
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFeasible bottom-up development of conjugated microporous polymers (CMPs) for boosting the deep removal of sulfur dioxide†
He
Li
a,
Hanqian
Pan
b,
Yijian
Li
b,
Shuaishuai
Shang
a,
Shihui
Huang
a,
Xili
Cui
 *b,
Jun
Hu
*a and
Honglai
Liu
a
*b,
Jun
Hu
*a and
Honglai
Liu
a
aState Key Laboratory of Chemical Engineering, School of Chemistry and Molecular Engineering, East China University of Science and Technology, 130 Meilong Road, Shanghai, 200237, China. E-mail: junhu@ecust.edu.cn
bKey Laboratory of Biomass Chemical Engineering of Ministry of Education, College of Chemical and Biological Engineering, Zhejiang University, Hangzhou, 310027, China. E-mail: cuixl@zju.edu.cn
First published on 17th July 2023
Abstract
A pain-point for material development is that computer-screened structures are usually difficult to realize in experiments. Herein, considering that linkages are crucial for building functional nanoporous polymers with diverse functionalities, we develop an efficient approach for constructing target-specific conjugated microporous polymers (CMPs) based on screening feasible polymerization pathways. Taking the deep removal of SO2 from a SO2/CO2 mixture as the specific target, we precisely screen the linkages and fabricate different CMPs by manipulating the porosity and hydrophobicity. Based on the optimized Buchwald–Hartwig amination, the obtained CMPs can achieve SO2/CO2 selectivity as high as 113 and a moderate Qst of 30 kJ mol−1 for feasible regeneration. Furthermore, the potential of CMPs for practical SO2/CO2 separation is demonstrated through continued breakthrough tests. The SO2 binding sites are consistent with the screening results and proved by in situ Fourier transform infrared spectroscopy and grand canonical Monte Carlo simulation, providing solid feasibility for synthesis realizability for future boosts of task-specific CMPs.
Introduction
Deep removal of SO2 is of great significance because of its serious environmental hazards.1,2 Especially when addressing the challenge of carbon neutrality, the presence of SO2 in flue gas is detrimental to CO2 capture and utilization (CCU) since acidic SO2 will preferentially bind with amine-based absorption solvents or adsorbents; meanwhile, even residual SO2 may cause the deactivation of metallic catalysts for CO2 chemical conversion.3–6 Compared with conventional wet and dry flue gas desulfurization processes such as alkaline slurry scrubbing and lime spray drying, reversible physical adsorption via porous materials such as zeolites and activated carbon is recognized as a sustainable energy-saving technology for SO2 removal.7–10 However, SO2 and CO2 are both acidic gases and have similar kinetic sizes (0.4 nm for SO2vs. 0.33 nm for CO2), making the selective adsorption of SO2 over CO2 full of challenges.Recent years have witnessed a surge in metal–organic frameworks (MOFs) with good SO2/CO2 separation performance.11–14 In these state-of-the-art MOFs, creating open metal sites,15–17 introducing strong host–guest interaction,18–20 as well as the gate-opening effect in MOFs21 have been applied to enhance the deep desulfurization performance. Nevertheless, most MOFs are unable to withstand strong corrosive SO2 or moisture, which usually results in irreversible structural collapse.15,19,22 Therefore, the stable skeleton becomes one of the most critical factors for long-term SO2 removal service during the design of porous materials. Alternatively, conjugated microporous polymers (CMPs) that are constructed using pure organic elements would exhibit promising characteristics of high acid tolerance and water resistance. Moreover, the combination of intrinsic microporous structures and designable task-specific binding sites would endow them with great potential for the efficient removal of SO2.23
Although thousands of CMPs have been developed, the one committing to deep desulfurization has been rarely reported. Considering that computer-screened structures of CMPs from billions of possible monomers may be invalid in realistic synthesis and trial-and-error experimental approaches are extremely chemically expensive and time-consuming,24 the efficient bottom-up development of CMPs for boosting the deep removal of SO2 is still full of challenges. From a realistic synthesis perspective, feasible coupling reactions for the CMPs' construction are limited, and if we screen from linkages extracted from the limited coupling reactions, rather than randomly from billions of monomers, the synthesis realizability of the screened CMPs can be guaranteed. Moreover, as each linkage corresponding to one specific coupling reaction can be formed by diverse monomers, we can further regulate the functionality and porous structure, simultaneously, to achieve task-specific applications.
Results and discussion
To this end, there are six typical coupling reactions for CMPs' construction that are Suzuki–Miyaura cross-coupling, Heck reaction, Sonogashira–Hagihara cross-coupling, Buchwald–Hartwig amination, cyclotrimerization, and thioether connection, resulting in the linkages of carbon–carbon single bonds (–C–C–), carbon–carbon double bonds (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–), carbon–carbon triple bonds (–C
C–), carbon–carbon triple bonds (–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–), secondary amine (–NH–), triazine (–C3N3–), and thioether (–S–), respectively. As shown in Fig. 1, the SO2 binding energies (BEs) of different linkages are in the order of –NH– (21.42 kJ mol−1) > –CC– (14.88 kJ mol−1) > –C3N3– (13.82 kJ mol−1) > –C–C– (11.76 kJ mol−1) > –CC– (11.52 kJ mol−1) > –S– (7.4 kJ mol−1). In comparison, the BEs of CO2 are much lower in the range of 5.78–7.92 kJ mol−1 due to its lower polarity (Fig. S1–S4†). Consequently, the binding energy difference between SO2 and CO2 (ΔBE(SO2/CO2)) is in the order of –NH– > –CC– > –C–C– > –C3N3– > –CC– > –S– (Table 1). As the selective separation of SO2 over CO2 depends much on ΔBE(SO2/CO2), this linkage screening approach could be a feasible method to effectively construct CMPs for the deep removal of SO2 from CO2.
C–), secondary amine (–NH–), triazine (–C3N3–), and thioether (–S–), respectively. As shown in Fig. 1, the SO2 binding energies (BEs) of different linkages are in the order of –NH– (21.42 kJ mol−1) > –CC– (14.88 kJ mol−1) > –C3N3– (13.82 kJ mol−1) > –C–C– (11.76 kJ mol−1) > –CC– (11.52 kJ mol−1) > –S– (7.4 kJ mol−1). In comparison, the BEs of CO2 are much lower in the range of 5.78–7.92 kJ mol−1 due to its lower polarity (Fig. S1–S4†). Consequently, the binding energy difference between SO2 and CO2 (ΔBE(SO2/CO2)) is in the order of –NH– > –CC– > –C–C– > –C3N3– > –CC– > –S– (Table 1). As the selective separation of SO2 over CO2 depends much on ΔBE(SO2/CO2), this linkage screening approach could be a feasible method to effectively construct CMPs for the deep removal of SO2 from CO2.
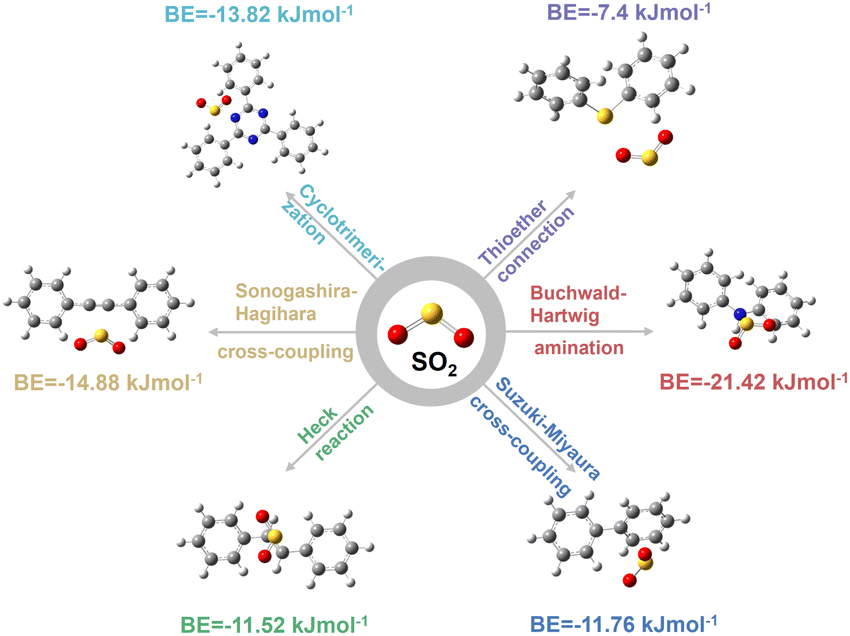 | ||
| Fig. 1 BEs between a SO2 molecule and different CMP linkages that were extracted from the typical coupling reactions of Suzuki–Miyaura cross-coupling, Heck reaction, Sonogashira–Hagihara cross-coupling, Buchwald–Hartwig amination, cyclotrimerization, and thioether connection. | ||
| linkage | –C–C– | –CC– |
–CC– |
–NH– | –C3N3– | –S– |
|---|---|---|---|---|---|---|
| a Unit: kJ mol−1. | ||||||
| SO2 | 11.8 | 11.5 | 14.9 | −21.4 | −13.82 | −11.8 |
| CO2 | −5.8 | −5.8 | −6.6 | −7.8 | −7.92 | −5.8 |
| ΔBE | 6 | 5.7 | 8.3 | 13.6 | 5.9 | 6 |
To make the screening results reliable, we synthesized a CMP of TMM–PD with the –NH– linkage through Buchwald–Hartwig amination between tetrakis(4-bromophenyl)methane (TMM) and p-phenylenediamine (PD). Meanwhile, TMM–BA containing –C–C– linkage was also constructed through Suzuki–Miyaura cross-coupling by employing the same TMM core but replacing the monomer with 1,4-phenylenebisboronic acid (BA) (Fig. 2a). The successful constructions of TMM–PD and TMM–BA are confirmed by Fourier transform infrared (FT-IR) spectra, with the absence of the characteristic peaks of amino group at 3383 cm−1 and 3318 cm−1 in the TMM–PD spectrum and the disappearance of the B–O peak at 640 cm−1 in the TMM–BA spectrum, respectively (Fig. S5†). Further evidence came from the appearance of the absorption peak of –NH– linkage at 399.7 eV and the C–N bond at 285.2 eV in the X-ray photoelectron spectroscopy (XPS) spectra (Fig. S6†). TMM–PD also has characteristic resonances at ∼62 ppm and ∼150 ppm in solid-state nuclear magnetic resonance (ssNMR) spectra, suggesting the successful Buchwald–Hartwig amination condensation (Fig. S7†). Determined using the N2 adsorption isotherms at 77 K, the Brunauer–Emmett–Teller (BET) surface areas of TMM–PD and TMM–BA are similar at 526 m2 g−1 and 567.59 m2 g−1, respectively (Fig. S8a†). However, due to the relatively soft –NH– linkage in TMM–PD in comparison with the rigid C–C linkage between two aromatic benzene rings in TMM–BA, the dominant micropore size in TMM–PD (1.1 nm) is smaller than that in TMM–BA (1.8 nm) and the pore volume of TMM–PD (0.11 cm3 g−1) is only half of that of TMM–BA (0.22 cm3 g−1) (Fig. S8b†). The microporosity is further verified by the transmission electron microscopy (TEM) images, but the morphologies of two-dimensional graphene-like TMM–PD and spherical TMM–BA are quite different (Fig. S9†).
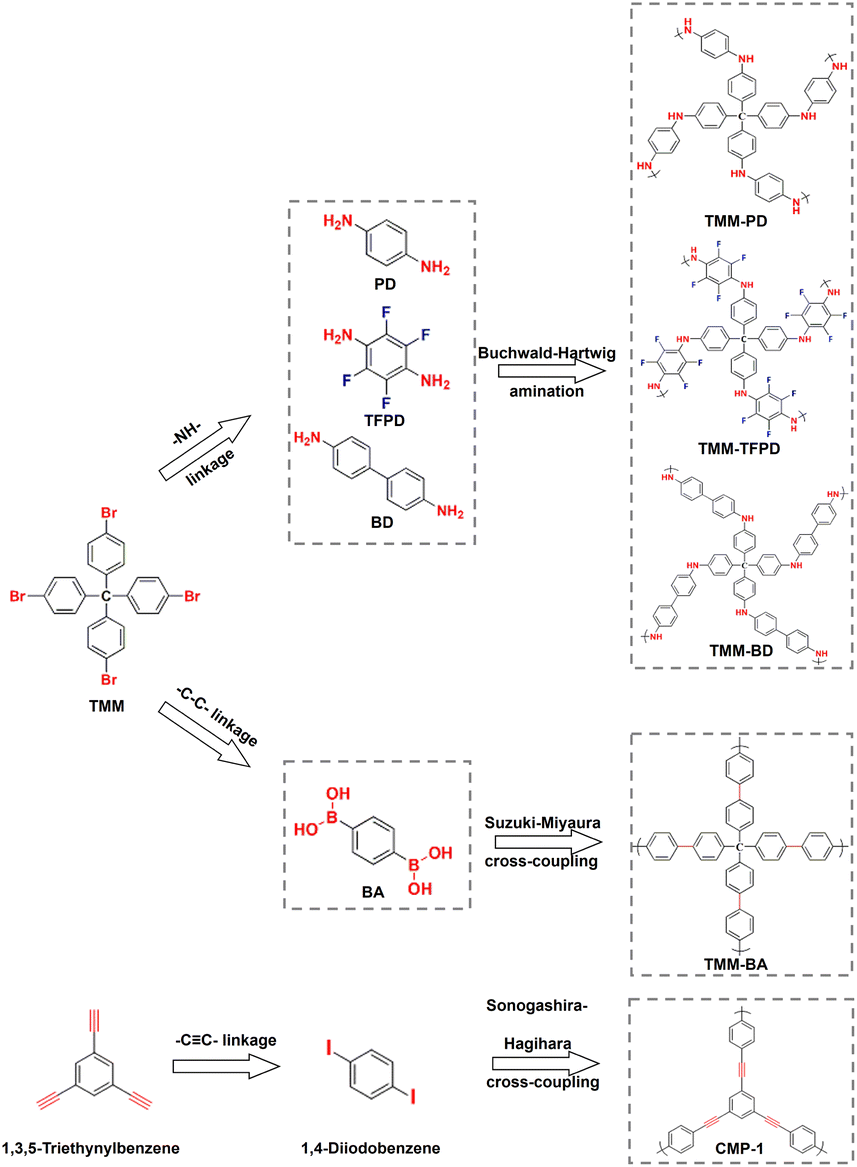 | ||
| Fig. 2 Schematic synthesis of TMM–PD, TMM–TFPD, TMM–BD, TMM–BA, and CMP-1 through Buchwald–Hartwig amination, Suzuki–Miyaura cross-coupling, and Sonogashira–Hagihara cross-coupling respectively (red color: condensation sites and blue color: functional groups). | ||
The SO2 and CO2 adsorption isotherms provide clear evidence of reversible desulfurization performances of the obtained CMPs. It turns out that at a high pressure of 1 bar, the SO2 capacities at 273 K of TMM–PD (11.34 mmol g−1) and TMM–BA (10.82 mmol g−1) are similar to each other due to their closed surface area and each shows a decline to 6.65 mmol g−1 and 7.54 mmol g−1 at 298 K, respectively (Fig. 3a and b). Meanwhile, the CO2 capacities of TMM–PD (2.6 and 1.7 mmol g−1) and of TMM–BA (2.1 and 1.2 mmol g−1) were much lower at 273 K and 298 K (Fig. S10†), demonstrating the great potential of CMPs for SO2/CO2 selective separation. Considering that SO2 is usually in a trace amount in the flue gas, the adsorption performance at low pressure is lethal for deep desulfurization. Comparing the adsorption performance at a low pressure of 0.1 bar, TMM–PD shows much higher SO2 capacity (3.2 mmol g−1) than TMM–BA (2.1 mmol g−1) (Fig. 3c), demonstrating the higher SO2 affinity of –NH– linkage compared to –C–C– linkage. We further used the Grand Canonical Monte Carlo (GCMC) simulation to vividly illustrate the adsorption processes (Fig. S11†). Consistent with the experimental results, both TMM–PD and TMM–BA have much higher SO2 capacities than CO2. Moreover, in comparison with TMM–BA, TMM–PD shows higher SO2 density, and most SO2 molecules stay around the –NH– linkages, proving that the –NH– linkages in CMPs are beneficial for SO2/CO2 separation.
 | ||
| Fig. 3 SO2 and CO2 adsorption isotherms of TMM–PD, TMM–TFPD and TMM–BD at (a) 273 K and (b) 298 K. (c) SO2 adsorption capacity of TMM–PD, TMM–TFPD, TMM–BD, TMM–BA, and CMP-1 at 0.1 bar and 298 K. (d) IAST selectivity of SO2/CO2 in TMM–PD, TMM–TFPD, TMM–BD, TMM–BA, and CMP-1 at 298 K. (e) Dynamic breakthrough curves of SO2 and CO2 from the mixed gas (2000 ppm SO2) with a flow rate of 20 mL min−1 at 298 K and 1 bar. (f) Cycling dynamic breakthrough curves for CO2/SO2 (2000 ppm SO2) separations in TMM–BD. | ||
Significantly, based on the ideal adsorbed solution theory (IAST), for the simulated flue gas with 0.2% SO2, the SO2/CO2 selectivity of TMM–PD was calculated to be as high as 113 using the dual site Langmuir–Freundlich model, much higher than 37 in TMM–BA (Fig. 3d). Furthermore, by fitting the SO2 adsorption isotherms at 273 K and 298 K through the virial-type expression, the adsorption heat (Qst) of SO2 and CO2 of TMM–PD and TMM–BA were calculated to be (31.7 vs. 13.4 kJ mol−1) and (27.9 vs. 29.3 kJ mol−1) at zero coverage, respectively (Fig. S12†). The higher Qst of SO2 and much lower Qst of CO2 confirmed that TMM–PD with –NH– linkage has greater potential for selective SO2 separation than TMM–BA with –C–C– linkage. Besides, this relatively low Qst of 31.7 kJ mol−1 also ensures an excellent reversible physical adsorption–desorption performance. By comparison, the excellent SO2 adsorption capacity of TMM–PD is much higher than that of most reported adsorbents, such as MOFs of MFM-305-CH3 (5 mmol g−1),25 SIFSIX-3-Ni (2.74 mmol g−1),19 Co-gallate (5.2 mmol g−1),11 organic polymer of P(TMGA-co-MBA) (4.06 mmol g−1),26 traditional porous materials of active carbon (3.3 mmol g−1),27 and zeolite 13X (2.7 mmol g−1 at 323 K and 0.4 bar),28 and is comparable with that of the benchmark materials SIFSIX-2-Cu-I (6.9 mmol g−1) and NOTT-300 (8.12 mmol g−1).2,19 Moreover, the SO2/CO2 IAST selectivity in TMM–PD is also higher than that of the most reported MOFs, such as the ELM type of ELM-12 (30-13.1),29 ECUT types of ECUT-111 (25.2-22.2)14 and ECUT-100 (27.5-26.9),30 MFM types of MFM-300(In) (33.6-11.6)31 and MFM-601 (67.5-36.2),32 and SIFSIX types of SIFSIX-1-Cu (70.7-54.1)19 and SIFSIX-2-Cu-i (89.4-87.1).19 The relatively low Qst values are comparable with those of other reported MOF adsorbents such as benchmark adsorbents of Mg-gallate (49 kJ mol−1),11 SIFSIX-2-Cu (36.1 kJ mol−1),19 and MFM types of MFM-300(In) (34.5 kJ mol−1),31 MFM-601 (38 kJ mol−1),32 and MFM-202a (35 kJ mol−1),33 suggesting the potential benefit of the temperature swing recycle usage.
To further evaluate the accuracy of the screening results, we synthesized –CC– linkage-based CMP-1 through Sonogashira–Hagihara cross-coupling by reacting 1,4-diiodobenzene with 1,3,5-triethynylbenzene (Fig. 2). Not surprisingly, CMP-1 exhibited favourable SO2 adsorption uptakes at 1 bar with 14.7 mmol g−1 at 273 K and 9.3 mmol g−1 at 298 K (Fig. S10†). However, compared with TMM–PD, CMP-1 showed lower SO2 uptakes at 0.1 bar (2.5 mmol g−1vs. 3.2 mmol g−1) and IAST selectivity (41 vs. 113) (Fig. 3c and d). Therefore, combining high SO2 capacity, high SO2/CO2 selectivity, and low-energy consumption for regeneration, TMM–PD constructed by –NH– linkage via Buchwald–Hartwig amination can be one of the first-class adsorbents for deep desulfurization, further proving the success of our approach for fabricating target-specific CMPs.
As the –NH– linkage via Buchwald–Hartwig amination can be achieved using various monomers with specific functional groups, to further demonstrate the feasibility of this novel screening strategy for preparing CMPs with high SO2/CO2 separation performance, we developed TMM–BD and TMM–TFPD by replacing PD with benzidine (BD) and 1,3-diamino-2,4,5,6-tetrafluorobenzene (TFPD) to regulate the porosity and hydrophobicity of the CMPs, respectively (Fig. 2a). The successful formation of –NH– linkage in CMPs is demonstrated by the appearance of peaks of the –NH– bond at 399.7 eV and C–N bond at 285.2 eV in the XPS spectra (Fig. S13†). Meanwhile, the new peak of the C–F bond at 687 eV suggests the incorporation of fluorine in TMM–TFPD. Moreover, ssNMR spectra also proved the successful preparation of TMM–BD and TMM–TFPD with the absence of characteristic resonances at ∼62 ppm and ∼150 ppm (Fig. S7†). With the pore volume doubled (0.22 cm3 g−1) that of TMM–PD (Fig. S14†), TMM–BD shows a higher SO2 capacity of 13.3 mmol g−1 at 273 K and 1 bar, whereas an almost unchanged SO2 capacity of about 2.5 mmol g−1 at 298 K and low pressure of 0.1 bar further demonstrates the dominant contribution of –NH– linkage to the desulfurization of the flue gas (Fig. 3c). Interestingly, although the BET surface area of TMM–TFPD (254.17 m2 g−1) decreases to a half less than that of TMM–PD (Fig. S14†), TMM–TFPD still shows an excellent SO2 capacity of 11.18 mmol g−1 at 273 K and 1 bar. It is worth mentioning that the presence of 5–7% water in flue gas would cause a significant decline in desulfurization performance.34 The hydrophobic modification makes the saturated water uptakes in TMM–TFPD decrease from 10 mmol g−1 in TMM–PD to 7 mmol g−1 at 298 K (Fig. S15†), suggesting that TMM–TFPD has potential to provide a good microenvironment for the desulfurization of flue gas in the presence of water.
To figure out the mechanisms of this extraordinary SO2 adsorption and the low porosity but high SO2 capacity of TMM–TFPD, we recorded the adsorption processes through the in situ FTIR spectra (Fig. S19†). During the adsorption of the 2000 ppm SO2 simulated flue gas, the absorption peaks of TMM–PD and TMM–TFPD changed significantly within 60 min. For TMM–PD, the peak of the –NH– linkage at 1143 cm−1 shows a redshift of 4 cm−1, suggesting that it is the active site for SO2 adsorption (Fig. 4c) whereas the S–O stretching peak at 1347 cm−1 in SO2 shows an obvious red shift of 5 cm−1, ascribed to the electrostatic attraction by the –NH– linkage (Fig. 4c). For TMM–TFPD, the characteristic peaks of –NH– linkage and the S–O bond show a much more complex behavior upon SO2 adsorption. Specifically, the peaks of –NH– linkage have a blue shift of 2 cm−1 during the initial 30 min (Fig. 4d). Subsequently, the peak splits into double ones at 40 min and combines into one peak again during the final 50–60 min (Fig. 4d). We assumed that the small surface area of TMM–TFPD may be attributed to the intramolecular hydrogen bonds between –NH– linkages and fluorine substituents (Fig. S17†) and the adsorption of SO2 at the –NH– linkage would break hydrogen bonds, further swelling TMM–TFPD for exposing more adsorption sites.35 This ratiocination is conducive to the complex changes of the –NH– linkage peak where the double splitting can be attributed to the competitive attractions between fluorine and SO2, and the reunion further demonstrated the strong affinity of –NH– linkage to SO2. Therefore, the relatively high Qst of SO2 adsorption of 62 kJ mol−1 in TMM–TFPD can be caused by the breakage of hydrogen bonds between –NH– and fluorine. To gain deep insights into this breathing phenomenon, we also performed a GCMC simulation of the SO2 adsorption in amorphous TMM–PD and TMM–TFPD cells (Fig. S18†). Notably, attributed to the strong electrostatic interaction between Oδ− of SO2 and Hδ+ of –NH– linkage, the distance between SO2 and –NH– linkage was similar at about 2.75 Å in TMM–PD and TMM–TFPD (Fig. 4), demonstrating that the –NH– linkage is the binding site for SO2 adsorption, whereas the distance between the –NH– and fluorine bond increased from 2.34 Å to 2.48 Å after SO2 adsorption in TMM–TFPD, which is consistent with the complex evolution of the –NH– peak in the in situ FTIR spectra, further verifying that the adsorption of SO2 can swell TMM–TFPD through breaking hydrogen bonds of –NH– and fluorine.
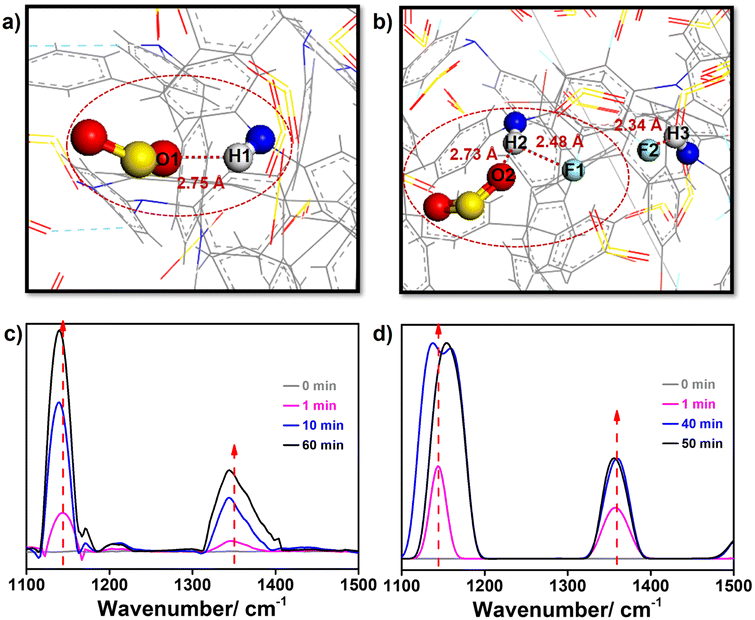 | ||
| Fig. 4 Visualization of SO2-binding sites in (a) TMM–PD and (b) TMM–TFPD. In situ FTIR spectra of (c) TMM–PD and (d) TMM–TFPD at various times within 60 min under 2000 ppm SO2 at 298 K. | ||
The dynamic breakthrough test was further conducted on the simulated flue gas of 2000 ppm SO2 and 99.8% CO2 to evaluate the practical implementation of deep desulfurization. At a flow rate of 20 mL min−1, the eluted CO2 concentration was as high as 99.99% and the elution time of SO2 was achieved as 180 min g−1, 185 min g−1, and 122 min g−1 on TMM–PD, TMM–BD, and TMM–TFPD, respectively (Fig. 3e). Accordingly, the dynamic adsorption capacities were calculated to be 0.47 mmol g−1, 0.4 mmol g−1, and 0.4 mmol g−1, in comparison with their static SO2 adsorption performance at 0.002 bar (corresponding to a SO2 content of 2000 ppm) (Fig. 3b). Therefore, under the guidance of the DFT calculations, the CMPs with –NH– linkage produced by Buchwald–Hartwig amination all showed excellent desulfurization performance for trace SO2 removal, superior to most state-of-the-art materials under similar operating conditions, such as polymers of 70 min g−1 in P(3DVB-[VEIm]Br),22 MOFs of 76 min g−1 in MFM-300 (Cr),36 152 min g−1 in Cage-U-Co-MOF,37 180 min g−1 in ECUT-111,30 60 min g−1 in MFM-601,19 and 28 min g−1 in MFM-600,19 and other traditional zeolites of NaX, FAU, LTA, and SBA-15,4,5,7,8 demonstrating the successful de novo design and screening of taking validated linkages into account for CMP preparation towards the specific deep desulfurization task (Table S2†). Furthermore, flue gas always contains other contaminants such as NO2 and HCl, which will result in a complicated system for practical separation.38 Based on our successful strategy, we further calculated the binding efficiency of the –NH– linkage with N2, HCl, and NO2 to provide more information about the potential use of CMPs for industrial desulfurization. As shown in Fig. S20,† the BEs between –NH– linkage with N2, HCl, and NO2 are lower than that of SO2 (−4.8 kJ mol−1, −13 kJ mol−1, and −7.2 kJ mol−1vs. −21.42 kJ mol−1), suggesting that the –NH– linkage has higher binding selectivity for SO2 over other contaminants.
Due to that SO2 is highly corrosive and few leading materials can be stable in the presence of SO2, we further performed cyclic breakthrough tests to evaluate the reusability of the synthesized CMPs. First, as shown in Fig. S21,† the desorption curves of SO2 showed a moderate degree of hysteresis because the molecular flexibility would cause open loop hysteresis due to swelling effects. Moreover, the breakthrough performance did not decline in TMM–BD during five cycles for a 0.2% SO2/99.8% CO2 mixture (Fig. 3f). Further PXRD patterns and N2 adsorption proved that TMM–BD can maintain the structure and porosity after SO2 adsorption, suggesting its excellent stability and reusability for desulfurization (Fig. S22†). Very recently, amine-based porous organic cages (POCs) have been proven to have great potential for SO2 capture with the sequence of tertiary amine > second amine > imine.9 In this system, Liu and co-authors indicated the importance of the basicity of the amine in SO2 capture. Even though the second amine has a high SO2 binding efficiency, the strong acid and corrosive nature of SO2 would destroy the POC structure after removal of the adsorbed SO2. While for CMPs that were synthesized by Buchwald–Hartwig amination, the electron density of the second amine was decoupled by two connected benzene rings, leading to the retention of the structure and porosity after SO2 treatment. Moreover, once the optimal linkage to fabricate CMPs for SO2 capture was identified, the high diversity of the starting monomers could benefit different properties in CMPs such as hydrophobicity, larger pore volume, and pore structure.
Conclusions
In summary, we developed a feasible approach by screening linkages extracted from the coupling reactions for CMPs' construction through calculating the affinity with SO2 and simulating the SO2 adsorption density. Under the guidance of the screening results, TMM–PD, TMM–TFPD, and TMM–BD were fabricated via Buchwald–Hartwig amination with different porosities and hydrophobicities which showed higher selective SO2 adsorption performance. In particular, the excellent dynamic SO2 removal performance of TMM–BD with an elution time of 185 min g−1 and a high CO2 purity of greater than 99.99% was achieved from the simulated flue gas containing 2000 ppm SO2, superior to the most reported state-of-the-art adsorbents. Moreover, the fluorine functionalized TMM–TFPD significantly increased the water resistance and maintained high SO2 capacity, providing a potential application for the practical flue gas desulfurization process. Through successfully bypassing the invalid synthesis of screened CMPs from billions of monomers, our work demonstrates the significance of linkage diversities on the intrinsic properties of porous materials and solves the dilemma between the implementability of computational screening and the synthesis of porous materials.Data availability
Should any raw data files be needed they are available from the corresponding author upon reasonable request.Author contributions
He Li, Xili Cui, Jun Hu, and Honglai Liu conceived and designed the project. He Li synthesized the materials and carried out all the characterization studies and computational simulations. Hanqian Pan and Yijian Li performed the breakthrough test. All authors participated in data analysis, discussion of the results and manuscript revision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for the financial support from the National Natural Science Foundation of China (No. 21878076 and 22278126), the Fundamental Research Funds for the Central Universites (2022ZFJH004), and Intergovernmental International Science and Technology Innovation Cooperation Key Project (2021YFE0112800).Notes and references
- E. Croiset and K. Thambimuthu, Fuel, 2001, 80, 2117–2121 CrossRef CAS.
- S. Yang, J. Sun, A. J. Ramirez-Cuesta, S. K. Callear, W. I. David, D. P. Anderson, R. Newby, A. J. Blake, J. E. Parker, C. C. Tang and M. Schroder, Nat. Chem., 2012, 4, 887–894 CrossRef CAS PubMed.
- H. Yi, H. Deng, X. Tang, Q. Yu, X. Zhou and H. Liu, J. Hazard. Mater., 2012, 203–204, 111–117 CrossRef CAS PubMed.
- X. Zhou, H. Yi, X. Tang, H. Deng and H. Liu, Chem. Eng. J., 2012, 200–202, 399–404 CrossRef CAS.
- A. Czyżewski, J. Kapica, D. Moszyński, R. Pietrzak and J. Przepiórski, Chem. Eng. J., 2013, 226, 348–356 CrossRef.
- D. Yang, M. Hou, H. Ning, J. Ma, X. Kang, J. Zhang and B. Han, ChemSusChem, 2013, 6, 1191–1195 CrossRef CAS PubMed.
- H. Deng, H. Yi, X. Tang, H. Liu and X. Zhou, Ind. Eng. Chem. Res., 2013, 52, 6778–6784 CrossRef CAS.
- L. Wei, Z. Gao and Y. Wang, Asia-Pac. J. Chem. Eng., 2017, 12, 660–670 CrossRef CAS.
- I. Ibarra, E. Martínez-Ahumada, D. He, V. Berryman, V. Jancik, V. Martis, M. A. Vera, E. Lima, D. J. Parker and A. I. Cooper, Angew. Chem., Int. Ed., 2021, 60, 17556–17563 CrossRef PubMed.
- X. Suo, Y. Yu, S. Qian, L. Zhou, X. Cui and H. Xing, Angew. Chem., 2021, 133, 7062–7067 CrossRef.
- F. Chen, D. Lai, L. Guo, J. Wang, P. Zhang, K. Wu, Z. Zhang, Q. Yang, Y. Yang, B. Chen, Q. Ren and Z. Bao, J. Am. Chem. Soc., 2021, 143, 9040–9047 CrossRef CAS PubMed.
- E. Martinez-Ahumada, M. L. Diaz-Ramirez, M. J. Velasquez-Hernandez, V. Jancik and I. A. Ibarra, Chem. Sci., 2021, 12, 6772–6799 RSC.
- S. Xing, J. Liang, P. Brandt, F. Schafer, A. Nuhnen, T. Heinen, I. Boldog, J. Mollmer, M. Lange, O. Weingart and C. Janiak, Angew. Chem., 2021, 60, 17998–18005 CrossRef CAS.
- M. J. Yin, X. H. Xiong, X. F. Feng, W. Y. Xu, R. Krishna and F. Luo, Inorg. Chem., 2021, 60, 3447–3451 CrossRef CAS PubMed.
- G. L. Smith, J. E. Eyley, X. Han, X. Zhang, J. Li, N. M. Jacques, H. G. W. Godfrey, S. P. Argent, L. J. McCormick McPherson, S. J. Teat, Y. Cheng, M. D. Frogley, G. Cinque, S. J. Day, C. C. Tang, T. L. Easun, S. Rudic, A. J. Ramirez-Cuesta, S. Yang and M. Schroder, Nat. Mater., 2019, 18, 1358–1365 CrossRef CAS PubMed.
- K. Tan, S. Zuluaga, H. Wang, P. Canepa, K. Soliman, J. Cure, J. Li, T. Thonhauser and Y. J. Chabal, Chem. Mater., 2017, 29, 4227–4235 CrossRef CAS.
- J. Yu, Y. Ma and P. B. Balbuena, Langmuir, 2012, 28, 8064–8071 CrossRef CAS PubMed.
- V. Chernikova, O. Yassine, O. Shekhah, M. Eddaoudi and K. N. Salama, J. Mater. Chem. A, 2018, 6, 5550–5554 RSC.
- X. Cui, Q. Yang, L. Yang, R. Krishna, Z. Zhang, Z. Bao, H. Wu, Q. Ren, W. Zhou, B. Chen and H. Xing, Adv. Mater., 2017, 29, 1606929 CrossRef PubMed.
- M. R. Tchalala, P. M. Bhatt, K. N. Chappanda, S. R. Tavares, K. Adil, Y. Belmabkhout, A. Shkurenko, A. Cadiau, N. Heymans, G. De Weireld, G. Maurin, K. N. Salama and M. Eddaoudi, Nat. Commun., 2019, 10, 1328 CrossRef CAS PubMed.
- P. Brandt, A. Nuhnen, S. Öztürk, G. Kurt, J. Liang and C. Janiak, Adv. Sustainable Syst., 2021, 5, 2000285 CrossRef CAS.
- L. Xia, Q. Cui, X. Suo, Y. Li, X. Cui, Q. Yang, J. Xu, Y. Yang and H. Xing, Adv. Funct. Mater., 2018, 28, 1704292 CrossRef.
- J.-S. M. Lee and A. I. Cooper, Chem. Rev., 2020, 120, 2171–2214 CrossRef CAS PubMed.
- S. Jiang, K. E. Jelfs, D. Holden, T. Hasell, S. Y. Chong, M. Haranczyk, A. Trewin and A. I. Cooper, J. Am. Chem. Soc., 2013, 135, 17818–17830 CrossRef CAS PubMed.
- L. Li, I. da Silva, D. I. Kolokolov, X. Han, J. Li, G. Smith, Y. Cheng, L. L. Daemen, C. G. Morris, H. G. W. Godfrey, N. M. Jacques, X. Zhang, P. Manuel, M. D. Frogley, C. A. Murray, A. J. Ramirez-Cuesta, G. Cinque, C. C. Tang, A. G. Stepanov, S. Yang and M. Schroder, Chem. Sci., 2019, 10, 1472–1482 RSC.
- L. Wu, D. An, J. Dong, Z. Zhang, B.-G. Li and S. Zhu, Macromol. Rapid Commun., 2006, 27, 1949–1954 CrossRef CAS.
- Y. Guo, Y. Li, T. Zhu and M. Ye, Fuel, 2015, 143, 536–542 CrossRef CAS.
- H. Deng, H. Yi, X. Tang, Q. Yu, P. Ning and L. Yang, Chem. Eng. J., 2012, 188, 77–85 CrossRef CAS.
- Y. Zhang, P. Zhang, W. Yu, J. Zhang, J. Huang, J. Wang, M. Xu, Q. Deng, Z. Zeng and S. Deng, ACS Appl. Mater. Interfaces, 2019, 11, 10680–10688 CrossRef CAS.
- L. J. Guo, X. F. Feng, Z. Gao, R. Krishna and F. Luo, Inorg. Chem., 2021, 60, 1310–1314 CrossRef CAS.
- M. Savage, Y. Cheng, T. L. Easun, J. E. Eyley, S. P. Argent, M. R. Warren, W. Lewis, C. Murray, C. C. Tang, M. D. Frogley, G. Cinque, J. Sun, S. Rudic, R. T. Murden, M. J. Benham, A. N. Fitch, A. J. Blake, A. J. Ramirez-Cuesta, S. Yang and M. Schroder, Adv. Mater., 2016, 28, 8705–8711 CrossRef CAS PubMed.
- J. H. Carter, X. Han, F. Y. Moreau, I. da Silva, A. Nevin, H. G. W. Godfrey, C. C. Tang, S. Yang and M. Schroder, J. Am. Chem. Soc., 2018, 140, 15564–15567 CrossRef CAS PubMed.
- S. Yang, L. Liu, J. Sun, K. M. Thomas, A. J. Davies, M. W. George, A. J. Blake, A. H. Hill, A. N. Fitch, C. C. Tang and M. Schroder, J. Am. Chem. Soc., 2013, 135, 4954–4957 CrossRef CAS PubMed.
- D. Wang, A. Bao, W. Kunc and W. Liss, Appl. Energy, 2012, 91, 341–348 CrossRef CAS.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS.
- L. Briggs, R. Newby, X. Han, C. G. Morris, M. Savage, C. P. Krap, T. L. Easun, M. D. Frogley, G. Cinque, C. A. Murray, C. C. Tang, J. Sun, S. Yang and M. Schröder, J. Mater. Chem. A, 2021, 9, 7190–7197 RSC.
- Y. Fan, M. Yin, R. Krishna, X. Feng and F. Luo, J. Mater. Chem. A, 2021, 9, 4075–4081 RSC.
- P. N. Chisholm and G. T. Rochelle, Ind. Eng. Chem. Res., 2000, 39, 1048–1060 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc02622a |
| This journal is © The Royal Society of Chemistry 2023 |