DOI:
10.1039/D3SC01995K
(Edge Article)
Chem. Sci., 2023,
14, 7569-7580
Stereoselective alkyl C-glycosylation of glycosyl esters via anomeric C–O bond homolysis: efficient access to C-glycosyl amino acids and C-glycosyl peptides†
Received
18th April 2023
, Accepted 18th June 2023
First published on 20th June 2023
Abstract
C-Glycosyl peptides possess excellent metabolic stability and therapeutic properties and thus play critical roles in biological studies as well as drug discoveries. However, the limited accessibility of C-glycosyl amino acids has significantly hindered the broader research of their structural features and mode of action. Herein, for the first time we disclose a novel visible-light-driven radical conjugate addition of 1,4-dihydropyridine (DHP)-derived glycosyl esters with dehydroalanine derivatives, generating C-glycosyl amino acids and C-glycosyl peptides in good yields with excellent stereoselectivities. Redox-active glycosyl esters, as readily accessible and bench-stable radical precursors, could be easily converted to glycosyl radicals via anomeric C(sp3)–O bond homolysis under mild conditions. Importantly, the generality and practicality of this transformation were fully demonstrated in >40 examples including 2-dexosugars, oligosaccharides, oligopeptides, and complex drug molecules. Given its mild reaction conditions, robust sugar scope, and high anomeric control and diastereoselectivity, the method presented herein could find widespread utility in the preparation of C(sp3)-linked sugar-based peptidomimetics.
Introduction
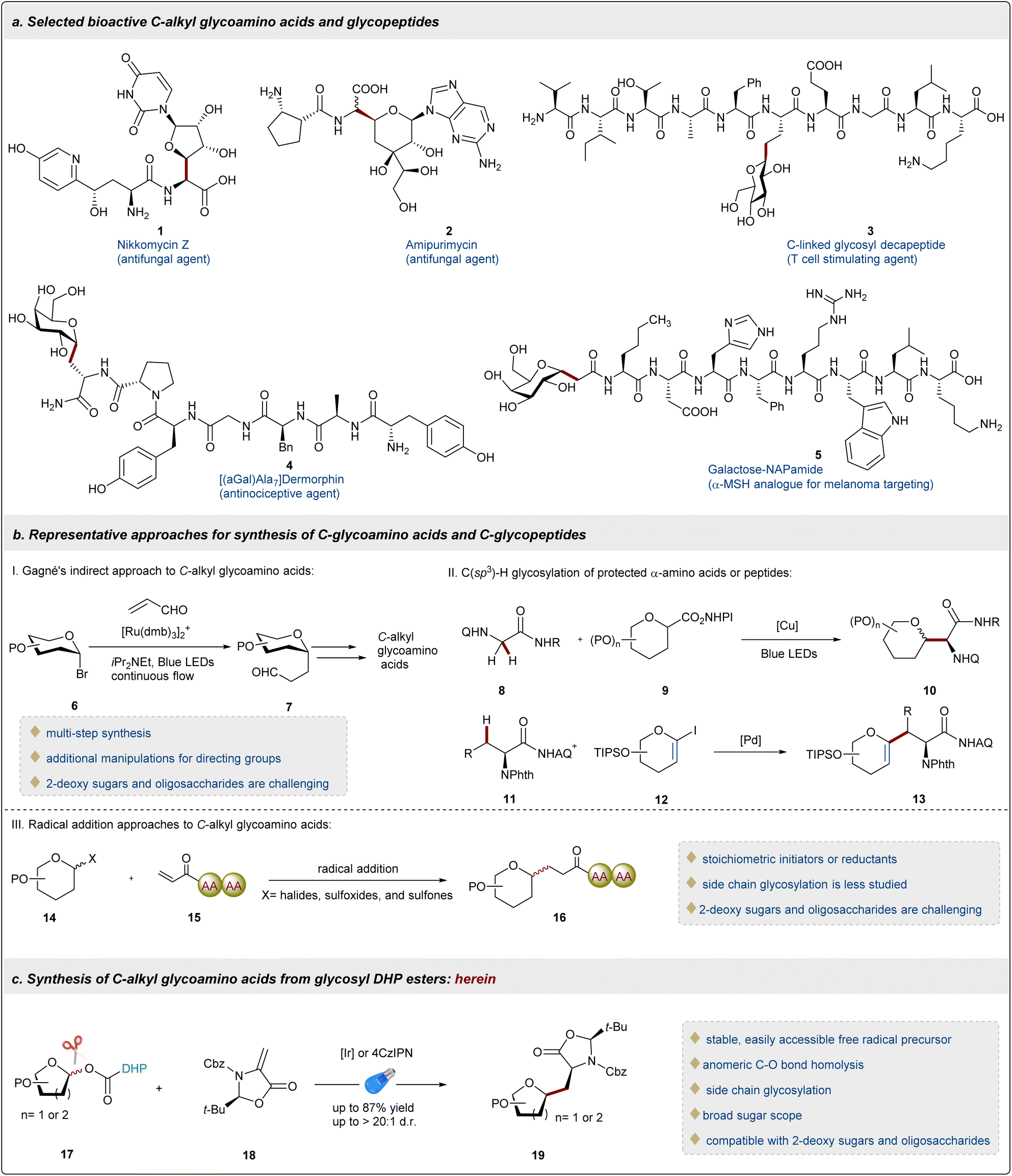
Glycoproteins are widely present on the cell surface and play irreplaceable roles in a wide range of biological processes, such as immune response, signal transduction, and cell adhesion.1,2 In nature, most of the glycopeptides/proteins bear O/N-glycosidic linkages, and the only known form of C-glycosidic linkage is found to be the Manα1-C2-Trp motif.3 While glycopeptides/proteins hold promise as novel therapeutic and diagnostic tools, the labile O/N-glycosidic bonds are susceptible to hydrolysis and enzymatic cleavage potentially hindering their widespread utility in drug discovery.4 As a result, it is of great interest to replace the hydrolysable O/N-glycosidic linkage with a more stable C-, S- or Se-linked analogue.5,6 Given their remarkable resistance to hydrolysis and enzymatic degradation and innate drug-like properties, C-glycosides have emerged as a robust surrogate of the native O/N-glycoconjugate with enhanced pharmacokinetic properties.7 Within this class, C-alkyl glycoamino acids and glycopeptides are becoming the focus of various studies of biological functions and disease models, as well as in the design of therapeutic peptides.8 For example, peptidyl nucleoside antibiotics, including Nikkomycin Z 19 and Amipurimycin 2,10 show excellent antimycotic activities. Furthermore, glycosylation as an effective synthetic strategy can significantly improve the bioavailability of endogenous peptides,11 for instance, C-linked glycosyl decapeptide 3,12 [(αGal)Ala7]dermorphin 4,13 and galactosylated-NAPamide 5 (ref. 14) (Fig. 1a).
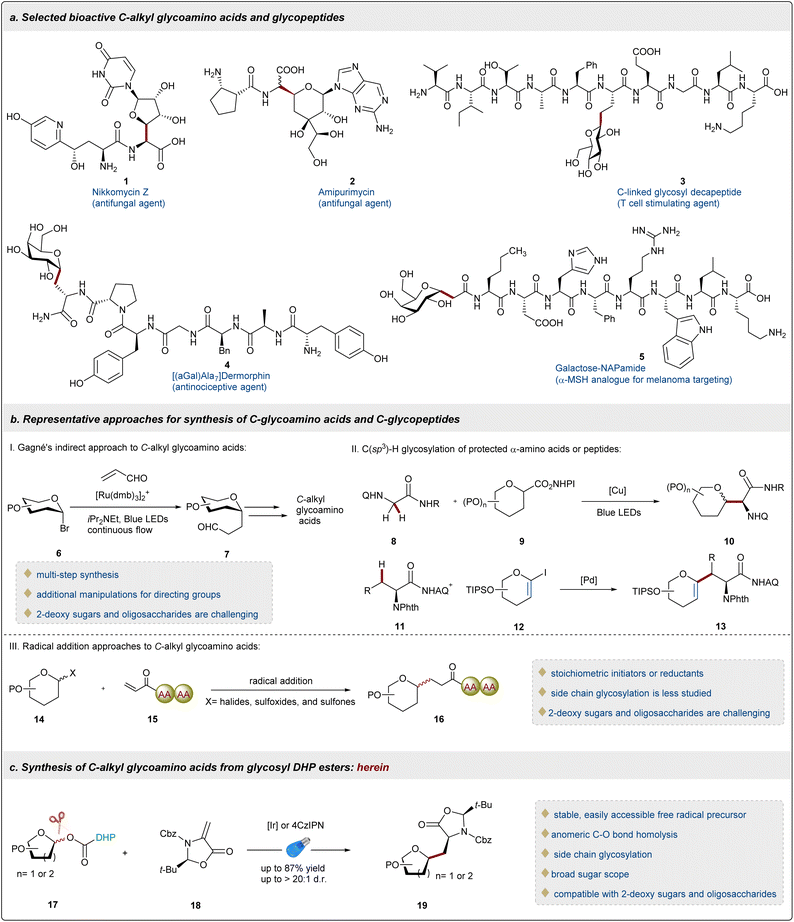 |
| | Fig. 1 Selected bioactive C-alkyl glycopeptides and their synthetic strategies. | |
In comparison to the well-developed O/N-glycosylation2,3,15 or S/Se-glycosylation16–19 of amino acids or peptides, C-glycosylation of amino acids or peptides is much more challenging and has lagged far behind.20 Although outstanding progress has been recently made in the synthesis of C(sp2)-linked glycoamino acids and glycopeptides,21–23 the precise preparation of C-alkyl analogues remains a longstanding challenge.20 Given the high-density functionalization of peptides and acidic nature of the α-position of amino acids, the conventional methods to prepare C-alkyl glycosides become frequently futile for C-alkyl glycoamino acids or glycopeptides.24,25 Gagné and co-workers reported an indirect approach to C-alkyl glycoamino acids using C-glycosyl aldehydes 6 as key intermediates (Fig. 1b),26 which promoted the research on the photoredox-mediated synthesis of C-glycoamino acids. Recently, significant efforts have been directed toward the preparation of C-glycosyl glycines 10, with key contributions from Wang,27 Wang,28 Xu,29 and Liang.30 Of particular note, these elegant approaches are applicable to C-glycosyl alanines 13. Notable advances have been achieved in Pd-catalyzed β-C(sp3)–H glycosylation of amino acids and peptides with 1-iodoglycals 12, as reported independently by Liu31 and Ackermann32 using triazolyldimethylmethyl triazole and aminoquinoline as directing groups, respectively (Fig. 1b). This powerful strategy may not be compatible with oligosaccharides due to the intrinsic instability of 1-iodoglycals 12, and the further hydroboration/oxidation reaction of C-alkyl-substituted glycals 13 obtaining sugars in a mixture of diastereomers.31 Recently, Niu33 and Koh34,35 have developed novel methods for the synthesis of C-alkyl glycopeptides through the Giese reaction of glycosyl radicals to peptides 15 on the N-terminus, utilizing novel glycosyl sulfoxides, sulfones and common glycosyl chlorides as glycosyl precursors 14, respectively. However, the use of stoichiometric amounts of radical initiators or reductants and the synthesis of 2-deoxy saccharides and oligosaccharides remain crucial issues. Therefore, a general and concise method to attain alkyl C-amino acids or glycopeptides from readily available starting materials under mild conditions is still a formidable challenge. Radical glycosylation has become a highly active area of research, owing to its excellent chemoselectivity and good anomeric control.5,36 Various unnatural glycosyl derivatives are extensively explored as glycosyl radical precursors, including glycosyl halides,37 xanthates,38 sulfones,39 glycosyl-based DHPs,40,41 glycosyl carboxylic acids,42,43 redox-active esters,27,44 and anomeric nucleophiles.45,46 It is worth noting that multi-step synthesis, inherent instability, special reaction conditions and narrow sugar scope remain as several of the key obstacles reducing the practical value of these unnatural glycosyl derivatives. Undoubtedly, the ideal generation of glycosyl radicals is the direct homolytic cleavage of anomeric C(sp3)–OH bonds of native carbohydrates. However, anomeric C(sp3)–O bond homolysis poses a significant challenge because the BDE of the anomeric C(sp3)–OH (∼99 kcal mol−1 for furanose and ∼101 kcal mol−1 for pyranose) is even higher than that of an alcohol (∼96 kcal mol−1).47 In general, various redox auxiliaries that include xanthates,48,49 phosphites,50 oxalates,51,52 carboxylates,53,54 and ethers55 have been employed to transform hydroxyl groups into activating groups to generate C-radical intermediates under electron transfer or light irradiation. Unfortunately, most of the above redox auxiliaries are not suitable for carbohydrate substrates, possibly due to the instability of the corresponding glycosyl esters at the anomeric position.33,45,46,56 Recently, dihydropyridine (DHP) as a versatile activating group has been successively applied in various radical glycosylation reactions via C–C bond cleavage at the C4 or C5 position of sugars.40,41,57 To homolytically cleave challenging anomeric C(sp3)–O bonds, DHPs were used as redox auxiliaries to obtain stable glycosyl esters and then C-aryl and C-acyl glycosides via sequential C–C and C–O bond cleavage under nickel/photoredox dual catalysis were successfully achieved.53,54 In comparison to most of the glycosyl radical precursors, the redox glycosyl DHP esters are very impressive because of their easy accessibility, stability, and compatibility with 2-deoxysugars. However, studies on C(sp3)-glycosylation of DHP-derived glycosyl esters and the reactivity and compatibility of DHP groups in complex oligosaccharides remain unexplored.
In order to provide a meaningful solution to these fundamental issues and in line with our interest on the precise preparation of carbohydrates and modification of complex peptides,36,58 we herein disclose the first example of photoredox-catalyzed radical conjugated addition of DHP-derived glycosyl esters 17 with highly reactive dehydroalanine (Dha) 18 for the synthesis of C(sp3)-glycosyl amino acids and peptides via anomeric C–O bond homolysis (Fig. 1c). This novel glycosylation method advances the development of catalytic methods applicable to complex and challenging carbohydrate substrates and complements the current state-of-the art methodologies.
Results and discussion
Reaction development
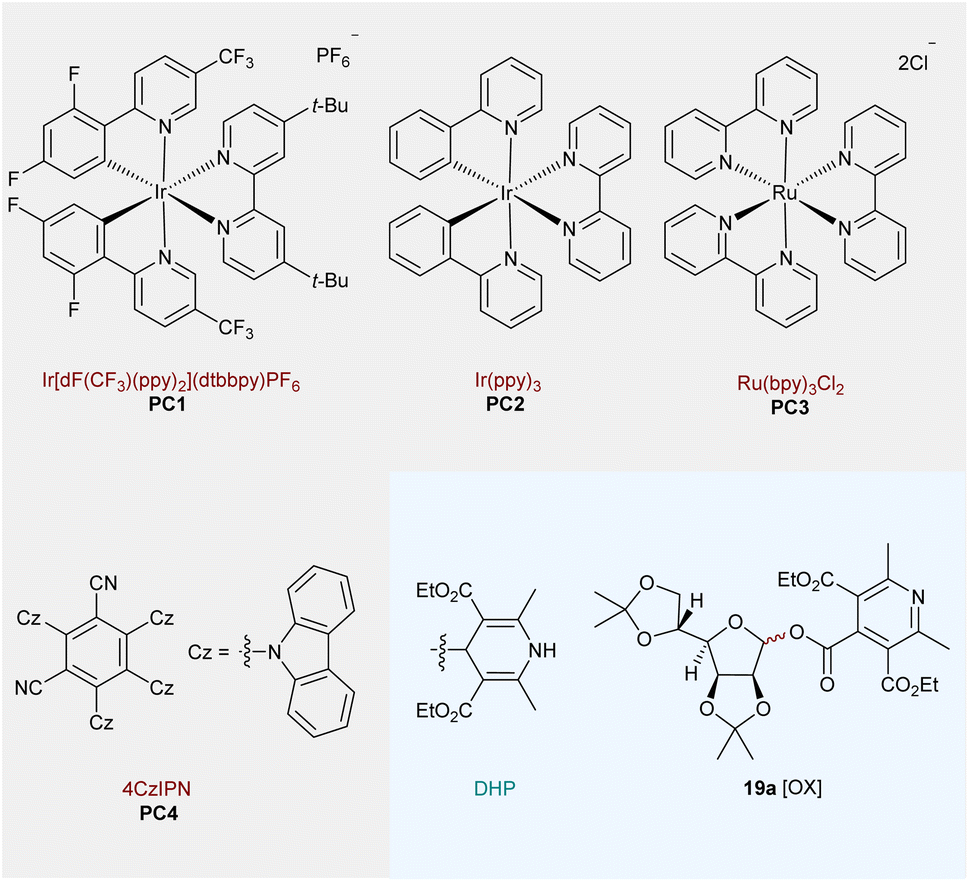
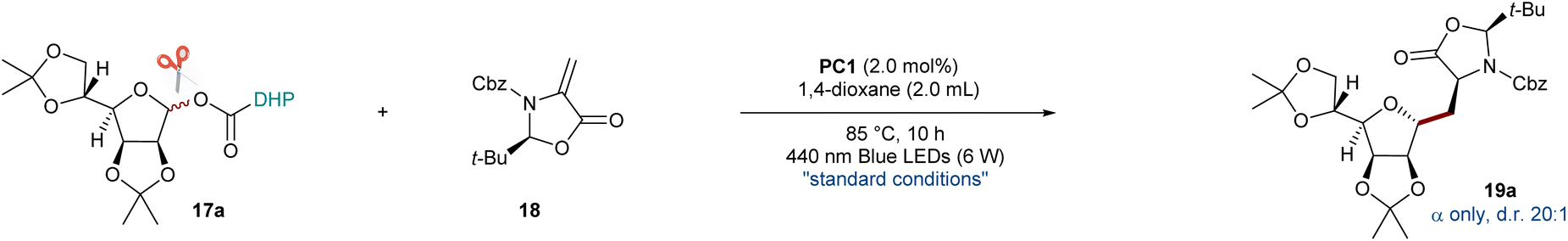
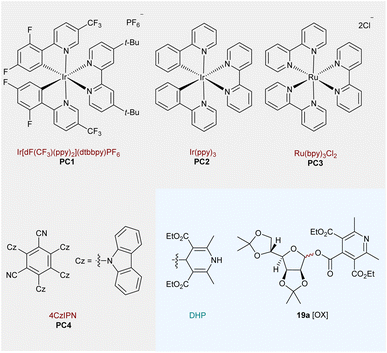
The visible-light-driven radical conjugate addition (RAC) of Dhas is a powerful tool to access nonproteinogenic amino acids, peptides, and proteins.59 In particular, diastereoselective radical addition to chiral Karady–Beckwith Dhas is becoming an attractive method for the precise preparation of α-amino acid derivatives.60 Based on the high diastereocontrol, the model reaction of O-mannofuranosyl ester 17a (1.5 equiv) with Karady–Beckwith Dha 18 (1.0 equiv) to afford C-alkyl glycoamino acid 19a was used to optimize reaction conditions (Table 1). To our delight, the desired C-alkyl glycoamino acid 19a was isolated in 69% yield with excellent anomeric control (only α isomer) and diastereoselectivity around the oxazolidinone auxiliary (d.r.>20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) after 10 h of irradiation with blue light emitting diodes (LEDs, 6 W) in the presence of a common photocatalyst Ir[dF(CF3)ppy]2(dtbbpy)PF6PC1 in 1,4-dioxane (entry 1). When MeCN or THF was used as an alternative solvent, the isolated yields were reduced to 52% and 33%, respectively (entries 2–3). Further modifications of the reaction conditions, such as the modified ratio of 17a:18 (entry 4), had only negative effects on the reaction yields. Similarly, reduced or elevated temperatures reduced the yield of 19a (entry 5). We hypothesize that lower temperature might not provide sufficient driving force for the sequential C–C and C–O bond cleavage of glycosyl DHP esters (confirmed by the recovery of 17a), whereas higher temperatures result in the oxidation of the glycosyl donor to 19a[ox] bearing a pyridine motif. We further probed the reaction conditions with different photocatalysts under blue LED irradiation (entries 6–8). While no reaction was observed using Ir(ppy)3 (PC2) and Ru(bpy)3Cl2 (PC3), about 7% of 19a was isolated when inexpensive 4CzIPN (PC4) was employed. To our delight, simply changing the solvent to MeCN led to a 64% yield of the desired product using PC4 as the photocatalyst (entry 9). Subsequently, increasing the amount of PC4 and extending the reaction time further improved the yields to 70% and 82%, respectively (entries 10 and 11). Extended exposure of the reaction to light radiation and changing the concentration to 0.67 M led to further improvement of the isolated yield of 19a to 85% (entry 12). Using the same strategy, the desired glycosyl amino acid 19a was obtained in an 80% yield using PC1 as a photocatalyst after 10 h of irradiation (entry 13). Notably, the radical addition reaction can also be conducted in a 3:1 mixture of MeCN and H2O with 78% isolated yield of 19a (for details, see the ESI†). Finally, control experiments demonstrated the necessity of a photocatalyst and visible light for the alkyl C-glycosylation reaction to take place (entry 14).
:1) after 10 h of irradiation with blue light emitting diodes (LEDs, 6 W) in the presence of a common photocatalyst Ir[dF(CF3)ppy]2(dtbbpy)PF6PC1 in 1,4-dioxane (entry 1). When MeCN or THF was used as an alternative solvent, the isolated yields were reduced to 52% and 33%, respectively (entries 2–3). Further modifications of the reaction conditions, such as the modified ratio of 17a:18 (entry 4), had only negative effects on the reaction yields. Similarly, reduced or elevated temperatures reduced the yield of 19a (entry 5). We hypothesize that lower temperature might not provide sufficient driving force for the sequential C–C and C–O bond cleavage of glycosyl DHP esters (confirmed by the recovery of 17a), whereas higher temperatures result in the oxidation of the glycosyl donor to 19a[ox] bearing a pyridine motif. We further probed the reaction conditions with different photocatalysts under blue LED irradiation (entries 6–8). While no reaction was observed using Ir(ppy)3 (PC2) and Ru(bpy)3Cl2 (PC3), about 7% of 19a was isolated when inexpensive 4CzIPN (PC4) was employed. To our delight, simply changing the solvent to MeCN led to a 64% yield of the desired product using PC4 as the photocatalyst (entry 9). Subsequently, increasing the amount of PC4 and extending the reaction time further improved the yields to 70% and 82%, respectively (entries 10 and 11). Extended exposure of the reaction to light radiation and changing the concentration to 0.67 M led to further improvement of the isolated yield of 19a to 85% (entry 12). Using the same strategy, the desired glycosyl amino acid 19a was obtained in an 80% yield using PC1 as a photocatalyst after 10 h of irradiation (entry 13). Notably, the radical addition reaction can also be conducted in a 3:1 mixture of MeCN and H2O with 78% isolated yield of 19a (for details, see the ESI†). Finally, control experiments demonstrated the necessity of a photocatalyst and visible light for the alkyl C-glycosylation reaction to take place (entry 14).
Table 1 Optimization of the reaction conditionsa
|

|
| Entry |
Variation from standard conditions |
Yield |
|
|
Standard reaction conditions: 17a (0.30 mmol), 18 (0.20 mmol), Ir[dF(CF3)(ppy)2](dtbbpy)PF6PC1 (2.0 mol%), 1,4-dioxane (3.0 mL), 440 nm blue LEDs (6 W), 85 °C, 10 h, N2, and isolated yields.
PC4 (2.5 mol%) was used.
MeCN (3.0 mL) was used; d.r. was determined by 1H NMR analysis of crude reaction mixtures.
|
| 1 |
None |
69 |
|
| 2 |
MeCN instead of 1,4-dioxane |
52 |
| 3 |
THF instead of 1,4-dioxane |
33 |
| 4 |
A ratio of 17a:18 (1:15) was used |
36 |
| 5 |
60 °C, 70 °C or 100 °C instead of 85 °C |
35–63 |
| 6 |
PC2 instead of PC1 |
n.d. |
| 7 |
PC3 instead of PC1 |
n.d. |
| 8 |
PC4 instead of PC1 |
7 |
| 9 |
PC4 and MeCN were used |
64 |
| 10 |
PC4 and MeCN were used |
70b |
| 11 |
PC4, MeCN and 20 h were used |
82b |
| 12 |
PC4, MeCN and 20 h were used |
85b,c |
| 13 |
3.0 mL of 1,4-dioxane instead of 2.0 mL |
80 |
| 14 |
No PC or light |
0 |
Scope of monosaccharides
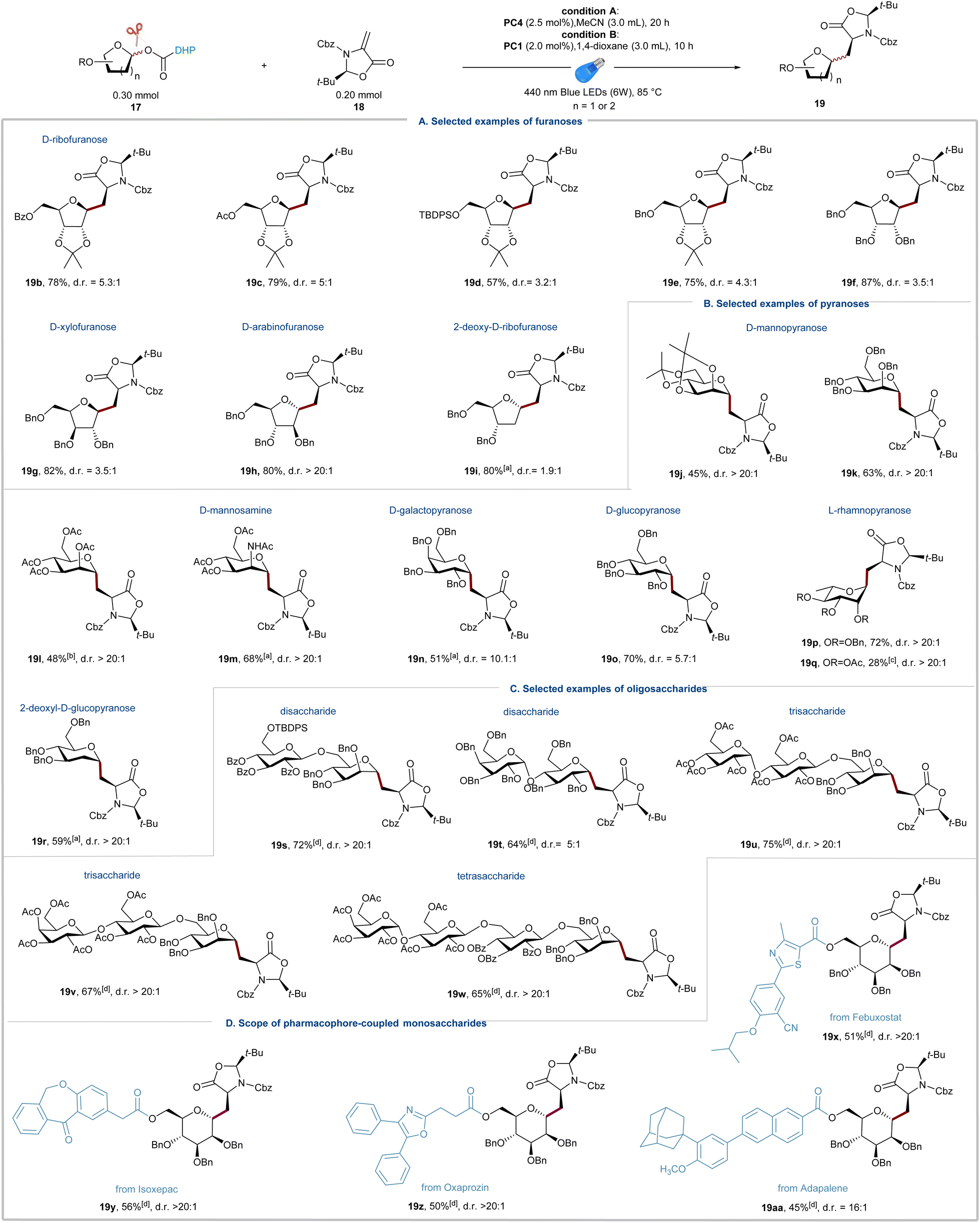
With the optimal reaction conditions established, we next focused on testing the scope of DHP-derived glycosyl esters. As shown in Fig. 2, a broad range of structurally diverse glycosyl DHP esters derived from various monosaccharides and oligosaccharides underwent the radical addition smoothly and produced the corresponding alkyl C-glycosylamino acids 19b–19w in moderate to good yields and excellent diastereoselectivity around the oxazolidinone auxiliary. D-Ribofuranoses with various common protecting groups, such as benzoyl (19b), acetyl (19c), silyl (19d), and benzyl (19e and 19f), readily underwent the alkyl C-glycosylation, affording 19b–19f in 57–87% isolated yields. Other glycosyl NHP esters derived from common furanoses, including D-xylofuranose (19g), D-arabinofuranose (19h), and 2-deoxy-D-ribofuranose (19i), produced the corresponding radical addition products (19g–19i) in good yields. The opposite anomeric stereoselectivity between D-arabinofuranose 19h and 2-deoxy-D-ribofuranose 19i reflects a dominating effect of the C2 substituent group on the stereochemical outcome because of the steric hindrance.34,54 These results stand in contrast to the reported methods on radical addition of glycosyl radicals,33–35 and 2-deoxy-D-ribofuranosyl NHP esters show good reactivity and selectivity under the standard conditions due to their excellent stability and generation of glycosyl radicals under mild photoredox conditions. Subsequently, a variety of common pyranoses, such as D-mannopyranose, D-mannosamine, D-galactopyranose, L-rhamnopyranose, and 2-deoxy-D-glucopyranose, were readily transformed into alkyl C-glycoamino acids 19j–19r with good to excellent stereoselectivities (Fig. 2b). The suboptimal yields of fully protected monosaccharides with disarmed protected groups, such as 19l, 19m and 19q, could be attributed to the low nucleophilicity of the corresponding glycosyl radicals. Importantly, 2-deoxy-D-glucopyranose, proved to be a competent substrate for the radical addition with high α-selectivity despite the lack of the controlling C2 substituent. The stereochemistry of all cross-coupling products was further confirmed by 2D COSY and HSQC analyses (see the ESI† for details). In general, the stereochemical outcome of desired products could be explained by a combination of the anomeric and steric effects.61,62
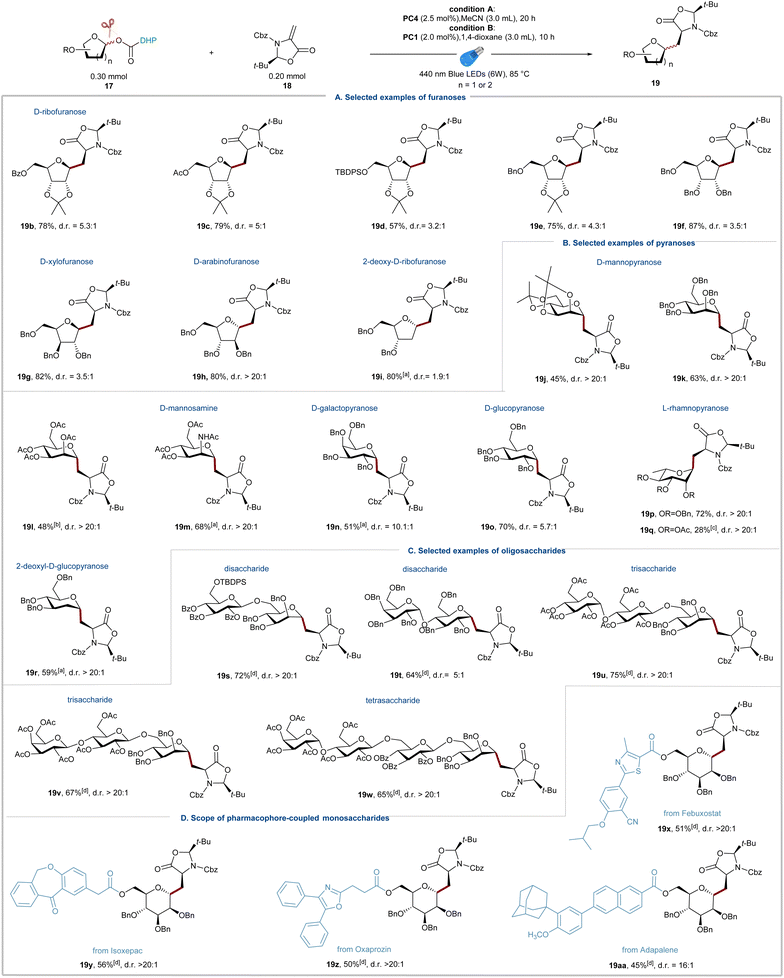 |
| | Fig. 2 Scope of C-alkyl glycosylation with DHP-derived glycosyl esters. (a) Scope of furanoses. (b) Scope of pyranoses. (c) Scope of oligosaccharides. (d) Scope of pharmacophore-coupled monosaccharides. 19b–19f, 19h, 19o, and 19p used conditions A; 19g, 19i–19n, and 19q–19aa used conditions B; a20 h was used; b24 h was used; c17 (0.15 mmol), 18 (0.10 mmol), 1,4-dioxane (1.5 mL), and 20 h was used; d17 (0.15 mmol), 18 (0.10 mmol), 1,4-dioxane (3.0 mL), and 24 h. d.r. was determined by 1H NMR analysis of crude reaction mixtures. | |
Scope of oligosaccharides
To further expand the scope of the alkyl C-glycosylation reaction, we then investigated the reactions of DHP esters derived from complex oligosaccharides (Fig. 2c). As mentioned above, most of the current methods for the formation of alkyl C-glycosides are limited to the reactions with monosaccharides, and the direct stereoselective C-glycosylation with free radical precursors of oligosaccharides and Michael acceptors is rare. These limitations could be attributed to the incompatibility of free radical precursors with oligosaccharide synthesis and the lack of effective methods to generate and stabilize oligosaccharide radicals. We found that oligosaccharide DHP esters could be easily prepared on a gram scale by simple coupling of DHP acids and C1-hemiacetals (24–53%) and remain bench stable for several months. To our delight, disaccharide DHP esters could be engaged in cross-coupling reactions without any significant modifications of the standard conditions, allowing for the formation of alkyl C-disaccharides 19s and 19t in 72% and 64% isolated yields with good to excellent stereoselectivity. The scope of the radical glycosylation could be further extended to more complex oligosaccharides and the radical C-glycosylation reactions forming alkyl C-trisaccharides (19u and 19v) and a tetrasaccharide (19w) produced the corresponding products in yields exceeding 65% with d.r. >20:1.
Scope of pharmacophore-coupled monosaccharides
Besides the well-known antibody–drug conjugates (ADCs), peptide–drug conjugates (PDCs) are gaining recognition as a novel modality for targeted drug delivery with improved efficacy and reduced side effects for cancer therapy.63 The linker between the peptide and drug plays a critical role in the circulation time of the conjugate and release of the drug for full activity at the target site. Therefore, the research to develop effective linkers for the design of PDCs is important and promising. Herein, we were intrigued by the viability of the visible-light driven radical C-glycosylation of pharmacophore-coupled glycosyl DHP esters and substrate 18 (Fig. 2d). To our delight, the bioconjugation reactions using sugars as linkers proved reliable and a series of glycosyl DHP esters derived from D-ribofuranose and D-mannopyranose could be coupled with commercial drugs, including Febuxostat, Isoxepac, Oxaprozin, and Adapalene, affording the corresponding products 19x–19aa in 45–56% yield. The above results revealed that our method exhibits remarkable potential application in rapid assembly of glycopeptide drug molecules.
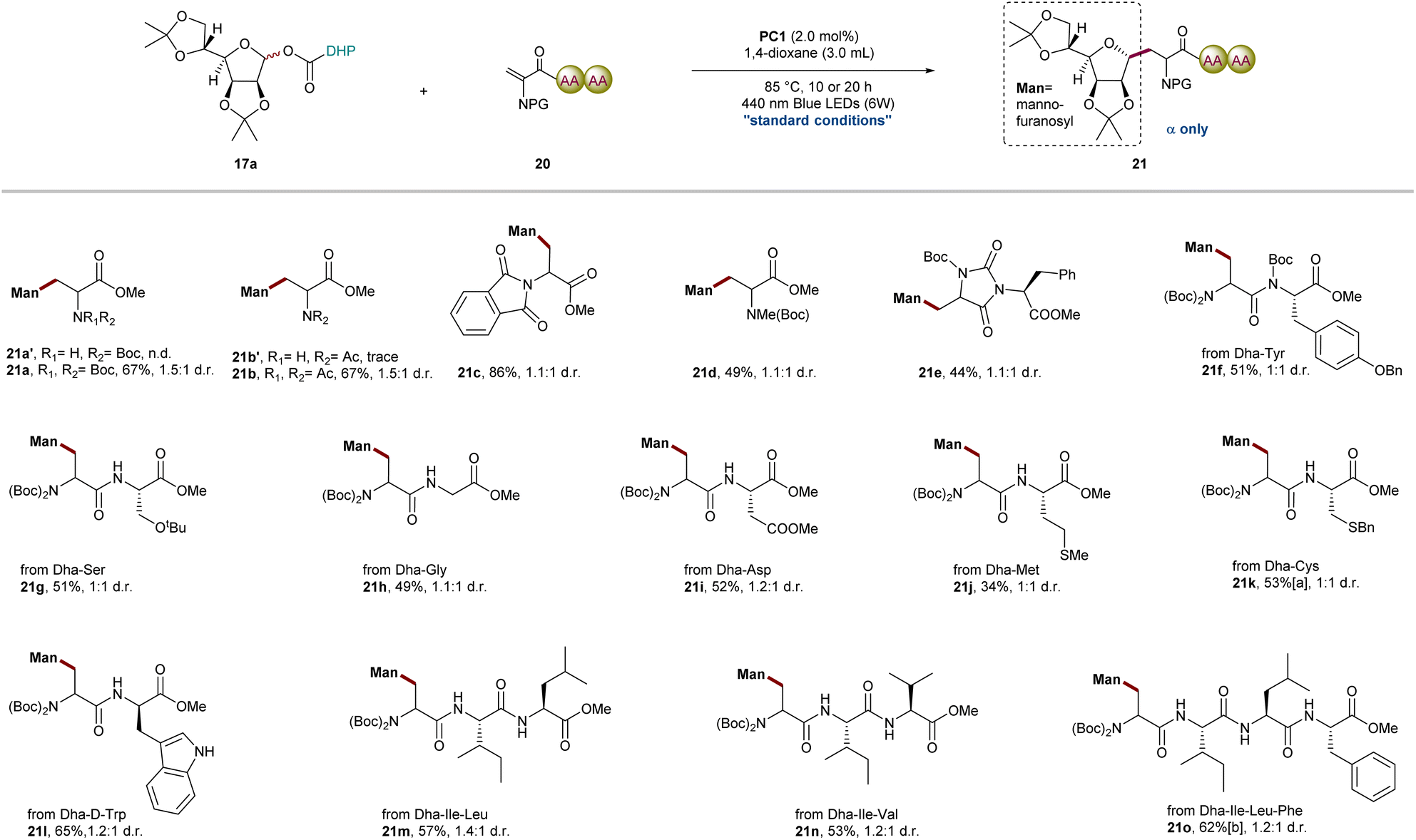
Scope of Dha amino acids and peptides
Encouraged by the above results, we next investigated the radical addition of O-mannofuranosyl esters 17a to various amino acids and Dha peptides (Fig. 3). We hypothesized that the efficiency of radical addition will be correlated with different protecting groups located on the nitrogen atom with the electron-withdrawing group promoting the addition of a nucleophilic radical. In addition to chiral Dha 18, bis-N-Boc-protected, bis-N-Ac-protected, Boc-N-methyl protected and bis-N-phthalimide protected Dha were all effectively transformed into the desired product in 49–86% isolated yields; however, trace product was detected when N-Boc-protected or N-Ac-protected Dha was used. This observation is consistent with the prior studies highlighting the importance of the torsional angle [ω(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–N–C)] between the plane of the olefin and the carbamate that might contribute to the disparate reactivity.64 The introduction of two bulky N-protected groups or a cyclic structure weakens the conjugation effect of the nitrogen lone pair into the adjoining π system and significantly decreases the electronic density of the terminus of the olefin to improve the efficiency of radical addition. Capitalizing on these results, we then studied a series of Dha-containing dipeptides and tripeptides, such as cyclic Dha–Phe 20e, Dha–Tyr 20f, Dha–Ser 20g, Dha–Gly 20h, Dha–Asp 20i, Dha–Met 20j, Dha–Cys 20k, Dha–D-Trp 20l, Dha–Ile-Leu 20m, and Dha–Ile-Val 20n, all of which smoothly deliver the corresponding mannofuranosyl peptides in acceptable yields (34–65%) with excellent anomeric selectivity and chemoselectivity. Importantly, this mild radical C-glycosylation between oligopeptides such as tetrapeptide(Dha–IIe-Leu–Phe) and sugar DHP ester was also conducted in 62% yield, which further highlighted the generality of this method in late-stage glycodiversification of complex oligopeptides.19,33 As we all know, radical additions to Dha derivatives often result in mixtures of diastereomers, presenting a significant challenge in peptide modification. In contrast to the highly stereoselective outcomes observed with chiral Karady-Beckwith Dha 18, peptides containing Dha exhibit moderate diastereoselectivity due to the limited steric hindrance and electronic effects. However, it is worth emphasizing that recent studies have demonstrated the potential of catalytic systems composed of transition metals and chiral ligands to achieve stereoselective radical additions to peptides containing Dha.65
C–N–C)] between the plane of the olefin and the carbamate that might contribute to the disparate reactivity.64 The introduction of two bulky N-protected groups or a cyclic structure weakens the conjugation effect of the nitrogen lone pair into the adjoining π system and significantly decreases the electronic density of the terminus of the olefin to improve the efficiency of radical addition. Capitalizing on these results, we then studied a series of Dha-containing dipeptides and tripeptides, such as cyclic Dha–Phe 20e, Dha–Tyr 20f, Dha–Ser 20g, Dha–Gly 20h, Dha–Asp 20i, Dha–Met 20j, Dha–Cys 20k, Dha–D-Trp 20l, Dha–Ile-Leu 20m, and Dha–Ile-Val 20n, all of which smoothly deliver the corresponding mannofuranosyl peptides in acceptable yields (34–65%) with excellent anomeric selectivity and chemoselectivity. Importantly, this mild radical C-glycosylation between oligopeptides such as tetrapeptide(Dha–IIe-Leu–Phe) and sugar DHP ester was also conducted in 62% yield, which further highlighted the generality of this method in late-stage glycodiversification of complex oligopeptides.19,33 As we all know, radical additions to Dha derivatives often result in mixtures of diastereomers, presenting a significant challenge in peptide modification. In contrast to the highly stereoselective outcomes observed with chiral Karady-Beckwith Dha 18, peptides containing Dha exhibit moderate diastereoselectivity due to the limited steric hindrance and electronic effects. However, it is worth emphasizing that recent studies have demonstrated the potential of catalytic systems composed of transition metals and chiral ligands to achieve stereoselective radical additions to peptides containing Dha.65
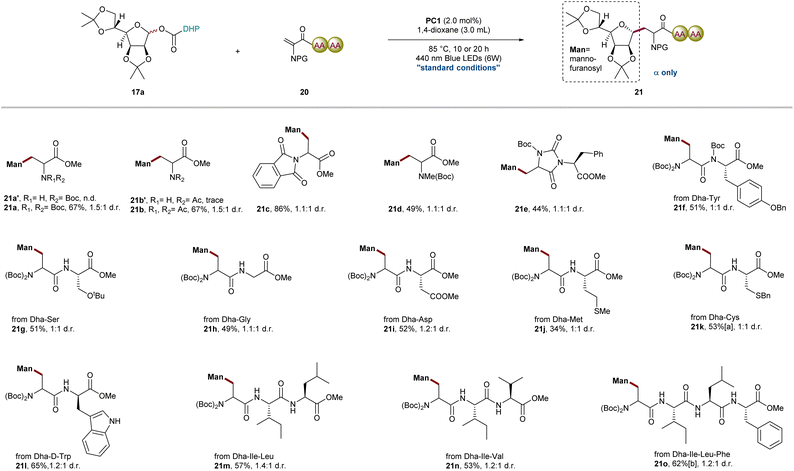 |
| | Fig. 3 Scope of Dha amino acids and peptides. General reaction conditions: 17a (0.30 mmol), 20 (0.20 mmol), PC1 (2.0 mol%), 1,4-dioxane (3.0 mL), blue LEDs (6 W), 85 °C, 10 h, N2, and isolated yields; a20h; b20o (0.10 mmol) and 24 h. d.r. was determined by 1H NMR analysis of crude reaction mixture. | |
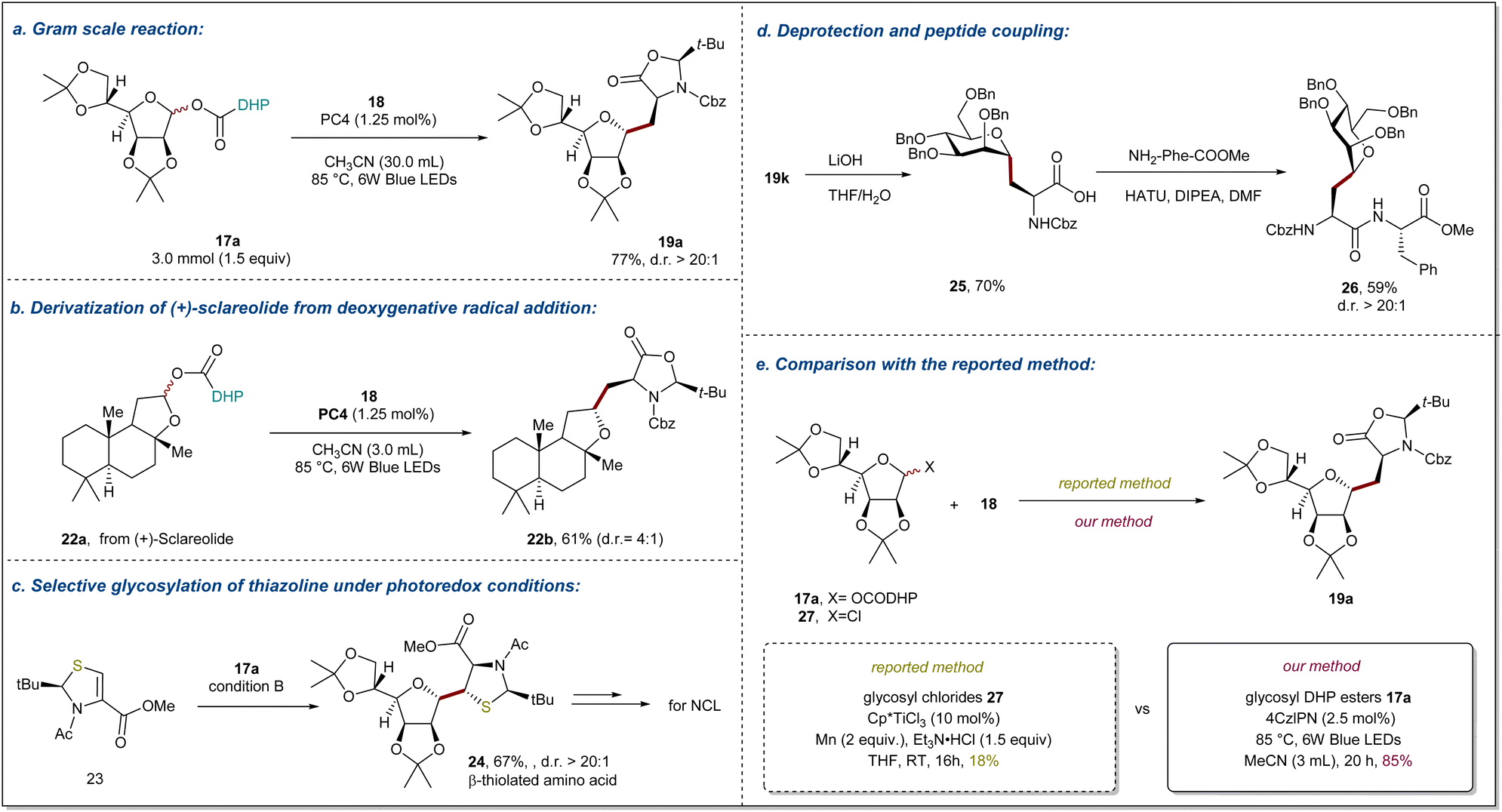
Application
The practicality of this method was demonstrated by a gram-scale synthesis of 19a (77% yield in a 3.0 mmol scale reaction) as shown in Fig. 4a. One of the objectives of this study is to provide an efficient method for the preparation of alkyl C-glycosyl amino acids/peptides and establish a general method for the late-stage functionalization of complex molecules. To demonstrate the generality of deoxygenative radical addition, we prepared DHP ester 22a derived from natural product (+)-sclareolide and engaged it in visible-light photoredox-catalyzed radical addition to afford 22b in 61% yield with 4:1 d.r (Fig. 4b). It is well known that β-thiolated amino acid derivatives not only play important roles in regulating the function and conformation of proteins, but also serve as important units for peptide design and bioconjugation. However, the difficulty in obtaining enantiopure β-thiolated amino acids with diverse structures is a major limitation to their widespread application. Recently, the Wang group developed an interesting photoredox-catalyzed Giese reaction of chiral thiazolidines to access enantiopure β-thiolated amino acids.66 Inspired by this, we wondered whether our method could be applied to prepare β-thiolated amino acids containing sugar units, which are difficult to obtain using conventional methods. Gratifyingly, new chiral thiazolidines 23 could be smoothly transformed into β-thiolated amino acids containing sugar units 24 (67%) with excellent diastereoselectivity (Fig. 4c). As illustrated in Fig. 4d, the deprotection of the cyclic group on compound 19k with LiOH provided glycosyl amino acids 25 in 70% yield, which can be easily converted into side chain C-glycosylated dipeptides 26 in 59% with >20:1 d.r. under standard peptide coupling conditions. Finally, to further illustrate the effective complementarity of our method to existing methods, the model reaction of glycosyl chlorides 27 using Ti-catalyzed conditions was conducted, providing the desired product in only 18% isolated yield (Fig. 4e).34,35
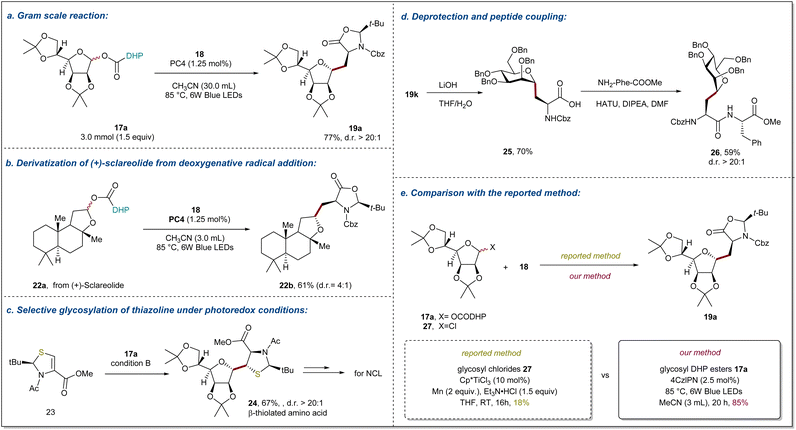 |
| | Fig. 4 Synthetic applications and downstream transformations. (a) Gram scale reaction of 17a. (b) Derivatization of (+)-sclareolide. (c) Radical glycosylation of thiazolines for preparing β-thiolated amino acids. (d) Deprotection of glycosyl amino acids and preparation of glycopeptides via a peptide coupling reaction. (e) Comparison of glycosyl chlorides and glycosyl DHP esters. Reaction details are shown in ESI notes 7.† | |
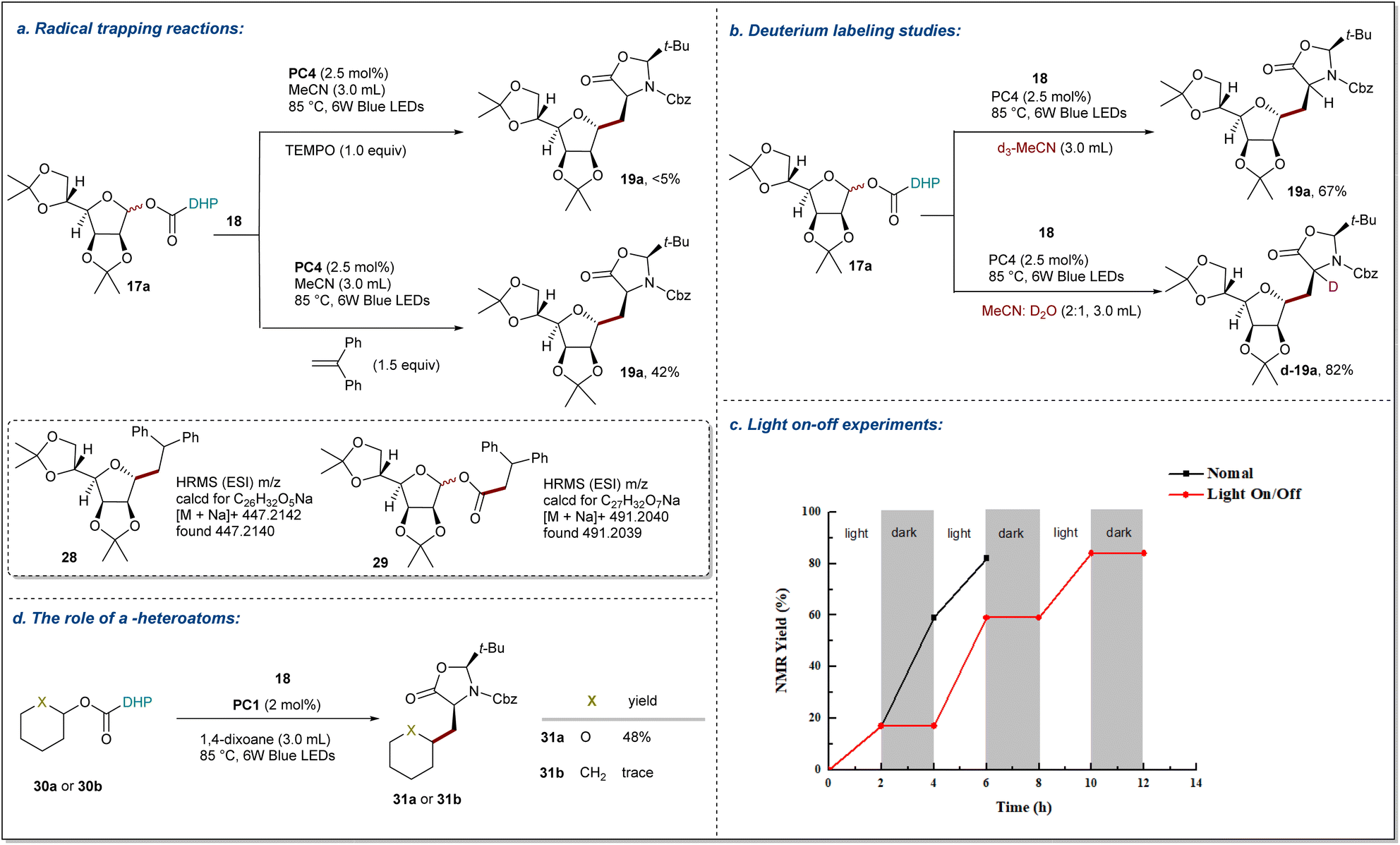
Mechanistic studies
To gain more insights into the reaction mechanism, several control experiments were conducted as outlined in Fig. 5. First, the model reactions were carried out in the presence of radical scavengers TEMPO (2,2,6,6-tetramethyl-1-piperidinyloxy) and DPE (1,1-diphenylethylene), which reduced the yield of 19a to <5% and 42%, respectively (Fig. 5a). In the radical trapping experiments with glycosyl radicals, Tempol glycosides were not detected due to their further decomposition under blue irradiation.67 Gratefully, the radical addition products of glycosyl radicals with DPE 28 and alkoxycarbonyl radicals with DPE 29 are detected by HRMS. Thus, these results suggest that the reaction path of this process is a radical pathway. Next, deuterium labeling experiments to probe the origin of the hydrogen source were performed (Fig. 5b). The model reaction with d3-MeCN as solvent was found to give product 19a in 67% yield with no deuterium incorporation, suggesting that any hydrogen atom transfer with solvent MeCN is less likely. Additionally, in a mixture of MeCN and D2O, d-19a was obtained in 82% yield with >99% deuterium incorporated at the α-site of the amino acid precursor, which indicates that a carbanion might be involved in the transformation. Furthermore, a light on-off experiment was carried out with 17a and 18 (Fig. 5c). We found that 19a was only obtained during the periods of constant irradiation.
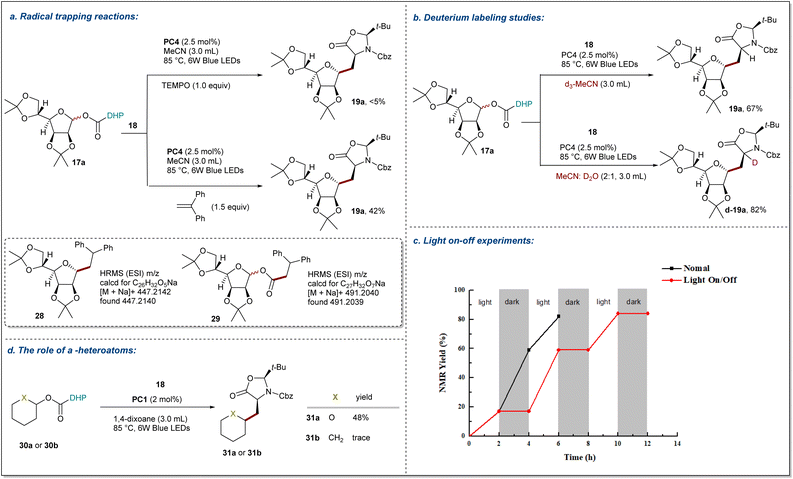 |
| | Fig. 5 Mechanistic studies. (a) Radical-trapping experiment. (b) Deuterium labeling studies. (c) Light on-off experiments. (d) Control experiment supporting the essential role of α-heteroatoms. Reaction details are shown in the ESI notes 8 Mechanistic studies.† | |
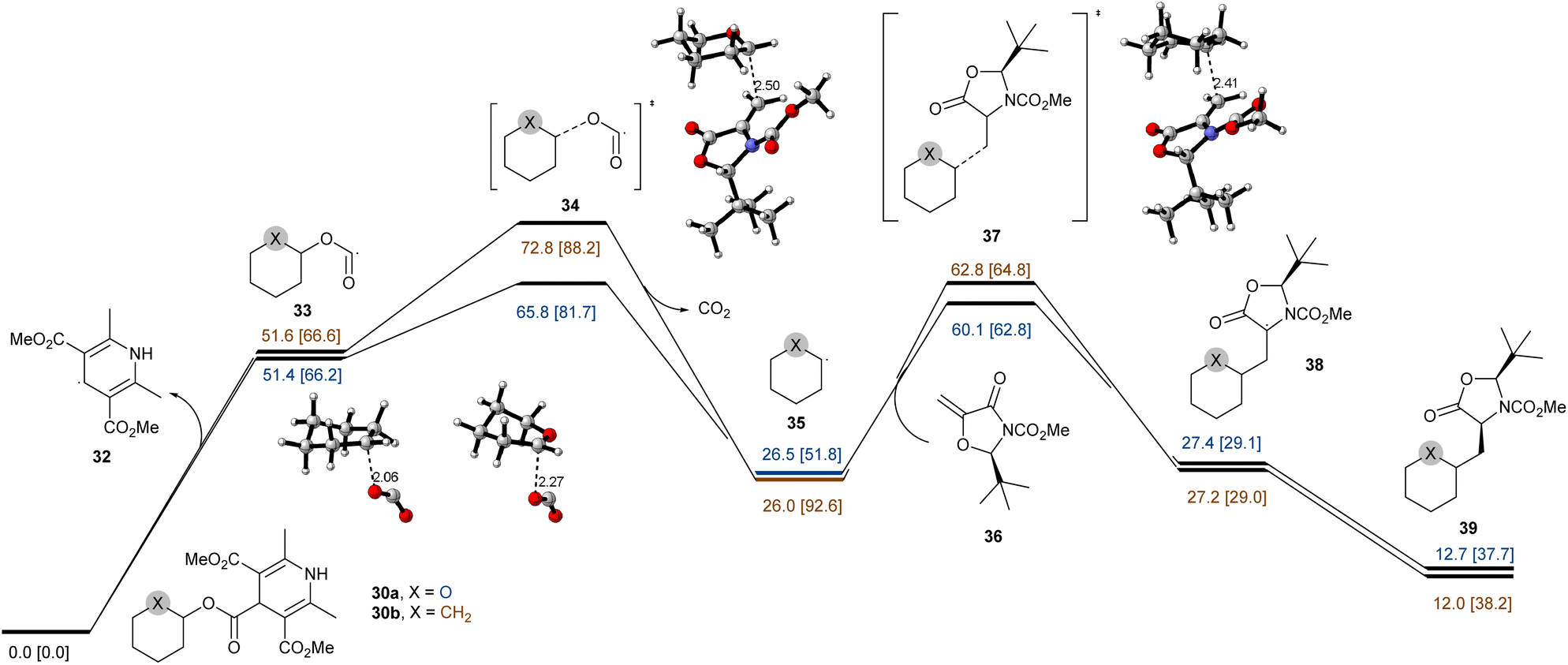
These results indicate that the reaction underwent a catalytic radical process rather than a radical chain pathway. Taken together, these data support the proposal that the NH group of DHP ester serves as a hydrogen atom source. Additionally, a pair of NHP esters 30a and 30b that differ only by the O atom vs. a CH2 group were prepared and subjected to the standard conditions (Fig. 5d). While 30a was smoothly converted to 31a in 48% isolated yield, the all-carbon analog 30b failed to deliver the Giese addition product 31b and was completely consumed due to decomposition. These findings demonstrate that α-oxygen atoms are significant to facilitate the C–O bond homolysis, and the resulting carbon radicals are stabilized by the adjacent ion pair of oxygen atoms.68 These observations were further supported by the DFT calculations for 30a and 30b summarized in Fig. 6. To reduce the computational cost and complexity, we simplify the process of producing free radicals by homolysis of C–O bonds without PC. While the homolytic cleavage of the DHP esters results in cyclohexane and tetrahydropyran radicals 33 of almost identical energies, the fragmentation of 33 is strongly controlled by the presence of a heteroatom which facilitates the expulsion of CO2 by 7.0 kcal mol−1. The resultant radicals show differential NBO atomic charges (0.132 and −0.100 for O and CH2 substituents, respectively) but comparable spin distributions (0.850 and 0.950, respectively), which translate into the fate of the subsequent reactions with a Michael acceptor. The conjugate addition with 36 shows ∼2.7 kcal mol−1 acceleration for the heteroatom-stabilized radicals, whereas the following steps yield structures 38 and 39 with comparable energies slightly favoring the carbocycle over the oxygen-substituted derivative. We attribute the overall efficiency of the Giese addition to the efficiency of the initial radial generation, which can then be engaged in a productive reaction with a competent acceptor.
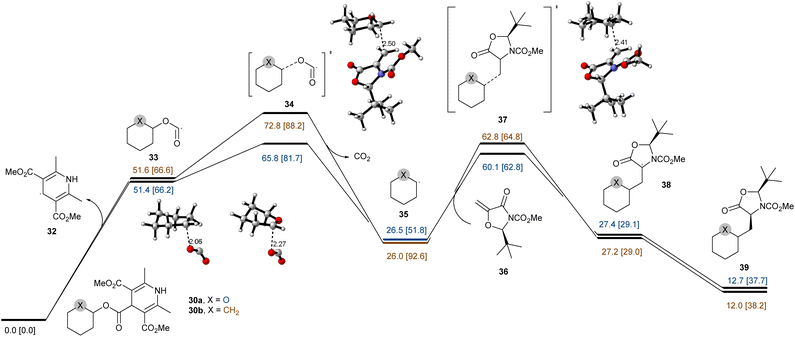 |
| | Fig. 6 Computational studies of the mechanistic aspects of the reaction. Computational analysis of addition reactions for cyclohexane (blue) and tetrahydropyran (yellow) DHP esters calculated at the ωB97XD/6-31+G(d,p)-SDM(dichloromethane)//ωB97XD/6-311+G(d,p)-SDM(dichloromethane) levels of theory. Gibbs free energy (298 K, 1 atm) and enthalpy (in brackets) are reported in kcal mol−1. | |
Based on the above-mentioned preliminary and literature reports,54 the following mechanism for this radical reaction mechanism is proposed (Fig. 7). Under blue LED irradiation, the photocatalyst [PC] is excited to its triple state [PC]*, which promotes the SET oxidation of A to B followed by deprotonation and radical fragmentation to generate the corresponding alkoxycarbonyl radical C and Hantzsch pyridine D. The subsequent decarboxylation is driven by the ejection of CO2 and generates the active glycosyl radical E. The addition of E to chiral Dha gives rise to the radical intermediate F. Finally, the resulting radical F undergoes SET with the reduced photocatalyst [PC]˙− to provide, after protonation, the desired product H and close the catalytic cycle.
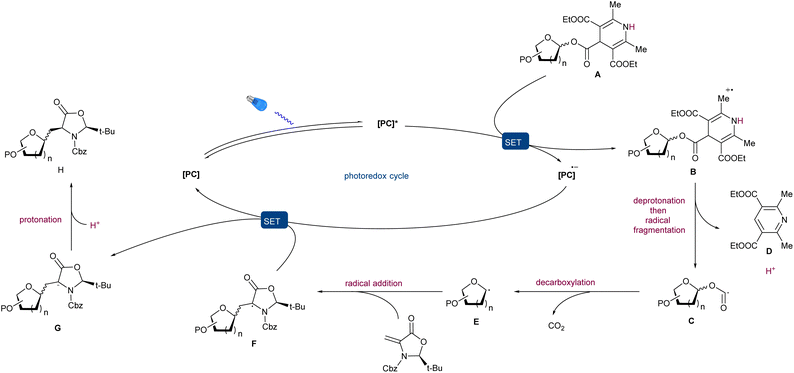 |
| | Fig. 7 Proposed reaction mechanism for alkyl C-glycosylation via C–O bond homolysis. | |
Conclusions
In summary, we have developed the first visible-light photoredox catalyzed C(sp3)-glycosylation of redox-active glycosyl DHP esters with dehydroalanine (Dha) derivatives via anomeric C–O bond homolysis for the concise synthesis of alkyl C-glycoamino acids and C-glycosyl peptides. By taking advantage of mild photocatalytic conditions, the novel alkyl C-glycosylation is compatible with a wide variety of carbohydrate substrates and Dha peptides, providing various C-glycoamino acids and C-glycopeptides in good yields with excellent anomeric control and good diastereoselectivity. Importantly, readily accessible and bench-stable glycosyl DHP esters smoothly participate in the radical glycosylation, allowing for efficient access to C-glycosyl alanine moieties involving challenging sugars, such as 2-deoxysugars and oligosaccharides. Therefore, this alkyl C-glycosylation of glycosyl esters via anomeric C–O bond homolysis not only promotes the development of radical glycosylation, but also provides a powerful tool for glycopeptide therapeutic discoveries. Furthermore, the integration of our method with the emerging technologies in peptide and protein chemistry, such as encoded libraries and direct bioconjugation, is currently ongoing in our laboratory.
Data availability
The authors declare that the data supporting the findings of this study, including experimental details and compound characterization, are available within the article and its ESI† file. All data are available from the corresponding author upon request.
Author contributions
F. Z. conceived and directed the project. A. C., S. Z., Y. H., and Z. Z. performed the experiments and analyzed the data. M. W. carried out computational studies. F. Z. and B. Y. wrote the manuscript with input from all the authors. All authors have read and approved the final version of the manuscript.
Conflicts of interest
A patent application by S. Z., L.-G. X., F. Z., A. C., and Y. H. detailing part of this research was filed through the Patent Office of the People's Republic of China (November 2022). S. Z., L.-G. X., F. Z., A. C., and Y. H. declare no other competing interests. The other authors declare no competing interests.
Acknowledgements
This work was sponsored by the Shanghai Pilot Program for Basic Research - Shanghai Jiao Tong University (Grant No. 21TQ1400210), Fundamental Research Funds for the Central Universities (Grant No. 22X010201631), Excellent Young Scientists Fund Program (Overseas), Natural Science Foundation of Shanghai (Grant No. 21ZR1435600), Shanghai Sailing Program (Grant No 21YF1420600), and start-up fundings from Shanghai Jiao Tong University (SJTU). Part of this study was supported by the National Science Foundation (grant CHE-1753225 to MW). Prof. Juntao Ye and Prof. Changkun Li are acknowledged for sharing basic instruments for this project.
Notes and references
- C. R. Bertozzi and L. L. Kiessling, Chemical Glycobiology, Science, 2001, 291, 2357–2364 CrossRef CAS PubMed.
- M. R. Pratt and C. R. Bertozzi, Synthetic Glycopeptides and Glycoproteins as Tools for Biology, Chem. Soc. Rev., 2005, 34, 58 RSC.
- H. Herzner, T. Reipen, M. Schultz and H. Kunz, Synthesis of Glycopeptides Containing Carbohydrate and Peptide Recognition Motifs, Chem. Rev., 2000, 100, 4495–4538 CrossRef CAS PubMed.
- S. Cecioni, A. Imberty and S. Vidal, Glycomimetics versus Multivalent Glycoconjugates for the Design of High Affinity Lectin Ligands, Chem. Rev., 2015, 115, 525–561 CrossRef CAS PubMed.
- Y. Yang and B. Yu, Recent Advances in the Chemical Synthesis of C-Glycosides, Chem. Rev., 2017, 117, 12281–12356 CrossRef CAS PubMed.
- É. Bokor, S. Kun, D. Goyard, M. Tóth, J.-P. Praly, S. Vidal and L. Somsák, C-Glycopyranosyl Arenes and Hetarenes: Synthetic Methods and Bioactivity Focused on Antidiabetic Potential, Chem. Rev., 2017, 117, 1687–1764 CrossRef PubMed.
- G. P. Hultin, Bioactive C-Glycosides from Bacterial Secondary Metabolism, Curr. Top. Med. Chem., 2005, 5, 1299–1331 CrossRef PubMed.
- T. P. Hogervorst, R. J. E. Li, L. Marino, S. C. M. Bruijns, N. J. Meeuwenoord, D. V. Filippov, H. S. Overkleeft, G. A. Van Der Marel, S. J. Van Vliet, Y. Van Kooyk and J. D. C. Codée, C-Mannosyl Lysine for Solid Phase Assembly of Mannosylated Peptide Conjugate Cancer Vaccines, ACS Chem. Biol., 2020, 15, 728–739 CrossRef CAS.
- R. Kovács, F. Nagy, Z. Tóth, A. Bozó, B. Balázs and L. Majoros, Synergistic effect of nikkomycin Z with caspofungin and micafungin against Candida albicans and Candida parapsilosis biofilms, Lett. Appl. Microbiol., 2019, 69, 271–278 CrossRef PubMed.
- K. Takahashi and A. Kubo, New Antibiotics, Saframycins A, B, C, D and E, J. Antibiot., 1977, 30, 1015–1018 CrossRef PubMed.
- S. V. Moradi, W. M. Hussein, P. Varamini, P. Simerska and I. Toth, Glycosylation, an effective synthetic strategy to improve the bioavailability of therapeutic peptides, Chem. Sci., 2016, 7, 2492–2500 RSC.
- U. Tedebark, M. Meldal, L. Panza and K. Bock, C-Linked Glycosyl Azido Acid in Novel Solid-Phase C-Glycopeptide Synthesis, Tetrahedron Lett., 1998, 39, 1815–1818 CrossRef CAS.
- L. Negri, R. Lattanzi, F. Tabacco, B. Scolaro and R. Rocchi, Glycodermorphins: Opioid Peptides with Potent and Prolonged Analgesic Activity and Enhanced Blood–Brain Barrier Penetration, Br. J. Pharmacol., 1998, 124, 1516–1522 CrossRef CAS PubMed.
- J.-P. Bapst, M. Calame, H. Tanner and A. N. Eberle, Glycosylated DOTA−α-Melanocyte-Stimulating Hormone Analogues for Melanoma Targeting: Influence of the Site of Glycosylation on in Vivo Biodistribution, Bioconjugate Chem., 2009, 20, 984–993 CrossRef CAS PubMed.
- D. P. Gamblin, E. M. Scanlan and B. G. Davis, Glycoprotein Synthesis: An Update, Chem. Rev., 2009, 109, 131–163 CrossRef CAS PubMed.
- N. Floyd, B. Vijayakrishnan, J. R. Koeppe and B. G. Davis, Thiyl Glycosylation of Olefinic Proteins: S-Linked Glycoconjugate Synthesis, Angew. Chem., Int. Ed., 2009, 48, 7798–7802 CrossRef CAS.
- F. Zhu, S. O'Neill, J. Rodriguez and M. A. Walczak, Stereoretentive Reactions at the Anomeric Position: Synthesis of Selenoglycosides, Angew. Chem., Int. Ed., 2018, 57, 7091–7095 CrossRef CAS PubMed.
- S. Zhu, G. Samala, E. T. Sletten, J. L. Stockdill and H. M. Nguyen, Facile triflic acid-catalyzed α-1,2-cis-thio glycosylations: scope and application to the synthesis of S-linked oligosaccharides, glycolipids, sublancin glycopeptides, and TN/TF antigens, Chem. Sci., 2019, 10, 10475–10480 RSC.
- L.-Q. Wan, X. Zhang, Y. Zou, R. Shi, J.-G. Cao, S.-Y. Xu, L.-F. Deng, L. Zhou, Y. Gong, X. Shu, G. Y. Lee, H. Ren, L. Dai, S. Qi, K. N. Houk and D. Niu, Nonenzymatic Stereoselective S-Glycosylation of Polypeptides and Proteins, J. Am. Chem. Soc., 2021, 143, 11919–11926 CrossRef CAS PubMed.
- A. Dondoni and A. Marra, Methods for Anomeric Carbon-Linked and Fused Sugar Amino Acid Synthesis: The Gateway to Artificial Glycopeptides, Chem. Rev., 2000, 100, 4395–4422 CrossRef CAS PubMed.
- F. Zhu, M. J. Rourke, T. Yang, J. Rodriguez and M. A. Walczak, Highly Stereospecific Cross-Coupling Reactions of Anomeric Stannanes for the Synthesis of C-Aryl Glycosides, J. Am. Chem. Soc., 2016, 138, 12049–12052 CrossRef CAS PubMed.
- Q. Wang, Y. Fu, W. Zhu, S. An, Q. Zhou, S.-F. Zhu, G. He, P. Liu and G. Chen, Total Synthesis of C-α-Mannosyl Tryptophan via Palladium-Catalyzed C–H Glycosylation, CCS Chem., 2021, 3, 1729–1736 CrossRef CAS.
- R. Mao, S. Xi, S. Shah, M. J. Roy, A. John, J. P. Lingford, G. Gäde, N. E. Scott and E. D. Goddard-Borger, Synthesis of C-Mannosylated Glycopeptides Enabled by Ni-Catalyzed Photoreductive Cross-Coupling Reactions, J. Am. Chem. Soc., 2021, 143, 12699–12707 CrossRef CAS.
- H. Gong and M. R. Gagné, Diastereoselective Ni-Catalyzed Negishi Cross-Coupling Approach to Saturated, Fully Oxygenated C-Alkyl and C-Aryl Glycosides, J. Am. Chem. Soc., 2008, 130, 12177–12183 CrossRef CAS PubMed.
- H. Gong, R. S. Andrews, J. L. Zuccarello, S. J. Lee and M. R. Gagné, Sn-Free Ni-Catalyzed Reductive Coupling of Glycosyl Bromides with Activated Alkenes, Org. Lett., 2009, 11, 879–882 CrossRef CAS PubMed.
- R. S. Andrews, J. J. Becker and M. R. Gagné, Intermolecular Addition of Glycosyl Halides to Alkenes Mediated by Visible Light, Angew. Chem., Int. Ed., 2010, 49, 7274–7276 CrossRef CAS PubMed.
- P. Ji, Y. Zhang, Y. Wei, H. Huang, W. Hu, P. A. Mariano and W. Wang, Visible-Light-Mediated, Chemo- and Stereoselective Radical Process for the Synthesis of C-Glycoamino Acids, Org. Lett., 2019, 21, 3086–3092 CrossRef CAS.
- X. Yan, F. Feng, L. Zhou, L. Chen, S. Tang, J. Liu, F. Cai and X. Wang, Stereodivergent synthesis of C-glycosamino acids via Pd/Cu dual catalysis, Sci. China Chem., 2021, 64, 552–557 CrossRef CAS.
- R. Qi, C. Wang, Z. Ma, H. Wang, Q. Chen, L. Liu, D. Pan, X. Ren, R. Wang and Z. Xu, Visible-Light-Promoted Stereoselective C(sp3)-H Glycosylation for the Synthesis of C-Glycoamino Acids and C-Glycopeptides, Angew. Chem., Int. Ed., 2022, 61 Search PubMed.
- Y.-N. Ding, N. Li, Y.-C. Huang, Y. An and Y.-M. Liang, Visible-Light-Induced Copper-Catalyzed Asymmetric C(sp3)−C(sp3)−H Glycosylation: Access to C-Glycopeptides, Org. Lett., 2022, 24, 4519–4523 CrossRef CAS.
- Y. Liu, Y. Wang, W. Dai, W. Huang, Y. Li and H. Liu, Palladium-Catalysed C(sp3)-H Glycosylation for the Synthesis of C-Alkyl Glycoamino Acids, Angew. Chem., Int. Ed., 2020, 59, 3491–3494 CrossRef CAS PubMed.
- J. Wu, N. Kaplaneris, S. Ni, F. Kaltenhäuser and L. Ackermann, Late-stage C(sp2)–H and C(sp3)–H glycosylation of C-aryl/alkyl glycopeptides: mechanistic insights and fluorescence labeling, Chem. Sci., 2020, 11, 6521–6526 RSC.
- W. Shang, S. N. Su, R. Shi, Z. D. Mou, G. Q. Yu, X. Zhang and D. Niu, Generation of Glycosyl Radicals from Glycosyl Sulfoxides and Its Use in the Synthesis of C-linked Glycoconjugates, Angew. Chem., Int. Ed., 2021, 60, 385–390 CrossRef CAS.
- Y. Jiang, Q. Wang, X. Zhang and M. J. Koh, Synthesis of C-Glycosides by Ti-Catalyzed Stereoselective Glycosyl Radical Functionalization, Chem, 2021, 7, 3377–3392 CAS.
- Q. Wang, B. C. Lee, T. J. Tan, Y. Jiang, W. H. Ser and M. J. Koh, Visible light activation enables desulfonylative cross-coupling of glycosyl sulfones, Nat. Synth., 2022, 1, 967–974 CrossRef.
- A. Chen, L. Xu, Z. Zhou, S. Zhao, T. Yang and F. Zhu, Recent advances in glycosylation involving novel anomeric radical precursors, J. Carbohydr. Chem., 2021, 40, 361–400 CrossRef CAS.
- B. Giese and J. Dupuis, Diastereoselective Syntheses of C-Glycopyranosides, Angew. Chem., Int. Ed. Engl., 1983, 22, 622–623 CrossRef.
- N. Kiya, Y. Hidaka, K. Usui and G. Hirai, Synthesis of CH2-Linked α(1,6)-Disaccharide Analogues by α-Selective Radical Coupling C-Glycosylation, Org. Lett., 2019, 21, 1588–1592 CrossRef CAS PubMed.
- N. Miquel, G. Doisneau and J.-M. Beau, Reductive Samariation of Anomeric 2-Pyridyl Sulfones with Catalytic Nickel: An Unexpected Improvement in the Synthesis of 1,2-trans-Diequatorial C-Glycosyl Compounds, Angew. Chem., Int. Ed., 2000, 39, 4111–4114 CrossRef CAS.
- A. Dumoulin, J. K. Matsui, Á. Gutiérrez-Bonet and G. A. Molander, Synthesis of Non-Classical Arylated C-Saccharides through Nickel/Photoredox Dual Catalysis, Angew. Chem., Int. Ed., 2018, 57, 6614–6618 CrossRef CAS PubMed.
- S. O. Badir, A. Dumoulin, J. K. Matsui and G. A. Molander, Synthesis of Reversed C-Acyl Glycosides through Ni/Photoredox Dual Catalysis, Angew. Chem., Int. Ed., 2018, 57, 6610–6613 CrossRef CAS PubMed.
- Y. Ma, S. Liu, Y. Xi, H. Li, K. Yang, Z. Cheng, W. Wang and Y. Zhang, Highly stereoselective synthesis of aryl/heteroaryl-C-nucleosides via the merger of photoredox and nickel catalysis, Chem. Commun., 2019, 55, 14657–14660 RSC.
- M. Zhu and S. Messaoudi, Diastereoselective Decarboxylative Alkynylation of Anomeric Carboxylic Acids Using Cu/Photoredox Dual Catalysis, ACS Catal., 2021, 11, 6334–6342 CrossRef CAS.
- I. C. S. Wan, M. D. Witte and A. J. Minnaard, From D- to L-Monosaccharide Derivatives via Photodecarboxylation−Alkylation, Org. Lett., 2019, 21, 7669–7673 CrossRef CAS PubMed.
- E. M. Miller and M. A. Walczak, Light-Mediated Cross-Coupling of Anomeric Trifluoroborates, Org. Lett., 2021, 23, 4289–4293 CrossRef CAS PubMed.
- D. Takeda, M. Yoritate, H. Yasutomi, S. Chiba, T. Moriyama, A. Yokoo, K. Usui and G. Hirai, β-Glycosyl Trifluoroborates as Precursors for Direct α-C-Glycosylation: Synthesis of 2-Deoxy-α-C-glycosides, Org. Lett., 2021, 23, 1940–1944 CrossRef CAS PubMed.
-
J. B. Pedley, R. D. Naylor, S. P. Kirby and P. Francis, Thermochemical Data of Organic Compounds, 2nd edn, 1987 Search PubMed.
- J. Wu, R. M. Bär, L. Guo, A. Noble and V. K. Aggarwal, Photoinduced Deoxygenative Borylations of Aliphatic Alcohols, Angew. Chem., Int. Ed., 2019, 58, 18830–18834 CrossRef CAS PubMed.
- T. Nanjo, T. Matsugasako, Y. Maruo and Y. Takemoto, Photocatalytic C–O Bond Cleavage of Alcohols Using Xanthate Salts, Org. Lett., 2022, 24, 359–363 CrossRef CAS PubMed.
- E. E. Stache, A. B. Ertel, T. Rovis and A. G. Doyle, Generation of Phosphoranyl Radicals via Photoredox Catalysis Enables Voltage–Independent Activation of Strong C–O Bonds, ACS Catal., 2018, 8, 11134–11139 CrossRef CAS PubMed.
- G. L. Lackner, K. W. Quasdorf and L. E. Overman, Direct Construction of
Quaternary Carbons from Tertiary Alcohols via Photoredox-Catalyzed Fragmentation of tert-Alkyl N-Phthalimidoyl Oxalates, J. Am. Chem. Soc., 2013, 135, 15342–15345 CrossRef CAS PubMed.
- Y. Ye, H. Chen, J. L. Sessler and H. Gong, Zn-Mediated Fragmentation of Tertiary Alkyl Oxalates Enabling Formation of Alkylated and Arylated Quaternary Carbon Centers, J. Am. Chem. Soc., 2019, 141, 820–824 CrossRef CAS PubMed.
- Y. Wei, J. Lam and T. Diao, Synthesis of C-acyl furanosides via the cross-coupling of glycosyl esters with carboxylic acids, Chem. Sci., 2021, 12, 11414–11419 RSC.
- Y. Wei, B. Ben-Zvi and T. Diao, Diastereoselective Synthesis of Aryl C-Glycosides from Glycosyl Esters via C−O Bond Homolysis, Angew. Chem., Int. Ed., 2021, 60, 9433–9438 CrossRef CAS PubMed.
- Z. Dong and D. W. C. Macmillan, Metallaphotoredox-enabled deoxygenative arylation of alcohols, Nature, 2021, 598, 451–456 CrossRef CAS PubMed.
- F. Zhu, S.-Q. Zhang, Z. Chen, J. Rui, X. Hong and M. A. Walczak, Catalytic and Photochemical Strategies to Stabilized Radicals Based on Anomeric Nucleophiles, J. Am. Chem. Soc., 2020, 142, 11102–11113 CrossRef CAS PubMed.
- Q. Wang, J. Duan, P. Tang, G. Chen and G. He, Synthesis of non-classical heteroaryl C-glycosides via Minisci-type alkylation of N-heteroarenes with 4-glycosyl-dihydropyridines, Sci. China Chem., 2020, 63, 1613–1618 CrossRef CAS.
- F. Zhu, W. C. Powell, R. Jing and M. A. Walczak, Organometallic AlaM reagents for umpolung peptide diversification, Chem. Catal., 2021, 1, 870–884 CrossRef CAS PubMed.
- X. Peng, K. Xu, Q. Zhang, L. Liu and J. Tan, Dehydroalanine modification sees the light: a photochemical conjugate addition strategy, Trends Chem., 2022, 4, 643–657 CrossRef CAS.
- R. A. Aycock, D. B. Vogt and N. T. Jui, A practical and scalable system for heteroaryl amino acid synthesis, Chem. Sci., 2017, 8, 7998–8003 RSC.
- H. Abe, S. Shuto and A. Matsuda, Highly α- and β-Selective Radical C-Glycosylation Reactions Using a Controlling Anomeric Effect Based on the Conformational Restriction Strategy. A Study on the Conformation−Anomeric Effect−Stereoselectivity Relationship in Anomeric Radical Reactions, J. Am. Chem. Soc., 2001, 123, 11870–11882 CrossRef CAS PubMed.
- F. Zhu and M. A. Walczak, Stereochemistry of Transition Metal Complexes Controlled by the Metallo-Anomeric Effect, J. Am. Chem. Soc., 2020, 142, 15127–15136 CrossRef CAS PubMed.
- M. Alas, A. Saghaeidehkordi and K. Kaur, Peptide–Drug Conjugates with Different Linkers for Cancer Therapy, J. Med. Chem., 2021, 64, 216–232 CrossRef CAS PubMed.
- J. Sim, M. W. Campbell and G. A. Molander, Synthesis of α-Fluoro-α-amino Acid Derivatives via Photoredox-Catalyzed Carbofluorination, ACS Catal., 2019, 9, 1558–1563 CrossRef CAS PubMed.
- X. Qi, S. Jambu, Y. Ji, K. M. Belyk, N. R. Panigrahi, P. S. Arora, N. A. Strotman and T. Diao, Late-Stage Modification of Oligopeptides by Nickel-Catalyzed Stereoselective Radical Addition to Dehydroalanine, Angew. Chem., Int. Ed., 2022, 61, e202213315 CAS.
- H. Yin, M. Zheng, H. Chen, S. Wang, Q. Zhou, Q. Zhang and P. Wang, Stereoselective and Divergent Construction of β-Thiolated/Selenolated Amino Acids via Photoredox-Catalyzed Asymmetric Giese Reaction, J. Am. Chem. Soc., 2020, 142, 14201–14209 CrossRef CAS PubMed.
- P. Wen and D. Crich, Blue Light Photocatalytic Glycosylation without Electrophilic Additives, Org. Lett., 2017, 19, 2402–2405 CrossRef CAS PubMed.
- H. Zipse, Radical Stability-A Theoretical Perspective, Top. Curr. Chem., 2006, 263, 163–189 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b,
Maciej A.
Walczak
*b,
Maciej A.
Walczak