
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControlling aromatic helix dimerization in water by tuning charge repulsions†
Binhao
Teng
a,
Pradeep K.
Mandal
 a,
Lars
Allmendinger
a,
Céline
Douat
a,
Yann
Ferrand
b and
Ivan
Huc
*a
a,
Lars
Allmendinger
a,
Céline
Douat
a,
Yann
Ferrand
b and
Ivan
Huc
*a
aDepartment of Pharmacy, Ludwig-Maximilians-Universität, Butenandtstr. 5–13, 81377, München, Germany. E-mail: ivan.huc@cup.lmu.de
bUniv. Bordeaux, CNRS, Bordeaux Institut National Polytechnique, CBMN UMR 5248, 2 rue Escarpit, 33600 Pessac, France
First published on 25th September 2023
Abstract
Several helically folded aromatic oligoamides were designed and synthesized. The sequences were all water-soluble thanks to the charged side chains borne by the monomers. Replacing a few, sometimes only two, charged side chains by neutral methoxy groups was shown to trigger the formation of various aggregates which could be tentatively assigned to head-to-head stacked dimers of single helices, double helical duplexes and a quadruplex, none of which would form in organic solvent with organic-soluble analogues. The nature of the aggregates was supported by concentration and solvent dependent NMR studies, 1H DOSY experiments, mass spectrometry, and X-ray crystallography or energy-minimized models, as well as analogies with earlier studies. The hydrophobic effect appears to be the main driving force for aggregation but it can be finely modulated by the presence or absence of a small number of charges to an extent that had no precedent in aromatic foldamer architectures. These results will serve as a benchmark for future foldamer design in water.
Introduction
Molecular aggregation in water often rests on a balance between hydrophobic effects that act as a driving force, and hydrophilic interactions mediated by polar groups without which molecules may not be water-soluble. When hydrophilic groups consist of ions, it is generally beneficial that a net total charge exists. Zwitterions may cancel each other's hydrophilic interactions and tend to be poorly water-soluble. Illustrations of this hydrophobic–hydrophilic balance can be found in objects as diverse as a simple charged surfactant and a polyanion such as DNA. In both cases, one may say that aggregation – micelle formation for the former and double helix association for the latter – occurs ‘despite’ electrostatic repulsions between charges. Tuning these repulsions may in turn result in changes in aggregation properties. For instance, upon screening electrostatic interactions, spherical micelles may transit to cylindrical micelles or bilayer phases.1 Conversely, DNA forms more stable duplexes with its neutral analogue PNA than with itself.2 Here, we show that minor changes in the hydrophobic–hydrophilic balance of helical aromatic foldamers result in great variations of their propensity to form different types of discrete aggregates in water.Aromatic helices are an important class of foldamers with aryl groups in their main chain. They have attracted attention for their ability to serve as scaffolds for molecular recognition either in their interior3–9 or at their surface,10–12 in ion13–15 and water16–18 transport through membranes, in molecular machinery,19,20 and for their charge transport21,22 and chiroptical properties.23,24 Aromatic helix folding may be driven by solvophobic effects,25–31 the presence of neutral or ionic guests,32–36 steric congestion combined with aryl–aryl interactions,37,38 or local conformational preferences.39–43 Aromatic helices may undergo different types of aggregation: stacking into continuous44 or discrete45 tubular objects, side-by-side bundling,46 or intertwining in doubly,9,47–54 triply,36,55,56 or quadruply57,58 stranded structures. Helix aggregation may contribute to the properties mentioned above. For example, double helices may also act as molecular containers.9,19,59,60 Double helix formation has been shown to depend on solvent52,53,61 and on the presence of guests.62,63 A few reports have also highlighted the role of interactions between side chains.50,51 However, aggregation has been investigated mostly in organic solvents. Reports on aggregation in water are few. They include head-to-head stacked dimers of single helices based on quinolinecarboxamide oligomers,45 as well as double helical dimers of some aromatic oligoamides and oligoresorcinols.52–54 Yet none of these systems made use of the hydrophobic–hydrophilic charge balance. We now report that removing as few as two or four out of fourteen positive charges from a helical oligoamide can drive its association into discrete aggregates tentatively assigned to stacked dimers or double helical dimers, respectively (Fig. 1). In a shorter sequence, removing four out of seven charges allows for the formation of a higher aggregate probably corresponding to a tetramer.
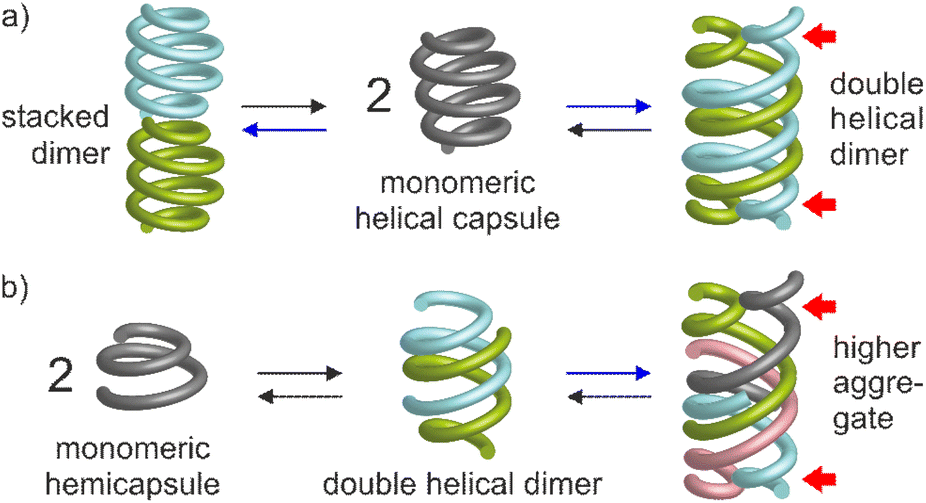 | ||
| Fig. 1 Aggregation modes of aromatic helical capsules (a) and hemicapsules (b) in water reported in this study. Blue arrows point to species found to be favored when decreasing electrostatic repulsions. Red arrows indicate pairs of adjacent bulky single helical segments, an arrangement not seen in organic solvents. | ||
Results and discussion
Design and synthesis
QR and QfR monomers bearing various solubilizing groups R in position 4 have been widely investigated as building blocks of helically folded oligoamides (Fig. 2a).4,39,45,46,53,58,64 In these sequences, folding is driven by local electrostatic attractions and repulsions between the amide NH or CO and neighboring quinoline endocyclic nitrogen atoms or exocyclic fluorine atoms that create a conformational bias at aryl–amide rotatable bonds, resulting in a well-defined curvature of the aromatic strand and eventually promoting a helical shape.41,42 These helices are stable in all kinds of solvents and are particularly stable in water due to hydrophobic effects associated with aromatic stacking.65 We recently introduced QhDap as an analogue of QfDap lacking the conformational control associated with the fluorine atom of Qf, which gives rise to a conformational equilibrium at the 7-carbonylamino-quinoline bond of Qh.66 Using sequence 1, we showed that this additional conformational degree of freedom hampers folding in DMSO but that hydrophobic effects still promote helix folding in water, thus favoring the s-trans conformation of each Qh unit. Sequence 1 comprises a central (QhDap)8 segment flanked with (QDap)3 segments. Qh, like Qf, was expected to generate a wider helix diameter possibly conducive of dimerization into multistranded helices through intertwining of the strands.53,58 In contrast, Qn oligomers possess a narrower diameter and do not form multiple helices. Also, they have been shown to prevent double helix formation of wider helical segments placed between them.4,67 Indeed, such a double helix would have two Qn segments at each end (one at each end of both strands), which would expectedly create steric hindrance and perturb folding.67 As expected, sequence 1 was shown to be a single helix in water.66 It may possess a cavity potentially capable of encapsulating guest molecules, as in designs that we and others have presented previously.4,7,30 Its molecular recognition properties will be described elsewhere. The structure of 1 in solution was elucidated using 1H NMR spectroscopy, but attempts to produce crystals suitable for X-ray diffraction analysis were unsuccessful. We hypothesized that the high charge density at the surface of single helical 1 was, at least in part, responsible for the difficulty to produce crystals. In the context of this work, new sequences 2–5 were thus synthesized as analogues of 1 in which two, four or seven charged side chains were replaced by neutral methoxy groups in the hope to obtain crystals. In the case of 5, the seven remaining charged side chains were all made anionic. As shown in the following, these variations gave rise to various types of unanticipated aggregation behavior (and also to one crystal structure).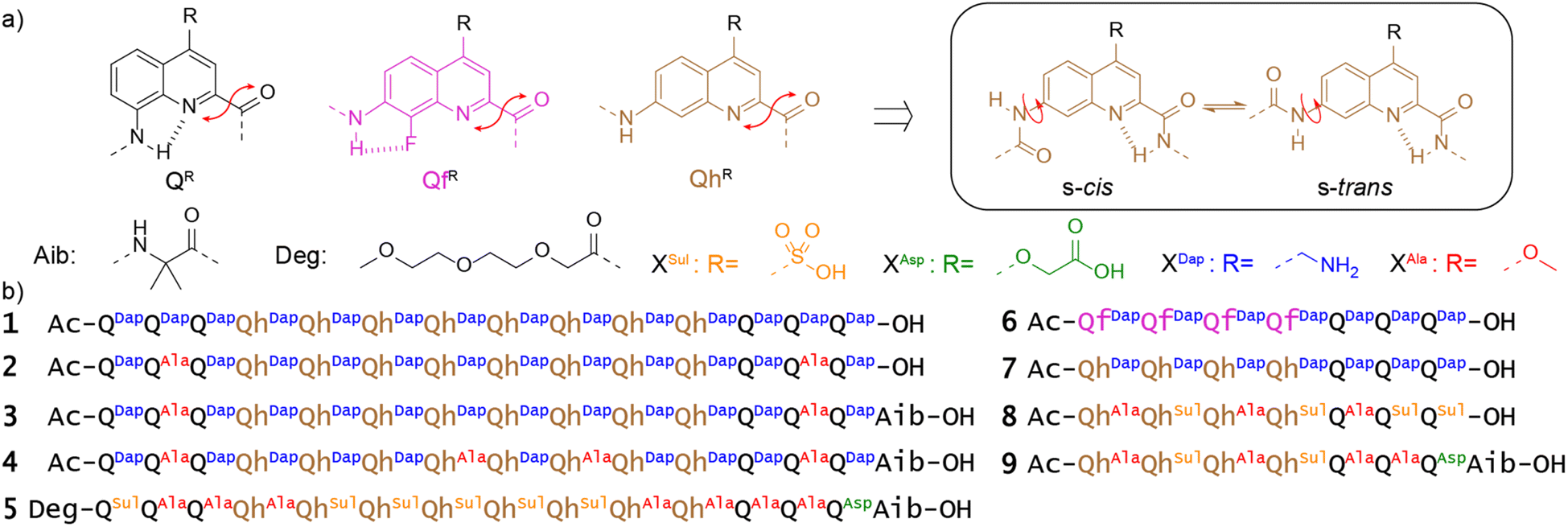 | ||
| Fig. 2 (a) Structure of the building blocks QR, QfR and QhR bearing R side chains in position 4 and composition of sequences 1–9 investigated in this study. Different quinoline monomers and side chains are highlighted with different colors. Monomers: Q: black, Qh: brown, Qf: purple; side chains: (Dap: blue, Ala: red, Sul: gold, Asp: green). XR refers to any QR, QhR or QhR quinoline unit bearing an R side chain. (b) Sequences prepared for this study. | ||
In the meantime, sequences 6–9 were prepared to purposely investigate possible double helix formation. Sequence 6 possesses Qf monomers that encode helicity regardless of the solvent. Analogues of 6 have been shown to form double helical dimers, including in water.53 Indeed, this sequence contains only one Q3 segment and may thus accommodate two of them in a head-to-tail double helical dimer. Because of their additional degree of conformational freedom, it was unknown whether Qh monomers would also undergo double helix formation. Sequence 7, was thus also prepared, that amounts to the seven C-terminal residues of 1. Sequences 8 and 9 are analogues of 7, bearing anionic residues and three or four methoxy side chains.
The preparation of these new sequences entailed the synthesis of two new monomers ready for oligoamide synthesis on solid phase, Fmoc-QhAla-OH and Fmoc-QhSul-OH. Details of their preparation is reported in the ESI (Schemes S1 and S2†). All sequences were synthesized on low-loading Wang or Cl-MPA Protide resins using either manual or automated protocols.68,69
Head-to-head dimerization of single helices
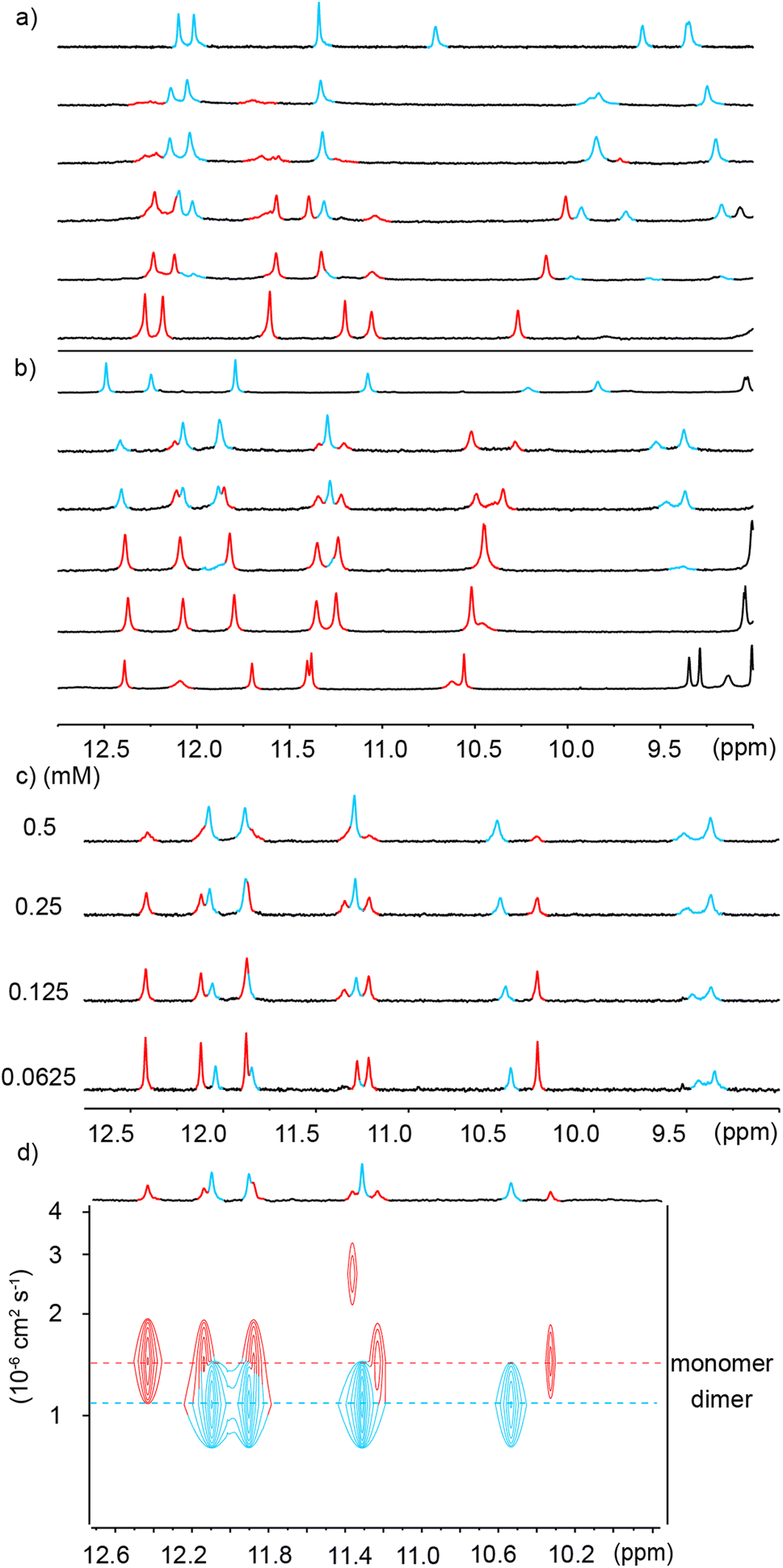
In order to reduce water solubility and promote crystal growth in water, oligomer 2 was produced as an analogue of 1 in which two charged aminomethyl side chains were replaced with neutral and isosteric methoxy groups near the ends. Crystals suitable for X-ray diffraction analysis could not be obtained. In contrast with 1, the 1H NMR spectrum of 2 in water showed two sets of sharp signals, the proportion of which varies with concentration (Fig. 3a), therefore demonstrating a process of reversible aggregation in slow exchange on the NMR timescale. The different sizes of the two species were highlighted by a 1H DOSY experiment clearly showing different hydrodynamic radii (Fig. 3c). The number of resonances indicated that the aggregate is symmetrical, i.e., its helical subcomponents are in the same environment. The signals of the aggregate are upfield-shifted, suggesting that aggregation involved aromatic stacking. Of note, all amide proton resonances shift upon aggregation. The dimeric species [2M + 4H]4+ and [2M + 5H]5+ were detected in the gas phase by ESI-MS (Fig. S1†) indicating that the aggregation is probably a dimerization. Upon integrating the NMR signals of the two species, a dimerization constant of 970 M−1 was calculated.‡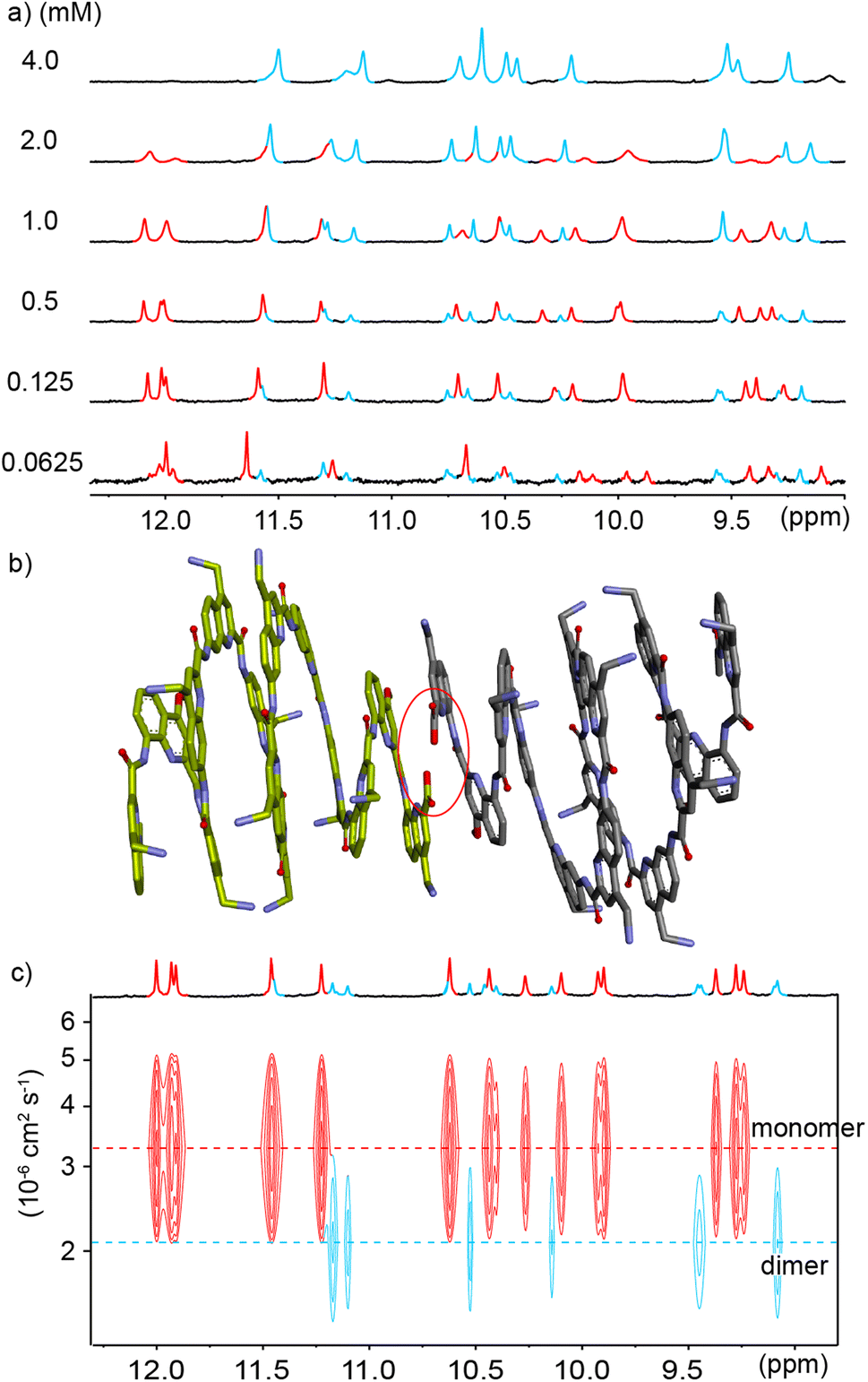 | ||
Fig. 3 (a) Excerpts of the 1H NMR (500 MHz) spectra of compound 2 at different concentrations in 45 mM sodium acetate aqueous buffer pH 4.0 (D2O/H2O, 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :90 v/v). Selected amide proton signals belonging to either the monomeric or dimeric form are highlighted in red and blue, respectively. (b) Energy minimized model (Maestro, MMFFs force field, water as implicit solvent)71 of a dimer of 2 formed by stacking two C-terminal cross-sections. The model is based on a similar head-to-head arrangement previously characterized by NMR and X-ray crystallography.45 The two distinct molecules engaged in binding are colored in light green and gray, respectively. The carboxylic acid and carboxylate involved in intermolecular hydrogen bonding at the C-termini are circled in red. (c) Amide region of the 1H DOSY NMR (500 MHz) spectrum of compound 2 (0.49 mM) in the same solvent as in (a). The two different hydrodynamic radii related to different levels of the signals in the 1H DOSY spectrum are indicated by dashed lines. Signals corresponding to the monomeric or dimeric form are highlighted in red and blue, respectively. :90 v/v). Selected amide proton signals belonging to either the monomeric or dimeric form are highlighted in red and blue, respectively. (b) Energy minimized model (Maestro, MMFFs force field, water as implicit solvent)71 of a dimer of 2 formed by stacking two C-terminal cross-sections. The model is based on a similar head-to-head arrangement previously characterized by NMR and X-ray crystallography.45 The two distinct molecules engaged in binding are colored in light green and gray, respectively. The carboxylic acid and carboxylate involved in intermolecular hydrogen bonding at the C-termini are circled in red. (c) Amide region of the 1H DOSY NMR (500 MHz) spectrum of compound 2 (0.49 mM) in the same solvent as in (a). The two different hydrodynamic radii related to different levels of the signals in the 1H DOSY spectrum are indicated by dashed lines. Signals corresponding to the monomeric or dimeric form are highlighted in red and blue, respectively. | ||
The observations described above are in principle compatible with either the formation of a symmetrical double helix,51,53,70 or with head-to-head stacking of two single helices (Fig. 1a, left) as reported for Qn oligomers bearing other residues.45 Indeed, it has been previously observed that even the stacked dimers give rise to NMR upfield shifts along the entire oligomer and not just for protons near the intermolecular contact area.45 Evidence in favor of a stacked dimer came from the behavior of 3, an analogue of 2 bearing a C-terminal Aib. The 1H NMR spectrum of 3, shows a single set of signals at chemical shift values very similar to those of the monomer of 2 (Fig. S2†) suggesting that the Aib has suppressed dimerization as it does for other oligomers.45 This C-terminal extension would not be expected to much alter double helix formation, but it is not compatible with the stacking of the C-terminal cross-sections of two helices which normally requires direct hydrogen bonding between a carboxylate and a carboxylic acid of the two strands involved (Fig. S3†).45 Furthermore, the ratio of monomer and dimer of 2 was shown to reach equilibrium within minutes after a dilution (Fig. S4†), whereas kinetics would be expected to be much slower – of the order of hours or days – for a long double helix as was previously highlighted for pyridinecarboxamide oligomers.70 Based on these experiments, a plausible energy-minimized model of a stacked dimer of 2 derived from an earlier crystal structure45 was constructed (Fig. 3b). In this model, the quinoline C4 bearing the methoxy residue of each helix lies in the vicinity (4–8 Å) of the C4 of three quinoline residues bearing cationic aminomethyl residues of the other helix. The QAla ⇒ QDap mutation thus removes six potential repulsive electrostatic interactions, explaining why dimerization of 1 is completely suppressed at concentrations appropriate for NMR measurements. These findings extend the formation of stacked Qn helix dimers to cationic sequences.
We also found that dimerization was enhanced by increasing buffer concentration, which we interpreted as the result of an enhanced polarity of the solvent that promotes aggregation via a stronger hydrophobic effects and better screening of charge repulsions. In an attempt to decrease charge repulsion by neutralizing some cationic residues, the pH was raised to 5.6 (sodium acetate buffer, 45 mM), but this led to precipitation and no NMR signals could be recorded.
Double helices may accommodate four Q3 segments
Using a C-terminal Aib extension to avoid head-to-head stacking of single helices, sequence 4 was designed to further reduce its water solubility and favor crystal growth. Thus, two additional positively charged side chains were replaced by two neutral methoxy groups. Unexpectedly, this change led to another kind of aggregation. Despite the Aib termination, the 1H NMR spectrum of compound 4 in water again revealed two sets of sharp signals whose proportions vary with concentration (Fig. 4a). 1H DOSY NMR confirmed that the aggregate – the species more abundant at high concentration – has a larger hydrodynamic radius (Fig. 4b), i.e. a lower diffusion coefficient. Furthermore, dimeric species [2M + 4H]4+ and [2M + 5H]5+ were detected in the gas phase by ESI-MS (Fig. S5†) hinting at a dimerization process again. Integration of the 1H NMR signals allowed for the calculation of a dimerization constant of 32500 M−1 at pH 4.0 in 13.5 mM sodium acetate buffer.‡ The dimerization of 4 was enhanced upon increasing buffer concentration more significantly than the dimerization of 2, (4)2 being formed quantitatively at 0.5 mM in 45 mM sodium acetate while it represents only 76% at the same concentration in 13.5 mM sodium acetate (Fig. 4a).
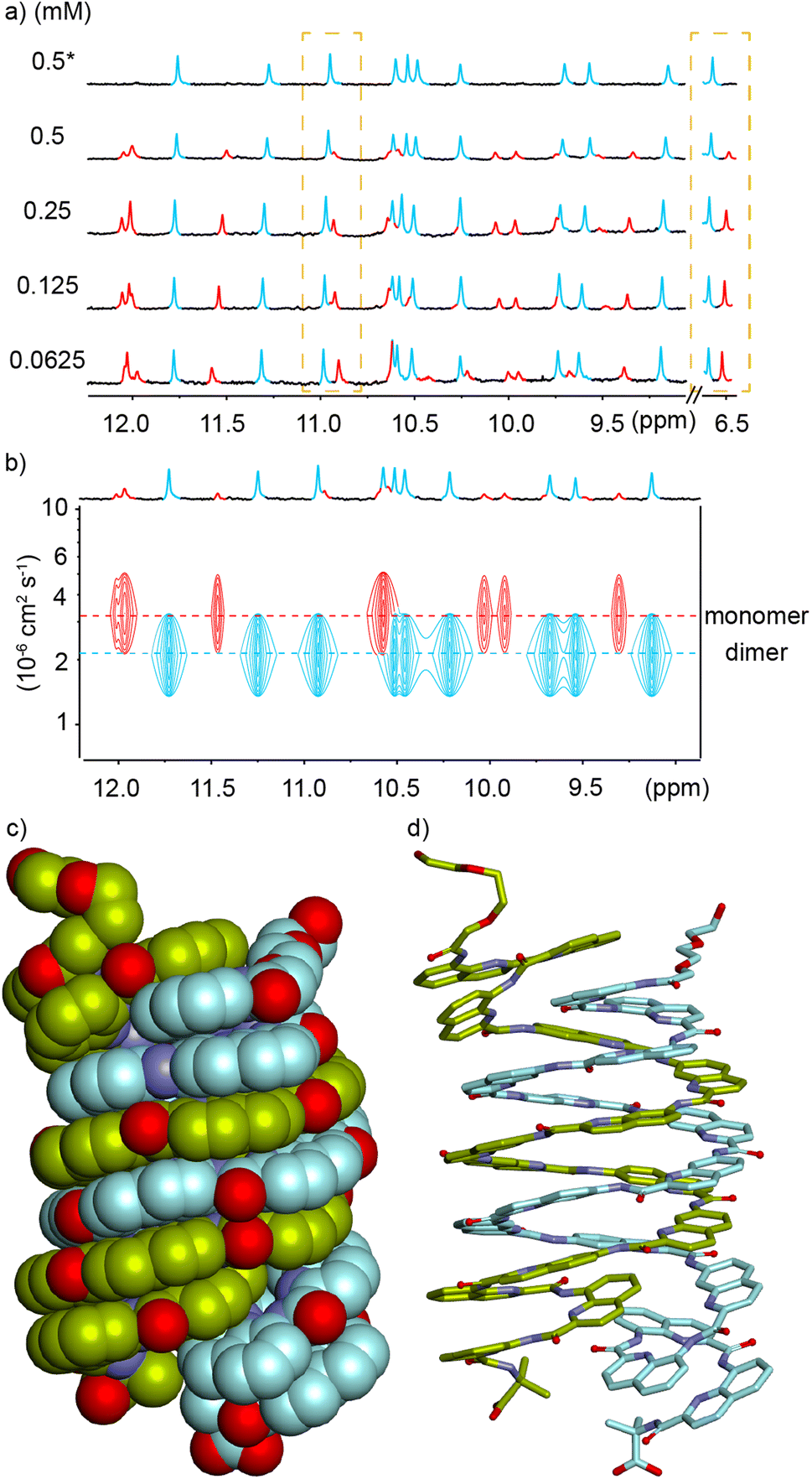 | ||
| Fig. 4 (a) Excerpts of the 1H NMR (500 MHz) spectra of compound 4 at different concentrations in 13.5 mM (* means in 45 mM) sodium acetate aqueous buffer pH 4.0 (D2O/H2O, 10:90 v/v). Selected amide proton signals belonging to either the monomeric or dimeric form are highlighted in red and blue, respectively. Dashed yellow boxes highlight the change of proportion. (b) Amide-region of the 1H DOSY NMR (500 MHz) spectrum of compound 4 (0.5 mM) in 13.5 mM sodium acetate aqueous buffer pH 4.0 (D2O/H2O, 10:90 v/v). The two different levels of signals related to different molecularities in the 1H DOSY spectrum are indicated by dashed lines. Signals corresponding to the monomeric or dimeric form are highlighted in red and blue. Solid-state structure of (5)2: side view shown in (c) CPK and (d) stick representation for different strands. In both representations, two distinct strands are colored in light green or cyan, respectively. Side chains have been omitted for clarity. | ||
The exchange between monomeric and dimeric 4 was found to be much slower than between monomeric and dimeric 2. After a dilution, several hours of incubation were required to reach equilibrium, i.e. until proportions stop evolving (Fig. S6†). This indicates a larger kinetic barrier between the two species, that is, a change in molecular structure more significant than just stacking two single helices. These features, especially the slow kinetics, apparently match with those of the numerous aromatic amide double helices that form double helices in organic solvents70 and may thus hint at a double helix in this case again. However, in all previously described helical aromatic oligoamides, adding a Q3 segment at both ends of the sequence resulted in a complete suppression of double helix formation.67 Furthermore, double helix formation with Qh units was unknown.
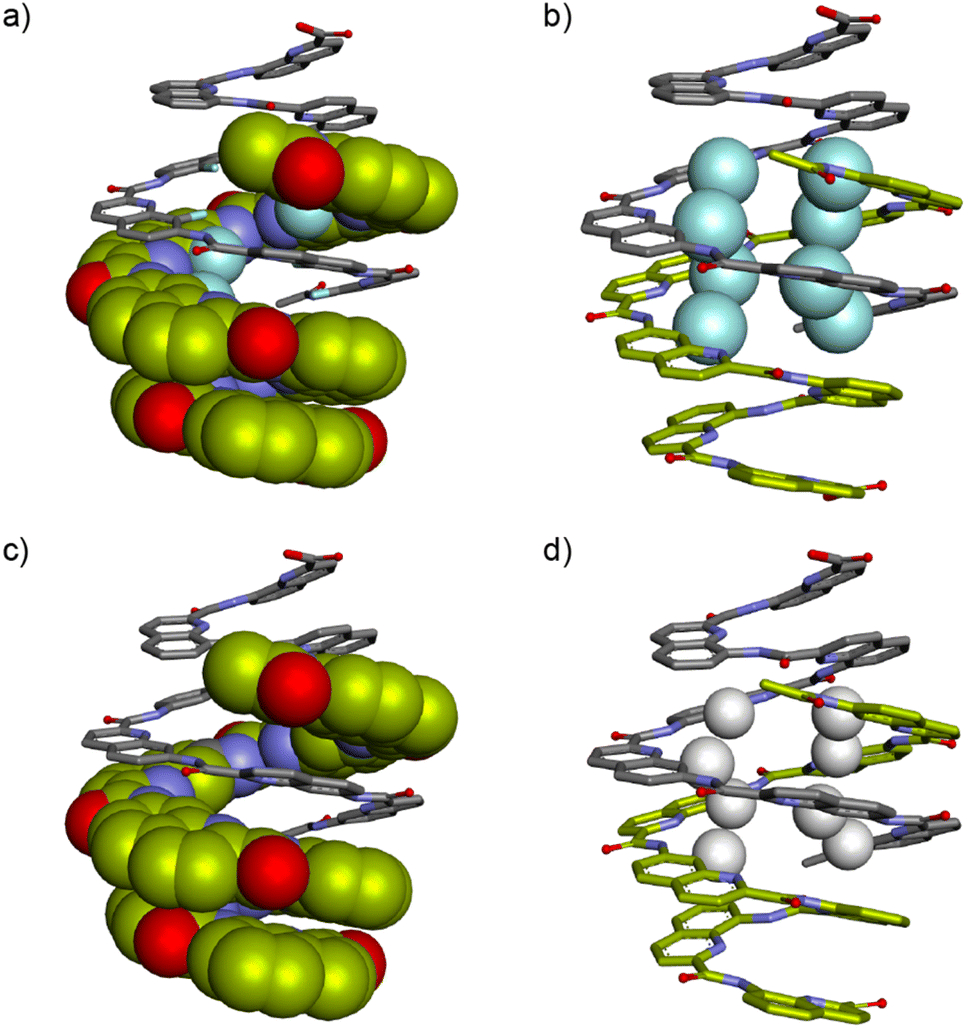
We tried to obtain crystals of 4 to elucidate its structure and aggregation mode but without success, as for all cationic oligomers in our hands so far. We therefore turned to anionic derivatives that have crystallized more readily in other systems68 and prepared oligomer 5. It is important to point out that, over the course of our experiments, this sequence was not conceived as a direct analogue of 4, as it contains more and differently located neutral methoxy side chains. It nevertheless provided a new paradigm to interpret the aggregation of 4. In solution, the mass spectrum indicated the presence of a dimer. The 1H NMR spectrum of 5 showed one species and did not change upon diluting down to 0.03 mM (Fig. S7†). Single crystals of 5 that diffracted at atomic resolution could be obtained, revealing a parallel double helical dimer that forms despite the presence of Q3 segments at each end of the two strands (Fig. 4c and S8†).
The double helix of (5)2 only involves the central Qh8 segments and spans two full turns. The two strands are offset by half a turn and engage in extensive intermolecular aromatic stacking. The four Q3 segments are all single helical, as expected, and the steric crowd that they create is somehow accommodated. For that, two outer Q3 segments are slightly tilted away from the duplex and overlap with two inner Q3 segments. This arrangement suggests that segments having more than three Q units may not be accommodated as easily, but this hypothesis was not tested. The structure also shows that terminal Aib units play no role in aggregation. A molecular model of the single helical monomer of 5 was built (Fig. S9†) and its solvent accessible surface was calculated to be 1790 Å2, compared to 2690 Å2 for the duplex solid state structure. Thus, the duplex surface amounts to about 75% of the surface of two monomers, giving a measure of how much is hidden from water upon dimerization. The structure of (5)2 shares a key design feature with 1–4 and with other helical capsules:4 it possesses a sizeable cavity surrounded by the helix backbone (350 Å3 in this case, see Fig. S8†). The solid state structure of (5)2 thus represents the first example of a water-soluble helical capsule crystal structure.
Despite the differences between 4 and 5, we think it reasonable to propose that the crystal structure of (5)2 informs about the aggregation mode of 4 in that it shows a type of dimerization previously thought to be disfavored and that is compatible with the solution behavior of 4 (Fig. 1a, right). Of note, the double helix (5)2 is parallel and lacks any symmetry. Its two strands are inequivalent. In contrast, the number of 1H NMR resonances of (4)2 suggests a symmetrical structure (Fig. 4a). Dimer (4)2 may instead be a symmetrical anti-parallel dimer, or perhaps its NMR spectrum represents an average between two degenerate parallel conformations.
To conclude this section, it is again remarkable that differences as small as those that exist between 3 and 4 – minus two charged residues – can trigger such a drastic change of aggregation behavior, presumably towards an unexpected double helical dimer. We also infer that the monomeric nature of 1 and 3 is not just the result of the bulky terminal Q3 segments, but also of charge repulsions within double helical dimers.
Hemicapsule dimerization
To explore further the modes of dimerization of helically folded aromatic oligoamides in water, several hemicapsule sequences were synthesized. The term hemicapsule holds for sequences such as 6–9 because they bear only one Q3 segment at one end of their sequence. Contrary to the capsules, which we initially expected to be monomeric, hemicapsules were expected to aggregate into antiparallel double helices with one bulky Q3 segment at each end of the duplex (Fig. 1b, left). Several such anti-parallel double helices of hemicapsules have been characterized in the solid state with organic-soluble sequences.73,74 The parallel duplexes where both Q3 segments would be at the same end have never been observed when this situation can be avoided. Furthermore, a sequence differing from 6 only by the length of the cationic side chains has already been shown to form a double helical dimer in water while it is monomeric in DMSO.53The behavior of 6 was consistent with these earlier results. Upon adding water to a DMSO-d6 solution of 6, a species assigned to a single helical monomer in DMSO is replaced by a new species assigned to a double helical dimer (Fig. 5a). In the gas phase, ESI-MS showed a substantial population of [2M + 2H]2+ and [2M + 3H]3+ dimers (Fig. S10†). The dimerization constant of 6 was too high to observe the monomer by 1H NMR upon diluting a solution in water (Fig. S11†). Also, given the multiplicity of NMR signals, one can infer that (6)2 has a symmetrical structure and propose a molecular model as shown in Fig. 6a. This model closely matches with a solid state structure we recently obtained for a related sequence in the context of another project and that unambiguously established the antiparallel nature of the duplex. This structure will be published elsewhere.
 | ||
| Fig. 5 Excerpts of the 1H NMR (500 MHz) spectra at 298 K of 6 (a) and 7 (b) in DMSO-d6/H2O mixtures. The percentage of DMSO is from top to bottom: (a) 0, 50, 66, 70, 76 and 100; and (b) 0, 33, 40, 50, 57 and 100. Note: 0% DMSO means the spectrum was recorded in H2O/D2O (90:10 v/v). Red and blue amide proton signals stand for either the monomeric or dimeric form, respectively. (c) Excerpts of the 1H NMR spectra of 7 at different concentrations in DMSO-d6/H2O (40:60 v/v). Selected amide proton signals belonging to either the monomeric or dimeric form are highlighted in red and blue, respectively. (d) Amide NH region of the 1H DOSY NMR (500 MHz) spectrum of 7 (0.32 mM) in DMSO-d6/H2O (40:60 v/v). The two different levels of signals related to different hydrodynamic radii are indicated by dashed lines. Signals corresponding to the monomeric or dimeric form are highlighted in red and blue. | ||
 | ||
| Fig. 6 Energy minimized models (Maestro, MMFFs force field, water as implicit solvent)71 of (6)2 (a and b) and (7)2 (c and d). In (a) and (c), one strand is shown in space filling representation and the other in stick representation. In (b) and (d), both strands are shown in stick representation but the fluorine atoms in position 8 of Qf units and the hydrogen atoms in position 8 of Qh units are shown in space filling representations and colored in cyan and white, respectively. In all representations two distinct strands are colored in light green or gray, respectively. Side chains and most hydrogen atoms have been omitted for clarity. | ||
Similar experiments were carried out using analogous sequence 7 in which Qf units were replaced by Qh units, a change that should provide additional degrees of conformational freedom. Thus, a similar transition as for 6 was observed upon adding water to a DMSO-d6 solution of 7 (Fig. 5b). That the two species correspond to an aggregation phenomenon was ascertained by a 1H DOSY NMR experiment and from the concentration dependence of the proportion of the two species (Fig. 5c and d) in DMSO-d6/H2O (40:60 v/v). A dimerization constant of 104 M−1 in this solvent could be calculated. ESI-MS confirmed the presence of [2M + 2H]2+ and [2M + 3H]3+ dimeric species in the gas phase (Fig. S12†). Again, no dissociation was observed upon diluting a solution of 7 in water (Fig. S13†). Nevertheless, less DMSO is required to disrupt (7)2 than to disrupt (6)2 (Fig. 5a and b). In short, Qh units also promote double helix formation, albeit slightly less efficiently than Qf monomers. The model shown in Fig. 6b can thus be proposed for the structure of (7)2. The dimeric segment involves eight Qh units, allowing some cooperative solvophobic folding to occur. In contrast, the four units of monomeric 7 cannot in principle undergo cooperative folding because they do not span a full helix turn. Estimations of the solvent-accessible surface of (7)2 and of different conformations of monomeric 7 suggest that the surface of a dimer amounts to 61 to 71% of the surface of two monomers (Fig. S14†), shedding light on the significant hydrophobic effects observed. Interestingly, when mixing 6 and 7 in water, only homodimers (6)2 and (7)2 were observed (Fig. S15 and S16†), indicating a sort of narcissistic self-sorting that contrasts with heterodimerization observed in other systems.67,72
Double helices may also aggregate
In view of the success at obtaining crystals of anionic sequences, oligomer 8 was introduced as an analogue of 7. Both compounds have the same Qh4Q3 main chain. In 8, the cationic side chains have been replaced by methoxy and sulfonic acid side chains. Some methoxy side chains were introduced because the exceedingly high water solubility of sequences bearing sulfonic acid chains on every residues has been shown to hamper crystallization.53 We found that the neutral methoxy groups did not alter aggregation properties and that 8 behaved like 7 in pure water (Fig. S17†) and in water/DMSO-d6 mixtures (Fig. S18†). [2M – 3H]3− and [2M – 2H]2− dimeric species could be observed by ESI-MS in the gas phase (Fig. S19†). Like all other Qf4Q3 and Qh4Q3 sequences, 8 dimerizes, a priori into an anti-parallel double helix. However, sequence 9 in which one more sulfonic acid has been replaced by a methoxy group exhibited a new behavior.§Unlike 6, 7 and 8, the 1H NMR spectrum of 9 in water showed two sets of signals (Fig. 7a). Upon adding water to a DMSO-d6 solution of 9, a first transition occurred near 30% of water, which we assigned to the expected monomer-to-double-helical-dimer transition, and a second transition occurred when reaching 100% water (Fig. 7a). All three species are in slow exchange on the NMR times scale. The proportion of the two species present in water was shown to depend on concentration: the relative intensity of the signals assigned to the double helical dimer decreases when concentration increases (Fig. 7b), indicating that the new species is an aggregate of higher molecularity than a dimer. The conversion is quantitative at 5 mM. 1H DOSY NMR confirmed that the higher aggregate has a larger hydrodynamic radius, i.e. a lower diffusion coefficient (Fig. 7c). Native ESI-MS analysis could only be performed at concentrations too low for the higher aggregate to be present in significant amount and only the [2M − 2H]2− dimeric species was detectable in the gas phase (Fig. S20†). MS thus did not allow to establish the molecularity of the higher aggregate. However, information may be inferred from the number of 1H NMR resonances. This number is double the number of resonances of (9)2, meaning the aggregate possesses two inequivalent strands in equal proportions. This rules out a possible trimer. Indeed, a triple helix may be fully parallel with three equivalent strands, or partly parallel with one strand antiparallel to the two others which results into three inequivalent strands. We have previously characterized these two states of triple helices using both NMR and crystallography for aggregates formed in organic solvents.55 The higher aggregate of 9 therefore possesses an even number of strands. The fact that quadruple aromatic amide helices have already been characterized in organic solvents and crystallized57,58 makes a tetramer (9)4 the most likely possibility, with two strands pointing in a direction and two others in the other direction (Fig. 1b, right).
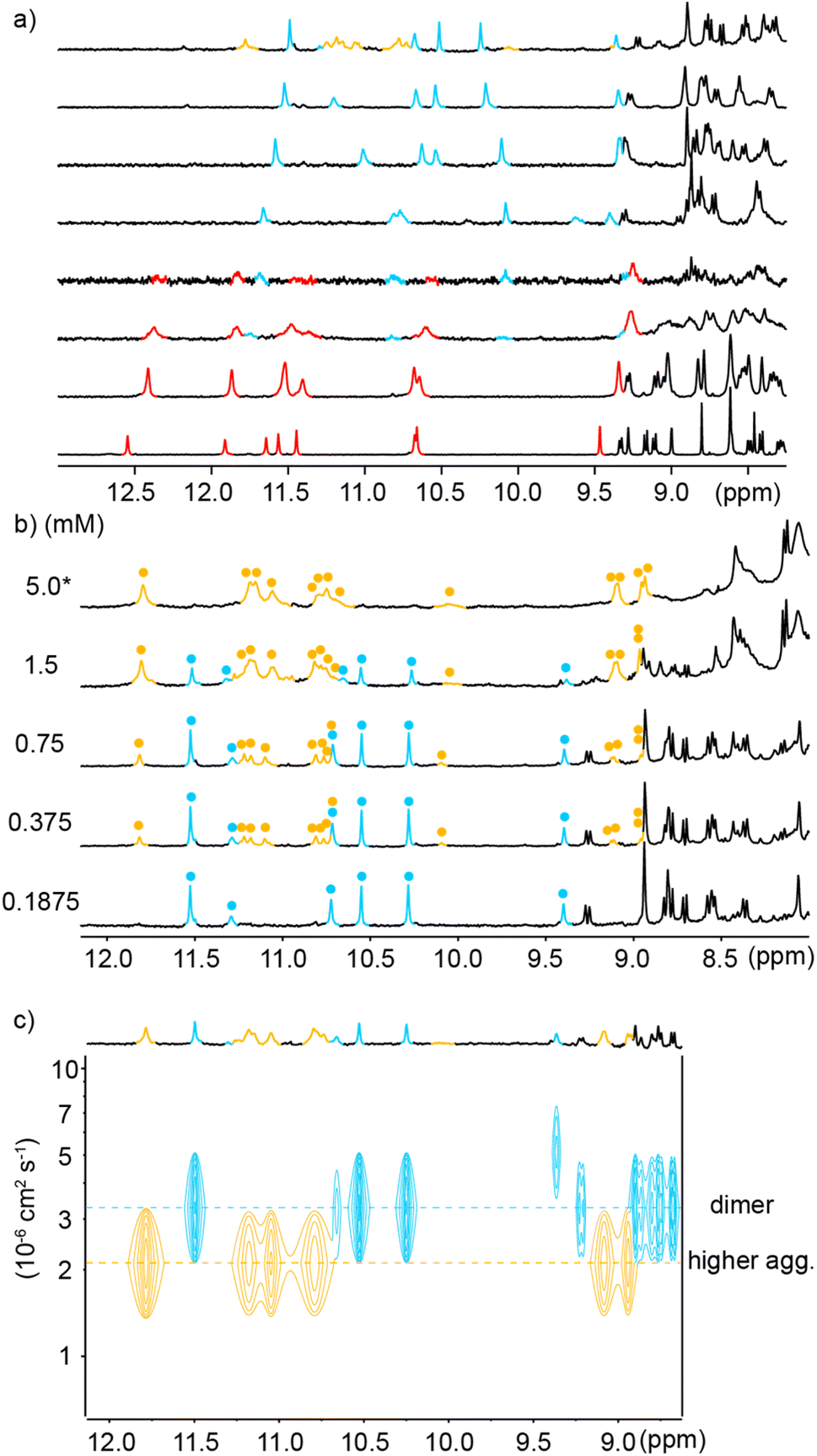 | ||
| Fig. 7 Excerpts of the 1H NMR (500 MHz) titration at 298 K of 9 (0.72 mM) in DMSO-d6/buffer mixtures in the following DMSO percentage (top to bottom): (a) 0, 16.7, 50, 66.6, 72.5, 76.9, 83.3 and 100. Note: buffer means 13 mM ammonium acetate aqueous buffer pH 8.5. 0% DMSO means 1H NMR (500 MHz) spectrum of 9 in buffer (buffer/D2O 90:10 v/v). Red, blue and yellow amide proton signals stand for the monomeric, dimeric and tetrameric form, respectively. (b) Excerpts of the 1H NMR (500 MHz) spectra of compound 8 at different concentrations in 11.7 mM (* means in 47.7 mM) ammonium acetate aqueous buffer pH 8.5. Selected amide proton signals belonging to either the dimeric or tetrameric form are highlighted in blue and yellow, respectively. (c) Amide-region of the 1H DOSY NMR (500 MHz) spectrum of compound 9 (1 mM) in 11.7 mM ammonium acetate aqueous buffer pH 8.5. The two different levels of signals related to different molecularities in the 1H DOSY spectrum are indicated by dashed lines. Signals corresponding to the dimeric or tetrameric form are highlighted in blue and yellow, respectively. | ||
The time required to form (9)4 after dissolving 9 in water was found to be of the order of hours (Fig. S21†), indicating that significant structural changes are required. A plausible molecular model for (9)4 was proposed based on the structures of quadruple helices crystallized from organic solvents57,58 and on the structure of (5)2 (Fig. 8) which shows how to accommodate two Q3 segments at the same end of a multiple helix. Thus, taking the C-terminal half of (5)2 (bottom part of Fig. 4c), a parallel duplex (9)2 can be proposed distinct from the anti-parallel duplex that prevails at lower concentration and that would be similar to the structure shown in Fig. 6c and d. Next, two of these parallel duplexes can be intertwined in an antiparallel fashion through their N-terminal Qh4 segments to produce the C2-symmetric quadruplex shown in Fig. S7† having two pairs of inequivalent strands. This model is of course speculative. It nevertheless allows pointing to some factors that could play a role: its solvent-accessible surface is smaller than that of two anti-parallel dimers (Fig. S22†); removing one charge from a monomer means removing four charges from the tetramer; two methoxy groups of 9 that replaced sulfonic acids of 8 are found to be buried between two hydrophobic aromatic surfaces pointing to the destabilization of such a structure in the case of 8. Besides electrostatic effects between charges, the fact that an electron withdrawing group in 8 has been replaced by a donor in 9 might also influence interactions between aryl rings but this is hard to estimate. Single crystals of 9 have been obtained that diffracted up to 1.5 Å but no structure could be solved so far.
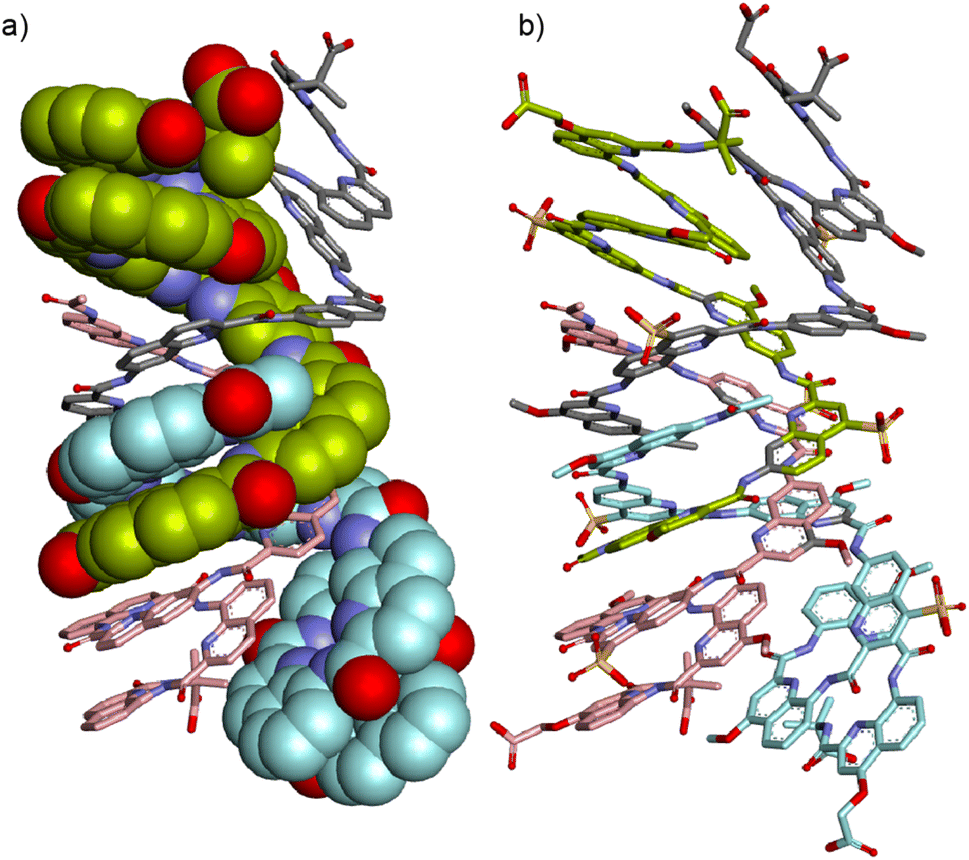 | ||
| Fig. 8 Energy minimized model (Maestro, MMFFs force field, water as implicit solvent)71 of (9)4: (a) side view with two strands in space-filling representation and two strands in stick representation. Side chains and hydrogen atoms have been omitted for clarity. (b) Same side view including the side chains with all four strands in stick representation. In all representations, four distinct strands are colored in light green, gray, cyan and pink, respectively. The symmetry of the structure is such that gray and cyan strands on one hand and light green and pink strands on the other hand, are equivalent. | ||
Conclusions
We have shown that the formation of at least three different types of discrete aggregates of helical aromatic foldamers can be triggered in water by minor changes in side chain properties. These changes always consisted in replacing a small number of charged residues, sometimes only one, by a neutral methoxy group. Specifically, a head-to-tail stacked dimer of two single helices, a dimer tentatively assigned to a double helical duplex with four terminal Q3 segments, and a higher aggregate tentatively assigned to a quadruplex were shown to form whereas related sequences containing a few more charged residues did not produce such objects. The last two aggregates are new in that they comprise two Q3 segments at each end of a multistranded object. All aggregates are kinetically stable. They are in slow exchange with their subcomponents on the NMR timescale. In the case of the double helix and the quadruplex, kinetic stability is high and equilibrium may take hours to be reached. Hydrophobic effects appear to be a major driving force for aggregation, but these effects are counterbalanced by the side chain charges. Earlier observations in organic solvents had shown that Q3 segments prevent the formation of some types of multistranded helices. We now have shown that this effect can be overcome in water unless sufficient charge repulsions are also at play. Overall, these results provide a foundation for future aromatic foldamer structure design in water. In addition, the monomeric and multimeric species presented here possess cavities in which guest molecules may be recognized, and secluded from the surrounding aqueous environment. These cavities, in particular those generated by more than one strand, may undergo rearrangements triggered by external stimuli and exhibit controlled release or guest swapping properties. Progress in this direction is being made and will be reported in due course.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Author contributions
BT synthesized the monomers. BT and CD synthesized the oligomeric sequences. LA and BT performed solution studies. PKM grew crystals, collected diffraction data and solved the solid state structure. YF and IH supervised the research. BT and IH wrote the manuscript. All authors proofread and improved the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the DFG (project HU 1766/6-1). B. Teng gratefully acknowledges financial support from the China Scholarship Council. We thank D. Gill and E. Merlet for contributing some synthetic precursors and G. Leonard (ID30B, ESRF) for assistance during data collection at the Synchrotron beamline.References
- D. Lombardo, M. A. Kiselev, S. Magazù and P. Calandra, Adv. Condens. Matter Phys., 2015, 2015, e151683 CrossRef.
- P. E. Nielsen, M. Egholm, R. H. Berg and O. Buchardt, Science, 1991, 254, 1497–1500 CrossRef CAS PubMed.
- R. B. Prince, S. A. Barnes and J. S. Moore, J. Am. Chem. Soc., 2000, 122, 2758–2762 CrossRef CAS.
- Y. Ferrand and I. Huc, Acc. Chem. Res., 2018, 51, 970–977 CrossRef CAS PubMed.
- H. Juwarker and K.-S. Jeong, Chem. Soc. Rev., 2010, 39, 3664–3674 RSC.
- J.-L. Hou, X.-B. Shao, G.-J. Chen, Y.-X. Zhou, X.-K. Jiang and Z.-T. Li, J. Am. Chem. Soc., 2004, 126, 12386–12394 CrossRef CAS PubMed.
- W. Wang, C. Zhang, S. Qi, X. Deng, B. Yang, J. Liu and Z. Dong, J. Org. Chem., 2018, 83, 1898–1902 CrossRef CAS PubMed.
- Y. Liu, F. C. Parks, E. G. Sheetz, C.-H. Chen and A. H. Flood, J. Am. Chem. Soc., 2021, 143, 3191–3204 CrossRef CAS PubMed.
- Y. Zhong, T. A. Sobiech, B. Kauffmann, B. Song, X. Li, Y. Ferrand, I. Huc and B. Gong, Chem. Sci., 2023, 14, 4759–4768 RSC.
- J. Ahmed, T. C. Fitch, C. M. Donnelly, J. A. Joseph, T. D. Ball, M. M. Bassil, A. Son, C. Zhang, A. Ledreux, S. Horowitz, Y. Qin, D. Paredes and S. Kumar, Nat. Commun., 2022, 13, 2273 CrossRef CAS PubMed.
- K. Ziach, C. Chollet, V. Parissi, P. Prabhakaran, M. Marchivie, V. Corvaglia, P. P. Bose, K. Laxmi-Reddy, F. Godde, J.-M. Schmitter, S. Chaignepain, P. Pourquier and I. Huc, Nat. Chem., 2018, 10, 511–518 CrossRef CAS PubMed.
- S. Kumar, A. Henning-Knechtel, I. Chehade, M. Magzoub and A. D. Hamilton, J. Am. Chem. Soc., 2017, 139, 17098–17108 CrossRef CAS PubMed.
- J. Shen, R. Ye, Z. Liu and H. Zeng, Angew. Chem., Int. Ed., 2022, 61, e202200259 CrossRef CAS PubMed.
- L. Zhang, C. Zhang, X. Dong and Z. Dong, Angew. Chem., Int. Ed., 2023, 135, e202214194 CrossRef.
- X. Wei, G. Zhang, Y. Shen, Y. Zhong, R. Liu, N. Yang, F. Y. Al-mkhaizim, M. A. Kline, L. He, M. Li, Z.-L. Lu, Z. Shao and B. Gong, J. Am. Chem. Soc., 2016, 138, 2749–2754 CrossRef CAS PubMed.
- Y. Shen, F. Fei, Y. Zhong, C. Fan, J. Sun, J. Hu, B. Gong, D. M. Czajkowsky and Z. Shao, ACS Cent. Sci., 2021, 7, 2092–2098 CrossRef CAS PubMed.
- A. Roy, J. Shen, H. Joshi, W. Song, Y.-M. Tu, R. Chowdhury, R. Ye, N. Li, C. Ren, M. Kumar, A. Aksimentiev and H. Zeng, Nat. Nanotechnol., 2021, 16, 911–917 CrossRef CAS PubMed.
- D.-D. Su and M. Barboiu, CCS Chem., 2022, 5, 279–291 CrossRef.
- X. Wang, B. Wicher, Y. Ferrand and I. Huc, J. Am. Chem. Soc., 2017, 139, 9350–9358 CrossRef CAS PubMed.
- M. Gauthier, V. Koehler, C. Clavel, B. Kauffmann, I. Huc, Y. Ferrand and F. Coutrot, Angew. Chem., Int. Ed., 2021, 60, 8380–8384 CrossRef CAS PubMed.
- A. Méndez-Ardoy, N. Markandeya, X. Li, Y.-T. Tsai, G. Pecastaings, T. Buffeteau, V. Maurizot, L. Muccioli, F. Castet, I. Huc and D. M. Bassani, Chem. Sci., 2017, 8, 7251–7257 RSC.
- X. Li, N. Markandeya, G. Jonusauskas, N. D. McClenaghan, V. Maurizot, S. A. Denisov and I. Huc, J. Am. Chem. Soc., 2016, 138, 13568–13578 CrossRef CAS PubMed.
- K. Moreno, E. Merlet, N. McClenaghan, T. Buffeteau, Y. Ferrand and C. Olivier, ChemPlusChem, 2021, 86, 496–503 CrossRef CAS PubMed.
- D. Zheng, L. Zheng, C. Yu, Y. Zhan, Y. Wang and H. Jiang, Org. Lett., 2019, 21, 2555–2559 CrossRef CAS PubMed.
- X. Hu, A. Schulz, J. O. Lindner, M. Grüne, D. Bialas and F. Würthner, Chem. Sci., 2021, 12, 8342–8352 RSC.
- J. C. Nelson, J. G. Saven, J. S. Moore and P. G. Wolynes, Science, 1997, 277, 1793–1796 CrossRef CAS PubMed.
- H. Goto, H. Katagiri, Y. Furusho and E. Yashima, J. Am. Chem. Soc., 2006, 128, 7176–7178 CrossRef CAS PubMed.
- K. P. de Carvasal, N. Aissaoui, G. Vergoten, G. Bellot, J.-J. Vasseur, M. Smietana and F. Morvan, Chem. Commun., 2021, 57, 4130–4133 RSC.
- Y. Zhao and J. S. Moore, in Foldamers, John Wiley & Sons, Ltd, 2007, pp. 75–108 Search PubMed.
- Y. Hua, Y. Liu, C.-H. Chen and A. H. Flood, J. Am. Chem. Soc., 2013, 135, 14401–14412 CrossRef CAS PubMed.
- K. P. Divya, S. Sreejith, C. H. Suresh, D. S. Philips and A. Ajayaghosh, Chem.–Asian J., 2013, 8, 1579–1586 CrossRef CAS PubMed.
- K.-J. Chang, B.-N. Kang, M.-H. Lee and K.-S. Jeong, J. Am. Chem. Soc., 2005, 127, 12214–12215 CrossRef CAS PubMed.
- H. Juwarker and K.-S. Jeong, Chem. Soc. Rev., 2010, 39, 3664–3674 RSC.
- V. R. Naidu, M. C. Kim, J. Suk, H.-J. Kim, M. Lee, E. Sim and K.-S. Jeong, Org. Lett., 2008, 10, 5373–5376 CrossRef CAS PubMed.
- E. A. John, C. J. Massena and O. B. Berryman, Chem. Rev., 2020, 120, 2759–2782 CrossRef CAS PubMed.
- C. J. Massena, D. A. Decato and O. B. Berryman, Angew. Chem., Int. Ed., 2018, 57, 16109–16113 CrossRef CAS PubMed.
- C. S. Hartley, Acc. Chem. Res., 2016, 49, 646–654 CrossRef CAS PubMed.
- S. Peddi, M. C. Bookout, G. N. Vemuri and C. S. Hartley, J. Org. Chem., 2022, 87, 3686–3690 CrossRef CAS PubMed.
- H. Jiang, J.-M. Léger and I. Huc, J. Am. Chem. Soc., 2003, 125, 3448–3449 CrossRef CAS PubMed.
- X. Yang, A. L. Brown, M. Furukawa, S. Li, W. E. Gardinier, E. J. Bukowski, F. V. Bright, C. Zheng, X. C. Zeng and B. Gong, Chem. Commun., 2003, 56–57 RSC.
- A. M. Abramyan, Z. Liu and V. Pophristic, Chem. Commun., 2015, 52, 669–672 RSC.
- Z. Liu, A. M. Abramyan and V. Pophristic, New J. Chem., 2015, 39, 3229–3240 RSC.
- L. Yuan, H. Zeng, K. Yamato, A. R. Sanford, W. Feng, H. S. Atreya, D. K. Sukumaran, T. Szyperski and B. Gong, J. Am. Chem. Soc., 2004, 126, 16528–16537 CrossRef CAS PubMed.
- V. Maurizot, J. Duhamel, I. Huc, B. Wicher, K. Lulic, J. Wang and A. Galan, Chem. Commun., 2023, 59, 5253–5256 RSC.
- D. Bindl, P. K. Mandal, L. Allmendinger and I. Huc, Angew. Chem., Int. Ed., 2022, 61, e202116509 CrossRef CAS PubMed.
- S. De, B. Chi, T. Granier, T. Qi, V. Maurizot and I. Huc, Nat. Chem., 2018, 10, 51–57 CrossRef CAS PubMed.
- F. C. Parks, Y. Liu, S. Debnath, S. R. Stutsman, K. Raghavachari and A. H. Flood, J. Am. Chem. Soc., 2018, 140, 17711–17723 CrossRef CAS PubMed.
- N. Ousaka, M. Itakura, A. Nagasaka, M. Ito, T. Hattori, D. Taura, T. Ikai and E. Yashima, J. Am. Chem. Soc., 2021, 143, 4346–4358 CrossRef CAS PubMed.
- A. Urushima, D. Taura, M. Tanaka, N. Horimoto, J. Tanabe, N. Ousaka, T. Mori and E. Yashima, Angew. Chem., Int. Ed., 2020, 59, 7478–7486 CrossRef CAS PubMed.
- N. Saito, R. Terakawa, M. Shigeno, R. Amemiya and M. Yamaguchi, J. Org. Chem., 2011, 76, 4841–4858 CrossRef CAS PubMed.
- D. Haldar, H. Jiang, J.-M. Léger and I. Huc, Angew. Chem., Int. Ed., 2006, 45, 5483–5486 CrossRef CAS PubMed.
- H. Goto, Y. Furusho, K. Miwa and E. Yashima, J. Am. Chem. Soc., 2009, 131, 4710–4719 CrossRef CAS PubMed.
- J. Shang, Q. Gan, S. J. Dawson, F. Rosu, H. Jiang, Y. Ferrand and I. Huc, Org. Lett., 2014, 16, 4992–4995 CrossRef CAS PubMed.
- Y. Wang, Y. He, Z. Yu, J. Gao, S. ten Brinck, C. Slebodnick, G. B. Fahs, C. J. Zanelotti, M. Hegde, R. B. Moore, B. Ensing, T. J. Dingemans, R. Qiao and L. A. Madsen, Nat. Commun., 2019, 10, 801 CrossRef PubMed.
- Y. Ferrand, A. M. Kendhale, J. Garric, B. Kauffmann and I. Huc, Angew. Chem., Int. Ed., 2010, 49, 1778–1781 CrossRef CAS PubMed.
- C. J. Massena, N. B. Wageling, D. A. Decato, E. Martin Rodriguez, A. M. Rose and O. B. Berryman, Angew. Chem., Int. Ed., 2016, 55, 12398–12402 CrossRef CAS PubMed.
- K. Zhang, C. Ma, N. Li, C. Lu, D. Li, S. Fu and Q. Gan, Chem. Commun., 2019, 55, 10968–10971 RSC.
- Q. Gan, C. Bao, B. Kauffmann, A. Grélard, J. Xiang, S. Liu, I. Huc and H. Jiang, Angew. Chem., Int. Ed., 2008, 47, 1715–1718 CrossRef CAS PubMed.
- Y. Liu, F. C. Parks, W. Zhao and A. H. Flood, J. Am. Chem. Soc., 2018, 140, 15477–15486 CrossRef CAS PubMed.
- E. A. John, C. J. Massena and O. B. Berryman, Chem. Rev., 2020, 120, 2759–2782 CrossRef CAS PubMed.
- H. Sugiura, Y. Nigorikawa, Y. Saiki, K. Nakamura and M. Yamaguchi, J. Am. Chem. Soc., 2004, 126, 14858–14864 CrossRef CAS PubMed.
- H. Li, L. Kou, L. Liang, B. Li, W. Zhao, X.-J. Yang and B. Wu, Chem. Sci., 2022, 13, 4915–4921 RSC.
- Q. Gan, Y. Ferrand, N. Chandramouli, B. Kauffmann, C. Aube, D. Dubreuil and I. Huc, J. Am. Chem. Soc., 2012, 134, 15656–15659 CrossRef CAS PubMed.
- C. Dolain, A. Grélard, M. Laguerre, H. Jiang, V. Maurizot and I. Huc, Chem.–Eur. J., 2005, 11, 6135–6144 CrossRef CAS PubMed.
- T. Qi, V. Maurizot, H. Noguchi, T. Charoenraks, B. Kauffmann, M. Takafuji, H. Ihara and I. Huc, Chem. Commun., 2012, 48, 6337–6339 RSC.
- B. Teng, J. Atcher, L. Allmendinger, C. Douat, Y. Ferrand and I. Huc, Org. Biomol. Chem., 2023, 21, 3525–3530 RSC.
- M. L. Singleton, G. Pirotte, B. Kauffmann, Y. Ferrand and I. Huc, Angew. Chem., Int. Ed., 2014, 53, 13140–13144 CrossRef CAS PubMed.
- X. Hu, S. J. Dawson, P. K. Mandal, X. de Hatten, B. Baptiste and I. Huc, Chem. Sci., 2017, 8, 3741–3749 RSC.
- V. Corvaglia, F. Sanchez, F. Menke, C. Douat and I. Huc, Chem.–Eur. J., 2023, 29, e202300898 CrossRef CAS PubMed.
- B. Baptiste, J. Zhu, D. Haldar, B. Kauffmann, J.-M. Léger and I. Huc, Chem.–Asian J., 2010, 5, 1364–1375 CAS.
- Maestro, Schrödinger, LLC, New York, NY, 2021, https://www.schrodinger.com/citations/ Search PubMed.
- C. Zhan, J.-M. Léger and I. Huc, Angew. Chem., Int. Ed., 2006, 45, 4625–4628 CrossRef CAS PubMed.
- N. Chandramouli, Y. Ferrand, B. Kauffmann and I. Huc, Chem. Commun., 2016, 52, 3939–3942 RSC.
- C. Bao, Q. Gan, B. Kauffmann, H. Jiang and I. Huc, Chem.–Eur. J., 2009, 15, 11530–11536 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic protocols, crystallographic studies and characterisation of new compounds. CCDC 2216788. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc02020g |
‡ Care should be taken during integration as the intensity of the 1H NMR signals of exchangeable NH protons may be affected by the water suppression. This effect is more pronounced at lower concentration and results in lower-than-expected intensities for protons that exchange most. Typically, these results in different intensities for signals belonging to the same species that would be expected to have the same integration. The N![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) resonances belonging to Q units found at highest chemical shift values typically exchange least and may be preferred for accurate integration. resonances belonging to Q units found at highest chemical shift values typically exchange least and may be preferred for accurate integration. |
| § Compound 9 was not produced as an analogue of 8 but as another candidate for crystal growth, hence the other differences between the two. Crystal structures, including that of (5)2, suggest that C-terminal Aib units, as in 9, do not influence aggregation. There is also no ground to invoke a role of QAsp as the seventh residue in 9 instead of QSul in 8. We thus assign the different aggregation behavior of these compounds to the QSul6QAla mutation. |
| This journal is © The Royal Society of Chemistry 2023 |