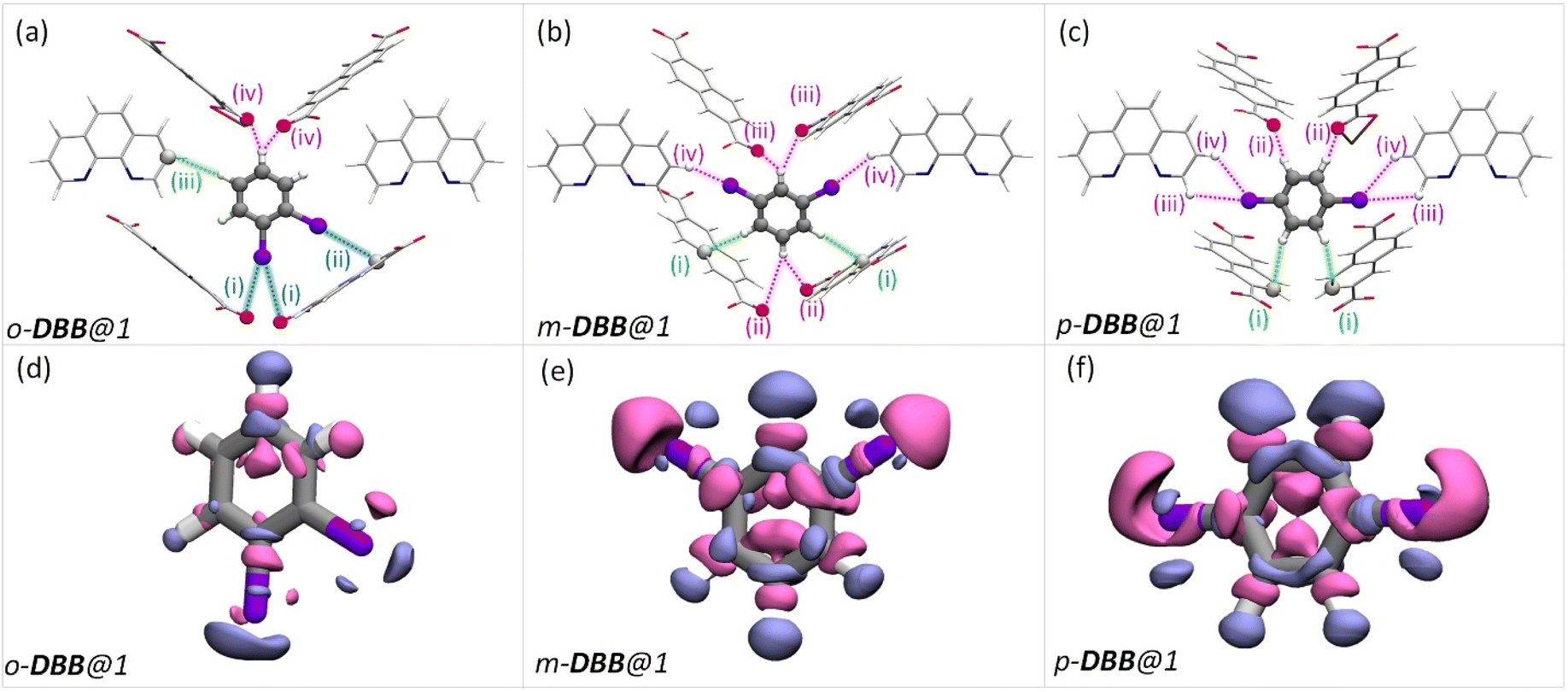 | ||
| Fig. 5 Dibromobenzene encapsulated cavities of 1. Each panel is analogous to the corresponding panels in Fig. 4(a–f), but bromine is in the place of chlorine atoms. Color schemes, the protocol for obtaining the structures, etc. are again identical to that in Fig. 4(a–f). | ||
We noted that due to the nonpolarizable nature of fluorine,55o-DFB does not participate in halogen bonding interactions with the PCP. Consequently, the position of o-DFB within the cavity is distinctively different from those of either o-DCB or o-DBB. On the other hand, the corresponding orientations of meta and para isomers within the PCP cavity are similar for all three dihalobenzenes, and so are the guest–framework interactions. However, differences in the binding energies among meta (or para) isomers of dihalobenzenes are significant for dihalobenzenes with different halogen substitutions (Table S17†). Halogen atoms of meta or para dihalobenzenes interact via hydrogen bonding with the framework. Due to the increase in polarizability with halogen size, the strength of hydrogen bonding interactions is in the order F < Cl < Br;55 thus the binding energies are in the order p-DFB < p-DCB < p-DBB (similarly for meta isomers) (ESI, Table S17†). In addition to differences in the hydrogen bonding strengths, other factors can also bring about differences in the binding energies. Thus, the PCP not only discriminates positional isomers of dihalobenzenes, but also on the basis of the halogen or, simply, based on the chemical nature.
Additional confirmation for the selectivity was obtained through differential scanning calorimetry by experimental quantification of the guest binding enthalpy (ΔH). We observed that the order of binding enthalpy is identical to the selectivity trend obtained in the in situ crystallization experiments. For the dihalobenzenes, from ortho to para, the binding enthalpy increased linearly. However, the energy difference between the different halogen isomers is negligible, e.g., the enthalpy values for the ortho DFB, DCB, and DBB are 69.53 ± 4.39, 76.86 ± 2.28 and 71.92 ± 1.14 J g−1 (see Fig. S20†). The difference due to the presence of different halogen atoms becomes clear by comparing the binding enthalpy values of the monohalobenzenes. These are 39.55 ± 1.81, 103.65 ± 2.35, and 116.90 ± 4.50 J g−1 for FB, CB, and BB, respectively (Table S16†). The order of binding enthalpies measured by DSC experiments and the binding energies obtained by periodic-DFT calculations are identical for various guest molecule encapsulations in the PCP.
Conclusion
In conclusion, we have demonstrated selective encapsulation of halobenzene isomers using a flexible porous coordination polymer. The current work showcases the prominence of concerted supramolecular interactions for the optimal and selective placement of guest molecules in a geometrically ordered position. This resulted from the directionally specific, short-range noncovalent interactions inside the nanospace. The adaptive nature of the PCP preferentially sorts para- > meta- > ortho dihalobenzene geometrical isomers and bromo-> chloro-> fluoro- in the case of monohalobenzenes. This preference is experimentally demonstrated by crystallization and separation experiments. All the experimental results were further corroborated by extensive computational studies such as ab initio cell optimizations, electron density difference mappings and periodic density functional theory (DFT) analyses. Additionally, theoretical binding energies and experimentally determined enthalpies unequivocally supported the order of selectivity obtained by the crystallization experiments. This unique preference of the supramolecular framework is attributed to the strength of the noncovalent interactions inside the pocket, but not to the type or number of interactions. The findings presented here can be a tool guide to design more advanced supramolecular systems, which can perform easy recognition and separation of challenging isomers and isotopes.Data availability
All associated data are in the ESI† or deposited with CCDC (2266788–2266798).Author contributions
R. J. and T. K. M. designed the concept of this work. R. J. performed major experiments and data analysis. S. L. assisted in the data analysis. N. D. performed all the theoretical calculations. A. H. assisted in the crystal structure refinements. R. H., S. B., and T. K. M. assisted in the writing and editing of the manuscript. All authors contributed to the preparation of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
R. J. acknowledges CSIR, Govt. of India, for a fellowship. T. K. M. thankfully acknowledges SERB, Dept. of Science and Technology (DST), Govt. of India, for financial support (Project no. SPR/2021/000592 and CRG/2019/005951). SAMat, ICMS, SSL research facility and the Sheikh Saqr senior fellowship (T. K. M) are also gratefully acknowledged. R. H. acknowledges intramural funds at TIFR Hyderabad, from the Department of Atomic Energy (DAE), India, under project identification number RTI 4007.References
- J. G. O'Connell-Danes, B. T. Ngwenya, C. A. Morrison and J. B. Love, Selective separation of light rare-earth elements by supramolecular encapsulation and precipitation, Nat. Commun., 2022, 13, 4497 CrossRef PubMed.
- E. Krieg, H. Weissman, E. Shirman, E. Shimoni and B. Rybtchinski, A recyclable supramolecular membrane for size-selective separation of nanoparticles, Nat. Nanotechnol., 2011, 6, 141–146 CrossRef CAS PubMed.
- J.-R. Li, R. J. Kuppler and H.-C. Zhou, Selective gas adsorption and separation in metal–organic frameworks, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- Y. Yang, P. Bai and X. Guo, Separation of Xylene Isomers: A Review of Recent Advances in Materials, Ind. Eng. Chem. Res., 2017, 56, 14725–14753 CrossRef CAS.
- Y. Wu and B. M. Weckhuysen, Separation and Purification of Hydrocarbons with Porous Materials, Angew. Chem., Int. Ed., 2021, 60, 18930–18949 CrossRef CAS PubMed.
- W. Guo, S. M. Mahurin, R. R. Unocic, H. Luo and S. Dai, Broadening the Gas Separation Utility of Monolayer Nanoporous Graphene Membranes by an Ionic Liquid Gating, Nano Lett., 2020, 20, 7995–8000 CrossRef CAS PubMed.
- L. F. Villalobos, D. J. Babu, K.-J. Hsu, C. Van Goethem and K. V. Agrawal, Gas Separation Membranes with Atom-Thick Nanopores: The Potential of Nanoporous Single-Layer Graphene, Acc. Mater. Res., 2022, 3, 1073–1087 CrossRef CAS PubMed.
- A. Knebel and J. Caro, Metal–organic frameworks and covalent organic frameworks as disruptive membrane materials for energy-efficient gas separation, Nat. Nanotechnol., 2022, 17, 911–923 CrossRef CAS PubMed.
- Y. Li, M. Karimi, Y.-N. Gong, N. Dai, V. Safarifard and H.-L. Jiang, Integration of metal-organic frameworks and covalent organic frameworks: Design, synthesis, and applications, Matter, 2021, 4, 2230–2265 CrossRef CAS.
- S. Kitagawa, R. Kitaura and S. Noro, Functional Porous Coordination Polymers, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed.
- D. Zhang, T. K. Ronson, Y.-Q. Zou and J. R. Nitschke, Metal–organic cages for molecular separations, Nat. Rev. Chem, 2021, 5, 168–182 CrossRef CAS PubMed.
- K. Geng, T. He, R. Liu, S. Dalapati, K. T. Tan, Z. Li, S. Tao, Y. Gong, Q. Jiang and D. Jiang, Covalent Organic Frameworks: Design, Synthesis, and Functions, Chem. Rev., 2020, 120, 8814–8933 CrossRef CAS PubMed.
- J.-S. M. Lee and A. I. Cooper, Advances in Conjugated Microporous Polymers, Chem. Rev., 2020, 120, 2171–2214 CrossRef CAS PubMed.
- T. Tozawa, J. T. A. Jones, S. I. Swamy, S. Jiang, D. J. Adams, S. Shakespeare, R. Clowes, D. Bradshaw, T. Hasell, S. Y. Chong, C. Tang, S. Thompson, J. Parker, A. Trewin, J. Bacsa, A. M. Z. Slawin, A. Steiner and A. I. Cooper, Porous organic cages, Nat. Mater., 2009, 8, 973–978 CrossRef CAS PubMed.
- L. Li, H.-M. Wen, C. He, R.-B. Lin, R. Krishna, H. Wu, W. Zhou, J. Li, B. Li and B. Chen, A Metal–Organic Framework with Suitable Pore Size and Specific Functional Sites for the Removal of Trace Propyne from Propylene, Angew. Chem., Int. Ed., 2018, 57, 15183–15188 CrossRef CAS PubMed.
- Z. Wang, N. Sikdar, S.-Q. Wang, X. Li, M. Yu, X.-H. Bu, Z. Chang, X. Zou, Y. Chen, P. Cheng, K. Yu, M. J. Zaworotko and Z. Zhang, Soft Porous Crystal Based upon Organic Cages That Exhibit Guest-Induced Breathing and Selective Gas Separation, J. Am. Chem. Soc., 2019, 141, 9408–9414 CrossRef CAS PubMed.
- S. Horike, S. Shimomura and S. Kitagawa, Soft porous crystals, Nat. Chem., 2009, 1, 695–704 CrossRef CAS PubMed.
- J. D. Evans, V. Bon, I. Senkovska, H.-C. Lee and S. Kaskel, Four-dimensional metal-organic frameworks, Nat. Commun., 2020, 11, 2690 CrossRef CAS PubMed.
- T. K. Maji, R. Matsuda and S. Kitagawa, A flexible interpenetrating coordination framework with a bimodal porous functionality, Nat. Mater., 2007, 6, 142–148 CrossRef CAS PubMed.
- N. Sikdar, A. Hazra, D. Samanta, R. Haldar and T. K. Maji, Guest-Responsive Reversible Electron Transfer in a Crystalline Porous Framework Supported by a Dynamic Building Node, Angew. Chem., Int. Ed., 2020, 59, 18479–18484 CrossRef CAS PubMed.
- N. Sikdar, S. Bonakala, R. Haldar, S. Balasubramanian and T. K. Maji, Dynamic Entangled Porous Framework for Hydrocarbon (C2–C3) Storage, CO2 Capture, and Separation, Chem. - Eur. J., 2016, 22, 6059–6070 CrossRef CAS PubMed.
- B. Manna, A. K. Chaudhari, B. Joarder, A. Karmakar and S. K. Ghosh, Dynamic Structural Behavior and Anion-Responsive Tunable Luminescence of a Flexible Cationic Metal–Organic Framework, Angew. Chem., Int. Ed., 2013, 52, 998–1002 CrossRef CAS PubMed.
- B. Manna, A. V Desai and S. K. Ghosh, Neutral N-donor ligand based flexible metal–organic frameworks, Dalton Trans., 2016, 45, 4060–4072 RSC.
- D. Tanaka, A. Henke, K. Albrecht, M. Moeller, K. Nakagawa, S. Kitagawa and J. Groll, Rapid preparation of flexible porous coordination polymer nanocrystals with accelerated guest adsorption kinetics, Nat. Chem., 2010, 2, 410–416 CrossRef CAS PubMed.
- S. Shimomura, M. Higuchi, R. Matsuda, K. Yoneda, Y. Hijikata, Y. Kubota, Y. Mita, J. Kim, M. Takata and S. Kitagawa, Selective sorption of oxygen and nitric oxide by an electron-donating flexible porous coordination polymer, Nat. Chem., 2010, 2, 633–637 CrossRef CAS PubMed.
- M. L. Foo, R. Matsuda, Y. Hijikata, R. Krishna, H. Sato, S. Horike, A. Hori, J. Duan, Y. Sato, Y. Kubota, M. Takata and S. Kitagawa, An Adsorbate Discriminatory Gate Effect in a Flexible Porous Coordination Polymer for Selective Adsorption of CO2 over C2H2, J. Am. Chem. Soc., 2016, 138, 3022–3030 CrossRef CAS PubMed.
- N. Yanai, K. Kitayama, Y. Hijikata, H. Sato, R. Matsuda, Y. Kubota, M. Takata, M. Mizuno, T. Uemura and S. Kitagawa, Gas detection by structural variations of fluorescent guest molecules in a flexible porous coordination polymer, Nat. Mater., 2011, 10, 787–793 CrossRef CAS PubMed.
- S. Ehrling, E. M. Reynolds, V. Bon, I. Senkovska, T. E. Gorelik, J. D. Evans, M. Rauche, M. Mendt, M. S. Weiss, A. Pöppl, E. Brunner, U. Kaiser, A. L. Goodwin and S. Kaskel, Adaptive response of a metal–organic framework through reversible disorder–disorder transitions, Nat. Chem., 2021, 13, 568–574 CrossRef CAS PubMed.
- C. K. Brozek, A. Ozarowski, S. A. Stoian and M. Dincă, Dynamic structural flexibility of Fe-MOF-5 evidenced by 57Fe Mössbauer spectroscopy, Inorg. Chem. Front., 2017, 4, 782–788 RSC.
- Y. Kim, R. Haldar, H. Kim, J. Koo and K. Kim, The guest-dependent thermal response of the flexible MOF Zn2(BDC)2(DABCO), Dalton Trans., 2016, 45, 4187–4192 RSC.
- D. Samanta, S. Roy, R. Sasmal, N. Das Saha, P. K. R., R. Viswanatha, S. S. Agasti and T. K. Maji, Solvent Adaptive Dynamic Metal-Organic Soft Hybrid for Imaging and Biological Delivery, Angew. Chem., Int. Ed., 2019, 58, 5008–5012 CrossRef CAS PubMed.
- Y. Takashima, V. M. Martínez, S. Furukawa, M. Kondo, S. Shimomura, H. Uehara, M. Nakahama, K. Sugimoto and S. Kitagawa, Molecular decoding using luminescence from an entangled porous framework, Nat. Commun., 2011, 2, 168 CrossRef PubMed.
- T. K. Maji, K. Uemura, H.-C. Chang, R. Matsuda and S. Kitagawa, Expanding and Shrinking Porous Modulation Based on Pillared-Layer Coordination Polymers Showing Selective Guest Adsorption, Angew. Chem., Int. Ed., 2004, 43, 3269–3272 CrossRef CAS PubMed.
- P. Kanoo, R. Haldar, S. K. Reddy, A. Hazra, S. Bonakala, R. Matsuda, S. Kitagawa, S. Balasubramanian and T. K. Maji, Crystal Dynamics in Multi-stimuli-Responsive Entangled Metal–Organic Frameworks, Chem. - Eur. J., 2016, 22, 15864–15873 CrossRef CAS PubMed.
- J.-M. Lehn, Supramolecular Chemistry, Science, 1993, 260, 1762–1763 CrossRef CAS PubMed.
- H.-J. Schneider and A. K. Yatsimirsky, Selectivity in supramolecular host–guest complexes, Chem. Soc. Rev., 2008, 37, 263–277 RSC.
- A. J. Varni, M. Kawakami, M. V. Bautista and K. J. T. Noonan, Polymerization Reactions via Cross Coupling. 2022, pp. 465-510.
- U. N. Rao, J. Maguire and E. Biehl, Effect of substituents and benzyne generating bases on the orientation to and reactivity of haloarynes, ARKIVOC, 2004, 2004, 88–100 Search PubMed.
- K. M. Cantor, P. Watts, Introduction to the Plastics Industry, Applied Plastics Engineering Handbook, 2011, xv-xvi Search PubMed.
- R. J. Rubio, G. T. S. Andavan, E. B. Bauer, T. K. Hollis, J. Cho, F. S. Tham and B. Donnadieu, Toward a general method for CCC N-heterocyclic carbene pincer synthesis: Metallation and transmetallation strategies for concurrent activation of three C–H bonds, J. Organomet. Chem., 2005, 690, 5353–5364 CrossRef CAS.
- D. R. Fahey and C. E. Ash, Mechanism of poly(p-phenylene sulfide) growth from p-dichlorobenzene and sodium sulfide, Macromolecules, 1991, 24, 4242–4249 CrossRef CAS.
- J. L. Wolk and A. A. Frimer, A Simple, Safe and Efficient Synthesis of Tyrian Purple (6,6′-Dibromoindigo), Molecules, 2010, 15, 5561–5580 CrossRef CAS PubMed.
- G. Chen, A. K. Mohanty and M. Misra, Progress in research and applications of Polyphenylene Sulfide blends and composites with carbons, Composites, Part B, 2021, 209, 108553 CrossRef CAS.
- E. Li, Y. Zhou, R. Zhao, K. Jie and F. Huang, Dihalobenzene Shape Sorting by Nonporous Adaptive Crystals of Perbromoethylated Pillararenes, Angew. Chem., Int. Ed., 2019, 58, 3981–3985 CrossRef CAS PubMed.
- D. S. Sholl and R. P. Lively, Seven chemical separations to change the world, Nature, 2016, 532, 435–437 CrossRef PubMed.
- H. Li, C. Li, Y. Wu, C. Wang, T. Guo, J. Zhang and L. Sun, Cross-linked γ-cyclodextrin metal-organic framework—a new stationary phase for the separations of benzene series and polycyclic aromatic hydrocarbons, Microchim. Acta, 2021, 188, 245 CrossRef CAS PubMed.
- K. J. Hartlieb, J. M. Holcroft, P. Z. Moghadam, N. A. Vermeulen, M. M. Algaradah, M. S. Nassar, Y. Y. Botros, R. Q. Snurr and J. F. Stoddart, CD-MOF: A Versatile Separation Medium, J. Am. Chem. Soc., 2016, 138, 2292–2301 CrossRef CAS PubMed.
- R. Haldar, R. Matsuda, S. Kitagawa, S. J. George and T. K. Maji, Amine-Responsive Adaptable Nanospaces: Fluorescent Porous Coordination Polymer for Molecular Recognition, Angew. Chem., Int. Ed., 2014, 53, 11772–11777 CrossRef CAS PubMed.
- S. Laha, R. Haldar, N. Dwarkanath, S. Bonakala, A. Sharma, A. Hazra, S. Balasubramanian and T. K. Maji, A Dynamic Chemical Clip in Supramolecular Framework for Sorting Alkylaromatic Isomers using Thermodynamic and Kinetic Preferences, Angew. Chem., Int. Ed., 2021, 60, 19921–19927 CrossRef CAS PubMed.
- A. Schneemann, V. Bon, I. Schwedler, I. Senkovska, S. Kaskel and R. A. Fischer, Flexible metal–organic frameworks, Chem. Soc. Rev., 2014, 43, 6062–6096 RSC.
- G. Lippert, J. Hutter and M. Parrinello, A hybrid Gaussian and plane wave density functional scheme, Mol. Phys., 1997, 92, 477–488 CrossRef CAS.
- J. VandeVondele, M. Krack, F. Mohamed, M. Parrinello, T. Chassaing and J. Hutter, Quickstep: Fast and accurate density functional calculations using a mixed Gaussian and plane waves approach, Comput. Phys. Commun., 2005, 167, 103–128 CrossRef CAS.
- T. D. Kühne, M. Iannuzzi, M. Del Ben, V. V Rybkin, P. Seewald, F. Stein, T. Laino, R. Z. Khaliullin, O. Schütt, F. Schiffmann, D. Golze, J. Wilhelm, S. Chulkov, M. H. Bani-Hashemian, V. Weber, U. Borštnik, M. Taillefumier, A. S. Jakobovits, A. Lazzaro, H. Pabst, T. Müller, R. Schade, M. Guidon, S. Andermatt, N. Holmberg, G. K. Schenter, A. Hehn, A. Bussy, F. Belleflamme, G. Tabacchi, A. Glöβ, M. Lass, I. Bethune, C. J. Mundy, C. Plessl, M. Watkins, J. VandeVondele, M. Krack and J. Hutter, CP2K: An electronic structure and molecular dynamics software package - Quickstep: Efficient and accurate electronic structure calculations, J. Chem. Phys., 2020, 152, 194103 CrossRef PubMed.
- J. Hutter, M. Iannuzzi, F. Schiffmann and J. VandeVondele, cp2k: atomistic simulations of condensed matter systems, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 15–25 CAS.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, The Halogen Bond, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- N. Dwarkanath and S. Balasubramanian, Gate Opening without Volume Change Triggers Cooperative Gas Interactions, Underpins an Isotherm Step in Metal–Organic Frameworks, Inorg. Chem., 2022, 61, 10810–10821 CrossRef CAS PubMed.
- N. Marzari, A. A. Mostofi, J. R. Yates, I. Souza and D. Vanderbilt, Maximally localized Wannier functions: Theory and applications, Rev. Mod. Phys., 2012, 84, 1419–1475 CrossRef CAS.
- R. C. Remsing and M. L. Klein, Halogen Bond Structure and Dynamics from Molecular Simulations, J. Phys. Chem. B, 2019, 123, 6266–6273 CrossRef CAS PubMed.
- A. Banerjee, A. Saha and B. K. Saha, Understanding the Behavior of π–π Interactions in Crystal Structures in Light of Geometry Corrected Statistical Analysis: Similarities and Differences with the Theoretical Models, Cryst. Growth Des., 2019, 19, 2245–2252 CrossRef CAS.
- M. Pitoňák, P. Neogrády, J.
![[R with combining breve]](https://www.rsc.org/images/entities/char_0052_0306.gif) ezáč, P. Jurečka, M. Urban and P. Hobza, Benzene Dimer: High-Level Wave Function and Density Functional Theory Calculations, J. Chem. Theory Comput., 2008, 4, 1829–1834 CrossRef PubMed.
ezáč, P. Jurečka, M. Urban and P. Hobza, Benzene Dimer: High-Level Wave Function and Density Functional Theory Calculations, J. Chem. Theory Comput., 2008, 4, 1829–1834 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2266788–2266798. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc03079b |
| This journal is © The Royal Society of Chemistry 2023 |