
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePalladium-catalyzed intramolecular asymmetric hydrocyclopropanylation of alkynes: synthesis of cyclopropane-fused γ-lactams†
Han-Ze
Lin‡
,
Zhuang
Qi‡
,
Qi-Min
Wu
,
Yong-Yu
Jiang
and
Jin-Bao
Peng
 *
*
School of Biotechnology and Health Sciences, Wuyi University, Jiangmen, Guangdong 529020, People's Republic of China. E-mail: pengjb_05@126.com
First published on 16th June 2023
Abstract
A palladium-catalyzed intramolecular asymmetric hydrocyclopropanylation of alkynes via C(sp3)–H activation has been developed for the synthesis of cyclopropane-fused γ-lactams. The presented strategy proceeds in a selective and 100% atom-economical manner. A range of cyclopropane-fused γ-lactams were prepared from readily available substrates in good yields and enantioselectivities with a chiral phosphoramidite ligand.
Introduction
Cyclopropane is the smallest carbocycle with considerable torsional and angular strains, and has constantly been of great interest to the organic community as a valuable synthetic building block.1 In addition, this unique structure is found in a number of natural products, chemical drugs, and agrochemicals.2 Various cyclopropane-containing natural products, spanning from terpenoids, fatty acid, pheromone, amino acid and other types of molecules, have been isolated from plants, fungus, and microorganisms. The introduction of a cyclopropane into drug molecules can increase their metabolic stability via spatial orientation of the substituents of the cyclopropyl subunit.In particularly, cyclopropanes fused to pyrrolidine units are found in many important biologically active agents as key structural features (Fig. 1). For example, boceprevir is a potent oral HCV-protease inhibitor.3 SUVN-911 is a potent neuronal nicotinic acetylcholine α4β2 receptor antagonist for the treatment of depression.4 Saxagliptin is a selective and reversible DPP4 inhibitor with IC50 of 26 nM.5
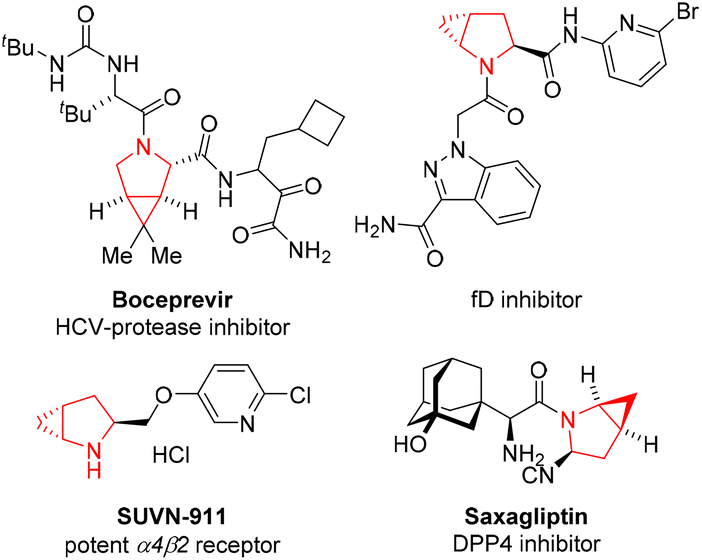 | ||
| Fig. 1 Representative natural products and pharmaceuticals containing cyclopropane-fused pyrrolidines. | ||
Due to their synthetic6 and pharmaceutical importance, substantial ongoing efforts have been devoted to develop efficient methodologies for building such an important family of cyclopropane-fused pyrrolidine scaffolds. Traditionally, these structures could be prepared via the Simmons–Smith reaction of N-protected dihydropyrroles7 or the direct cyclopropanation of pyrrole derivatives employing electrophilic metallocarbenoids (Scheme 1a).8 Other methods from non-pyrrole starting materials have also been reported.9–11 For example, the group of Yang11a and then the group of Bower11b constructed this structure utilizing a cascade aza-Heck cyclization/cyclopropanation strategy which proceeds via the intramolecular aza-palladation of an alkene followed by a C–H palladation-initiated cyclopropanation (Scheme 1b). Despite advantages, these methods suffer from several drawbacks, such as harsh reaction conditions, regio- or/and stereoselectivity issues, and the use of toxic and unstable materials. Accordingly, developing direct and flexible methods for these structures that use readily available and stable starting materials in a stereoselective manner is highly desirable.
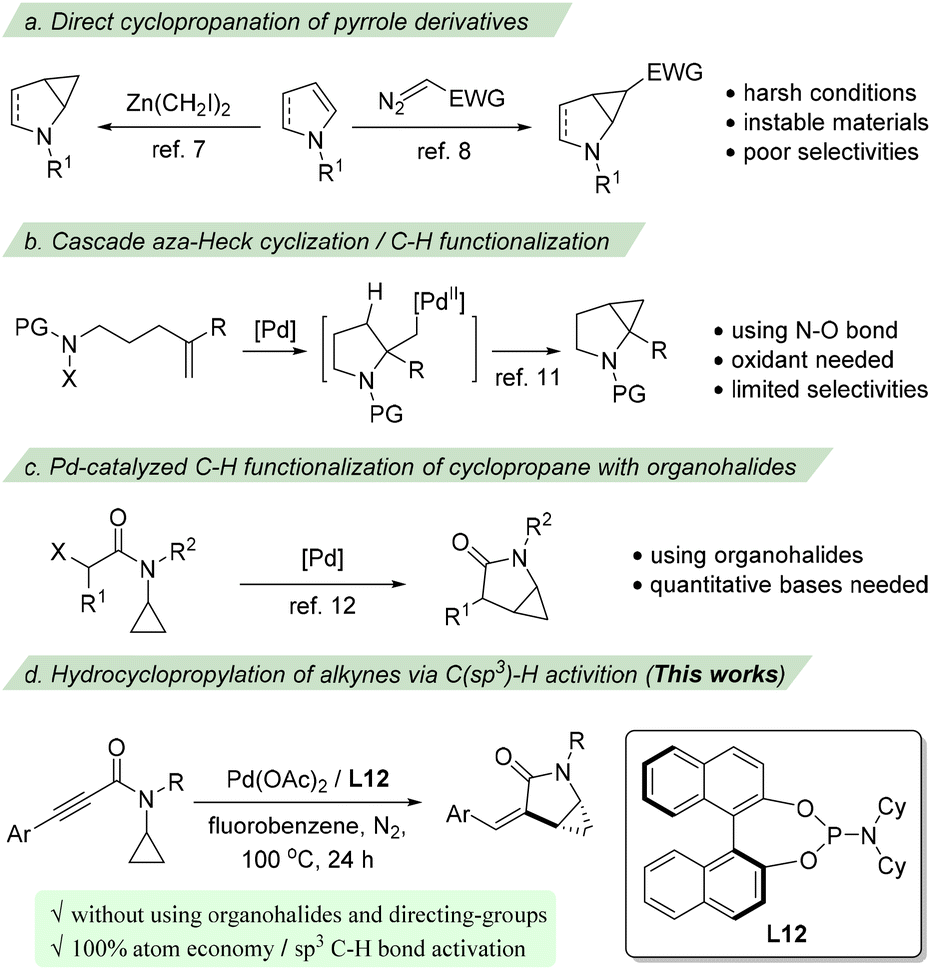 | ||
| Scheme 1 Strategies for the synthesis of cyclopropane-fused pyrrolidines. (a) Direct cyclopropanation of pyrrole derivatives. (b) Cascade aza-Heck cyclization/C–H functionalization. (c) Pd-catalyzed C–H functionalization of cyclopropane with organohalides. (d) Hydrocyclopropylation of alkynes via C(sp3)–H activition. | ||
On the other hand, with the rapid development of C–H bond functionalization, transition metal catalyzed functionalization of cyclopropane has emerged as one of the promising ways to construct cyclopropane-containing compounds.12 In 2015, Cramer reported a palladium catalyzed enantioselective C–H functionalization of chloroacetamide substrates to access cyclopropane-fused γ-lactams (Scheme 1c).13a Later, two cases of intramolecular alkenylation of cyclopropane using bromoalkenes to synthesize α-alkylidene-γ-lactams were reported by Baudoin and co-workers.13b,c Since organohalides were used as substrates, both procedures required quantitative amounts of bases. We assume that an intramolecular hydrocyclopropanylation of an appropriate π-system would provide an efficient and 100% atom-economic procedure to access cyclopropane-fused γ-lactams. However, despite some elegant examples of C(sp3)–H bond alkenylation with alkynes having been disclosed via the directing group assisted C–H activation14 or radical processes,15 the direct hydroalkylation of alkynes without using halides and other functional groups16 has been rarely reported. Herein, we report a palladium-catalyzed intramolecular hydrocyclopropanylation of alkynes via C(sp3)–H activation for the synthesis of cyclopropane-fused γ-lactams (Scheme 1d). A range of cyclopropane-fused γ-lactams were prepared from readily available substrates in good yields and enantioselectivities.
Results and discussion
Initially, we commenced our study by employing N-cyclopropyl-3-phenylpropiolamide 1a as the model substrate to test our assumption (Table 1). To our delight, when a toluene solution of 1a was treated with the Pd(OAc)2/DPPF catalyst system at 100 °C, the expected intramolecular hydrocyclopropanylation reaction proceeded successfully and produced cyclopropane-fused γ-lactam 2a in 52% yield (entry 1). Other palladium catalysts such as Pd(acac)2 and Pd2(dba)3 were less active and provided 2a in 17–32% yields (entries 2 and 3). However, Pd(PPh3)4 was found to be ineffective and no desired product 2a was obtained (entry 4). Subsequently, a series of ligands were examined and it was found that the ligand played an important role in this reaction. When the bidentate phosphine ligand with a smaller bite angle DPPP was used as a ligand, 2a was obtained in an excellent yield of 95% (entry 5). Large bite angle ligands such as DPEphos and xantphos were less effective (entries 6 and 7). Monodentate phosphine ligands were also active and electron-rich trialkylphosphines were found to be more effective. The yield of 2a was improved to 90% and 96% when PCy3 and BuPAd2 were used as the ligands, respectively (entries 9 and 10). The screening of the solvent revealed that toluene is the optimal. When the reaction was conducted in non-polar solvents such as THF and dioxane, the desired product 2a was obtained in 85% and 88% yields, respectively (entries 11 and 12). However, when MeCN was used as the solvent, the reaction was totally inhibited and only a trace amount of 2a was detected (entry 13). Strongly polar solvents like DMSO and DMF also led to decreased yields (entries 14 and 15).|
|
||||
|---|---|---|---|---|
| Entry | Cat. | L. | Sol. | Yieldb (%) |
| a Reaction conditions: 1a (0.2 mmol), [Pd] (5 mol%), ligand (10 mol% for monodentate ligands, 5 mol% for bidentate ligands), solvent (2 mL), 100 °C, 24 h. b Isolated yields. | ||||
| 1 | Pd(OAc)2 | DPPF | Toluene | 52 |
| 2 | Pd(acac)2 | DPPF | Toluene | 17 |
| 3 | Pd2(dba)3 | DPPF | Toluene | 23 |
| 4 | Pd(PPh3)4 | DPPF | Toluene | NR |
| 5 | Pd(OAc)2 | DPPP | Toluene | 95 |
| 6 | Pd(OAc)2 | DPEPhos | Toluene | 27 |
| 7 | Pd(OAc)2 | XantPhos | Toluene | NR |
| 8 | Pd(OAc)2 | PPh3 | Toluene | 35 |
| 9 | Pd(OAc)2 | PCy3 | Toluene | 90 |
| 10 | Pd(OAc) 2 | BuPAd 2 | Toluene | 96 |
| 11 | Pd(OAc)2 | BuPAd2 | THF | 85 |
| 12 | Pd(OAc)2 | BuPAd2 | Dioxane | 88 |
| 13 | Pd(OAc)2 | BuPAd2 | MeCN | Trace |
| 14 | Pd(OAc)2 | BuPAd2 | DMF | 44 |
| 15 | Pd(OAc)2 | BuPAd2 | DMSO | 36 |
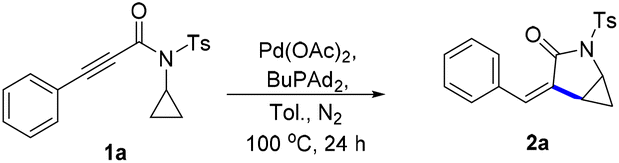
With the optimized reaction conditions in hand, we began exploration of the substrate scope by varying the substituents of N-cyclopropyl-3-phenylpropiolamide 1 (Scheme 2). First, the influence of the substitution on the benzene ring of N-cyclopropyl-3-phenylpropiolamide 1 was investigated. The electronic properties of the substituents played a minor role in this reaction. Both electron-donating (2e–2g) and electron-withdrawing group (2h–2j) substituted substrates underwent this reaction and produced the cyclopropane-fused γ-lactam products in good to excellent yields. However, the steric effect of the substituents influenced the yield significantly. When substrates with an ortho-substitution were used in this reaction, the yield dropped dramatically to 15% (2d). The configuration of product 2f was determined by X-ray crystallography analysis.17 Various N-substitutions of the N-cyclopropyl-3-phenylpropiolamide 1 were tolerated. When benzenesulfonamide 1k was subjected to the standard conditions, the desired product 2k was obtained in 79% yield. Fluoro- and nitro-substituted benzenesulfonamides aslo produced the corresponding products in 73% and 83% yields, respectively (2l and 2m). In addition, N-benzoyl and N-methyl substituted substrates were tolerated as well, generating the desired products 2n and 2o in high yields. Notably, when a methyl ester group was attached on the cyclopropyl group, the corresponding products (2p, 2s–2u) were obtained in good to excellent yields. The steric effect of the ester affected the yields significantly. When ethyl and isopropyl ester group substituted substrates were used in this reaction, a higher reaction temperature was needed and the yields dropped to 82% and 57%, respectively (2q and 2r). The N-cyclopropyl amide was found to be critical for this reaction. The cyclopropyl 3-phenylpropiolate as well as the N-isopropyl and N-cyclobutyl amides failed in this reaction.
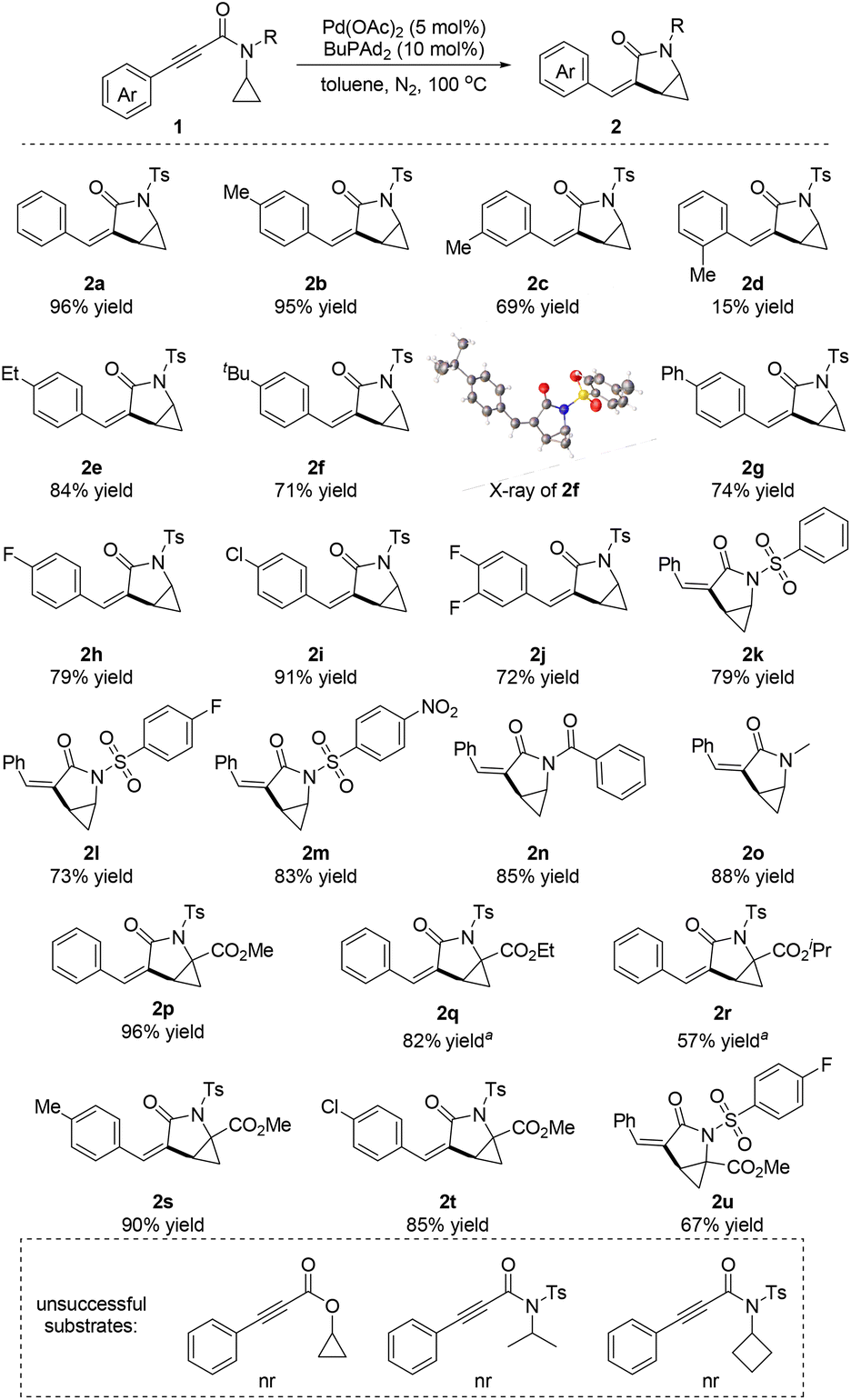 | ||
| Scheme 2 Substrate scope. Reaction conditions: 1 (0.2 mmol), Pd(OAc)2 (5 mol%), BuPAd2 (10 mol%), toluene (2 mL), 100 °C, 24 h, isolated yields. a120 °C. | ||
Since two adjacent stereogenic carbon centers are generated in the cyclopropane-fused γ-lactam product, we envisaged that an enantioselective hydrocyclopropanylation could be realized with an appropriate chiral ligand. Thus, we re-evaluated the reaction parameters with various nitrogen and phosphine chiral ligands (Scheme 3, see details in ESI†). After a thorough screening, phosphoramidite ligand L12 was found out to be optimal. Enantio-enriched (R,R)-2a was obtained in good yield and enantioselectivity with the replacement of toluene with fluorobenzene. Axially chiral biphenyl ligands such as BINAP (L1), H8-BINAP (L2) and SEGPHOS (L3) were ineffective, no or only trace amounts of product were detected. Bidentate phosphines with alkyl substitutions led to good yields but low levels of enantiocontrol (Scheme 3, L4–L6). When monophosphorus ligands such as BIDIME (L7), iPr-BIDIME (L8) and AntPhos (L9) were used, lower yields and moderate selectivities were obtained. Phosphoramidite ligands were found to be effective for this reaction (Scheme 3, L10–L12, see details in ESI†). The desired product 2a was produced in 76% yield and 85% ee when phosphoramidite ligand L12 was used. Having identified L12 as the optimal ligand, we examined the generality of this asymmetric protocol. All the tested substrates proceeded smoothly and provided the desired products in good yields and enantioselectivities. A total of fourteen compounds were prepared with variations on the aryl group with alkyl groups and halides. The absolute configuration of the products was determined based on the X-ray crystallography analysis of (R,R)-2i as a representative example.17 Notably, when methyl ester attached substrates (1p, 1s–1u) were used in this asymmetric protocol, excellent enantioselectivities of ≥90% ee were obtained by using BIDIME (L7) as the ligand.
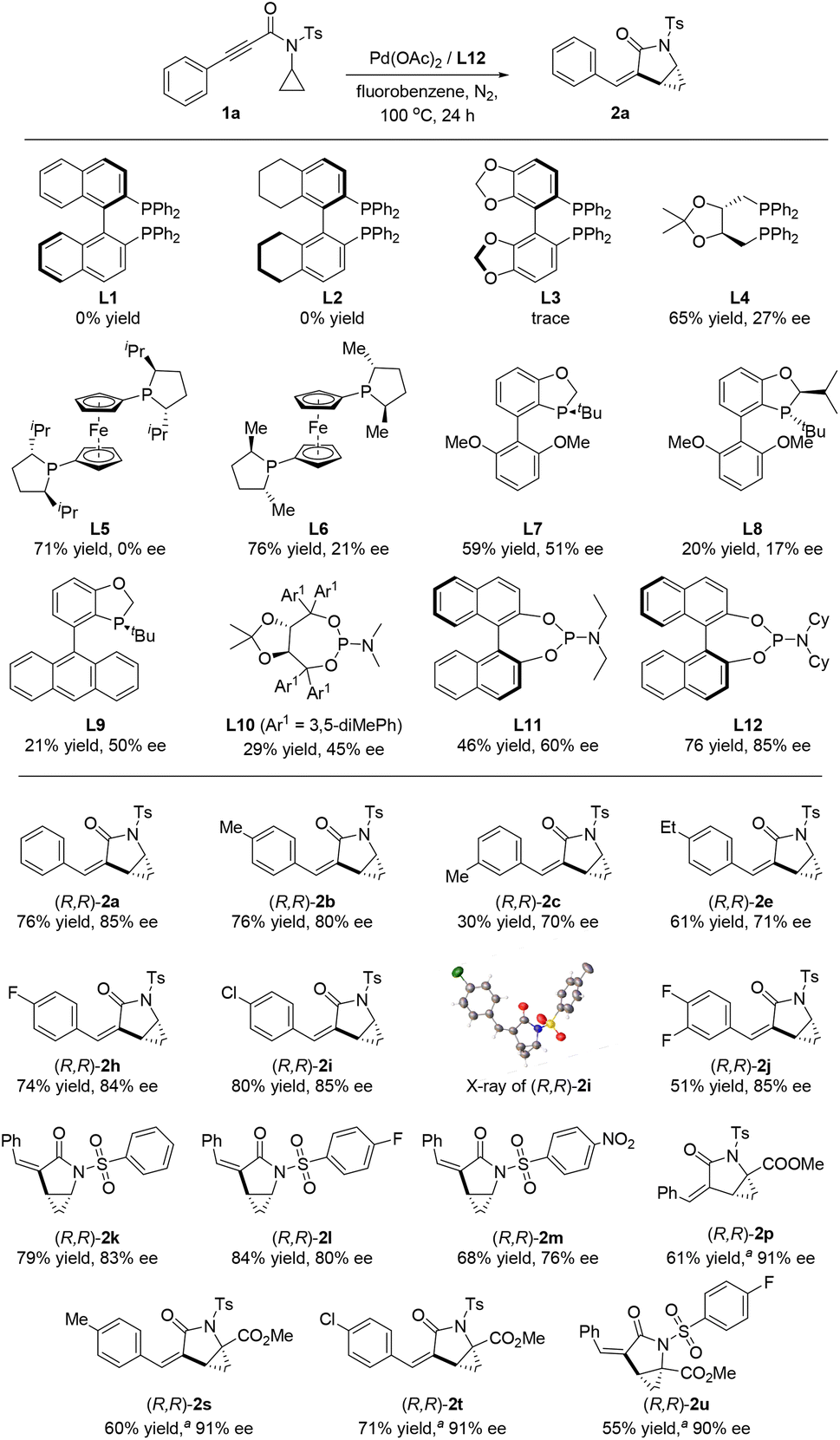 | ||
| Scheme 3 Screening of chiral ligands. Reaction conditions: 1 (0.2 mmol), Pd(OAc)2 (5 mol%), L* (10 mol%), toluene (2 mL), 100 °C, 24 h, isolated yields. aL7 instead of L12, 120 °C. | ||
A plausible reaction pathway for this asymmetric hydrocyclopropanylation of alkynes is proposed based on the present results and previous literature18 (Scheme 4). Initially, the oxidative ligation of the alkyne bond of 1 to the in situ generated Pd(0) generates an Pd(II)–complex B. Then, intramolecular ligand exchange of the C–H bond of the cyclopropyl group followed by hydrogen transfer affords the cyclopalladium complex Dvia the intermediacy of C. Finally, reductive elimination of intermediate D releases the desired product 2 and meanwhile regenerates Pd(0) for the next catalytic cycle.
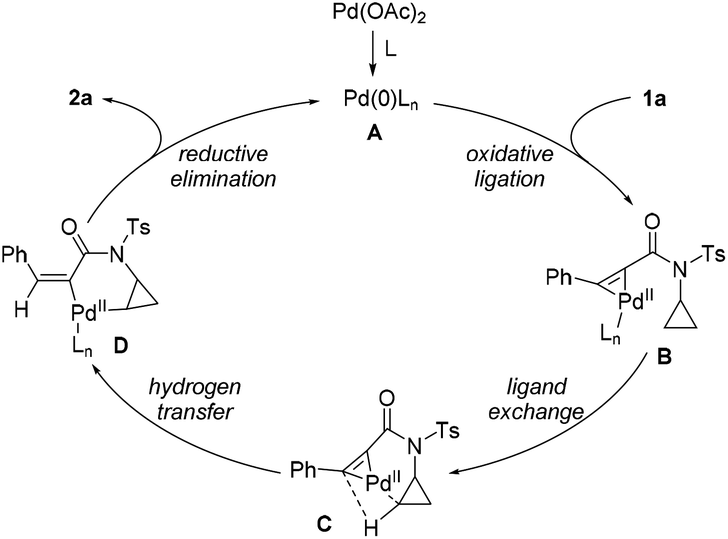 | ||
| Scheme 4 A plausible reaction pathway. | ||
Conclusions
In summary, we have developed a palladium-catalyzed intramolecular asymmetric hydrocyclopropanylation of alkynes via C(sp3)–H activation for the synthesis of cyclopropane-fused γ-lactams. The presented strategy proceeds in a selective and 100% atom-economical manner. A range of cyclopropane-fused γ-lactams were prepared from readily available substrates in good yields and enantioselectivities with a chiral phosphoramidite ligand.Data availability
All experimental data and detailed procedures are available in the ESI.†Author contributions
J.-B. P. conceived and directed the project. H.-Z. L. and Z. Q. performed the experiments. Q.-M. W. and Y.-Y. J. participated in substrates synthesis and discussions. H.-Z. L. and J.-B. P. wrote the manuscript and ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the NSFC (21801225), the Wuyi University (2018TP018, 202211349003), the Guangdong Province Universities and Colleges Pearl River Scholar Funded Scheme (2020AL015), and the Department of Education of Guangdong Province (2020KCXTD036) is gratefully acknowledged.Notes and references
-
(a)
O. G. Kulinkovich, in Cyclopropanes in Organic Synthesis, John Wiley & Sons, Hoboken, NJ, 2015 CrossRef
; (b) W. A. Donaldson, Tetrahedron, 2001, 57, 8589–8627 CrossRef CAS
- L. A. Wessjohann, W. Brandt and T. Thiemann, Chem. Rev., 2003, 103, 1625–1648 CrossRef CAS PubMed
-
(a) B. R. Bacon, S. C. Gordon, E. Lawitz, P. Marcellin, J. M. Vierling, S. Zeuzem, F. Poordad, Z. D. Goodman, H. L. Sings, N. Boparai, M. Burroughs, C. A. Brass, J. K. Albrecht and R. Esteban, N. Engl. J. Med., 2011, 364, 1207–1217 CrossRef CAS
- R. Nirogi, A. R. Mohammed, A. K. Shinde, S. R. Ravella, N. Bogaraju, R. Subramanian, V. R. Mekala, R. C. Palacharla, N. Muddana, J. B. Thentu, G. Bhyrapuneni, R. Abraham and V. Jasti, J. Med. Chem., 2020, 63, 2833–2853 CrossRef CAS
- D. J. Augeri, J. A. Robl, D. A. Betebenner, D. R. Magnin, A. Khanna, J. G. Robertson, A. Wang, L. M. Simpkins, P. Taunk, Q. Huang, S.-P. Han, A.-O. Benoni, C. Michael, X. Li, T. Li, T. Effie, E. W. Gustav, M. E. Donald, M. Jovita, Y. C. Shu, A. B. Scott, S. K. Mark, A. P. Rex and L. G. Hamann, J. Med. Chem., 2005, 48, 5025–5037 CrossRef CAS PubMed
- C. M. Sonnleitner, S. Park, R. Eckl, T. Ertl and O. Reiser, Angew. Chem., Int. Ed., 2020, 59, 18110–18115 CrossRef CAS
-
(a) J. Dong, Y. Gong, J. Liu, X. Chen, X. Wen and H. Sun, Bioorg. Med. Chem., 2014, 22, 1383 CrossRef CAS
-
(a) J. Fu, N. Wurzer, V. Lehner, O. Reiser and H. M. L. Davies, Org. Lett., 2019, 21, 6102–6106 CrossRef CAS
- M. A. Ischay, M. K. Takase, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2013, 135, 2478–2481 CrossRef CAS
- H.-L. Teng, Y. Luo, M. Nishiura and Z. Hou, J. Am. Chem. Soc., 2017, 139, 16506–16509 CrossRef CAS
-
(a) W. Du, Q. Gu, Z. Li and D. Yang, J. Am. Chem. Soc., 2015, 137, 1130–1135 CrossRef CAS PubMed
-
D. S. Roman and A. B. Charette, in Top. Organomet. Chem., ed. P. H. Dixneuf and H. Doucet, Springer, Switzerland, 2016, pp. 91–114 Search PubMed
-
(a) J. Pedroni and N. Cramer, Angew. Chem., Int. Ed., 2015, 54, 11826–11829 CrossRef CAS PubMed
-
(a) B. Liu, T. Zhou, B. Li, S. Xu, H. Song and B. Wang, Angew. Chem., Int. Ed., 2014, 53, 4191–4195 CrossRef CAS PubMed
- H.-P. Deng, X.-Z. Fan, Z.-H. Chen, Q.-H. Xu and J. Wu, J. Am. Chem. Soc., 2017, 139, 13579–13584 CrossRef CAS PubMed
-
(a) N. A. Till, R. T. Smith and D. W. C. MacMillan, J. Am. Chem. Soc., 2018, 140, 5701–5705 CrossRef CAS
- CCDC 2257065 (2f) and CCDC 2257688 ((R,R)-2i) contain the supplementary crystallographic data for this paper.
-
(a) Y. Shi, S. M. Peterson, W. W. Haberaecker III and S. A. Blum, J. Am. Chem. Soc., 2008, 130, 2168–2169 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data, and copies of NMR spectra. CCDC 2257065 and 2257688. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc02168h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |