
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCalculation-assisted regioselective functionalization of the imidazo[1,2-a]pyrazine scaffold via zinc and magnesium organometallic intermediates†
Agonist
Kastrati‡
 a,
Alexander
Kremsmair‡
a,
Alisa S.
Sunagatullina
a,
Vasilii
Korotenko
a,
Yusuf C.
Guersoy
a,
Saroj K.
Rout
a,
Fabio
Lima
b,
Cara E.
Brocklehurst
b,
Konstantin
Karaghiosoff
a,
Hendrik
Zipse
*a and
Paul
Knochel
*a
a,
Alexander
Kremsmair‡
a,
Alisa S.
Sunagatullina
a,
Vasilii
Korotenko
a,
Yusuf C.
Guersoy
a,
Saroj K.
Rout
a,
Fabio
Lima
b,
Cara E.
Brocklehurst
b,
Konstantin
Karaghiosoff
a,
Hendrik
Zipse
*a and
Paul
Knochel
*a
aDepartment Chemie, Ludwig-Maximilians-Universität München, Butenandtstrasse 5-13, Haus F, 81377 München, Germany. E-mail: zipse@cup.uni-muenchen.de; knoch@cup.uni-muenchen.de
bGlobal Discovery Chemistry, Novartis Institutes for BioMedical Research, 4057 Basel, Switzerland
First published on 28th September 2023
Abstract
Straightforward calculations such as determinations of pKa values and N-basicities have allowed the development of a set of organometallic reactions for the regioselective functionalization of the underexplored fused N-heterocycle imidazo[1,2-a]pyrazine. Thus, regioselective metalations of 6-chloroimidazo[1,2-a]pyrazine using TMP-bases (TMP = 2,2,6,6-tetramethylpiperidyl) such as TMPMgCl·LiCl and TMP2Zn·2MgCl2·2LiCl provided Zn- and Mg-intermediates, that after quenching with various electrophiles gave access to polyfunctionalized imidazopyrazine heterocycles. Additionally, the use of TMP2Zn·2MgCl2·2LiCl as base for the first metalation allowed an alternative regioselective metalation. Nucleophilic additions at position 8 as well as selective Negishi cross-couplings complete the set of methods for selectively decorating this heterocycle of the future.
Introduction
N-heterocycles are ubiquitous scaffolds in pharmaceutical and agrochemical research.1 They possess unique physicochemical and medicinal properties in addition to favorable pharmacokinetics, which make their efficient further functionalization an attractive research goal.2 Besides standard N-heterocycles such as pyridines, diazines and indoles for which functionalization methods are well established, there is a current trend to investigate less common N-heterocycles as pharmaceutical scaffolds.3 Recently, we have reported the decoration of a range of new fused N-heterocycles such as pyrazolo[1,5-a]pyrimidines (1) and imidazo[1,2-b]pyridazines (2).4 From these studies, we were anticipating useful properties of the imidazo[1,2-a]pyrazine ring system (3, Scheme 1a). This structural motif has already found pharmaceutical applications as the kinase inhibitor 45 and the AKT inhibitor 56 (Scheme 1a).7 However, most previous synthetic approaches towards the functionalization of the imidazo[1,2-a]pyrazine scaffold involve either electrophilic bromination,8 followed by Suzuki–Miyaura cross-couplings, or transition metal-catalyzed direct C–H arylations.9 As a result, there are significant limitations in scope for preparing more complex and multifunctionalized derivatives. Combining theoretical calculations and extensive experimental knowledge in the functionalization of N-heterocycles has now allowed us to develop a rational approach towards the decoration of this relatively underexplored heterocyclic scaffold. Thus, we calculated the pKa values of all ring hydrogens of 34a,b in order to predict the metalation regioselectivity with TMP-bases (TMP = 2,2,6,6-tetramethylpiperidyl).10 We have found very similar pKa values of position 3 and 5 as well as prohibitively high values for metalations in position 6 and 8 for the unsubstituted scaffold 3. In fact, metalation of 3 with TMPMgCl·LiCl (11) in THF (−60 °C, 0.5 h) produced after deuterolysis quench a 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40 mixture of C3/C5 deuterated products.11 This data let us consider the introduction of an electronegative substituent, which would lower all the pKa values and introduce a larger pKa difference between positions 3 and 5. A chlorine substituent at position 6 was an ideal solution to these problems since it can be readily removed or substituted.12 A web-search showed that 6-chloroimidazo[1,2-a]pyrazine (6) is easily prepared13 and commercially available. Calculations of the pKa values of 6 confirmed our hypothesis (Scheme 1b). Furthermore, thanks to extensive calculations, we were able to get a deeper and more accurate insight into the metalation regioselectivities of various substituted imidazo[1,2-a]pyrazines of type 6. We realized that the relative energies of the organometallic intermediates and the inclusion of additives such as LiCl in the calculations provided a basis for the rationalization of observed regioselectivities.
:40 mixture of C3/C5 deuterated products.11 This data let us consider the introduction of an electronegative substituent, which would lower all the pKa values and introduce a larger pKa difference between positions 3 and 5. A chlorine substituent at position 6 was an ideal solution to these problems since it can be readily removed or substituted.12 A web-search showed that 6-chloroimidazo[1,2-a]pyrazine (6) is easily prepared13 and commercially available. Calculations of the pKa values of 6 confirmed our hypothesis (Scheme 1b). Furthermore, thanks to extensive calculations, we were able to get a deeper and more accurate insight into the metalation regioselectivities of various substituted imidazo[1,2-a]pyrazines of type 6. We realized that the relative energies of the organometallic intermediates and the inclusion of additives such as LiCl in the calculations provided a basis for the rationalization of observed regioselectivities.
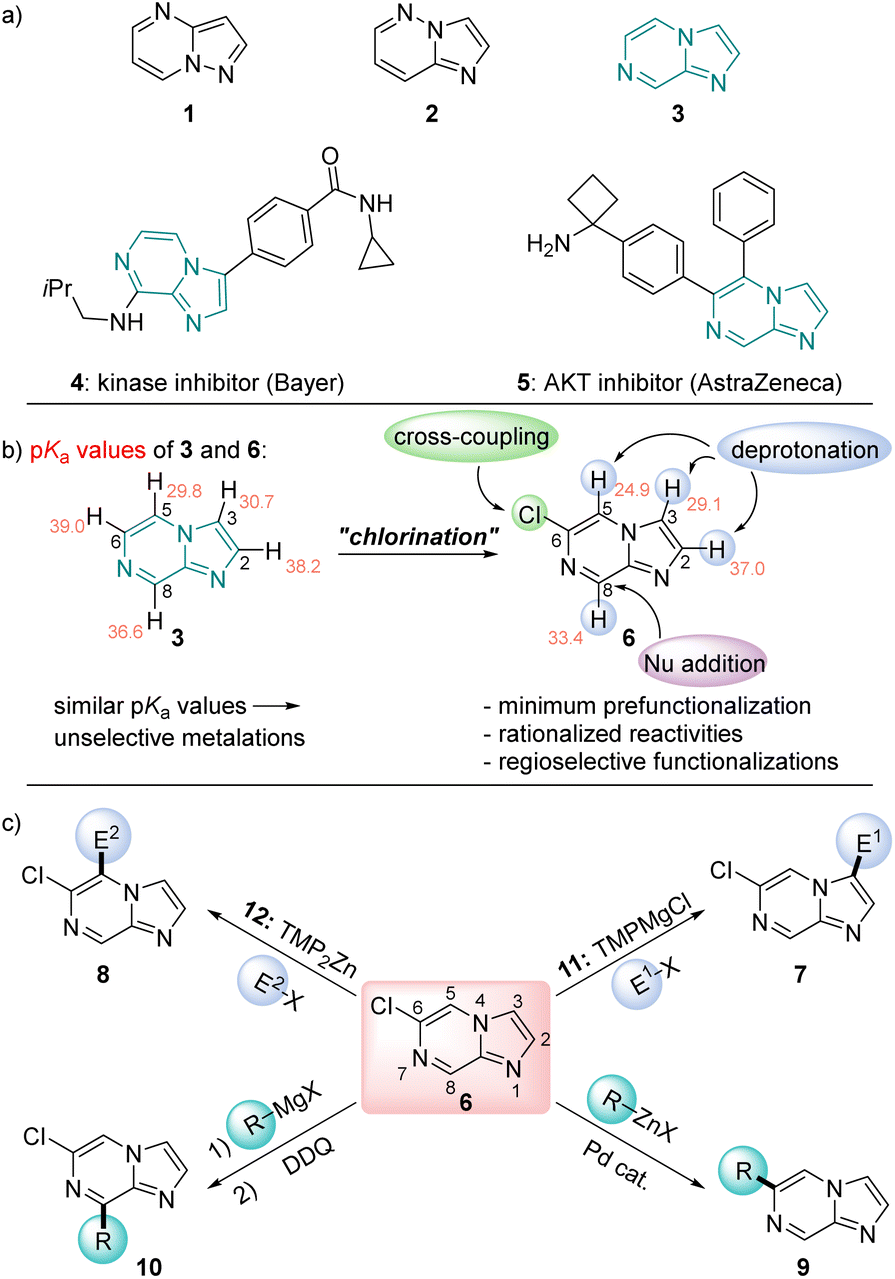 | ||
| Scheme 1 (a) Heterocycles of the future 1, 2 and 3.3 (b) Calculated pKa values indicated that a chlorination led to enhanced pKa differences. (c) Reactivity pattern of 6 producing imidazo[1,2-a]pyrazines of type 7–10. Salts are omitted for clarity. | ||
Herein, we report a versatile selective functionalization of this scaffold using regioselective metalations, nucleophilic additions and cross-couplings. As bases we have used TMP-bases10 of Mg14 and Zn15 such as TMPMgCl·LiCl14a (11), and TMP2Zn·2MgCl2·2LiCl15a (12, Scheme 1c).16
Results and discussion
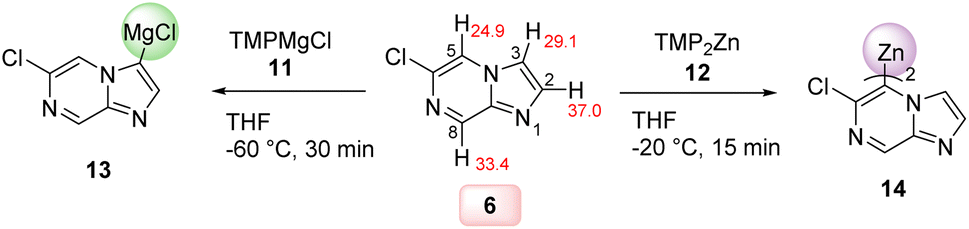
Thus, we have found that the metalation of 6-chloroimidazo[1,2-a]pyrazine (6) with TMPMgCl·LiCl (11) in THF led to a selective magnesiation at position 3 at −60 °C within 30 min affording magnesium derivatives of type 13.17 The quenching of 13 with various electrophiles gave 3,6-disubstituted imidazo[1,2-a]pyrazines 7a–g in 40–78% yield (Scheme 2).18 Iodolysis of 13 gave the corresponding iodide 7a in 78% yield. Whereas, treatment of 13 with allyl bromide or acyl chlorides (25 °C, 2 h) in the presence of catalytic amounts of CuCN·2LiCl19 (20–50 mol%) furnished allylated derivative 7b in 56% yield and ketones 7c and 7d in 40–56% yield. The scope was further increased by treating Mg-intermediate 13 with commercially available TsCN, ethyl cyanoformate or freshly prepared PhSO2SMe20 affording cyano-derivative 7e, ester derivative 7f and thioether 7g in 48–70% yield. The structure of 7a was confirmed by single crystal X-ray diffraction.11 DFT calculations confirmed the observed metalation regioselectivity at position 3 and clearly showed that the intermediate 13 was the most stable organometallic intermediate. In addition, we found that the NBO charge at C3 was the most negative among all carbon atoms in heterocycle 6.11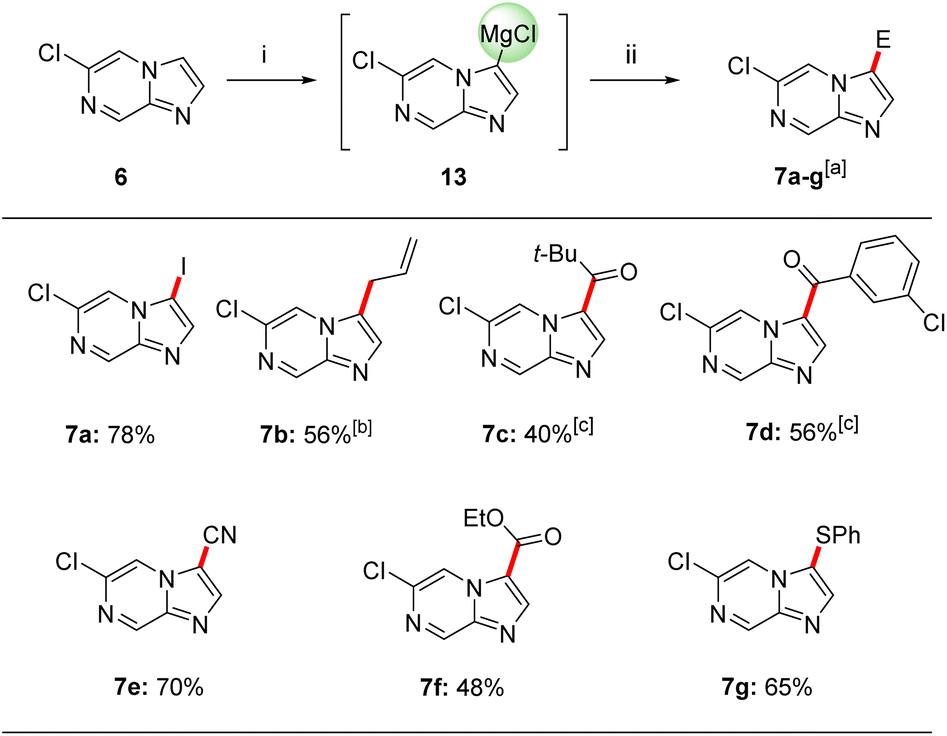 | ||
| Scheme 2 Regioselective magnesiation of 6-chloroimidazo[1,2-a]pyrazine (6) using TMPMgCl·LiCl (11) followed by quenching with electrophiles. Reagents and conditions: (i) TMPMgCl·LiCl (11, 1.2 equiv.), THF, −60 °C, 30 min; (ii) E-X, THF, 25 °C, 0.15–2 h; aisolated yield of analytically pure products; bthe allylation was mediated by CuCN·2LiCl (50 mol%), THF, 25 °C, 1.5 h; cthe acylation was mediated by CuCN·2LiCl (20–50 mol%), THF, 25 °C, 2 h. | ||
Interestingly, the treatment of 6-chloroimidazo[1,2-a]pyrazine (6) with TMP2Zn·2MgCl2·2LiCl (12) in THF led to a complete regioselectivity switch affording diheteroarylzinc derivatives of type 14 within 15 min at −20 °C.17 Subjecting this intermediate to various electrophiles gave 5,6-disubstituted imidazo[1,2-a]pyrazine 8a–g in 52–85% isolated yield (Scheme 3).
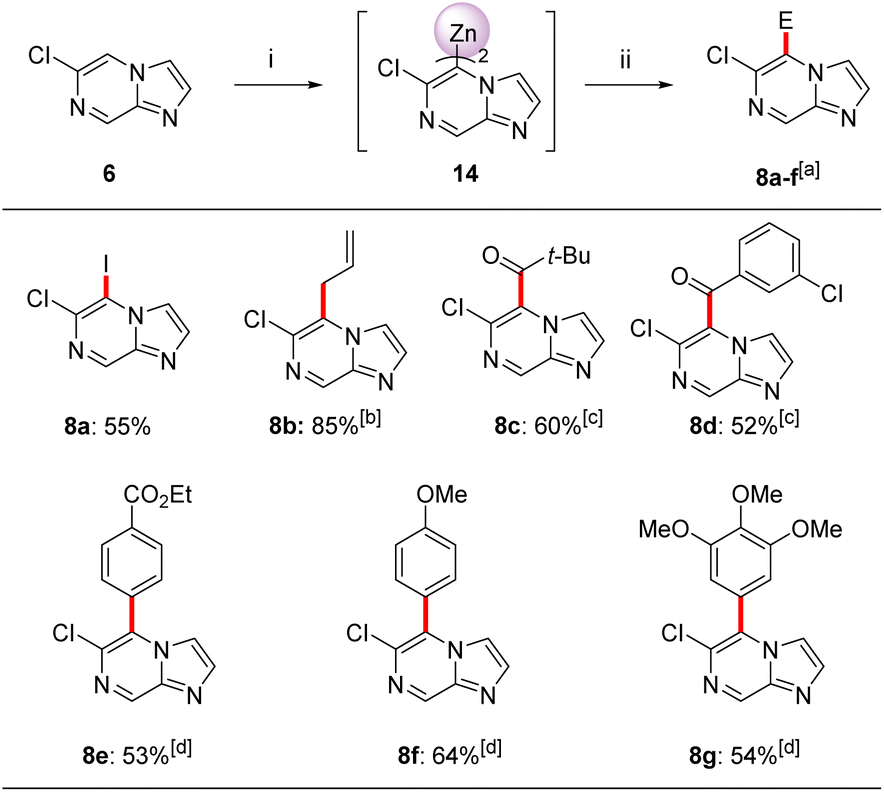 | ||
| Scheme 3 Regioselective zincation of 6-chloroimidazo[1,2-a]pyrazine (6) using TMP2Zn·2MgCl2·2LiCl (12) followed by quenching with electrophiles. Reagents and conditions: (i) TMP2Zn·2MgCl2·2LiCl (12, 0.6 equiv), THF, −20 °C, 15 min; (ii) E-X, THF, 25 °C, 2 h. aIsolated yield of analytically pure products; bthe allylation was mediated by CuCN·2LiCl (20 mol%), THF, 25 °C, 1.5 h; cthe acylation was mediated by CuCN·2LiCl (20 mol%), THF, 25 °C, 2 h. dPd(PPh3)4 (5 mol%), THF, 50 °C, 2 h. | ||
Thus, iodolysis of 14 gave the corresponding iodide 8a in 55% yield. Whereas, treatment of 14 with CuCN·2LiCl (20 mol%) followed by addition of allyl bromide or different acyl chlorides (25 °C, 1.5–2 h) furnished the allylated derivative 8b in 85% yield and ketones 8c and 8d in 52–60%. Negishi cross-couplings21 of the diheteroarylzinc derivative 14 with aryl iodides in the presence of catalytic amounts of Pd(PPh3)4 (5 mol%, 50 °C, 2 h) gave the arylated N-heterocycles 8e–8g in 53–64% isolated yield. The structure of 8b was confirmed by single crystal X-ray diffraction.11
The rationalization of these complementary regioselectivites may be tentatively done based on relative basicities. Thus, DFT calculations indicated that the C5-metalated intermediate 14 was preferred for thermochemical reasons, possibly due to attractive intramolecular CH⋯Cl interactions.11 The Zn-base 12 (bearing a N–Zn bond) is less reactive than the Mg-base 11 which is bearing a more ionic N–Mg bond. The temperatures −60 °C in the case of 13 and −20 °C in the case of 1 have been optimized to achieve the best yields.22 The observed regioselectivity reflects according to our calculations the relative stability of the produced organometallic intermediates 13 or 14 and overcomes complex-inducing proximity effects (Scheme 4).23
 | ||
| Scheme 4 Thermodynamically driven regioselectivity switch in the metalation of 6-chloroimidazo[1,2-a]pyrazine using either TMPMgCl·LiCl (11) or TMP2Zn·2MgCl2·2LiCl (12). Salts are omitted for clarity. | ||
A second zincation of 3,6-disubstituted imidazo[1,2-a]pyrazine (7e) using TMP2Zn·2MgCl2·2LiCl (12) was also possible (Scheme 5). Thus, treatment of imidazo[1,2-a]pyrazine-3-carbonitrile (7e) with 12 (0.6 equiv., THF, −20 °C, 15 min) provided diheteroarylzinc intermediate 15, which was successfully quenched with electrophiles providing trisubstituted imidazo[1,2-a]pyrazines 16a–e in 33–72% yield. Thus, iodolysis of 15 afforded iodide 16a in 52% yield. Whereas, treatment of 15 with CuCN·2LiCl (20 mol%) followed by addition of allyl bromide or pivaloyl chloride (25 °C, 2 h) furnished allyl derivative 16b in 50% yield and ketone 16c in 33% yield. Negishi cross-couplings of the diheteroarylzinc derivative 15 with aryl iodides in the presence of catalytic amounts of Pd(PPh3)4 (5 mol%, 50 °C, 2 h) gave the arylated N-heterocycles 16d–e in 55–72% yield. The metalation regioselectivity at position 2 of 7e was investigated by DFT calculations showing that C2 metalated intermediate 15 was stabilized by the neighbouring LiCl and MgCl2via non-covalent interactions. In this case, we have also considered an equilibrium between various Zn-TMP bases which may contain TMP2Zn as well as TMPZnCl. In addition, we have found that the cyano group was responsible for unfavourable steric effects onto the C5 position.11
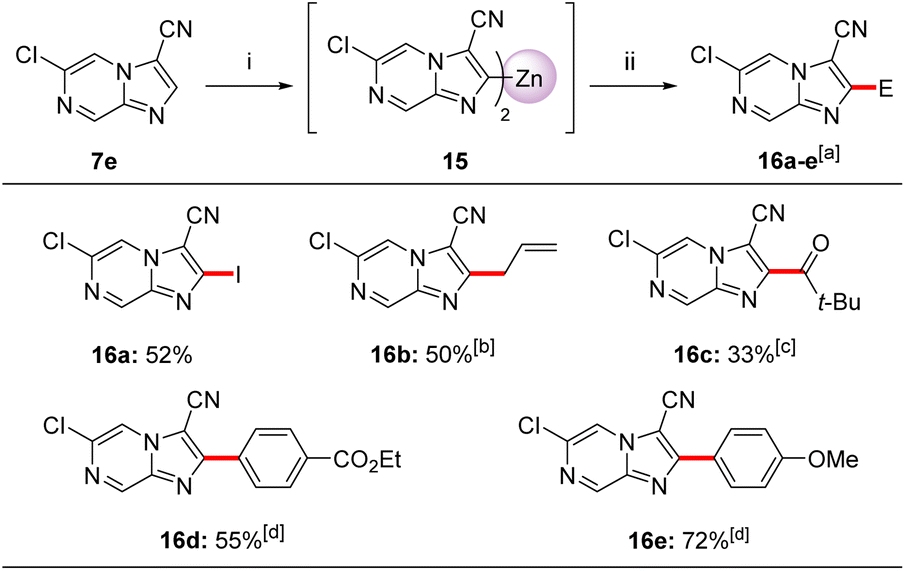 | ||
| Scheme 5 Selective zincation of N-heterocycle 7e using TMP2Zn·2MgCl2·2LiCl (12) followed by quenching with electrophiles affording trisubstituted N-heterocycles 16a–e. Reagents and conditions: (i) TMP2Zn·2MgCl2·2LiCl (12, 0.55 equiv.), THF, −20 °C, 15 min; (ii) E-X, THF, 25 °C, 2 h. aIsolated yield of analytically pure products; bthe allylation was mediated by CuCN·2LiCl (20 mol%), THF, 25 °C, 1.5 h; cthe acylation was mediated by CuCN·2LiCl (20 mol%), THF, 25 °C, 2 h; dPd(PPh3)4 (5 mol%), THF, 50 °C, 2 h. | ||
A further magnesiation of 2,3-disubstituted 6-chloroimidazo[1,2-a]pyrazine derivative 16e was possible (Scheme 6). Thus, treating the trisubstituted heterocycle 16e with TMPMgCl·LiCl (11) in THF at −40 °C within 30 min afforded the magnesiated species 17. This regioselectivity can be rationalized with calculations including LiCl, which formed the very stable mixed Li,Mg-intermediate 17 depicted in Scheme 6.11 Its quenching with electrophiles gave access to tetrasubstituted derivatives 18a–e in 39–69% yield. Thus, iodolysis of 17 provided the iodide 18a in 68% yield. Treatment of 17 with PhSO2SMe afforded the desired sulfide 18b in 39% yield. After transmetalation with a solution of ZnCl2 (1 M in THF, 1.2 equiv.) for 15 min the resulting heteroarylzinc species was subjected to Negishi cross-couplings with aryl iodides in the presence of catalytic amounts of Pd(PPh3)4 (5 mol%, THF, 50 °C, 2 h) giving the arylated N-heterocycles 18c–e in 46–69% yield. The structure of 18a was confirmed by single crystal X-ray diffraction.11
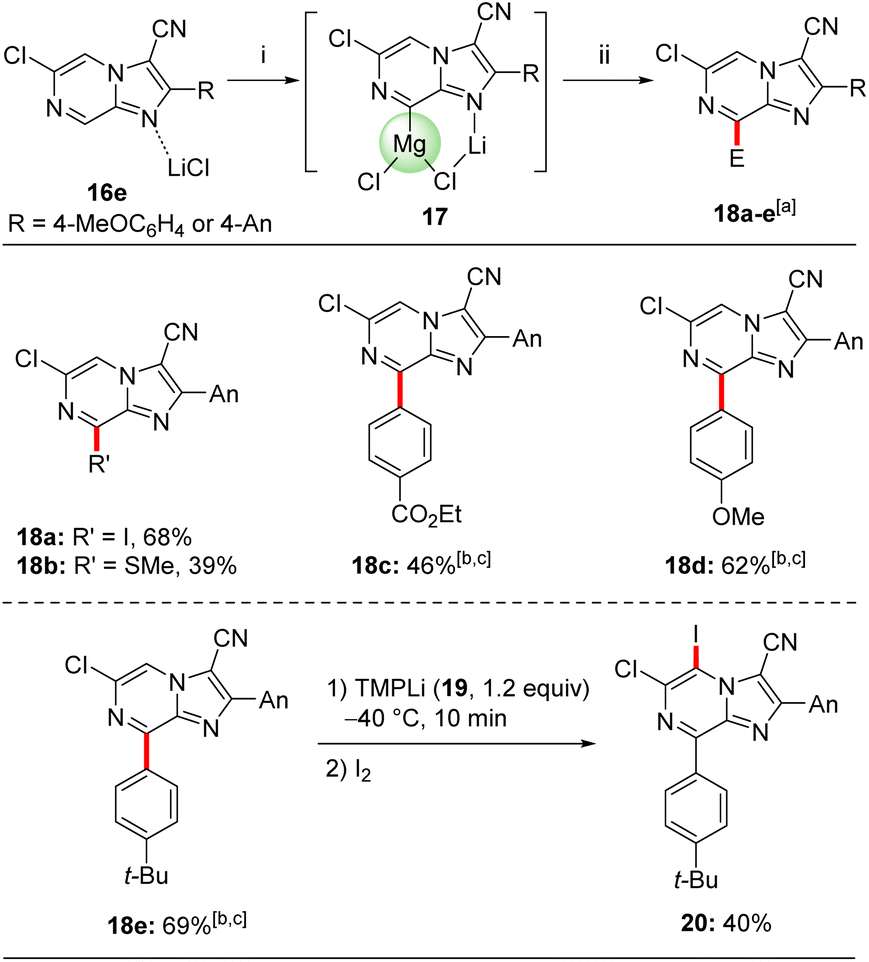 | ||
| Scheme 6 Magnesiation of the trisubstituted imidazopyrazine 16e using TMPMgCl·LiCl (11) followed by various quenching reactions with electrophiles. Reagents and conditions: (i) TMPMgCl·LiCl (11, 1.2 equiv.), THF, −40 °C, 30 min; (ii) E-X, THF, 25 °C, 0.15–2 h. aIsolated yield of analytically pure products; btransmetalation with ZnCl2 (1 M solution in THF, 1.2 equiv.), 15 min. cPd(PPh3)4 (5 mol%) THF, 50 °C, 2 h. | ||
Lithiation of the trisubstituted N-heterocycle 18e using TMPLi (19, 1.2 equiv.) at −40 °C, 10 min led to a regioselective lithiation at position 5 and subsequent trapping with iodine gave the fully functionalized N-heterocycle 20 in 40% yield (Scheme 6).24 The presence of chlorine- and iodine-substituents now opens the way for diverse further functionalizations. Concerning the replacement of the chlorine substituent, this was demonstrated by submitting 6-chloroimidazo[1,2-a]pyrazine (6) to cross-couplings with functionalized organozinc reagents of type 2125 in the presence of 5 mol% of Pd-PEPPSI-iPr26 or Pd-PEPPSI-iPent27 affording functionalized 6-substituted imidazo[1,2-a]pyrazines 22a–e in up to 98% yield (Scheme 7). Thus, cross-coupling of 6 with arylzinc reagents 21a or 21b bearing a sensitive ester25c or cyano25a functionality led to 6-arylated imidazo[1,2-a]pyrazines 22a-b in 90–93% yield. Interestingly, alkylzinc reagents such as 21c25d or 21d25d showed superior results when the more sterically hindered catalyst Pd-PEPPSI-iPent (5 mol%)27 was used resulting in the N-heterocycles 22c–d in up to 98% yield. Finally, functionalized benzylic zinc reagent 21e25b was an excellent substrate for this cross-coupling leading to the benzoylated heterocycle 22e in 95% yield. Related Suzuki–Miyaura cross-couplings can be performed using 7e and arylboronic acid esters in the presence of Pd(PPh3)4 (10 mol%), albeit in lower yields (up to 50% isolated yield) and under harsher reaction conditions (100 °C, 5 h).11
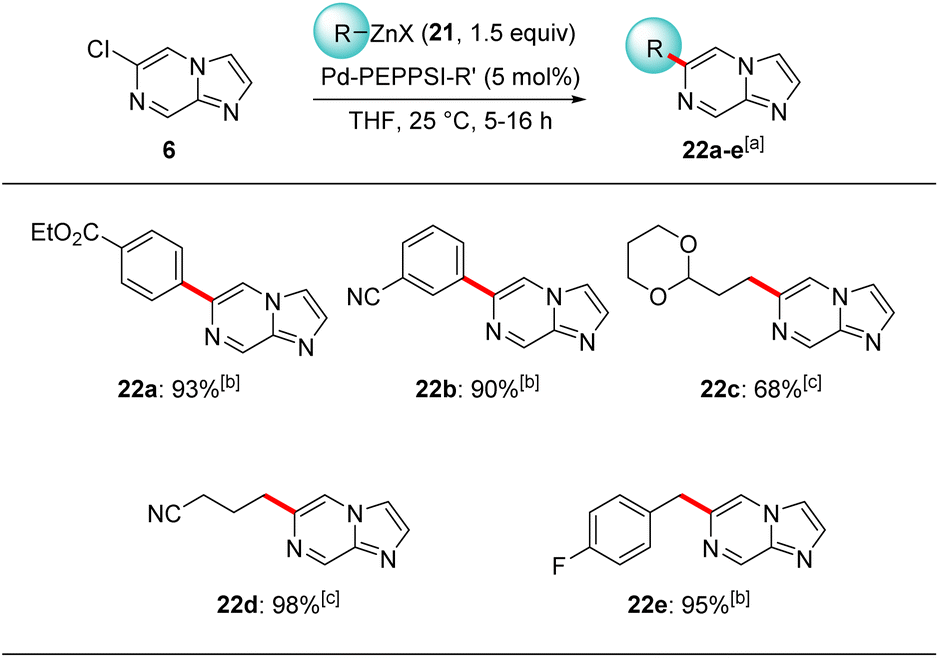 | ||
| Scheme 7 Palladium-catalyzed Negishi cross-couplings of 6 using functionalized aryl, alkyl and benzylic zinc reagents of type 21 leading to functionalized 6-substituted imidazo[1,2-a]pyrazines 22a–e. Reagents and conditions: X = Cl·MgICl·LiCl, Cl·MgBrCl·LiCl or Cl·MgCl2·LiCl; R = Alk, Ar, Bz; R′ = iPr or iPent. aIsolated yield of analytically pure products; bthe cross-coupling was catalyzed by Pd-PEPPSI-iPr (5 mol%), THF, 25 °C, 5 h; cthe cross-coupling was catalyzed by Pd-PEPPSI-iPent (5 mol%), THF, 25 °C, 16 h. | ||
In addition, based on previously described nucleophilic addition reactions to pyrazolo[1,5-a]pyrimidine (1, Scheme 1)4c we have examined the addition of Grignard reagents triggered by a coordination at the most basic nitrogen N1 (see 24). Thus, we have treated 6-chloroimidazo[1,2-a]pyrazine (6) with various organomagnesium halides complexed with lithium chloride of type 2328 (Scheme 8). Indeed, the reaction of 6 with 4-methoxyphenylmagnesium bromide·lithium chloride (23a)28a gave an intermediate magnesium species 25 which after water quenching provided dihydroimidazo[1,2-a]pyrazine 26a in 63% yield. Due to instability of such partially saturated heterocycles, these compounds of type 26 were, after an aqueous work-up, directly oxidized with DDQ (DDQ = 2,3-dichloro-5,6-dicyano-1,4-benzoquinone), furnishing 8-arylated imidazo[1,2-a]pyrazines 27a–c in 37–78% yield. This reaction sequence was also extended to the addition of alkylmagnesium reagents such as 23d-e resulting in the alkylated N-heterocycles 27d-e in 67–95% yield. Trapping of 25a with PhSO2SPh followed by rearomatization with DDQ gave disubstituted imidazo[1,2-a]pyrazine 27f in 33% isolated yield.
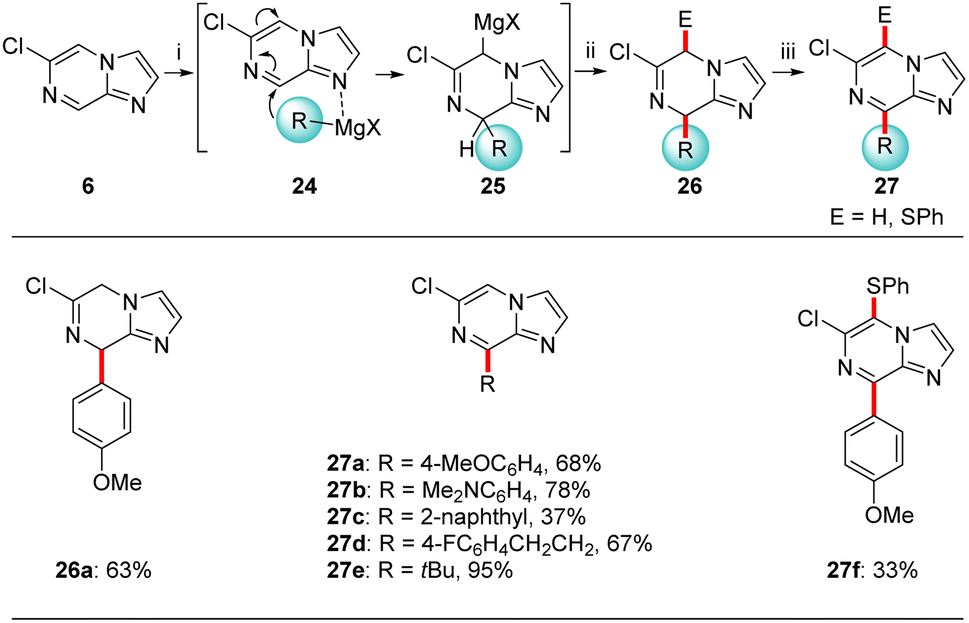 | ||
| Scheme 8 Nucleophilic addition of Grignard reagents of type 23 to 6-chloroimidazo[1,2-a]pyrazine (6). Reactions and conditions: (i) RMgX·LiCl (23, 1.2 equiv.), THF, 40 °C, 10 min; (ii) aq. workup or E-X (1.2 equiv.), THF, 25 °C, 2 h; (iii) DDQ (1.2 equiv.), THF, 25 °C, 16 h. | ||
Conclusions
In summary, with the help of theoretical calculations such as pKa determinations, N-basicity evaluations, and thermochemical analysis of the various Zn or Mg organometallic intermediates, we were able to fully rationalize the observed metalation selectivities of the underexplored fused N-heterocycle scaffold 6. Despite the general usefulness of pKa values and N-basicities, we find the relative stabilities of the Zn- or Mg-intermediates were key be more helpful for the rationalization of experimental results. In addition, we found that the coordination of additives such as LiCl to the N1 position were crucial for directing the metalation to the C2 or C8 positions due to stabilizing chelating interactions as shown for intermediate 17 in Scheme 6. This “chelate”-effect is equally possible in zinc intermediate 15 (Scheme 5).11 We have successively metalated all positions of the 5,6-fused bicyclic N-heterocycle 6-chloroimidazo[1,2-a]pyrazine (6) using a combination of different bases such as TMPMgCl·LiCl (11) and/or TMP2Zn·2MgCl2·2LiCl (12) as well as TMPLi (19). The resulting metalated species were then quenched with different electrophiles giving access to a broad scope of functionalized imidazopyrazine derivatives. Furthermore, 6-chloroimidazo[1,2-a]pyrazine (6) underwent Pd-catalyzed cross-couplings using aryl, alkyl and benzylic zinc reagents allowing further functionalizations of this important heterocycle and a nucleophilic addition of Grignard reagents was also possible giving an access to partially saturated heterocycle (26a) with favorable solubilities. This calculation-assisted functionalization of heterocyclic scaffolds should be applicable to further uncommon heterocycles of pharmaceutical (or agrochemical) interest and deserve further exploration.Data availability
Crystallographic data for compounds 7a, 8b, 16a and 18a has been deposited at the CCDC under 2258909–2258912. The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
A. K., A. K., A. S., Y. C. G. and S. K. R. performed and analyzed the experiments. K. K. measured and analyzed X-ray crystal structures. V. K. and H. Z. performed DFT calculations. A. K., A. K. and P. K. designed the experiments. A. K., H. Z. and P. K. prepared the manuscript with contributions of all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the LMU Munich and the DFG for financial support. We thank Albemarle (Hoechst, Germany) for the generous gift of chemicals.Notes and references
- (a) R. D. Taylor, M. MacCoss and A. D. G. Lawson, J. Med. Chem., 2014, 57, 5845–5859 CrossRef CAS PubMed; (b) E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed; (c) D. C. Blakemore, L. Castro, I. Churcher, D. C. Rees, A. W. Thomas, D. M. Wilson and A. Wood, Nat. Chem., 2018, 10, 383–394 CrossRef CAS PubMed.
- (a) E. J. Corey, B. Czakó and L. Kürti, Molecules and Medicine, John Wiley & Sons, Hoboken, 2007 Search PubMed; (b) l. D. Quin and J. A. Tyrell, Fundamentals of Heterocyclic Chemistry: Importance in Nature and in the Synthesis of Pharmaceuticals, John Wiley & Sons, Hoboken, 2010 Search PubMed; (c) L. Guillemard, N. Kaplaneris, L. Ackermann and M. J. Johansson, Nat. Rev. Chem., 2021, 5, 522–545 CrossRef CAS PubMed.
- W. R. Pitt, D. M. Parry, B. G. Perry and C. R. Groom, J. Med. Chem., 2009, 52, 2952–2963 CrossRef CAS PubMed.
- (a) K. Snégaroff, T. T. Nguyen, N. Marquise, Y. S. Halauko, P. J. Harford, T. Roisnell, V. E. Matulis, O. A. Ivashkevich, F. Chevallier, A. E. H. Wheatley, P. C. Gros and F. Mongin, Chem.–Eur. J., 2011, 17, 13284–13297 CrossRef PubMed; (b) M. Balkenhohl, H. Jangra, I. S. Makarov, S.-M. Yang, H. Zipse and P. Knochel, Angew. Chem. Int. Ed., 2020, 59, 14992–14999 ( Angew. Chem. , 2020 , 132 , 15102–15109 ) CrossRef CAS PubMed; (c) S. K. Rout, A. Kastrati, H. Jangra, K. Schwärzer, A. S. Sunagatullina, M. Garny, F. Lima, C. Brocklehurst, K. Karaghiosoff, H. Zipse and P. Knochel, Chem.–Eur. J., 2022, 28, e202200733 CrossRef PubMed.
- K.-i. Kusakabe, N. Ide, Y. Daigo, T. Itoh, T. Yamamoto, H. Hashizume, K. Nozu, H. Yoshida, G. Tadano, S. Tagashira, K. Higashino, Y. Okano, Y. Sato, M. Inoue, M. Iguchi, T. Kanazawa, Y. Ishioka, K. Dohi, Y. Kido, S. Sakamoto, S. Ando, M. Maeda, M. Higaki, Y. Baba and Y. Nakamura, J. Med. Chem., 2015, 58, 1760–1775 CrossRef CAS PubMed.
- J. G. Kettle, S. Brown, C. Crafter, B. R. Davies, P. Dudley, G. Fairley, P. Faulder, S. Fillery, H. Greenwood, J. Hawkins, M. James, K. Johnson, C. D. Lane, M. Pass, J. H. Pink, H. Plant and S. C. Cosulich, J. Med. Chem., 2012, 55, 1261–1273 CrossRef CAS PubMed.
- (a) R. C. X. Brys, P. Edwards, W. Schmidt, M. J. I. Andrews, M. S. Chambers, J. A. Clase, V. Birault, K. L. Hirst, A. Macleod, G. Bar, C. J. Harris and P. Huxley, WIPO (PCT), WO2007131991A1, 2007; (b) T. Yu, D. B. Belanger, A. D. Kerekes, Z. Meng, J. R. Tagat, S. J. Esposite, A. K. Mandal, Y. Xiao, B. A. Kulkarni, Y. Zhang, P. J. Curran, R. Doll and M. A. Siddiqui, WIPO (PCT), WO2008156614A2, 2008; (c) M. Koppitz, U. Klar, R. Jautelat, D. Kosemund, R. Bohlmann, B. Bader and P. Lienau, WIPO (PCT), WO2012080229A1, 2011.
- Y. Jin, P. Bu, Q. He, J. Lan, F. Zhou, L. Zhang and X. He, WIPO (PCT), WO2016050165, 2016.
- (a) I. Hyodo, M. Tobisu and N. Chatani, Chem.–Asian J., 2012, 7, 1357–1365 CrossRef CAS PubMed; (b) S. K. Guchhait, S. Kandekar, M. Kashyap, N. Taxak and P. V. Bharatam, J. Org. Chem., 2012, 77, 8321–8328 CrossRef CAS PubMed; (c) V. Gembus, J.-F. Bonfanti, O. Querolle, P. Jubault and C. Hoarau, Org. Lett., 2012, 14, 6012–6015 CrossRef CAS PubMed; (d) B. M. Jonson and M. P. Huestis, Eur. J. Org Chem., 2014, 1589–1593 CrossRef; (e) B. Zhang, W. Zhang, J. Wang, Q. Wang, N. Kambe and R. Qiu, Org. Lett., 2022, 24, 7918–7923 CrossRef CAS PubMed.
- (a) B. Haag, M. Mosrin, I. Hiriyakkanavar, V. Malakhov and P. Knochel, Angew. Chem. Int. Ed., 2011, 50, 9794–9824 ( Angew. Chem. , 2011 , 123 , 9968–9999 ) CrossRef CAS PubMed; (b) A. Kremsmair, J. H. Harenberg, K. Schwärzer, A. Hess and P. Knochel, Chem. Sci., 2021, 12, 6011–6019 RSC; (c) A. Kremsmair, A. Hess, B. Heinz and P. Knochel, Chem.–Eur. J., 2022, 28, e202103269 CrossRef CAS PubMed.
- For further information, see ESI.†.
- (a) K. Reddy, G. Martinez Botella, A. M. Griffin and E. Brian, Praxis Precision Medicines, Inc., WO2018098500A1, 2018; (b) S. B. Hoyt, C. J. Thomas, D. T. Starczynowski, J. S. Rosenbaum and G. Gracia Maldonado, Kurome Therapeutics Inc., WO2022140647A1, 2022.
- W. C. Lumma Jr., W. C. Randall, E. L. Cresson, J. R. Huff, R. D. Hartman and T. F. Lyon, J. Med. Chem., 1983, 26, 357–363 CrossRef CAS PubMed.
- (a) A. Krasovskiy, V. Krasovskaya and P. Knochel, Angew. Chem. Int. Ed., 2006, 45, 2958–2961 ( Angew. Chem. , 2006 , 118 , 3024–3027 ) CrossRef CAS PubMed; (b) G. C. Clososki, C. J. Rohbogner and P. Knochel, Angew. Chem. Int. Ed., 2007, 46, 7681–7684 ( Angew. Chem. , 2007 , 119 , 7825–7828 ) CrossRef CAS PubMed; (c) A. Hess, A. Kremsmair and P. Knochel, Encyclopedia of Reagents for Organic Synthesis, Wiley VCH, 2021, DOI:10.1002/047084289X.rn02373.
- (a) S. H. Wunderlich and P. Knochel, Angew. Chem. Int. Ed., 2007, 46, 7685–7688 ( Angew. Chem. , 2007 , 119 , 7829–7832 ) CrossRef CAS PubMed; (b) M. Mosrin and P. Knochel, Org. Lett., 2009, 11, 1837–1840 CrossRef CAS PubMed; (c) T. Klatt, P. Knochel and M. Mosrin, Encyclopedia of Reagents for Organic Synthesis, Wiley VCH, 2015, DOI:10.1002/047084289X.rn01806.
- (a) A. Hess, J. P. Prohaska, S. B. Doerrich, F. Trauner, F. H. Lutter, S. Lemaire, S. Wagschal, K. Karaghiosoff and P. Knochel, Chem. Sci., 2021, 12, 8424–8429 RSC; (b) A. Kremsmair, S. Graßl, C. J. B. Seifert, E. Godineau and P. Knochel, Synthesis, 2021, 53, 4068–4074 CrossRef CAS; (c) A. S. Sunagatullina, F. H. Lutter and P. Knochel, Angew. Chem. Int. Ed., 2022, 61, e202116625 ( Angew. Chem. , 2022 , 134 , e202116625 ) CrossRef CAS PubMed; (d) A. Kremsmair, A. S. Sunagatullina, L. J. Bole, P. Mastropierro, S. Graßl, H. R. Wilke, E. Godineau, E. Hevia and P. Knochel, Angew. Chem. Int. Ed., 2022, 61, e202210491 ( Angew. Chem. , 2022 , 134 , e202210491 ) CrossRef CAS PubMed.
- No organometallic intermediates have been isolated or characterized. Their structures are reasonable based on the obtained products and precedent in the literature as found in ref. 10.
- Other regioisomers are, if any, formed in negligible amounts (less than 2%). However, these functionalized heterocycles show moderate solubility, which may be at least partly responsible for low overall yields.
- P. Knochel, M. C. P. Yeh, S. C. Berk and J. Talbert, J. Org. Chem., 1988, 53, 2390–2392 CrossRef CAS.
- K. Fujiki, N. Tanifuji, Y. Sasaki and T. Yokoyama, Synthesis, 2002, 343–348 CrossRef CAS.
- E.-i. Negishi, Angew. Chem. Int. Ed, 2011, 50, 6738–6764 ( Angew. Chem. , 2011 , 123 , 6870–6897 ) CrossRef CAS PubMed.
- Deprotonation at position 5 requires higher temperature because a weaker base such as TMP2Zn·2MgCl2·LiCl is used compared to a stronger TMPMgCl·LiCl base capable of deprotonation at lower temperature. In addition, intermediates generated when using TMPZn-bases are usually more stable at elevated temperature.
- M. C. Whisler, S. MacNeil, V. Snieckus and P. Beak, Angew. Chem. Int. Ed, 2004, 43, 2206–2225 ( Angew. Chem. , 2004 , 116 , 2256–3227 ) CrossRef CAS PubMed.
- Quenching with other electrophiles such as an aldehyde led to only traces of isolated product.
- (a) A. Krasovskiy and P. Knochel, Angew. Chem. Int. Ed., 2004, 43, 3333–3336 ( Angew. Chem. , 2004 , 116 , 3396–3399 ) CrossRef CAS PubMed; (b) A. Metzger, F. M. Piller and P. Knochel, Chem. Commun., 2008, 5824–5826 RSC; (c) V. Dhayalan and P. Knochel, Synthesis, 2015, 3246–3256 CAS; (d) F. H. Lutter, L. Grokenberger, P. Spieß, J. M. Hammann, K. Karaghiosoff and P. Knochel, Angew. Chem. Int. Ed., 2020, 59, 5546–5550 ( Angew. Chem. , 2020 , 132 , 5591–5595 ) CrossRef CAS PubMed.
- M. G. Organ, S. Avola, I. Dubovyk, N. Hadei, E. A. B. Kantchev, C. J. O'Brien and C. Valente, Chem.–Eur. J., 2006, 12, 4749–4755 CrossRef CAS PubMed.
- (a) M. G. Organ, S. Calimsiz, M. Sayah, K. H. Hoi and A. J. Lough, Angew. Chem. Int. Ed., 2009, 48, 2383–2387 ( Angew. Chem. , 2009 , 121 , 2419–2423 ) CrossRef CAS PubMed; (b) J. Skotnitzki, A. Kremsmair, D. Keefer, Y. Gong, R. de Vivie-Riedle and P. Knochel, Angew. Chem. Int. Ed., 2020, 59, 320–324 ( Angew. Chem. , 2020 , 132 , 328–332 ) CrossRef CAS PubMed.
- (a) A. Kremsmair, H. R. Wilke, M. M. Simon, Q. Schmidt, K. Karaghiosoff and P. Knochel, Chem. Sci., 2022, 13, 44–49 RSC; (b) A. Kremsmair, H. R. Wilke, J. H. Harenberg, B. R. G. Bissinger, M. M. Simon, N. Alandini and P. Knochel, Angew. Chem. Int. Ed., 2023, 62, e202214377 ( Angew. Chem. , 2023 , 135 , e202214377 ) CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2258909–2258912. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc02893c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |