DOI:
10.1039/D3SC01874A
(Edge Article)
Chem. Sci., 2023,
14, 8327-8337
Regulating tumor glycometabolism and the immune microenvironment by inhibiting lactate dehydrogenase with platinum(IV) complexes†
Received
11th April 2023
, Accepted 10th July 2023
First published on 10th July 2023
Abstract
Lactate dehydrogenase (LDH) is a key enzyme involved in the process of glycolysis, assisting cancer cells to take in glucose and generate lactate, as well as to suppress and evade the immune system by altering the tumor microenvironment (TME). Platinum(IV) complexes MDP and DDP were prepared by modifying cisplatin with diclofenac at the axial position(s). These complexes exhibited potent antiproliferative activity against a panel of human cancer cell lines. In particular, DDP downregulated the expression of LDHA, LDHB, and MCTs to inhibit the production and influx/efflux of lactate in cancer cells, impeding both glycolysis and glucose oxidation. MDP and DDP also reduced the expression of HIF-1α, ARG1 and VEGF, thereby disrupting the formation of tumor vasculature. Furthermore, they promoted the repolarization of macrophages from the tumor-supportive M2 phenotype to the tumor-suppressive M1 phenotype in the TME, thus enhancing the antitumor immune response. The antitumor mechanism involves reprogramming the energy metabolism of tumor cells and relieving the immunosuppressive TME.
Introduction
Cancer cells metabolize differently from normal ones by shifting energy metabolism from oxidative phosphorylation (OXPHOS) to aerobic glycolysis.1,2 This is characterized by the increased utilization of glucose and efflux of lactate, i.e. the Warburg effect,3,4 which promotes cancer cells to proliferate and avoid apoptosis through anabolism. Monocarboxylate transporters (MCTs) participate in the transport of lactate to facilitate metabolic reprogramming during tumor progression.5 MCT1 has a high affinity for lactate and acts as a cellular influx or efflux transporter according to the concentration gradient of lactate; while MCT4 mainly facilitates the efflux of lactate in highly glycolytic cells.6 The lactate produced by glycolysis creates an acidic tumor microenvironment (TME),7 which suppresses immune activity and allows cancer cells to escape from antitumor immunity.8 For example, lactate induces alternative polarization of macrophages through hypoxia-inducible factor 1α (HIF-1α) stabilization, increases the production of vascular endothelial growth factor (VEGF) and L-arginine-metabolizing enzyme arginase (ARG), thereby forming an immunosuppressive TME;9,10 furthermore, it stimulates tumor angiogenesis by directly acting on endothelial cells and indirectly promoting M2-like tumor-associated macrophages (TAMs) programming.11
Lactate dehydrogenase (LDH), a tetrameric enzyme comprising LDHA and LDHB, is the rate-limiting enzyme catalyzing the transformation of pyruvate to lactate in glycolysis.12 LDHA is upregulated in human cancers and associated with the aggressiveness of cancer cells, while LDHB is consistently expressed in all living cells.13 LDH is a favorable target for inhibiting the generation of lactate because it is the key enzyme for the final step of lactate production and cannot be bypassed. The “reverse Warburg effect (RWE)” is a newly identified mechanism for energy metabolism, which suggests that lactate is produced and excreted via MCT4 by stromal cells and taken in by cancer cells via MCT1 for ATP production.6,14 The RWE supplies pyruvate to LDH, allowing tumor cells to evade drug action and achieve rapid proliferation. Therefore, inhibiting LDH or MCTs may bring therapeutic benefits by reducing lactate production. Several LDHA inhibitors have been described, though their potency and selectivity are modest;15–18 however, LDHB inhibitors are rarely reported.
Platinum anticancer drugs are used in more than 40% of chemotherapy regimens in clinical practice.19 However, drug resistance and systemic toxicity hinder their efficacy and applications.20,21 Platinum(IV) complexes exhibit greater kinetic inertness than platinum(II) complexes and offer additional sites for functional modifications; they usually exert cytotoxic effects after reduction to PtII species in cancer cells.22 Recent studies suggest that nonsteroidal anti-inflammatory drug diclofenac (DCF) may have potential use in cancer treatment, including impairing the Warburg effect by inhibiting LDH to decrease glucose uptake and lactate secretion23 or lowering lactate secretion of cancer cells to improve T cell activation, viability, and effector functions.8,24 DCF also inhibits MCT1 and MCT4 to diminish lactate influx/efflux.23 DCF-modified platinum complexes have demonstrated enhanced cytotoxicity or inhibition to glycolysis. For example, DCF was coordinated to PtII through the carboxylic group to obtain a PtII complex with more potent cytotoxicity and inhibition to glycolysis and lactate transport than parental cisplatin (CDDP);25 DCF was also used as an axial ligand(s) to modify PtIV complexes, resulting in enhanced antiproliferative activity and blockage of the glycolytic process.26,27 However, DCF-modified platinum complexes are rarely used to impair the Warburg effect or RWE through inhibiting LDH or MCTs, let alone to ameliorate the immunosuppressive TME.
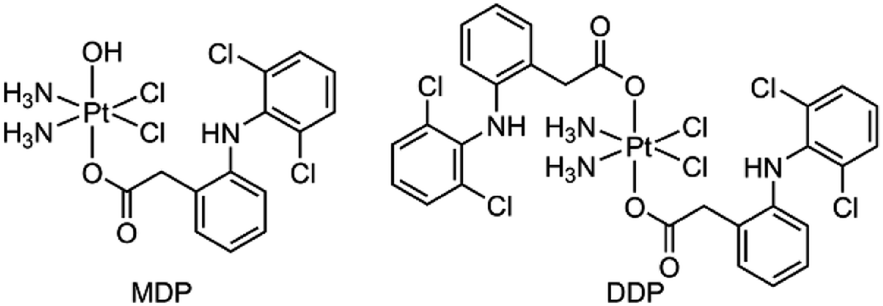
We herein report the biological properties of two DCF-modified PtIV complexes (Fig. 1), c,c,t-[Pt(NH3)2Cl2(OCOCH2C6H4NHC6H3Cl2)(OH)] (MDP) and c,c,t-[Pt(NH3)2Cl2(OCOCH2C6H4NHC6H3Cl2)2] (DDP). The former is a known compound that has been shown to possess stronger antiproliferative activity than CDDP,26 while the latter is a new compound. They exhibited excellent DNA binding and cytostatic activities; in particular, DDP altered the energy metabolism of cancer cells by inhibiting the activity of LDH and expression of MCTs, thereby decreasing the influx/efflux of lactate. Moreover, DDP reshaped the TME by restraining the expression of VEGF and ARG1 in macrophages and inducing the polarization of TAM from the protumoral M2 phenotype to the tumoricidal M1 phenotype. As a result, the synergy between chemotherapy and immunomodulation significantly boosted the antitumor activity.
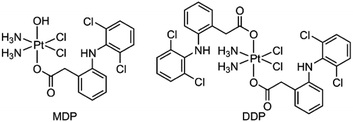 |
| | Fig. 1 Chemical structures of MDP and DDP. | |
Results and discussion
Synthesis and characterization
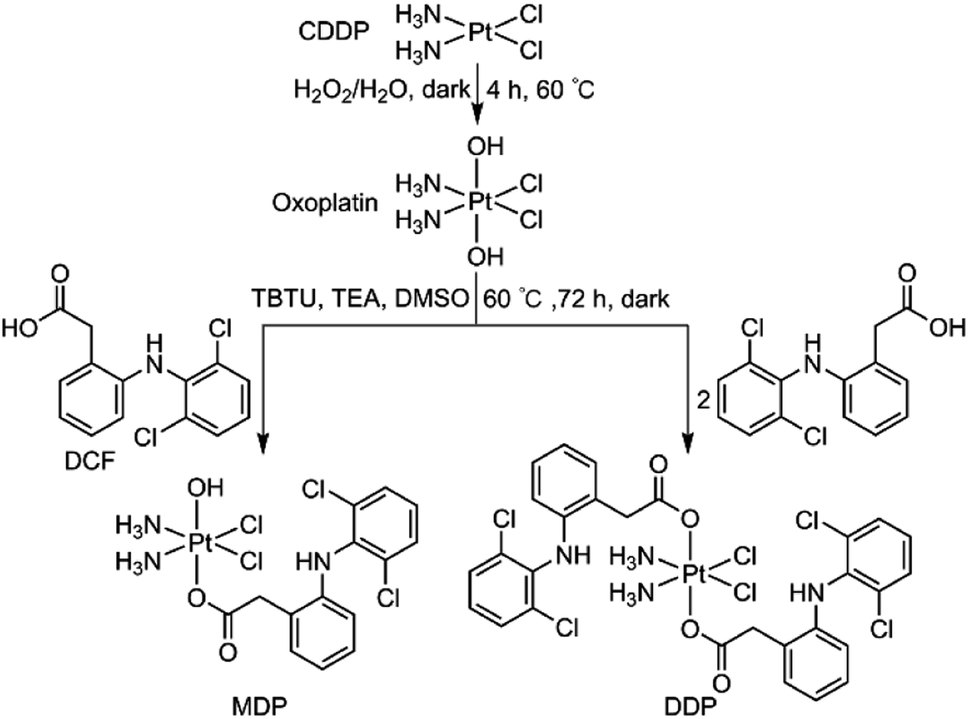
MDP and DDP were synthesized by linking one or two DCF moieties to CDDP as axial ligand(s). Briefly, CDDP was oxidized with hydrogen peroxide according to the literature to obtain oxoplatin,28 which was then coupled with DCF in the presence of O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium tetrafluoroborate (TBTU) and triethylamine (TEA) to obtain the target complexes. The synthetic routes are summarized in Scheme 1. MDP could also be prepared by a different method as reported in the literature.26 MDP and DDP were fully characterized by 1H-, 13C-, 195Pt NMR, and HR-ESI-MS (Fig. S1 and S2†). The major peak at m/z 635.9515 in the HR-ESI-MS spectrum was assignable to [M + Na]+ of MDP, while that at m/z 912.9543 was assignable to [M + Na]+ of DDP. The existence of PtIV in MDP and DDP was ascertained by the 195Pt NMR signal at ca. δ 1083.48 and 1220.58 ppm, respectively. The amino groups in the complexes were indicated by a broad peak ranging from δ 7.54 to 7.63 in the 1H NMR spectra. All the HR-ESI-MS and NMR data prove that DCF was successfully tethered to the PtIV center of the complexes. MDP and DDP are soluble in a range of organic solvents such as methanol, dimethylformamide (DMF), and dimethylsulfoxide (DMSO), but are almost insoluble in water.
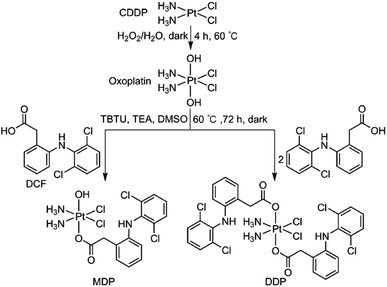 |
| | Scheme 1 Synthetic routes to MDP and DDP. | |
Reducibility and lipophilicity
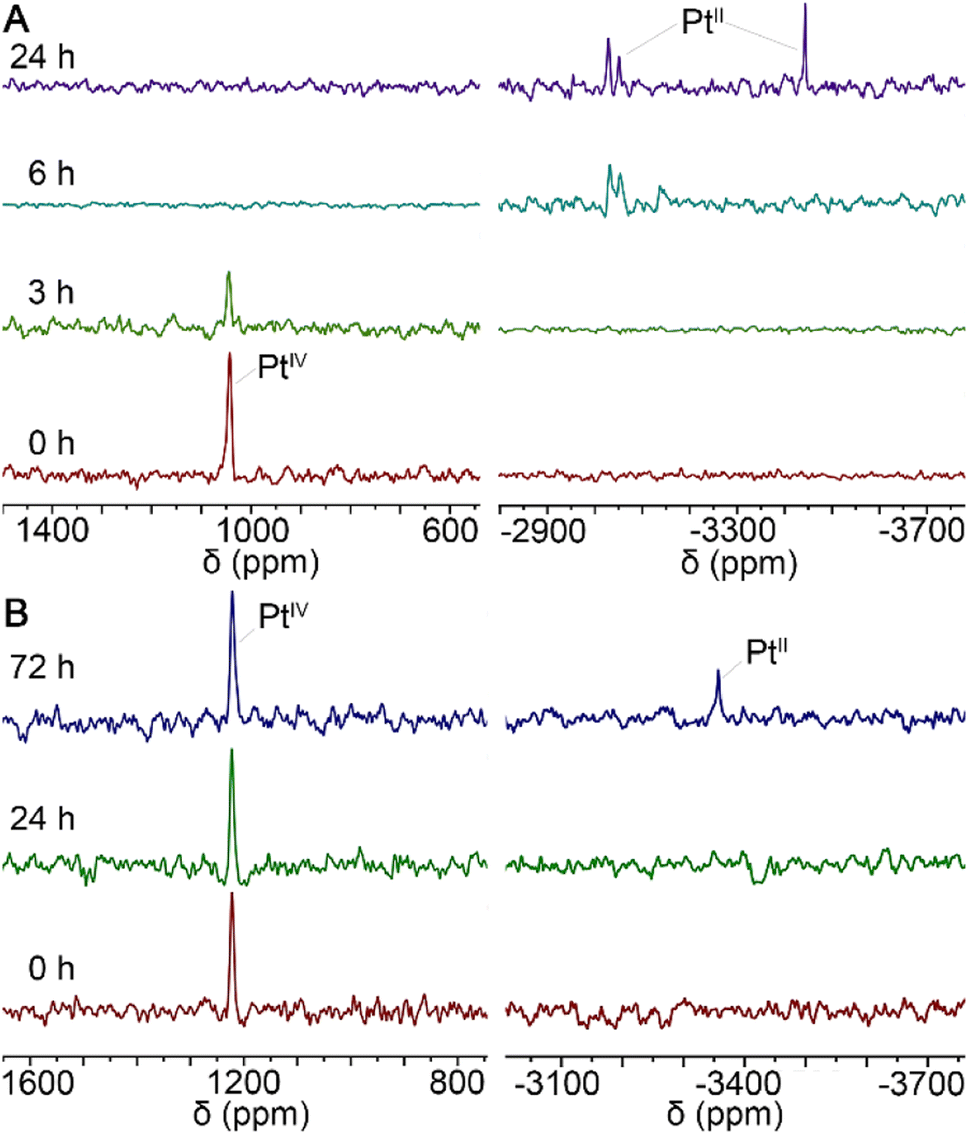
In general, PtIV complexes can be reduced into PtII species and axial ligands by intracellular reductants such as ascorbic acid (AsA) or glutathione, thus enriching biological activities.29 Therefore, the status of MDP and DDP in the presence of AsA in 90% DMSO/10% D2O solutions was monitored by 195Pt NMR spectrometry at 37 °C in the dark for 3 days. As shown in Fig. 2, the reduction of MDP (A) was quite faster than that of DDP (B), in that the former was completed within 24 h, while the latter was just started at 72 h. This may be due to that the presence of hydroxyl groups at the axial position of PtIV complexes can facilitate the transfer of electrons from the ascorbate to the PtIV center.30 The results indicate that the complexes could be reduced in the tumor cellular environment and thus are potential prodrugs.
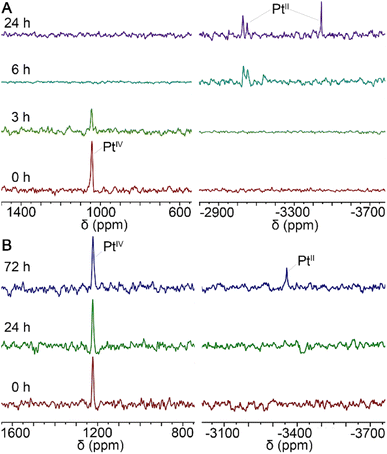 |
| | Fig. 2
195Pt NMR spectra for the reduction of MDP (A) and DDP (B) with 10 equiv. of AsA in 90% DMSO/10% D2O solution at 37 °C in the dark. | |
Lipophilicity (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) PO/W) is an important parameter to predict the membrane permeability of a compound,31 which was measured by the shaking flask method. The lipophilicity of MDP and DDP was −0.13 and −0.046, respectively (Table S1†), higher than that of CDDP (−2.35),28 indicating that they are more lipophilic than CDDP. DCF seems to be conducive to the lipophilicity of MDP and DDP. Since the cellular membrane is a bilayer of phospholipids, a compound with higher lipophilicity is more likely to be absorbed by the cells.
PO/W) is an important parameter to predict the membrane permeability of a compound,31 which was measured by the shaking flask method. The lipophilicity of MDP and DDP was −0.13 and −0.046, respectively (Table S1†), higher than that of CDDP (−2.35),28 indicating that they are more lipophilic than CDDP. DCF seems to be conducive to the lipophilicity of MDP and DDP. Since the cellular membrane is a bilayer of phospholipids, a compound with higher lipophilicity is more likely to be absorbed by the cells.
Antiproliferative activity
The antiproliferative activity of MDP and DDP was evaluated in the human breast cancer MCF-7, human cervical cancer HeLa, human colon cancer SW480, and human ovarian cancer SKOV-3 cell lines by the MTT assay. CDDP, physical mixtures of CDDP plus DCF at 1:1 and 1:2 molar ratios, and DCF were included as references. The detailed cytotoxic data are presented in Fig. S3.† The half maximal inhibitory concentrations (IC50) are summarized in Table 1. Generally, MDP and DDP exhibited higher antiproliferative activity than the reference compounds towards all the tested cancer cells, following an order of DDP > MDP > CDDP. DDP was more potent than MDP, possibly due to the two DCF moieties that potentiated the antiproliferative activity. Considering the difference in incubation time (48 vs. 72 h), the antiproliferative activity (IC50) of MDP against MCF-7 and SKOV-3 cells was comparable to the reported data (2.46 ± 0.22 and 1.70 ± 0.30 μM).26 DCF was almost nontoxic to these cancer cells (IC50 > 64 μM). The mixture of CDDP and DCF showed less activity than CDDP, indicating that free DCF hardly contributed to the cytotoxicity of CDDP and even has an antagonistic effect on it. According to these data, we chose the most sensitive SKOV-3 cells to perform the following biological assays.
Table 1 IC50 (μM) of MDP, DDP, CDDP, and mixtures of CDDP plus DCF (1:1 and 1:2) at 48 h against different cell lines. Data are the average of three measurements
| Compounds |
MCF-7 |
HeLa |
SW480 |
SKOV-3 |
| MDP |
3.02 ± 0.04 |
4.45 ± 0.08 |
2.66 ± 0.52 |
2.72 ± 0.75 |
| DDP |
0.78 ± 0.15 |
0.78 ± 0.29 |
1.14 ± 0.19 |
0.23 ± 0.02 |
| CDDP |
7.91 ± 0.20 |
2.38 ± 0.37 |
9.51 ± 1.06 |
7.00 ± 1.62 |
| CDDP + DCF |
16.36 ± 2.81 |
8.00 ± 0.32 |
24.49 ± 1.77 |
8.62 ± 0.13 |
| CDDP + 2DCF |
35.40 ± 2.06 |
9.55 ± 2.01 |
42.45 ± 1.53 |
19.14 ± 2.60 |
| DCF |
>64 |
>64 |
>64 |
>64 |
Cellular accumulation and DNA binding
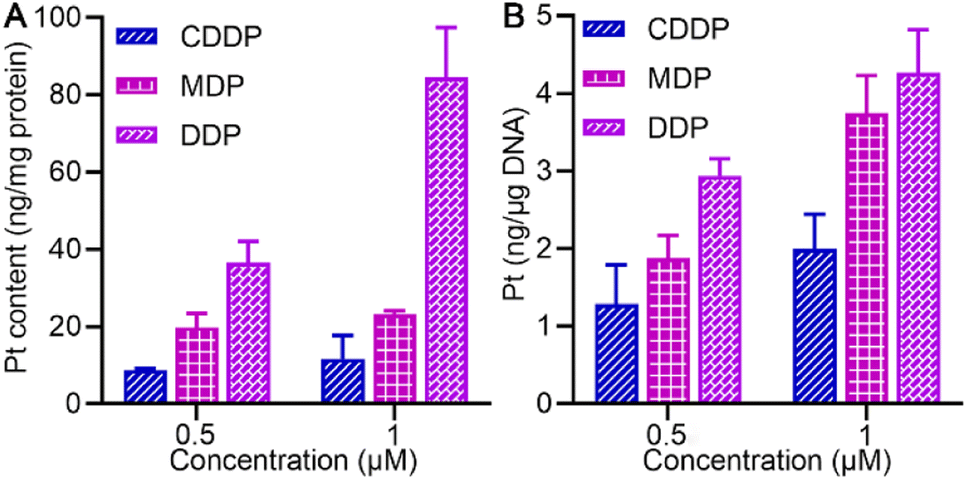
The cellular accumulation of MDP and DDP in the SKOV-3 cells in terms of Pt was evaluated by ICP-MS. As shown in Fig. 3A, the cellular uptake of MDP and DDP at 1 μM was about 2- and 7-fold higher than that of CDDP, respectively, which is positively associated with their lipophilicity. The accumulation of DDP in cells was especially high due to its higher lipophilicity, indicating that it efficiently entered the cells. The result corroborated the favorable effect of lipophilicity on the cellular uptake of the complexes.
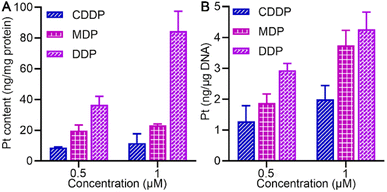 |
| | Fig. 3 Pt accumulation (A) and platination of cellular DNA (B) in SKOV-3 cells after treatment with 0.5 or 1 μM (0.5‰ v/v DMSO) of MDP, DDP, and CDDP, respectively, for 24 h. | |
Most PtIV complexes exert anticancer activity via binding to cellular DNA to form Pt–DNA adducts, which impede DNA replication and transcription in cancer cells. Hence, we investigated the interactions of MDP and DDP with calf thymus DNA (CT-DNA) in the absence and presence of AsA by circular dichroism (CD) spectrometry. There are two peaks in the CD spectrum—a positive peak at 275 nm representing the accumulation of bases and a negative one at 245 nm representing the right-handed helical structure of CT-DNA.32 In the absence of AsA, MDP and DDP did not react with CT-DNA; however, in the presence of AsA, the maximum ellipticity of both the positive and negative bands increased (Fig. S4†). The results imply that DDP and MDP destroyed the configuration of DNA when reduced to PtII species.
We further explored the impact of MDP and DDP on cellular DNA in SKOV-3 cells. As shown in Fig. 3B, the level of DNA platination is dependent on the initial concentration and cellular accumulation of the complex, following an order of DDP > MDP > CDDP, which is in line with their antiproliferative activities. Additionally, DDP showed a higher affinity for cellular DNA than MDP and CDDP, suggesting that it could induce more damage to cellular DNA, thus leading to higher toxicity to cancer cells. Although MDP was reduced much faster than DDP, it induced less DNA platination and lower antiproliferative activity in cancer cells. Therefore, the reduction kinetics of a PtIV complex did not necessarily determine the platination of cellular DNA and cytotoxicity. Of note, the cellular uptake of DDP was about 7-fold higher than that of CDDP and the DNA platination was only enhanced by about 2-fold, suggesting that cellular uptake is not consistent with the DNA damage and other factors may account for the higher bioactivity of DDP.
Cell cycle arrest and cell death mode
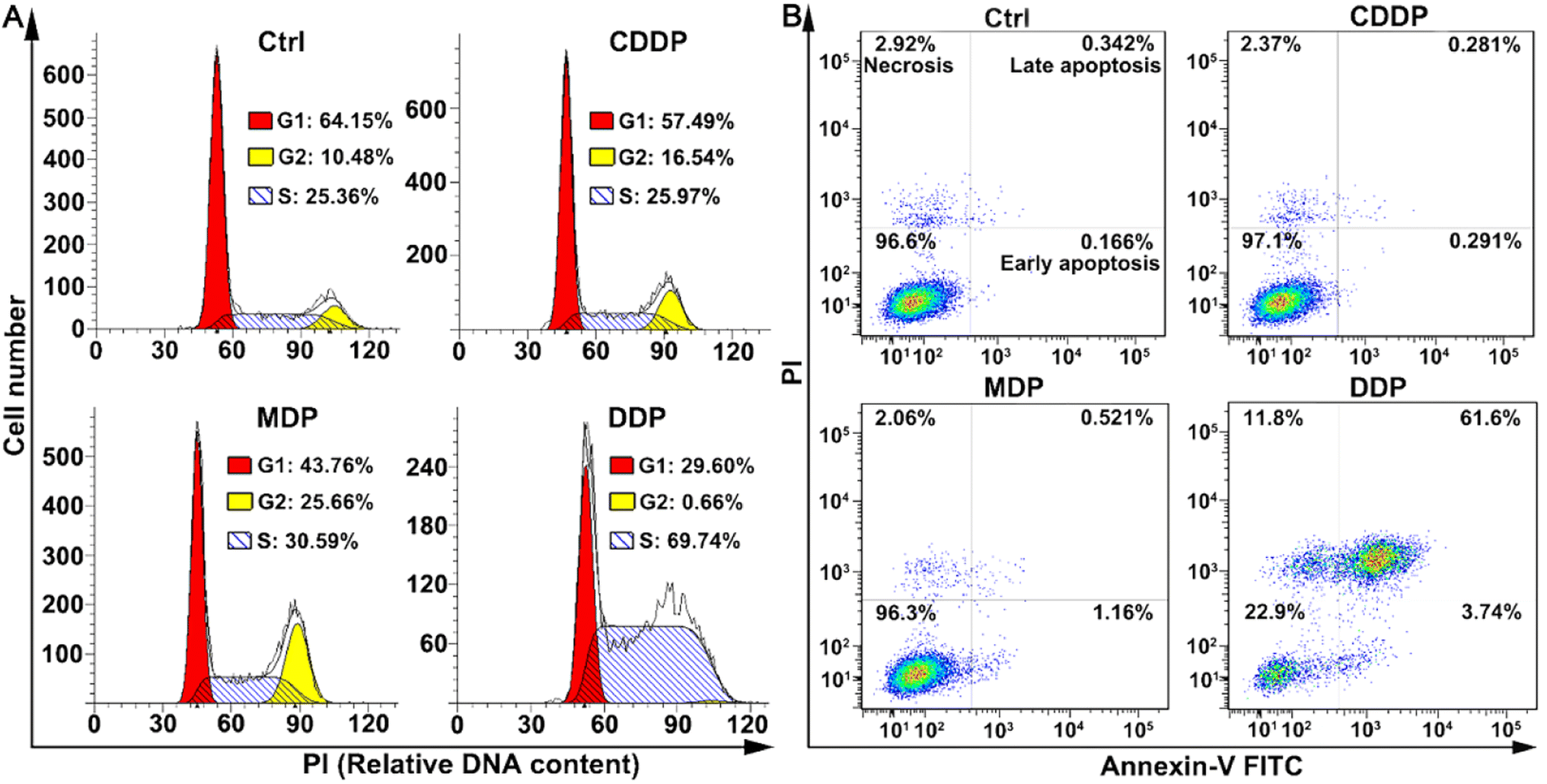
The cell cycle of SKOV-3 cells was analyzed by flow cytometry after staining DNA with propidium iodide (PI). As shown in Fig. 4A, DDP arrested the cell cycle mainly in the S phase, with the cells increasing from 25.36% to 69.74%; while MDP arrested the cell cycle mainly in the G2 and weakly in the S phases. CDDP weakly arrested the cell cycle in the G2 phase as reported.33 The arrest of the cell cycle in the S phase indicates that DNA replication was affected by DDP due to the damage to DNA in cancer cells.
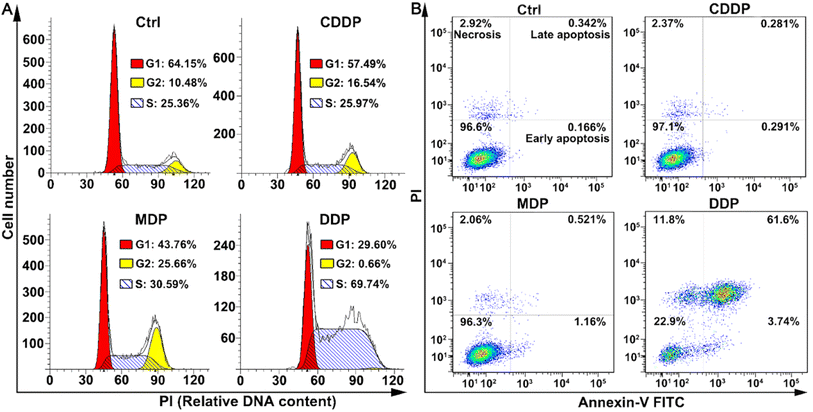 |
| | Fig. 4 Cell cycle arrest (A, 24 h) and distribution of SKOV-3 cells (B, 48 h) determined by flow cytometry after treatment with CDDP, MDP, and DDP (0.5 μM containing 0.5‰ DMSO), respectively, and staining with annexin V-FITC and/or PI. | |
The death mode of SKOV-3 cells induced by MDP and DDP was investigated by flow cytometry along with annexin V-FITC/PI double staining. As shown in Fig. 4B, DDP induced 61.6% of the cells to undergo late apoptosis, while MDP and CDDP did not cause apparent apoptosis under the same conditions, thus confirming the strong proapoptotic ability of DDP, which is consistent with its potent antiproliferative activity. Interestingly, the apoptosis and necrosis of SKOV3 cells induced by DDP are about 26-fold more than those induced by CDDP, while the DNA platination induced by DDP is only about 2-fold higher than that induced by CDDP, thus proving that DNA damage is not the determinant of cell death in this case and some other mechanism may be involved in the apoptosis.
Inhibition of LDH and lactate production
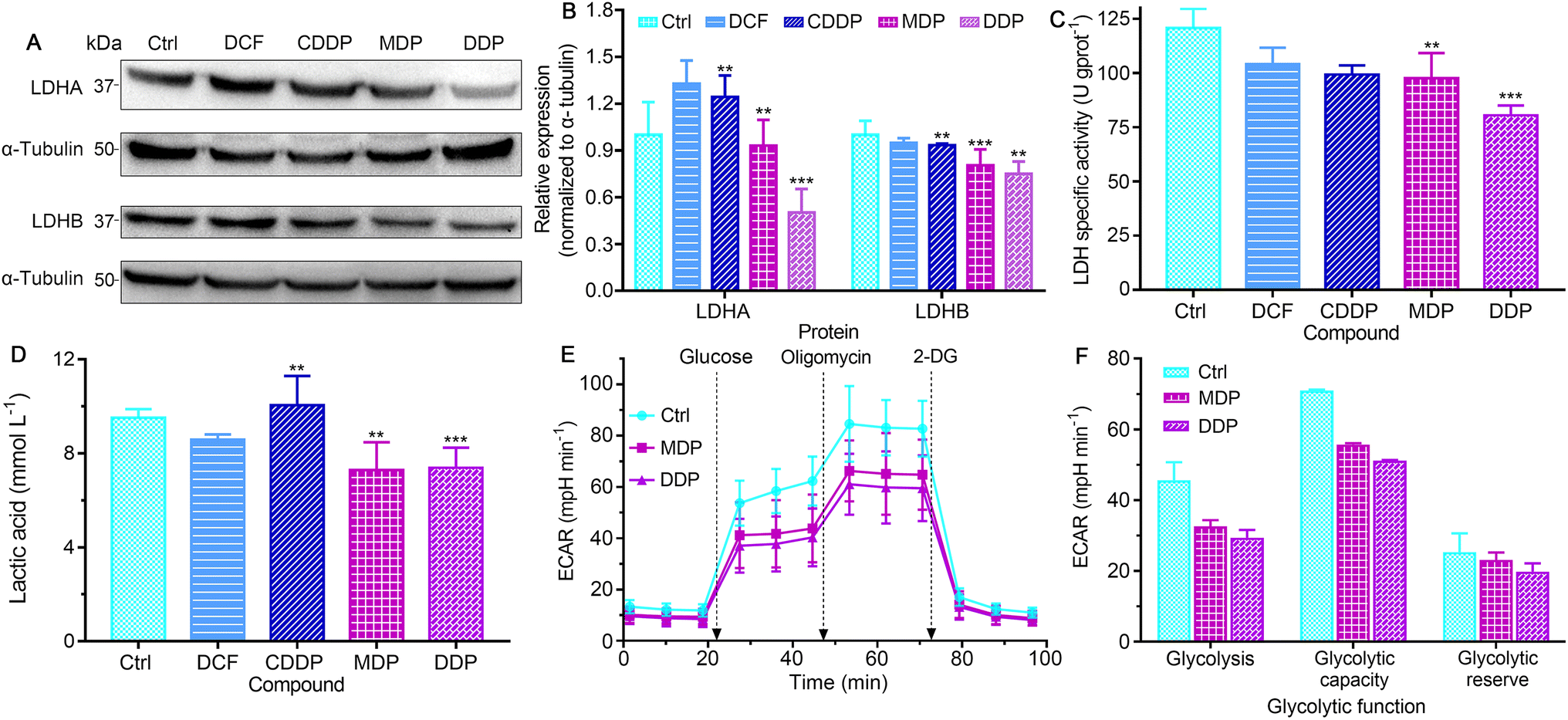
LDH constitutes a major checkpoint in anaerobic glycolysis by catalyzing the reduction of pyruvate into lactate.34 The inhibition of LDH could significantly decrease the tumorigenic potential of cancers.12 The expression of LDHA and LDHB in SKOV-3 cells was first examined by western blotting. As shown in Fig. 5A, B and S5,† DDP markedly suppressed the expression of LDHA as compared to the control, DCF and CDDP, and MDP moderately inhibited the expression of LDHA. MDP and DDP also reduced the expression of LDHB by 10% and 25%, respectively, while CDDP only decreased by 7%. The impact of DCF and CDDP on LDH expression is not substantial as compared with that of MDP and DDP, because they are more hydrophilic molecules (vide ante, Fig. 3) that cannot enter cancer cells readily to act on LDH. It is known that LDHA catalyzes the interconversion of pyruvate and lactate in human cancers and plays an important role in the development, invasion and metastasis of malignancies,15 and LDHB mediates the conversion of lactate that is re-absorbed in tumor cells to pyruvate for OXPHOS in the TME.12 Thus, the suppression of LDHA suggests that DDP could retard the production of lactate and occurrence of tumors, and the inhibition of LDHB implies that DDP could prevent tumor cells from allocating energy efficiently, that is, hampering the “RWE”. The effect of MDP and DDP on the activity of LDH was further examined by biochemical analysis. As shown in Fig. 5C, the LDH activity in untreated SKOV-3 cells is high (120.72 U g per prot), which may contribute to the acidic TME. DDP reduced the activity by 33.4%, while MDP, CDDP, and DCF only mildly decreased the activity (<19.1%) at the same concentration. It is noteworthy that most LDH inhibitors only show a limited effect on LDHA,35,36 and inhibitors for both LDHA and LDHB are uncommon. In short, the inhibition of LDH suggests that DDP could decrease the level of lactate in SKOV-3 cells and relieve the acidic TME, which may provide a new strategy for cancer treatment.
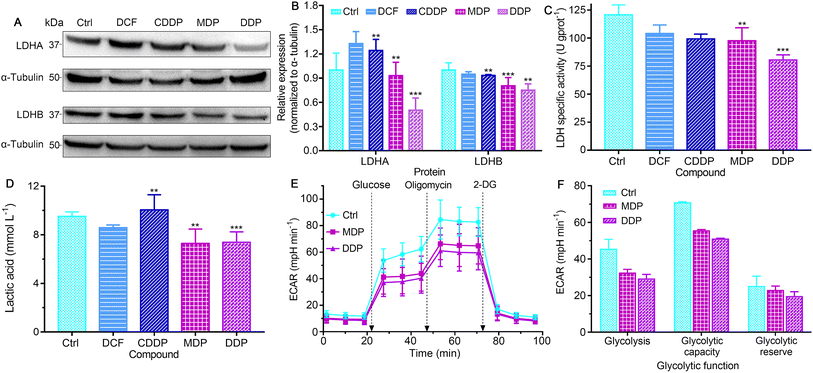 |
| | Fig. 5 The expressions (A), and the corresponding protein content of LDHA and LDHB relative to α-tubulin (B); LDH activity (C) and the level of lactate (D) in SKOV-3 cells after treatment with CDDP, MDP, DDP (0.6 μM containing 0.6‰ DMSO), and DCF (1.2 μM containing 0.1‰ DMSO), respectively, for 36 h; glycolytic profiles of the SKOV-3 cells treated with each complex (0.6 μM containing 0.2‰ DMSO) at 18 h (E), and key energy-profiling data related to the process (F). Four replicate experiments were performed independently, ECARglycolysis = ECARglucose − ECAR2-DG, ECARglycolytic capacity = ECARoligomycin − ECAR2-DG, ECARglycolytic reserve = ECARoligomycin − ECARglucose. **P < 0.01 and ***P < 0.001, statistical significance compared to the control. | |
The level of lactate in the supernatant of SKOV-3 cells was determined by colorimetry after treatment with the complexes. As shown in Fig. 5D, MDP and DDP decreased the lactate level by about 30% relative to the control. DCF was less effective than MDP and DDP, and CDDP even increased the level of lactate. The production of lactate was further determined by measuring the extracellular acidification rate (ECAR) on a Seahorse XFe24 cell bioanalyzer, which reflects the glycolytic capability of the cells. As shown in Fig. 5E and F, the ECAR was increased after injecting glucose into the complex-treated SKOV-3 cells, with the DDP-treated cells increasing the least. Oligomycin (an inhibitor of mitochondrial ATP synthase) triggered further lactate production, but the glycolytic capacity of the DDP-treated cells is obviously lower than that of the control. Addition of 2-deoxy-D-glucose (2-DG) to the cells induced a rapid decline in the ECAR, manifesting the reduction of glycolytic flux. These observations suggest that DDP greatly intervened in the glycolysis and energetic metabolism of SKOV-3 cells.
Inhibition of transporters and oncogenes
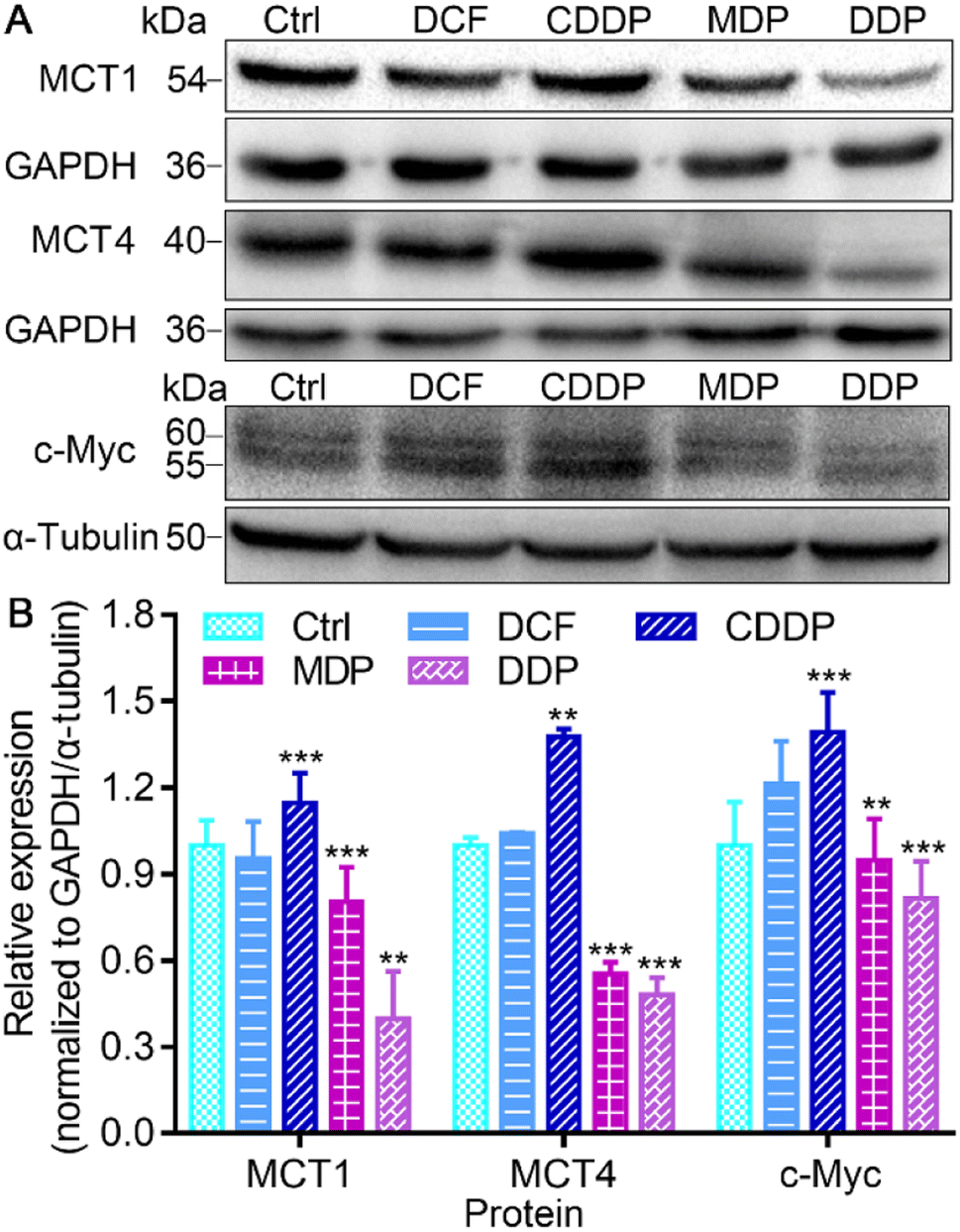
MCT1 and hypoxia-inducible MCT4 mediate the release of lactate assisted by LDH into interstitial space, leading to a decrease of pH in the TME.37 Oncogenic transcription factor c-Myc targets LDHA and its activation is directly linked to the Warburg effect.15 The conversion of pyruvate to lactate in the cytoplasm and the transport to the extracellular matrix rely on LDHA and MCT4, while the transport of extracellular lactate into cells and conversion to pyruvate for oxidation rely on MCT1 and LDHB.12 Thus the expressions of MCT1, MCT4, and c-Myc in SKOV-3 cells were investigated by immunoblotting after treatment with the complexes. As shown in Fig. 6 and S5,† DDP significantly suppressed the expression of MCT1, MCT4 and c-Myc, and MDP moderately inhibited MCT1 and MCT4. In contrast, CDDP upregulated the expression of these proteins. The downregulation of MCT1 and MCT4 could inhibit the influx or efflux of lactate in tumor cells, and the inhibition of c-Myc could affect the activity of LDHA, restraining the Warburg effect. Since lactate can promote the OXPHOS activity of mitochondria, resulting in sufficient energy supply for proliferation of cancer cells,6,38 the inhibition of lactate production suggests that DDP could inhibit the RWE, thus providing an additional pathway for suppressing cancer cells.
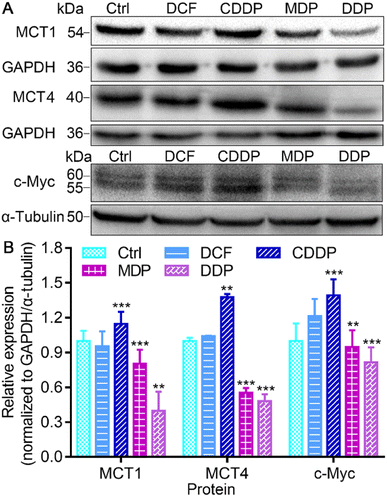 |
| | Fig. 6 Expressions of MCT1, MCT4, and c-Myc (A), and the corresponding protein content relative to GAPDH or α-tubulin (B) in SKOV-3 cells after treatment with different compounds (0.6 μM containing 0.6‰ DMSO) for 36 h. **P < 0.01 and ***P < 0.001, statistical significance compared to the control. | |
Effect on mitochondrial function
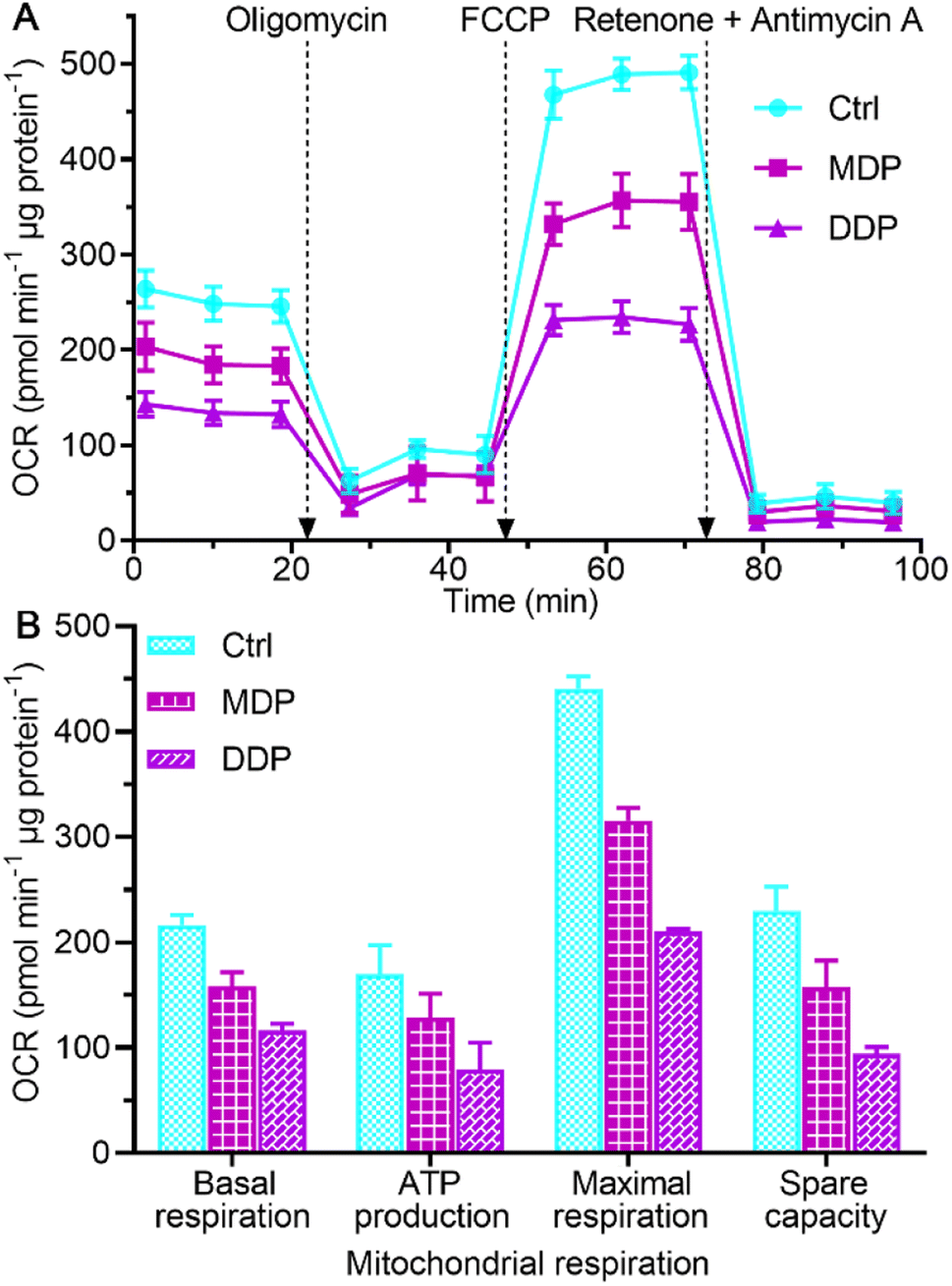
The inhibition on LDHA activity and aerobic lactate production would decrease the oxygen consumption and OXPHOS activity.39 The impact of MDP and DDP on the oxygen consumption rate (OCR) of SKOV-3 cells was measured using the Seahorse XFe24 cell bioanalyzer, which reflects the status of mitochondrial OXPHOS. As shown in Fig. 7, both MDP and DDP reduced the basal OCR levels, indicating a loss in total mitochondrial mass. As oligomycin was added to the cells, the OCR was further reduced, suggesting that the ATP production was decreased. When carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP, an uncoupler of mitochondrial OXPHOS) was injected into the media, the increase in the OCR was significantly deterred by MDP and DDP, implying that the maximal respiratory capacity was inhibited. Apparently, the stimulation of mitochondrial respiration by FCCP was substantially weakened by MDP and DDP. After injection of mitochondrial complex I inhibitor rotenone and complex III inhibitor antimycin A, the OCR was dramatically reduced, suggesting that respiration was almost completely inhibited. These results demonstrate that MDP and DDP can inhibit the mitochondrial OXPHOS besides blocking glycolysis; thus the production of ATP in tumor cells was effectively restrained. Since tumor cells with reserved respiratory function can obtain energy to meet the growth requirements through the RWE, the double inhibition on both the Warburg effect and RWE implies a radical cutoff of energy supply for the tumor growth.
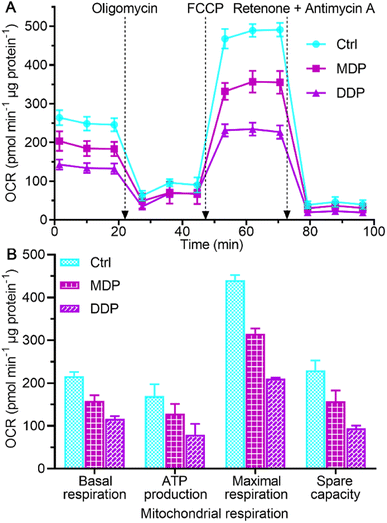 |
| | Fig. 7 Respiratory profiles of SKOV-3 cells in response to the complexes (0.6 μM containing 0.2‰ DMSO) at 18 h (A) and key energy-profiling data involved in the process (B). Four replicate samples prepared from a single-cell culture were measured independently. OCRbasal respiration = OCRinitial − OCRantimycin A/rotenone, OCRATP production = OCRbasal − OCRoligomycin, OCRmaximal respiration = OCRFCCP − OCRantimycin A/rotenone, and OCRspare capacity = OCRmaximal respiration − OCRbasal respiration.40 | |
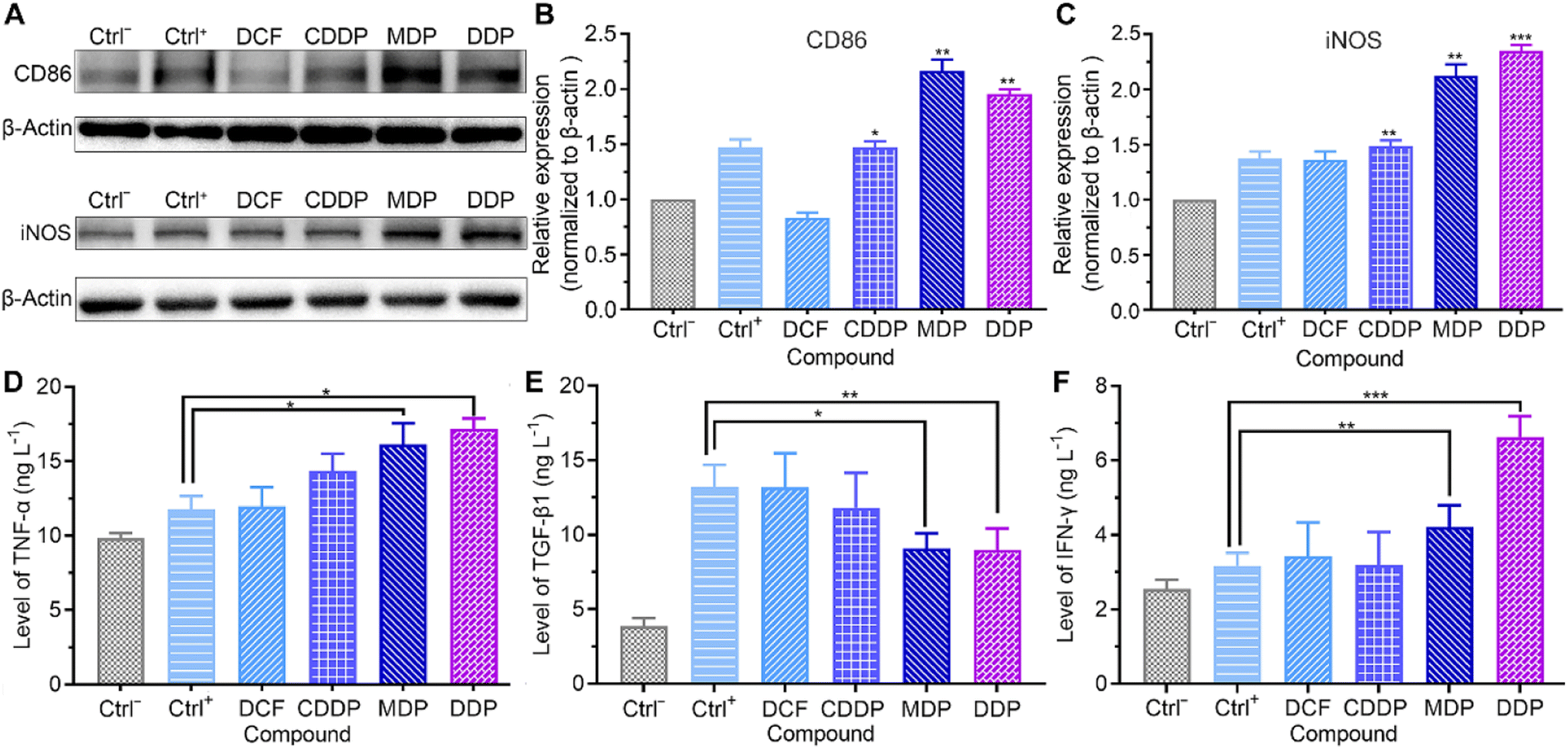
Effect on macrophages
Macrophages are the most abundant immune cells in the TME. Extracellular lactate can be sensed by macrophages, triggering intracellular signalling to fine tune cell behavior and influence the TME. The influence of the complexes on the markers of macrophage phenotypes was analyzed by western blotting and ELISA. Macrophages were derived from the human acute monocytic leukemia THP-1 cells under differentiation induced by phorbol ester (PMA), which mimic the primary human macrophages.41 As shown in Fig. 8A–E and S6,† the MDP- and DDP-treated SKOV-3 cell-conditioned culture supernatant increased the expression of M1 markers CD86 and inducible nitric oxide synthase (iNOS), promoted the secretion of tumor necrosis factor-alpha (TNF-α), and inhibited the production of transforming growth factor-beta 1 (TGF-β1). The immunosuppressive M2 macrophages are profoundly implicated in tumor initiation and progression.42 The results suggest that MDP and DDP promoted the repolarization of THP-1 macrophages from the M2 to the M1 phenotype. Low pH reduces the expression of iNOS and TNF-α in M1 macrophages, while increases the expression of M2 macrophage markers in the TME.10,43 Since MDP and DDP inhibited the production of lactate, the phenotype of macrophages was reversed. Strikingly, the MDP- and DDP-treated SKOV-3 cell-conditioned culture supernatant also increased the secretion of γ interferon (IFN-γ) in THP-1 macrophages (Fig. 8F). IFN-γ has been acknowledged to mediate immune responses and license immune cells to exert toxicity.44 The principal source of IFN-γ in the human immune response is believed to be T and natural killer (NK) cells; however, it can also be produced in other cell types, including macrophages.45,46 IFN-γ enhances antigen presentation through inducing specific gene expression programs in the TME, thereby increasing the phagocytic and killing abilities of macrophages to tumor cells.47 The results indicate that DDP stimulated the production of IFN-γ in THP-1 macrophages and participated in the immune response by modulating the TME. THP-1 macrophages almost remained intact under the test conditions (Fig. S7†).
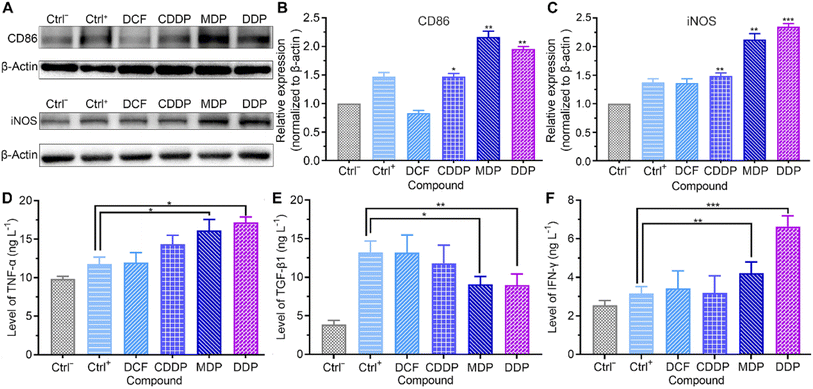 |
| | Fig. 8 Expressions of CD86 and iNOS (A), relative density of CD86 (B) and iNOS (C) to β-actin, and levels of TNF-α (D), TGF-β1 (E), and IFN-γ (F) in THP-1 cells stimulated with PMA (10 ng mL−1) for 48 h and incubated with compound-treated SKOV-3 cell-conditioned culture supernatant (0.6 μM containing 0.6‰ DMSO) for 36 h. Ctrl−: THP-1 cells stimulated with PMA (10 ng mL−1) for 48 h and cultured with an RPMI-1640 growth medium; Ctrl+: THP-1 cells stimulated with PMA (10 ng mL−1) for 48 h and cultured with SKOV-3 cell-conditioned culture supernatant without compound treatment. *P < 0.05, **P < 0.01 and ***P < 0.001, statistical significance compared to the Ctrl+ group. | |
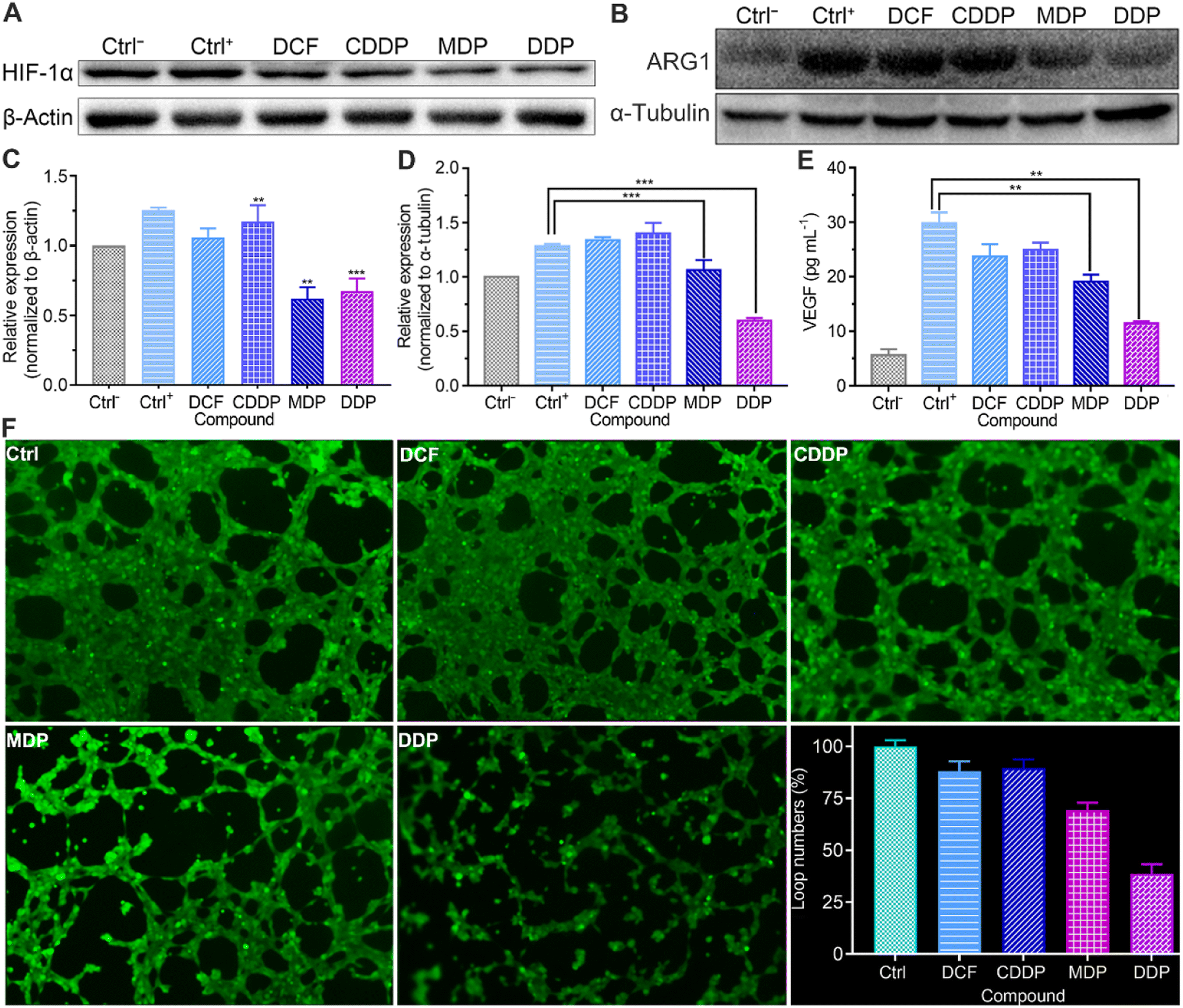
Repression of angiogenesis
Tumor angiogenesis is crucial for the survival and development of tumors. Lactate serves as a proangiogenic agent by increasing VEGF content in macrophages and promotes tumor cell proliferation by enhancing the expression of ARG1 in macrophages, thus enhancing the immunosuppression on T lymphocytes.48,49 HIF-1α promotes tumor angiogenesis not only by activating proangiogenic genes, but also by inhibiting anti-angiogenic genes under hypoxic conditions.50 The upregulation of HIF-1α regulates the transcription of VEGF and ARG1 in macrophages, thereby supporting tumor growth by inducing neovascularization and providing substrates for cancer cell proliferation.51 Therefore, we detected the expressions of HIF-1α and ARG1 and the level of VEGF by western blotting and using a human VEGF ELISA kit respectively. As shown in Fig. 9A–D and S6,† the expressions of HIF-1α and ARG1 were dramatically increased in the control once macrophages were stimulated with tumor-conditioned media. In the presence of MDP or DDP, the expressions of HIF-1α and ARG1 were downregulated obviously and VEGF was reduced by 36% and 62%, respectively (Fig. 9E). The blocking of VEGF suggests that MDP and DDP have the potential to inhibit tumor angiogenesis and growth.52 Since IFN-γ can interfere with the proliferation and survival of endothelial cells and impede angiogenesis in the TME,43 the above enhanced IFN-γ by DDP may also contribute to the repression of angiogenesis.
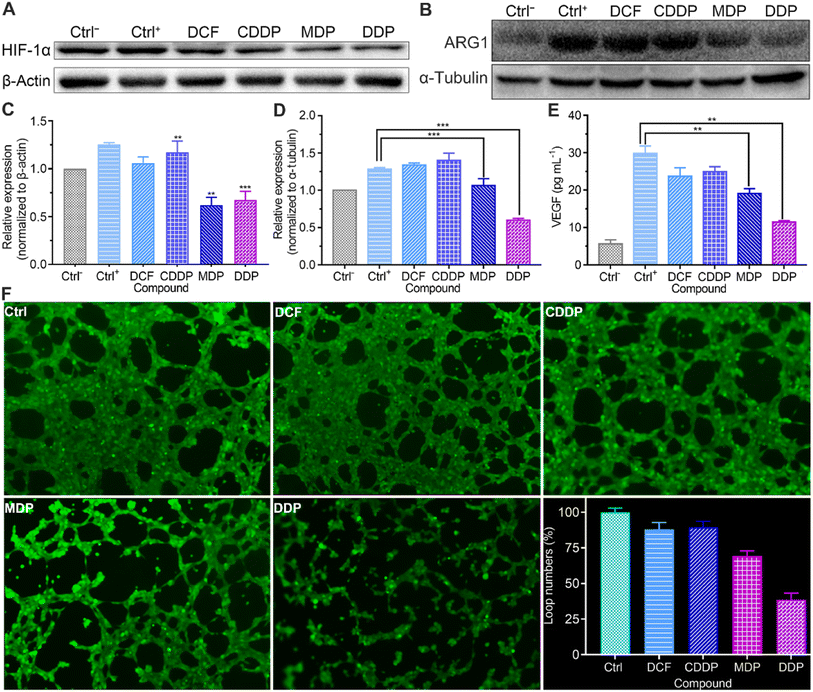 |
| | Fig. 9 Expressions of HIF-1α (A) and ARG1 (B), relative density of HIF-1α to β-actin (C), ARG1 to α-tubulin (D), and the secretion of VEGF (E) in THP-1 cells stimulated with PMA (10 ng mL−1) for 48 h and incubated with the compound-treated SKOV-3 cell-conditioned culture supernatant (0.6 μM containing 0.6‰ DMSO) for 36 h, and the effect of DCF (0.4 μM containing 0.4‰ DMSO), CDDP, MDP, and DDP (0.2 μM containing 0.2‰ DMSO) on the tube formation of HUV-EC-C after incubation for 6 h (F). Image magnification: 100×; Ctrl−: THP-1 cells stimulated with PMA (10 ng mL−1) for 48 h and cultured with an RPMI-1640 growth medium; Ctrl+: THP-1 cells stimulated with PMA (10 ng mL−1) for 48 h and cultured with SKOV-3 cell-conditioned culture supernatant without compound treatment. **P < 0.01 and ***P < 0.001, statistical significance compared to the Ctrl+ group. | |
The acidic environment formed by high concentrations of lactate is conducive to the angiogenesis of tumor cells.11 The lactate released from tumor cells through MCT4 may be taken up by endothelial cells via the MCT1 transporter and stimulate angiogenesis through multiple signaling pathways.53 The effect of the complexes on the vasculature network formed by HUV-EC-C endothelial cells was checked at a nonlethal concentration (Table S2†). As shown in Fig. 9F, plenty of capillary-like tubes were formed in the control and DCF- and CDDP-treated cells; however, the loop numbers and tube length (Fig. S8†) were decreased by 62% and 34%, respectively, in the DDP-treated HUV-EC-C cells. MDP showed a moderate inhibition effect with the loop numbers and tube length being declined 31% and 30%, respectively. The results prove that DDP could inhibit the formation of new blood vessels to some extent, which may be attributed to the reduction of lactate and elevation of IFN-γ.
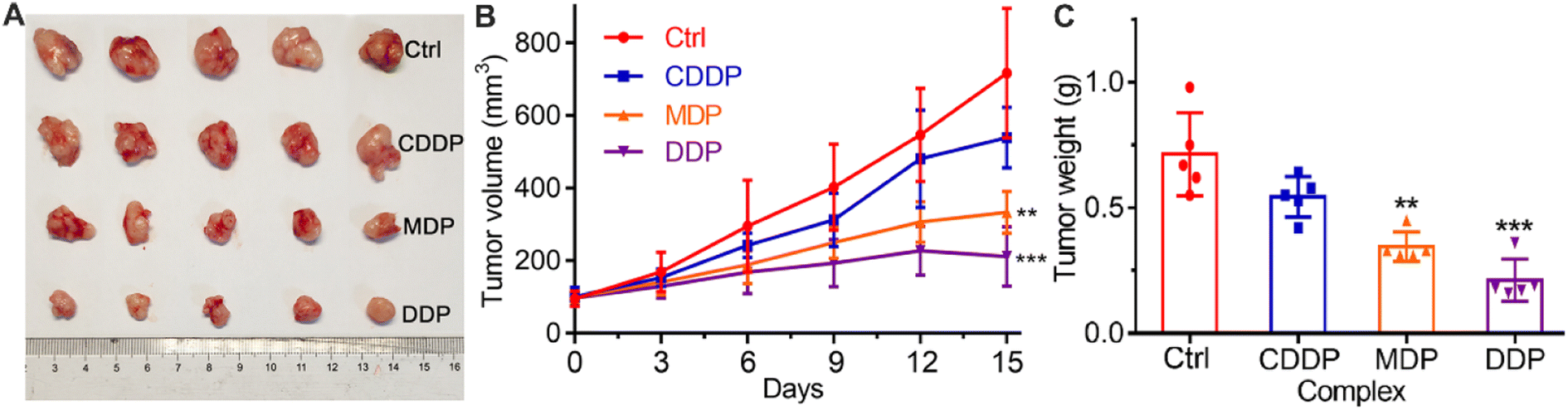
In vivo antitumor activity
The in vivo antitumor activity of the complexes was investigated on female Balb/C mouse models bearing SKOV-3 tumors. The tumor growth was evaluated after treatment with CDDP, MDP, and DDP, respectively, for 15 days, and the results are shown in Fig. 10. MDP and DDP reduced the tumor volume by 53.4 and 70.4%, respectively, while CDDP only reduced the tumor volume by 27.4% (A and B). The tumor weight was also reduced significantly as compared to CDDP (C). The body weight of the mice was almost unchanged during the therapy (Fig. S9†). Histological images with hematoxylin and eosin (H&E) staining showed no obvious damage in the major organs of mice (Fig. S10†). These results indicate that MDP and DDP displayed high antitumor activity in vivo with low systemic toxicity.
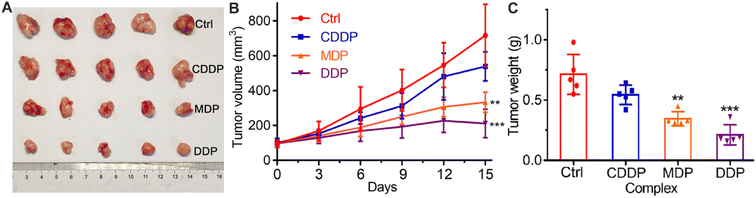 |
| | Fig. 10
In vivo antitumor activity of CDDP, MDP, and DDP (1.5 mg Pt per kg) in Balb/C nude mice bearing SKOV-3 xenograft tumors (n = 5), PBS was used as a control. (A) Representative tumor images after treatment for 15 days, (B) time-dependent tumor volume during 15 days, and (C) tumor weight after treatment for 15 days. **P < 0.01 and ***P < 0.001, statistical significance compared to the control. | |
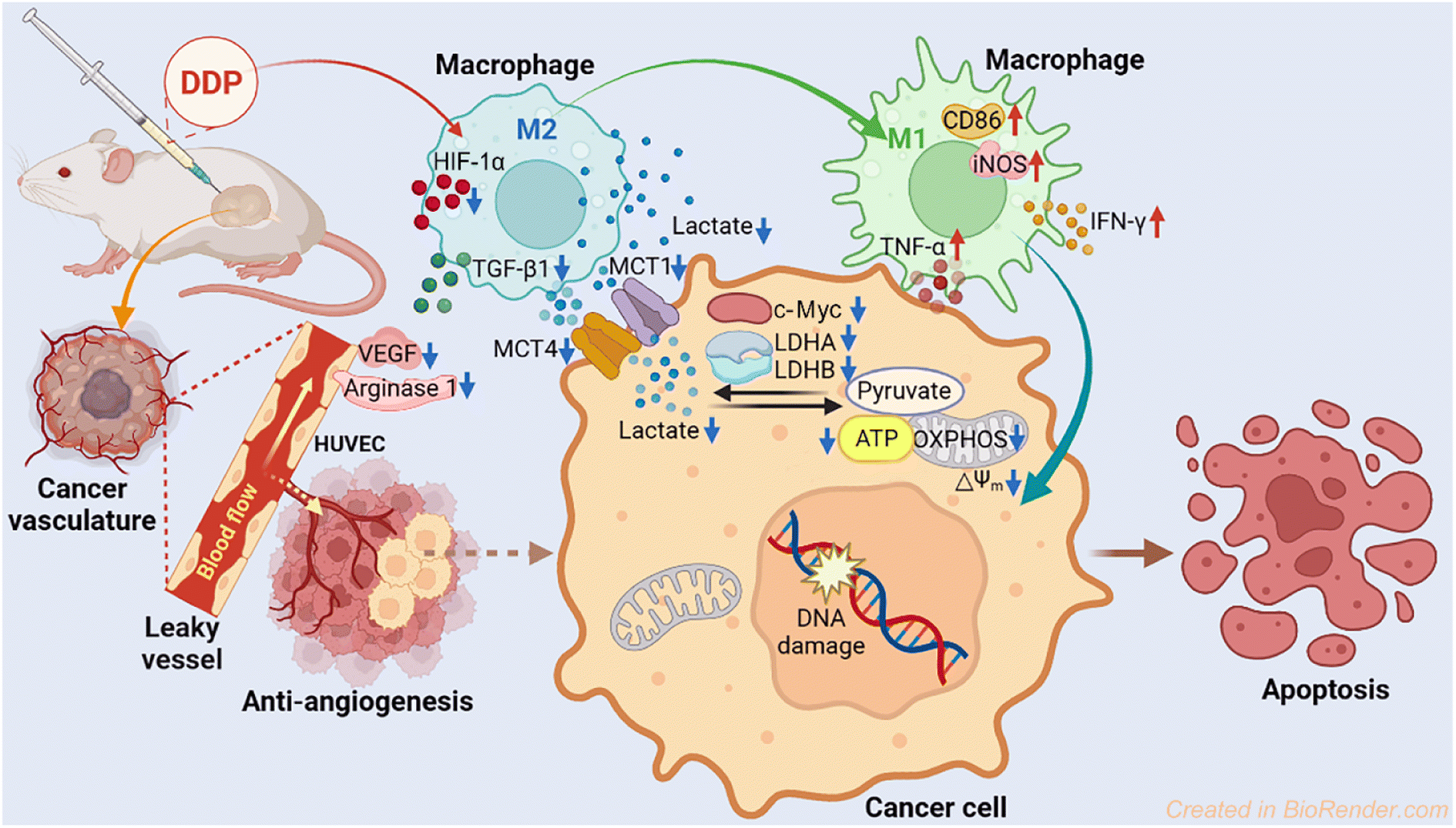
Mechanism of action
The antitumor mechanism of current platinum drugs involves reactions with nuclear DNA and induction of apoptosis by inhibiting DNA replication and gene transcription.54 Here we demonstrate that aside from damaging DNA, modulating energy metabolism of cancer cells and relieving immunosuppression of the TME are also effective pathways to curb the development of tumors. The parallel mechanism mainly includes suppressing LDH, modulating macrophages, and blocking angiogenesis. In particular, DDP downregulated the expression of LDHA, LDHB, MCT1, MCT4, and c-Myc, thus inhibiting the conversion of pyruvate to lactate and lowering the energy metabolism in tumor cells. Furthermore, DDP promoted the polarization of macrophages from the tumor-promoting M2 phenotype to the tumor-inhibiting M1 phenotype via reducing the acidic TME, leading to an increase of iNOS, TNF-α and IFN-γ in M1 macrophages and a decrease of TGF-β1 in M2 macrophages. Finally, DDP restrained the angiopoiesis by downregulating the expression of HIF-1α and ARG1 and impeding the secretion of VEGF, which could alleviate the tumor immunosuppression of the TME. The antitumor mechanism of DDP is illustrated in Fig. 11.
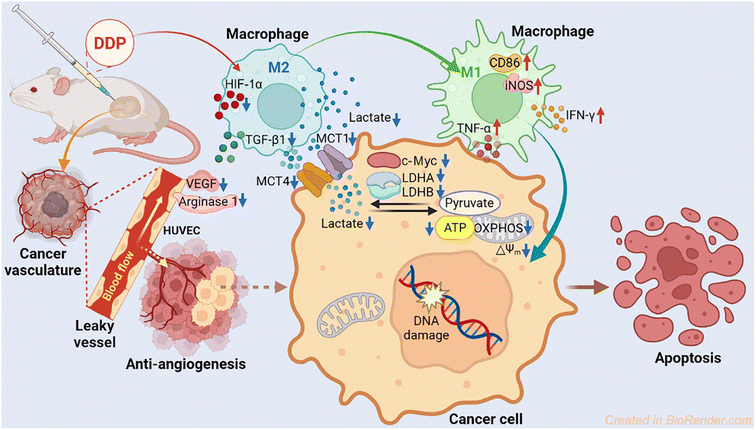 |
| | Fig. 11 Proposed mechanism of action for DDP. | |
Conclusion
Targeting tumor glycolysis and the immune microenvironment is attractive for chemotherapy and immunotherapy. Lactate not only serves as a key metabolite responsible for glycolysis, but also as a regulator for the tumor microenvironment and immune cell functions. In addition, the lactate originated from tumor cells stimulates angiogenesis by acting on endothelial cells and tumor-associated macrophages. The generation of lactate from pyruvate catalyzed by LDH is a key event for anaerobic glycolysis, which plays a pivotal role in regulating diverse biological processes, such as macrophage polarization and tumor immune surveillance. Lactate levels and fluxes reflect the adaptation extent of tumor cells to survival and proliferation. The acidic tumor microenvironment formed by excess lactate is an adverse factor affecting tumor immune surveillance and immune escape. Inhibiting LDH activity can suppress the production of lactate, hence relieving the immunosuppression as well as restraining the energy metabolism. Drug resistance is the major barrier limiting the efficacy of platinum anticancer drugs. This study provides a feasible strategy to overcome the shortcoming by regulating tumor glycometabolism or lactate metabolism and reshaping the tumor microenvironment. In a word, targeting energy metabolism and the immune microenvironment is a new approach for the development of antitumor drugs.
Data availability
The data that support the findings of this study are available in the ESI† of this article.
Author contributions
Suxing Jin performed the biological experiments and wrote the draft; Enmao Yin designed and synthesized the complexes; Chenyao Feng and Yuewen Sun performed the cellular experiments; Tao Yang and Hao Yuan provided the technical support; Zijian Guo contributed to the formation of concepts; Xiaoyong Wang edited the manuscript and supervised the research.
Ethical statement
All animal experiments were performed in accord with the guidelines for the care and use of laboratory animals approved by the Animal Ethical and Welfare Committee of NJU (Nanjing, China).
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
We thank the National Natural Science Foundation of China (Grants 22107050, 91953201, 92153303, and 21877059) and the Natural Science Foundation of Jiangsu Province (BK20202004).
References
- M. G. Vander Heiden, L. C. Cantley and C. B. Thompson, Science, 2009, 324, 1029–1033 CrossRef CAS PubMed.
- D. Hanahan and R. A. Weinberg, Cell, 2011, 144, 646–674 CrossRef CAS PubMed.
- W. H. Koppenol, P. L. Bounds and C. V. Dang, Nat. Rev. Cancer, 2011, 11, 325–337 CrossRef CAS PubMed.
- L. S. Silva, G. Poschet, Y. Nonnenmacher, H. M. Becker, S. Sapcariu, A. C. Gaupel, M. Schlotter, Y. Wu, N. Kneisel, M. Seiffert, R. Hell, K. Hiller, P. Lichter and B. Radlwimmer, EMBO Rep., 2017, 18, 2172–2185 CrossRef CAS.
- V. Miranda-Goncalves, M. Honavar, C. Pinheiro, O. Martinho, M. M. Pires, C. Pinheiro, M. Cordeiro, G. Bebiano, P. Costa, I. Palmeirim, R. M. Reis and F. Baltazar, Neuro Oncol., 2013, 15, 172–188 CrossRef CAS PubMed.
- A. N. Chen, Y. Luo, Y. H. Yang, J. T. Fu, X. M. Geng, J. P. Shi and J. Yang, Front. Immunol., 2021, 12, 688910 CrossRef CAS PubMed.
- C. Corbet and O. Feron, Nat. Rev. Cancer, 2017, 17, 577–593 CrossRef CAS PubMed.
- G. Multhoff and P. Vaupel, Signal Transduction Targeted Ther., 2021, 6, 171 CrossRef CAS PubMed.
- R. Haas, D. Cucchi, J. Smith, V. Pucino, C. E. Macdougall and C. Mauro, Trends Biochem. Sci., 2016, 41, 460–471 CrossRef CAS PubMed.
- M. Certo, C. H. Tsai, V. Pucino, P. C. Ho and C. Mauro, Nat. Rev. Immunol., 2021, 21, 151–161 CrossRef CAS PubMed.
- M. De Palma, D. Biziato and T. V. Petrova, Nat. Rev. Cancer, 2017, 17, 457–474 CrossRef CAS PubMed.
- Z. E. Stine, Z. T. Schug, J. M. Salvino and C. V. Dang, Nat. Rev. Drug Discovery, 2022, 21, 141–162 CrossRef CAS PubMed.
- C. Hayes, C. L. Donohoe, M. Davern and N. E. Donlon, Cancer Lett., 2021, 500, 75–86 CrossRef CAS.
- E. C. Nakajima and B. Van Houten, Mol. Carcinog., 2013, 52, 329–337 CrossRef CAS PubMed.
- A. Le, C. R. Cooper, A. M. Gouw, R. Dinavahi, A. Maitra, L. M. Deck, R. E. Royer, D. L. Vander Jagt, G. L. Semenza and C. V. Dang, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 2037–2042 CrossRef CAS PubMed.
- Y. Yu, J. A. Deck, L. A. Hunsaker, L. M. Deck, R. E. Royer, E. Goldberg and D. L. Vander Jagt, Biochem. Pharmacol., 2001, 62, 81–89 CrossRef CAS PubMed.
- C. Granchi, S. Roy, C. Giacomelli, M. Macchia, T. Tuccinardi, A. Martinelli, M. Lanza, L. Betti, G. Giannaccini, A. Lucacchini, N. Funel, L. G. Leon, E. Giovannetti, G. J. Peters, R. Palchaudhuri, E. C. Calvaresi, P. J. Hergenrother and F. Minutolo, J. Med. Chem., 2011, 54, 1599–1612 CrossRef CAS PubMed.
- A. D. Moorhouse, C. Spiteri, P. Sharma, M. Zloh and J. E. Moses, Chem. Commun., 2011, 47, 230–232 RSC.
- K. D. Mjos and C. Orvig, Chem. Rev., 2014, 114, 4540–4563 CrossRef CAS PubMed.
- T. C. Johnstone, K. Suntharalingam and S. J. Lippard, Chem. Rev., 2016, 116, 3436–3486 CrossRef CAS.
- K. M. Deo, D. L. Ang, B. McGhie, A. Rajamanickam, A. Dhiman, A. Khoury, J. Holland, A. Bjelosevic, B. Pages, C. Gordon and J. R. Aldrich-Wright, Coord. Chem. Rev., 2018, 375, 148–163 CrossRef CAS.
- X. H. Wang, X. Y. Wang, S. X. Jin, N. Muhammad and Z. J. Guo, Chem. Rev., 2019, 119, 1138–1192 CrossRef CAS PubMed.
- E. Gottfried, S. A. Lang, K. Renner, A. Bosserhoff, W. Gronwald, M. Rehli, S. Einhell, I. Gedig, K. Singer, A. Seilbeck, A. Mackensen, O. Grauer, P. Hau, K. Dettmer, R. Andreesen, P. J. Oefner and M. Kreutz, PLoS One, 2013, 8, e66987 CrossRef CAS PubMed.
- K. Renner, C. Bruss, A. Schnell, G. Koehl, H. M. Becker, M. Fante, A. N. Menevse, N. Kauer, R. Blazquez, L. Hacker, S. M. Decking, T. Bohn, S. Faerber, K. Evert, L. Aigle, S. Amslinger, M. Landa, O. Krijgsman, E. A. Rozeman, C. Brummer, P. J. Siska, K. Singer, S. Pektor, M. Miederer, K. Peter, E. Gottfried, W. Herr, I. Marchiq, J. Pouyssegur, W. R. Roush, S. Ong, S. Warren, T. Pukrop, P. Beckhove, S. A. Lang, T. Bopp, C. U. Blank, J. L. Cleveland, P. J. Oefner, K. Dettmer, M. Selby and M. Kreutz, Cell Rep., 2019, 29, 135–150 CrossRef CAS PubMed.
- F. P. Intini, J. Zajac, V. Novohradsky, T. Saltarella, C. Pacifico, V. Brabec, G. Natile and J. Kasparkova, Inorg. Chem., 2017, 56, 1483–1497 CrossRef CAS PubMed.
- D. V. Spector, K. G. Pavlov, R. A. Akasov, A. N. Vaneev, A. S. Erofeev, P. V. Gorelkin, V. N. Nikitina, E. V. Lopatukhina, A. S. Semkina, K. Y. Vlasova, D. A. Skvortsov, V. A. Roznyatovsky, N. V. Ul’yanovskiy, I. I. Pikovskoi, S. A. Sypalov, A. S. Garanina, S. S. Vodopyanov, M. A. Abakumov, Y. L. Volodina, A. A. Markova, A. S. Petrova, D. M. Mazur, D. A. Sakharov, N. V. Zyk, E. K. Beloglazkina, A. G. Majouga and O. O. Krasnovskaya, J. Med. Chem., 2022, 65, 8227–8244 CrossRef CAS.
- H. Kostrhunova, B. S. McGhie, L. Markova, O. Novakova, J. Kasparkova, J. R. Aldrich-Wright and V. Brabec, J. Med. Chem., 2023, 66, 7894–7908 CrossRef CAS PubMed.
- S. X. Jin, N. Muhammad, Y. W. Sun, Y. H. Tan, H. Yuan, D. F. Song, Z. J. Guo and X. Y. Wang, Angew. Chem., Int. Ed., 2020, 59, 23313–23321 CrossRef CAS PubMed.
- I. Romero-Canelon and P. J. Sadler, Inorg. Chem., 2013, 52, 12276–12291 CrossRef CAS PubMed.
- J. Z. Zhang, E. Wexselblatt, T. W. Hambley and D. Gibson, Chem. Commun., 2012, 48, 847–849 RSC.
- A. J. Leo, Chem. Rev., 1993, 93, 1281–1306 CrossRef CAS.
- S. X. Jin, Y. Guo, D. F. Song, Z. Z. Zhu, Z. Q. Zhang, Y. W. Sun, T. Yang, Z. J. Guo and X. Y. Wang, Inorg. Chem., 2019, 58, 6507–6516 CrossRef CAS PubMed.
- D. Wang and S. J. Lippard, Nat. Rev. Drug Discovery, 2005, 4, 307–320 CrossRef CAS PubMed.
- H. Xie, J. Hanai, J. G. Ren, L. Kats, K. Burgess, P. Bhargava, S. Signoretti, J. Billiard, K. J. Duffy, A. Grant, X. Wang, P. K. Lorkiewicz, S. Schatzman, M. Bousamra 2nd, A. N. Lane, R. M. Higashi, T. W. Fan, P. P. Pandolfi, V. P. Sukhatme and P. Seth, Cell Metab., 2014, 19, 795–809 CrossRef CAS PubMed.
- R. Arseneault, A. Chien, J. T. Newington, T. Rappon, R. Harris and R. C. Cumming, Cancer Lett., 2013, 338, 255–266 CrossRef CAS.
- Z. Y. Wang, T. Y. Loo, J. G. Shen, N. Wang, D. M. Wang, D. P. Yang, S. L. Mo, X. Y. Guan and J. P. Chen, Breast Cancer Res. Treat., 2012, 131, 791–800 CrossRef CAS PubMed.
- V. L. Payen, E. Mina, V. F. Van Hee, P. E. Porporato and P. Sonveaux, Mol. Metab., 2020, 33, 48–66 CrossRef CAS PubMed.
- S. Pavlides, D. Whitaker-Menezes, R. Castello-Cros, N. Flomenberg, A. K. Witkiewicz, P. G. Frank, M. C. Casimiro, C. Wang, P. Fortina, S. Addya, R. G. Pestell, U. E. Martinez-Outschoorn, F. Sotgia and M. P. Lisanti, Cell Cycle, 2009, 8, 3984–4001 CrossRef CAS PubMed.
- V. R. Fantin, J. St-Pierre and P. Leder, Cancer Cell, 2006, 9, 425–434 CrossRef CAS PubMed.
- J. Zhang, E. Nuebel, D. R. Wisidagama, K. Setoguchi, J. S. Hong, C. M. Van Horn, S. S. Imam, L. Vergnes, C. S. Malone, C. M. Koehler and M. A. Teitell, Nat. Protoc., 2012, 7, 1068–1085 CrossRef CAS PubMed.
- T. Yang, S. R. Zhang, H. Yuan, Y. Wang, L. X. Cai, H. H. Chen, X. Y. Wang, D. F. Song, X. H. Wang, Z. J. Guo and X. Y. Wang, Angew. Chem., Int. Ed., 2023, 62, e202213337 CAS.
- O. R. Colegio, N. Q. Chu, A. L. Szabo, T. Chu, A. M. Rhebergen, V. Jairam, N. Cyrus, C. E. Brokowski, S. C. Eisenbarth, G. M. Phillips, G. W. Cline, A. J. Phillips and R. Medzhitov, Nature, 2014, 513, 559–563 CrossRef CAS.
- K. Yang, J. J. Xu, M. Fan, F. Tu, X. H. Wang, T. Z. Ha, D. L. Williams and C. F. Li, Front. Immunol., 2020, 11, 587913 CrossRef CAS PubMed.
- M. A. Kursunel and G. Esendagli, Cytokine Growth Factor Rev., 2016, 31, 73–81 CrossRef PubMed.
- S. Gessani and F. Belardelli, Cytokine Growth Factor Rev., 1998, 9, 117–123 CrossRef CAS.
- L. Darwich, G. Coma, R. Peña, R. Bellido, E. J. J. Blanco, J. A. Este, F. E. Borras, B. Clotet, L. Ruiz, A. Rosell, F. Andreo, R. M. E. Parkhouse and M. Bofill, Immunology, 2008, 126, 386–393 CrossRef PubMed.
- C. M. Robinson, D. O'Dee, T. Hamilton and G. J. Nau, J. Innate Immun., 2010, 2, 56–65 CrossRef CAS PubMed.
- J. Ding, J. E. Karp and A. Emadi, Cancer Biomarkers, 2017, 19, 353–363 CAS.
- C. I. Chang, J. C. Liao and L. Kuo, Cancer Res., 2001, 61, 1100–1106 CAS.
- X. J. Jiang, J. Wang, X. Y. Deng, F. Xiong, S. S. Zhang, Z. J. Gong, X. Y. Li, K. Cao, H. Deng, Y. He, Q. J. Liao, B. Xiang, M. Zhou, C. Guo, Z. Y. Zeng, G. Y. Li, X. L. Li and W. Xiong, J. Exp. Clin. Cancer Res., 2020, 39, 204 CrossRef PubMed.
- B. Z. Qian and J. W. Pollard, Cell, 2010, 141, 39–51 CrossRef CAS.
- R. S. Apte, D. S. Chen and N. Ferrara, Cell, 2019, 176, 1248–1264 CrossRef CAS PubMed.
- F. Végran, R. Boidot, C. Michiels, P. Sonveaux and O. Feron, Cancer Res., 2011, 71, 2550–2560 CrossRef PubMed.
- Y. Guo, S. X. Jin, H. Yuan, T. Yang, K. Wang, Z. J. Guo and X. Y. Wang, J. Med. Chem., 2022, 65, 520–530 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Enmao
Yin‡
a,
Chenyao
Feng
a,
Yuewen
Sun
a,
Tao
Yang
cd,
Hao
Yuan
c,
Zijian
Guo
ab,
Enmao
Yin‡
a,
Chenyao
Feng
a,
Yuewen
Sun
a,
Tao
Yang
cd,
Hao
Yuan
c,
Zijian
Guo