
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile preparation of organosilanes from benzylboronates and gem-diborylalkanes mediated by KOtBu†
Man
Tang
a,
Wenyan
Zhu
a,
Huaxing
Sun
a,
Jing
Wang
a,
Su
Jing
 *a,
Minyan
Wang
*b,
Zhuangzhi
Shi
b and
Jiefeng
Hu
*a
*a,
Minyan
Wang
*b,
Zhuangzhi
Shi
b and
Jiefeng
Hu
*a
aSchool of Chemistry and Molecular Engineering, Nanjing Tech University, Nanjing, 211816, China. E-mail: iasjfhu@njtech.edu.cn
bState Key Laboratory of Coordination Chemistry, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing, 210023, China
First published on 13th June 2023
Abstract
Methods to efficiently synthesize organosilanes are valuable in the fields of synthetic chemistry and materials science. During the past decades, boron conversion has become a generic and powerful approach for constructing carbon–carbon and other carbon–heteroatom bonds, but its potential application in forming carbon–silicon remains unexplored. Herein, we describe an alkoxide base-promoted deborylative silylation of benzylic organoboronates, geminal bis(boronates) or alkyltriboronates, allowing for straightforward access to synthetically valuable organosilanes. This selective deborylative methodology exhibits operational simplicity, broad substrate scope, excellent functional group compatibility and convenient scalability, providing an effective and complementary platform for the generation of diversified benzyl silanes and silylboronates. Detailed experimental results and calculated studies revealed an unusual mechanistic feature of this C–Si bond formation.
Introduction
Organosilanes not only play pivotal roles in medicinal chemistry1 and material science,2 but they are also highly functionalized and versatile building blocks for further transformation in synthetic chemistry.3 In the past few decades, considerable attention has been placed on developing synthetic strategies to construct C(sp3)–Si bonds by cross-coupling reactions in the presence of transition metals, such as palladium, nickel, and copper salts, as catalysts (Fig. 1A).4 Highly reactive organometallic species, including Grignard reagents, organolithiums, organozincs or ganoaluminiums, are emerging as the most attractive substrates for the construction of organosilanes.5,6 Although these transformations are synthetically useful, these organometallic reagents exhibit a high basicity and strong nucleophilicity, making them incompatible with many functional groups. Organohalides are often used as precursors to organometallic reagents; therefore, substantial progress has been recently achieved to access silicon-containing molecules by cross-coupling from these abundant and structurally diverse feedstocks to replace preformed organometallic reagents.7,8 Despite these advances, a method to easily prepare organosilanes without reactive coupling partners and metal catalysts is more attractive. | ||
| Fig. 1 Background and discovery. (A) Classic synthesis methods of organosilanes; (B) boron conversion; (C) deborylative silylation. | ||
Interconversion of the main group elements has been a long-standing goal in organic chemistry.9 In this area, organoboron compounds have become valuable building blocks in organic synthesis because of their flexibility, as they can be synthesized from multiple different functional groups.10 Significant developments have been achieved for the interconversion of carbon–boron bonds to carbon–carbon and carbon–heteroatom (F, Cl, Br, I, O, N, S …) bonds (Fig. 1B).11,12 In this context, the conversion of C–Si bonds into C–B bonds has been developed through the reaction with boron trihalides (BCl3, BBr3).13 At the outset of this project, no inverse approach from C–B bonds to C–Si bonds had been developed.
Because of the diagonal relationship between boron and silicon in the periodic table, the boryl and silyl groups exhibit related properties and reactivity for further functionalization, but the latter groups are more stable during storage and handling.14 Furthermore, silicon reagents exhibit significant differences in functionality in materials, which are important precursors to commercial polymers and copolymers.15 Thus, developing a method to prepare organosilanes from related boronates is an attractive goal because it fills the gap in borane–silane interconversion. Here, we develop the first deborylative silylation of benzylic boronate esters or multiboronates to organosilanes mediated by an alkoxide base under transition metal-free conditions (Fig. 1C). In this surprisingly simple approach, the in situ formation of boron–ate complexes can react with chlorosilanes through a concerted process to access valuable benzyl silanes and silylboronates.
Results and discussion
We commenced our studies using commercially available 2-benzyl-4,4,5,5-tetramethyl-1,3,2-dioxaborolane 1a and chlorotriisopropylsilane 2a as model substrates. Based on a systematic examination of reaction conditions, we determined that the following settings were optimal: KOtBu (2 equiv.) as a base in tetrahydrofuran (THF) at 100 °C. As a result, benzyltriisopropylsilane 3a was obtained in 84% isolated yield (Table 1, entry 1). KOtBu was removed from standard conditions, and the target product could not be detected by GC-MS (Table 1, entry 2). While 3a was formed in 21% yield with KOMe (Table 1, entry 3), other commonly used bases, such as NaOtBu, LiOtBu, KOH and K3PO4, were ineffective for this cross-coupling reaction (Table 1, entries 4–7). These results demonstrated that potassium cations and alkoxide anions are indispensable for the high reactivity of this transformation. When the reaction was conducted in toluene or noctane with KOtBu as a base, lower yields were obtained (Table 1, entries 8 and 9). However, other polar solvents, including MeCN and DMF, failed to generate silylated products (Table 1, entries 10–11). Lowering the temperature led to a lower yield with incomplete conversion of the benzyl boron esters, and byproduct tert-butoxytriisopropylsilanes (tBuO-Si(iPr)3) were detected by GC-MS (Table 1, entry 12).|
|
||
|---|---|---|
| Entry | Variation from standard conditions | Yieldb (%) |
| a Reaction conditions: 1a (0.4 mmol), 2a (0.8 mmol), 2 equiv. of KOtBu in THF (2 mL), 5 h, at 100 °C under Ar. b The yields were determined by GC-MS analysis using an internal standard. c Isolated yield. | ||
| 1 | None | 92 (84c) |
| 2 | Without KOtBu | 0 |
| 3 | KOMe instead of KOtBu | 21 |
| 4 | NaOtBu instead of KOtBu | Trace |
| 5 | LiOtBu instead of KOtBu | 0 |
| 6 | KOH instead of KOtBu | 0 |
| 7 | K3PO4 instead of KOtBu | 0 |
| 8 | Toluene as the solvent | 69 |
| 9 | n Octane as the solvent | 58 |
| 10 | MeCN as the solvent | 0 |
| 11 | DMF as the solvent | Trace |
| 12 | At 80 °C | 64 |

With the optimized conditions in hand, we then evaluated the generality of this method. First, we examined a scope of alkylborons using chlorotriisopropylsilanes or chlorotriethylsilane as coupling partners (Table 2). To our delight, benzyl boronates 1b–1m reacted smoothly with chlorosilanes 2a under standard conditions, affording benzyl silanes 3b–3m in 49–90% yields. For example, secondary boronates 1b and 1c participated in the reactions to obtain corresponding products 3b and 3c in 87% and 68% yields, respectively. Naphthalene substrate 1d was shown to exhibit high levels of reactivity. The products with OMe and OCF3 groups were formed in acceptable yields (3e–3g). It should be mentioned that halo substituents, such as F, Cl, Br and I, on the substrates (1h–1k) did not influence the reaction, indicating their potential application in subsequent cross-coupling transformations. Furthermore, 1l and 1m bearing phosphine or ketone substituents on the phenyl group performed well, affording the corresponding products in 83% and 85% yields (3l and 3m, respectively). Remarkably, this strategy could be successfully applied to the synthesis of geminal silylboronates,16 which exhibit unique reactivity characteristics that allow them to participate in a variety of complexity-generating procedures. A range of carbon chain 1,1-bis(pinacolboronate) esters were tolerated under modified conditions, affording valuable silicated boronates in 70–79% yields (3n–3t). In addition, boronates 1u underwent cross-coupling with chlorotriethylsilane to form highly congested α-quaternary carbon centres 3u in 77% yield. Although oxygen-containing substrates participated in this reaction, their yields were low (42–46%; 3v–3x). Substrates possessing fluorine, chlorine, bromine, alkenyl and naphthyl groups were well tolerated in this system (3y–3ad). Delightedly, the common heterocyclic cores could favourably carry out these deboronative cross-coupling transformations (3ae–3ah). Selective functionalization of 1,2-bis(boronate esters) is a challenging topic in organic synthesis.17 Therefore, we tried the selective cross-coupling reaction with these bis(boronate esters) and chlorosilanes under standard conditions. We found that this cross-coupling exclusively occurs at the benzyl boron, providing 1,2-silylboronates in moderate to good yields (3ai–3ak). Subsequently, 1,1,1-alkyltriboronates and 1,1,2-alkyltriboronates were selectively coupled with chlorosilanes to afford the corresponding products 3al–3ap in 67–89% yield. Most of these silylboronate compounds are difficult to synthesize by known methods. Primary alkylboronates were also examined, but only trace amounts of silanes 3aq were detected by GC-MS.
| a Reaction conditions: 1 (0.4 mmol), 2a (0.8 mmol), 2 equiv. of KOtBu in THF (2 mL), 5 h, at 100 °C under Ar. b Reaction conditions: 1 (0.4 mmol), 2b (0.8 mmol), 2 equiv. of KOtBu in noctane (2 mL), 5 h, at 100 °C under Ar; yields given refer to isolated yields of product. c The reaction was carried out in THF; yields given refer to isolated yields of product. |
|---|

|
Next, we investigated the scope of chlorosilanes for cross-coupling with germinal boronates 1aa. Substrates with different carbon chains were amenable to the cross-coupling reaction as well (3ar–3au). Notably, chlorosilane with a bulky substituent, such as a tert-butyl, showed lower yields (3av). The reaction of aryl chlorosilanes could afford target products 3aw, 3ax and 3ay in satisfactory yields as well. Chlorodisilanes were also feasible substrates with good yields (3az). Additionally, the use of a chloro-containing example afforded product 3aaa in 53% yield. Finally, the presence of alkenyl substituents was tolerated (3aab and 3aac).
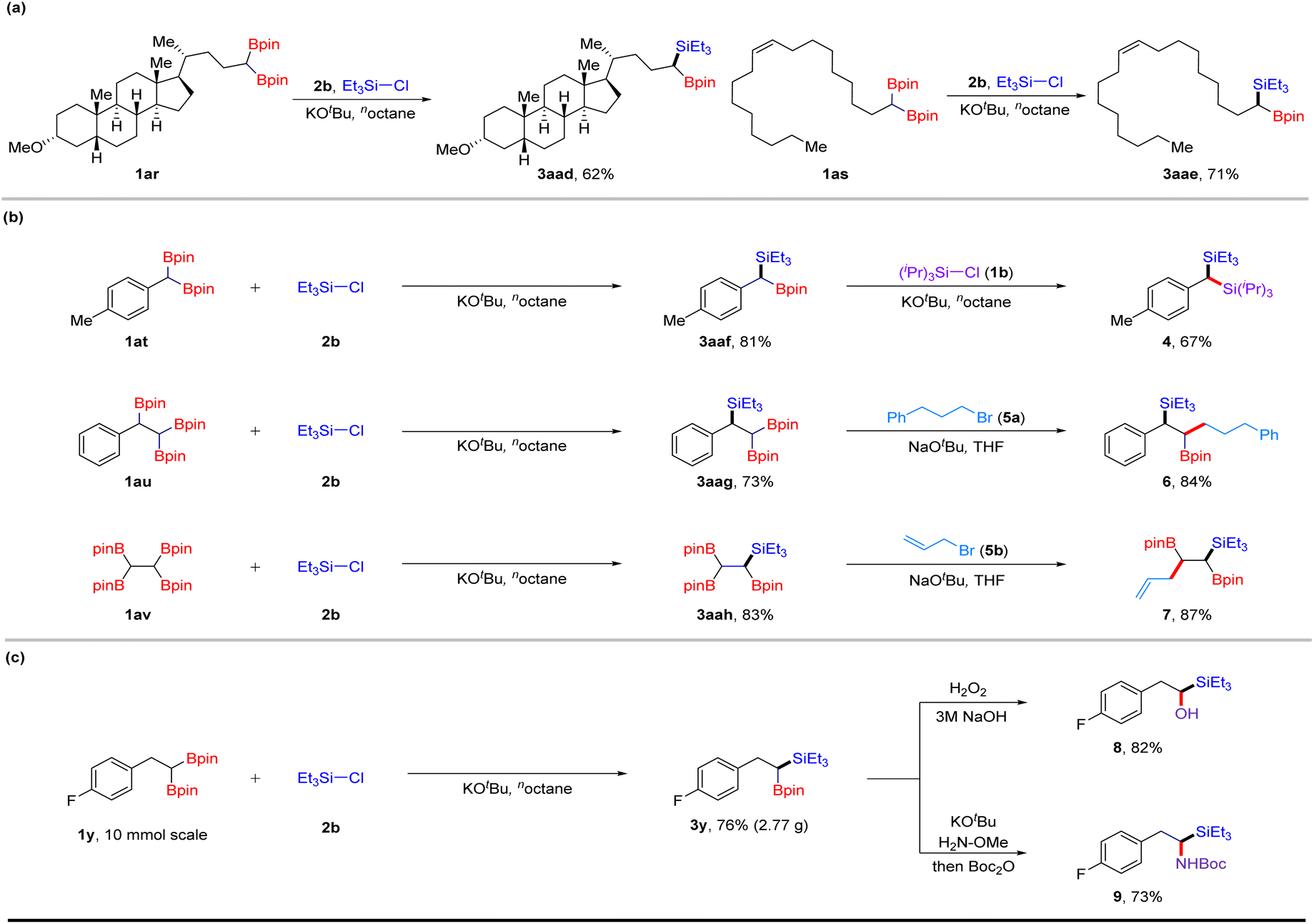
To showcase the synthetic applications of this robust deboronative platform, we performed late-stage modification of complex molecules and a series of sequential transformations (Scheme 1). Lithocholic acid derivative 1ar reacted with Et3SiCl (2b) under optimized conditions, affording product 3aad in 62% yield. Oleic acid derivative 1as bearing a trans-alkene group provided silylboronate 3aae in 71% yield. Interestingly, by continuously treating 1at with chlorosilanes and KOtBu, two deborylative silylations occurred, affording geminal disilanes 4 in 67% yield. When using 1,1,2-alkyltriboronate 1au with two reactive sites as a substrate, the selective process of silylation was observed to give triethyl(1-phenyl-2,2-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)ethyl) silane 3aag, which can further undergo cross-coupling with alkyl bromide 5a under the condition of sodium alkoxide (NaOtBu),18 providing 1,2-silylboronate 6 in 84% yield. Moreover, successive silylation and allylation led to the desired (1,2-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pent-4-en-1-yl)triethylsilane 7 in 87% yield. These results demonstrate the excellent chemoselectivity of this method. A gram–scale reaction proceeded with 1y, affording a 76% isolated yield of silylated product 3y, which can then undergo oxidation or amination processes to afford hydroxyl- or amino-products (8, 9) in good yields, respectively.
 | ||
| Scheme 1 Synthetic applications of the deboronative silylation. | ||
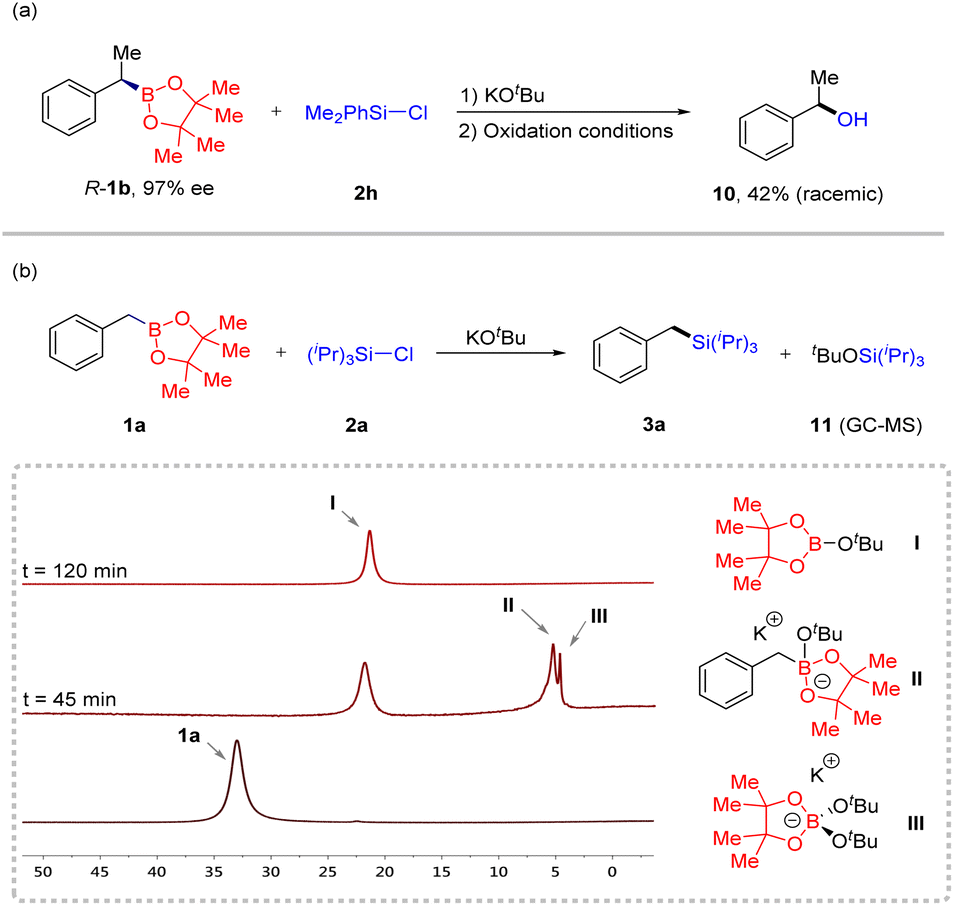
To determine the reaction mechanism, we performed some experiments (Scheme 2). First, when R-1b (97% ee) was reacted with Me2PhSi-Cl 2h under our conditions, racemic compound 10 was observed in 42% yield after oxidation treatment. GC-MS was used to detect the course of the reaction, and we found that byproduct 11 formed. Subsequently, in situ11B NMR of the reaction process was performed using benzyl boron ester 1a and chlorosilane 2a as substrates, and three new resonances (I, II, and III) appeared at 45 minutes. The resonance at 5.1 ppm (II) is assigned to an “ate”complex.19 Over time, this characteristic peak gradually disappeared. Based on these observations, we speculate that this transformation involves two processes. The first step was the generation of a carbanion through a deborylative pathway of an “ate” complex II. The second step was the nucleophilic reaction of the formed alkyl anion with chlorosilane to generate the final product.
 | ||
| Scheme 2 Mechanistic experiments. | ||
The proposed mechanism was further supported by a detailed density functional theory (DFT) study, as shown in Fig. 2. Initially, the boron atom of 1a is coordinated with the oxygen of KOtBu solvated by five tetrahydrofuran molecules, and then the oxygen of 1a replaces one molecule of THF at potassium to generate INT1A with an exothermic energy of 18.3 kcal mol−1. In INT1A, the boron centre is more electron-rich than that in 1a, and the C–B distance is elongated to 1.66 Å from 1.57 Å, decreasing the bond dissociation energy of the C–B bond. Subsequent C–B bond cleavage proceeds smoothly through transition state TS2A with an energy barrier of 6.7 kcal mol−1, in which the potassium cation binds to the aromatic ring of 1a to stabilize the developing negative charge centre and simultaneously release one molecule of THF. The direct cleavage of the C–B bond via transition state TS2B without the assistance of potassium requires a higher activation free energy (25.3 kcal mol−1). The negative charge centre in intermediate INT2A undergoes nucleophilic attack to chlorotriisopropylsilane 2avia transition state TS3A with an activation free energy of 28.9 kcal mol−1, producing the desired alkyl silane 3a and byproduct INT3A. The calculational results indicate that the nucleophilic attack step is the rate-determining step, and the overall activation free energy for this transformation is 31.0 kcal mol−1. The nucleophilic attack step for the alkyl anion released from INT2A must overcome an overall free energy of 35.7 kcal mol−1 through transition state TS3B, which is 4.7 kcal mol−1 higher than that of TS3A. This explains why the deborylative silylation can be promoted by KOMe but failed with sodium alkoxide or lithium alkoxide (Table 1, entries 3–5). Therefore, both the alkoxide anion and potassium cation are indispensable in this transformation, as these entities participate in the formation of a more stable intermediate species, lowering the energy barrier of C–B bond cleavage and C–Si bond formation.
 | ||
| Fig. 2 The calculated energy profiles for the alkoxide-promoted cross-coupling of 1a and 2a. | ||
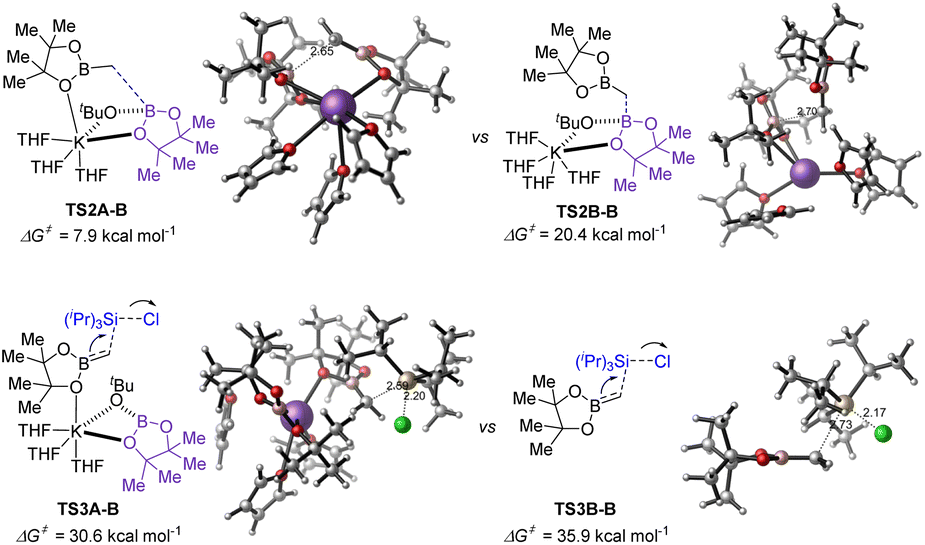
For substrate germinal boronates, the pathways for C–B bond cleavage through transition TS2A-B and the subsequent C–Si bond formation viaTS3A-B have the lower energies (Fig. 3). Comparing the energy barrier of transition state TS3A-B in the rate-determining step, the stepwise SN2 pathway through TS3B-B was found to be disfavoured kinetically and ruled out. Overall, DFT analysis further reveals that the transition states TS2A-B and TS3A-B are substantially stabilized by the coordination between the potassium cation and oxygen of the reserved boronate (see ESI† for details).
 | ||
| Fig. 3 The additional stabilization for substrate germinal boronates. | ||
Conclusions
In summary, we have established a general and practical method for synthesizing alkyl silane derivatives from readily available benzylic boronates, geminal bis(boronates) or alkyltriboronates. This transition-metal-free transformation features excellent chemoselectivity, broad substrate scope, versatility, and scalability. These borylated silanes can readily be further functionalized by well-established organoboron chemistry to enhance the molecular complexity. In view of these features, this transformation should have high synthetic value in the field of materials and pharmaceuticals.Data availability
The synthetic procedures, characterization, and spectral data supporting this article have been uploaded as part of the ESI.†Author contributions
J. H. conceived the project and directed the research. S. J. and Z. S. supervised the mechanistic study. J. H. and Z. S. wrote the paper. M. T., W. Z., J. W. and H. S. performed the experiments. M. W. performed the DFT calculations.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We are grateful for the financial support from the National Natural Science Foundation of China (21901114), the Start-up Grant from Nanjing Tech University (38274017102) and Tang Scholar. We are grateful to the High Performance Computing Center of Nanjing University for doing the numerical calculations in this paper on its blade cluster system.Notes and references
- (a) A. K. Franz and S. O. Wilson, J. Med. Chem., 2013, 56, 388–405 CrossRef CAS PubMed; (b) R. Ramesh and D. S. Reddy, J. Med. Chem., 2018, 61, 3779–3798 CrossRef CAS PubMed.
- (a) R. Pietschnig and S. Spirk, Coord. Chem. Rev., 2016, 323, 87–106 CrossRef CAS; (b) E. A. Marro and R. S. Klausen, Chem. Mater., 2019, 31, 2202–2211 CrossRef CAS.
- (a) F. Zhao, S. Zhang and Z. Xi, Chem. Commun., 2011, 47, 4348–4357 RSC; (b) S. E. Denmark and A. Ambrosi, Org. Process Res. Dev., 2015, 19, 982–994 CrossRef CAS PubMed; (c) T. Komiyama, Y. Minami and T. Hiyama, ACS Catal., 2017, 7, 631–651 CrossRef CAS.
- S. Bähr, W. Xue and M. Oestreich, ACS Catal., 2019, 9, 16–24 CrossRef.
- (a) M. Lacout-Loustalet, J. Dupin, F. Metras and J. Valade, J. Organomet. Chem., 1971, 31, 187–204 CrossRef CAS; (b) P. J. Lennon, D. P. Mack and Q. E. Thompson, Organometallics, 1989, 8, 1121–1122 CrossRef CAS; (c) A. P. Cinderella, B. Vulovic and D. A. Watson, J. Am. Chem. Soc., 2017, 139, 7741–7744 CrossRef CAS PubMed; (d) B. Vulovic, A. P. Cinderella and D. A. Watson, ACS Catal., 2017, 7, 8113–8117 CrossRef CAS PubMed; (e) S. Mallick, E.-U. Würthwein and A. Studer, Org. Lett., 2020, 22, 6568–6572 CrossRef CAS PubMed.
- (a) K. Tamao, A. Kawachi and K. Ito, J. Am. Chem. Soc., 1992, 114, 3989–3990 CrossRef CAS; (b) K. Tamao and A. Kawachi, Adv. Organomet. Chem., 1995, 38, 1–58 CrossRef CAS; (c) C. K. Chu, Y. Liang and G. C. Fu, J. Am. Chem. Soc., 2016, 138, 6404–6407 CrossRef CAS PubMed; (d) W. Xue, R. Shishido and M. Oestreich, Angew. Chem., Int. Ed., 2018, 57, 12141–12145 CrossRef CAS PubMed; (e) R. Chandrasekaran, F. T. Pulikkottil and K. S. Elama, Chem. Sci., 2021, 12, 15719–15726 RSC.
- (a) W. Xue, Z.-W. Qu, S. Grimme and M. Oestreich, J. Am. Chem. Soc., 2016, 138, 14222–14225 CrossRef CAS PubMed; (b) W. Xue and M. Oestreich, Angew. Chem., Int. Ed., 2017, 56, 11649–11652 CrossRef CAS PubMed; (c) Y. Takeda, K. Shibuta, S. Aoki, N. Tohnai and S. Minakata, Chem. Sci., 2019, 10, 8642–8647 RSC; (d) J. Scharfbier, B. M. Gross and M. Oestreich, Angew. Chem., Int. Ed., 2020, 59, 1577–1580 CrossRef CAS PubMed.
- (a) J. Duan, K. Wang, G.-L. Xu, S. Kang, L. Qi, X.-Y. Liu and X.-Z. Shu, Angew. Chem., Int. Ed., 2020, 59, 23083–23088 CrossRef CAS PubMed; (b) L. Zhang and M. Oestreich, Angew. Chem., Int. Ed., 2021, 60, 18587–18590 CrossRef CAS PubMed.
- (a) D. A. Petrone, J. Ye and M. Lautens, Chem. Rev., 2016, 116, 8003–8104 CrossRef CAS PubMed; (b) K. Ouyang, W. Hao, W.-X. Zhang and Z. Xi, Chem. Rev., 2015, 115, 12045–12090 CrossRef CAS PubMed; (c) L.-J. Cheng and N. P. Mankad, Chem. Soc. Rev., 2020, 49, 8036–8064 RSC; (d) R. Sharma and M. R. Yadav, Org. Biomol. Chem., 2021, 19, 5476–5500 RSC; (e) F. Mo, D. Qiu, L. Zhang and J. Wang, Chem. Rev., 2021, 121, 5741–5829 CrossRef CAS PubMed; (f) A. Varenikov, E. Shapiro and M. Gandelman, Chem. Rev., 2021, 121, 412–484 CrossRef CAS PubMed; (g) S. Huang, M. Wang and X. Jiang, Chem. Soc. Rev., 2022, 51, 8351–8377 RSC.
- (a) A. J. L. Lennox and G. C. Lloyd-Jones, Chem. Soc. Rev., 2014, 43, 412–443 RSC; (b) A. Fawcett, J. Pradeilles, Y. Wang, T. Mutsuga, E. L. Myers and V. K. Aggarwal, Science, 2017, 357, 283–286 CrossRef CAS PubMed; (c) J. Hu, Y. Zhao and Z. Shi, Nat. Catal., 2018, 1, 860–869 CrossRef CAS; (d) X. Liu, W. Ming, Y. Zhang, A. Friedrich and T. B. Marder, Angew. Chem., Int. Ed., 2019, 58, 18923–18927 CrossRef CAS PubMed; (e) J. Li, H. Wang, Z. Qiu, C. Y. Huang and C.-J. Li, J. Am. Chem. Soc., 2020, 142, 13011–13020 CrossRef CAS PubMed.
- From boron to other main group elements: (a) C. M. Crudden, B. W. Glasspoole and C. J. Lata, Chem. Commun., 2009, 6704–6716 RSC; (b) D. Leonori and V. K. Aggarwal, Angew. Chem., Int. Ed., 2015, 54, 1082–1096 CrossRef CAS PubMed; (c) T. C. Wilson, T. Cailly and V. Gouverneur, Chem. Soc. Rev., 2018, 47, 6990–7005 RSC.
- From other main group elements to boron: (a) E. C. Neeve, S. J. Geier, I. A. I. Mkhalid, S. A. Westcott and T. B. Marder, Chem. Rev., 2016, 116, 9091–9161 CrossRef CAS PubMed; (b) M. Wang and Z. Shi, Chem. Rev., 2020, 120, 7348–7398 CrossRef CAS PubMed; (c) J. Hu, M. Ferger, Z. Shi and T. B. Marder, Chem. Soc. Rev., 2021, 50, 13129–13188 RSC.
- (a) W. Haubold, J. Herdtle, W. Gollinger and W. Einholz, J. Organomet. Chem., 1986, 315, 1–8 CrossRef CAS; (b) A. A. Kolomeitsev, A. A. Kadyrov, J. Szczepkowska-Sztolcman, M. Milewska, H. Koroniak, G. Bissky, J. A. Bartene and G. RÖschenthaler, Tetrahedron Lett., 2003, 44, 8273–8277 CrossRef CAS; (c) D. L. Crossley, J. Cid, L. D. Curless, M. L. Turner and M. J. Ingleson, Organometallics, 2015, 34, 5767–5774 CrossRef CAS PubMed; (d) A. Lik, L. Fritze, L. Müller and H. Helten, J. Am. Chem. Soc., 2017, 139, 5692–5695 CrossRef CAS PubMed; (e) C. You, M. Sakai, C. G. Daniliuc, K. Bergander, S. Yamaguchi and A. Studer, Angew. Chem., Int. Ed., 2021, 60, 21697–21701 CrossRef CAS PubMed; (f) J. Yao, L. Yu, W. Duan and C.-J. Li, Org. Chem. Front., 2023, 10, 524–530 RSC.
- C. Cheng and J. F. Hartwig, Chem. Rev., 2015, 115, 8946–8975 CrossRef CAS PubMed.
- A. M. Muzafarov, Silicon Polymers, Springer, Heidelberg, 2011 Search PubMed.
- (a) A. A. Szymaniak, C. Zhang, J. R. Coombs and J. P. Morken, ACS Catal., 2018, 8, 2897–2901 CrossRef CAS PubMed; (b) Z. Cheng, J. Guo, Y. Sun, Y. Zheng, Z. Zhou and Z. Lu, Angew. Chem., Int. Ed., 2021, 60, 22454–22460 CrossRef CAS PubMed; (c) Y. You and S. Ge, Angew. Chem., Int. Ed., 2021, 60, 20684–20688 CrossRef CAS PubMed; (d) S. Jin, K. Liu, S. Wang and Q. Song, J. Am. Chem. Soc., 2021, 143, 13124–13134 CrossRef CAS PubMed; (e) W. Sun, L. Xu, Y. Qin and C. Liu, Nat. Synth., 2023, 2, 413–422 CrossRef.
- (a) S. N. Mlynarski, C. H. Schuster and J. P. Morken, Nature, 2014, 505, 386–390 CrossRef CAS PubMed; (b) R. D. Dewhurst and T. B. Marder, Nat. Chem., 2014, 6, 279–280 CrossRef CAS PubMed; (c) L. Xu, S. Zhang and P. Li, Chem. Soc. Rev., 2015, 44, 8848–8858 RSC.
- K. Hong, X. Liu and J. P. Morken, J. Am. Chem. Soc., 2014, 136, 10581–10584 CrossRef CAS PubMed.
- B. Lee and P. J. Chirik, J. Am. Chem. Soc., 2020, 142, 2429–2437 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc02461j |
| This journal is © The Royal Society of Chemistry 2023 |