
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect hydrogen selenide (H2Se) release from activatable selenocarbamates†
Turner D.
Newton‡
 a,
Keyan
Li‡
a,
Jyoti
Sharma
b,
Pier Alexandre
Champagne
*b and
Michael D.
Pluth
*a
a,
Keyan
Li‡
a,
Jyoti
Sharma
b,
Pier Alexandre
Champagne
*b and
Michael D.
Pluth
*a
aDepartment of Chemistry and Biochemistry, Materials Science Institute, Knight Campus for Accelerating Scientific Impact, Institute of Molecular Biology, University of Oregon, Eugene, Oregon 97403-1253, USA. E-mail: pluth@uoregon.edu
bDepartment of Chemistry and Environmental Science, New Jersey Institute of Technology, Newark, New Jersey 07103, USA. E-mail: pier.a.champagne@njit.edu
First published on 20th June 2023
Abstract
Hydrogen selenide (H2Se) is a possible bioregulator, potential gasotransmitter, and important precursor in biological organoselenium compound synthesis. Early tools for H2Se research have benefitted from available mechanistic understanding of analogous small molecules developed for detecting or delivering H2S. A now common approach for H2S delivery is the use of small molecule thiocarbamates that can be engineered to release COS, which is quickly converted to H2S by carbonic anhydrase. To expand our understanding of the chemical underpinnings that enable H2Se delivery, we investigated whether selenocarbamates undergo similar chemistry to release carbonyl selenide (COSe). Using both light- and hydrolysis-activated systems, we demonstrate that unlike their lighter thiocarbamate congeners, selenocarbamates release H2Se directly with concomitant isocyanate formation rather than by the intermediate release of COSe. This reaction mechanism for direct H2Se release is further supported by computational investigations that identify a ΔΔG‡ ∼ 25 kcal mol−1 between the H2Se and COSe release pathways in the absence of protic solvent. This work highlights fundamentally new approaches for H2Se release from small molecules and advances the understanding of reactivity differences between reactive sulfur and selenium species.
Introduction
Small molecule bioregulators play important roles in diverse biological processes. Of such molecules, small polyatomic examples, such as NO, H2S, and CO have joined a class of compounds often referred to as ‘gasotransmitters’ that play key roles in physiological function, disease progression, and signaling pathways.1–6 Each of these gasotransmitters has attracted significant work aimed at developing chemical tools for detection and delivery of these small molecules in complex biological environments.7–14 More recently, hydrogen selenide (H2Se) has attracted increased attention as a possible bioregulator, potential gasotransmitter, and important precursor in biological organoselenium compound synthesis.15–18 Moreover, H2Se is a likely reactive intermediate in initial reduction of dietary selenium sources to form selenophosphate, en route to downstream selenium-containing biomolecules including selenocysteinyl-tRNASec, selenouridine, and selenosugars.19,20Initial tools for H2Se delivery have benefited from earlier work on H2S donor development that provided a strong mechanistic background for understanding how to engineer small molecules to release H2Se in different environments (Fig. 1).9,10,21 For example, building from the analogous GYY4137![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 22 and FW125623 platforms that rely on P
22 and FW125623 platforms that rely on P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S hydrolysis for H2S delivery, we developed the PSe motifs TDN104224 and 2AP-PSe25 that are hydrolyzed to release H2Se. Similarly, Yi and co-workers adapted arylthioamides, which release H2S upon reaction with cysteine, to develop arylselenoamides that release H2Se upon activation by cysteine.19 Recent examples of H2Se donors have also developed new chemistry not based on prior H2S donor platforms, including work by Yi and co-workers on cysteine-mediated H2Se release from selenocyclopropenes19 and by Lukesh and co-workers on the base-mediated H2Se release from γ-ketoselenides.26 Taken together, these prior examples of H2Se delivery highlight that H2Se donor are poised to not only leverage previous sulfur-based chemistry but also identify new reactivity that differs from its lighter chalcogen congener.
S hydrolysis for H2S delivery, we developed the PSe motifs TDN104224 and 2AP-PSe25 that are hydrolyzed to release H2Se. Similarly, Yi and co-workers adapted arylthioamides, which release H2S upon reaction with cysteine, to develop arylselenoamides that release H2Se upon activation by cysteine.19 Recent examples of H2Se donors have also developed new chemistry not based on prior H2S donor platforms, including work by Yi and co-workers on cysteine-mediated H2Se release from selenocyclopropenes19 and by Lukesh and co-workers on the base-mediated H2Se release from γ-ketoselenides.26 Taken together, these prior examples of H2Se delivery highlight that H2Se donor are poised to not only leverage previous sulfur-based chemistry but also identify new reactivity that differs from its lighter chalcogen congener.
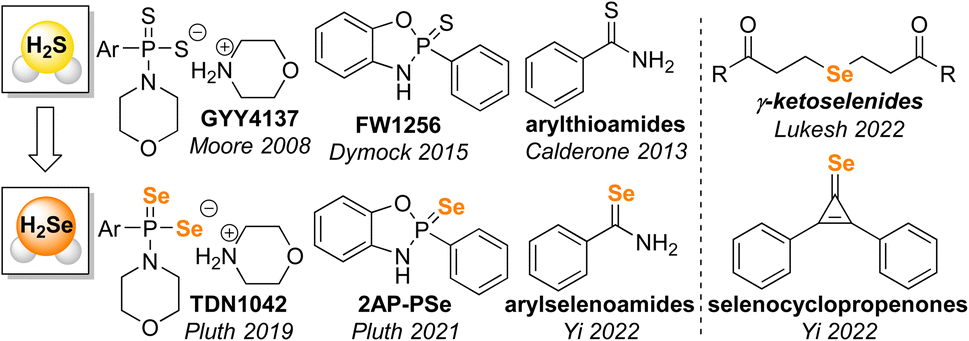 | ||
| Fig. 1 Overview of selected H2Se donors inspired by prior H2S releasing motifs and associated H2Se donors demonstrated to release H2Se directly. | ||
A recent area of H2S donor expansion has grown from the discovery that small molecules that release carbonyl sulfide (COS) can function as H2S donors due to the rapid conversion of COS to H2S by the ubiquitous enzyme carbonic anhydrase (CA).27 One benefit of this class of COS-based H2S donors is that many of the scaffolds can be engineered to only release COS/H2S in response to a specific trigger. Selected examples of such prior triggering motifs include reactive oxygen species,28–31 light,32–34 changes in pH,35,36 and enzymatic activation.37–39 Building from the potential analogous reactivity between S- and Se-containing donor platforms, we wanted to determine whether common motifs used to release COS/H2S could be repurposed to release COSe/H2Se by substitution of S for Se atom. In addition to advancing the fundamental chemistry of reactive selenium species (RSeS), if such donors were to release COSe and/or H2Se they would significantly expand the versatility of triggered RSeS release. Our work here, which is supported by both experimental and computational data, demonstrates that activatable selenocarbamates release H2Se directly rather than through intermediate COSe generation, which not only highlights fundamental differences between S and Se chemistry, but also enables a direct and modifiable approach for H2Se delivery (Fig. 2).
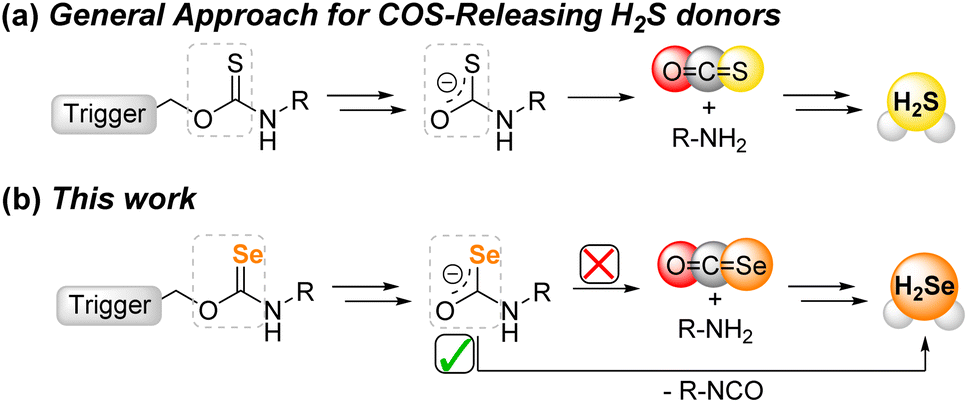 | ||
| Fig. 2 (a) General approach for COS release from thiocarbamates as precursor for H2S delivery. (b) Summary of this work highlighting that selenocarbamates release H2Se directly rather than requiring intermediate COSe formation. | ||
Results and discussion
Our first goal was to prepare a selenocarbamate (SeCM) that was stable until activated by a specific external stimulus. Based on our prior work with light-activated thiocarbamates containing the photocleavable o-nitrobenzyl group as COS/H2S donors,32 we focused on the analogous selenocarbamate motifs. Using a similar synthetic approach as for the thiocarbamates, we treated the p-fluorophenyl isoselenocyanate with 2-nitrobenzyl alcohol in THF at 0 °C in the presence of NaH to afford the desired selenocarbamate product (PhotoSeCM, Scheme 1). The 77Se NMR spectrum of the product showed a major peak at 278 ppm, which is consistent with formation of the O-alkyl selenocarbamate. We note that earlier work with the parent PhNCSe starting material showed either partial or full isomerization of the selenocarbamate product to the Se-alkyl selenocarbamate upon purification by SiO2 chromatography, which was supported by a shift in the 77Se NMR spectrum from 281.2 ppm to 462.2 ppm. This downfield resonance is consistent with previously reported Se-alkyl selenocarbamate 77Se shifts and suggests that these compounds may undergo a Newman–Kwart40,41 rearrangement promoted by silica gel. To avoid using mixtures of the O- and Se-alkyl selenocarbamates, we optimized the purification and isolation procedure of PhotoSeCM to include recrystallization rather than chromatography and allow for isolation of a single isomer.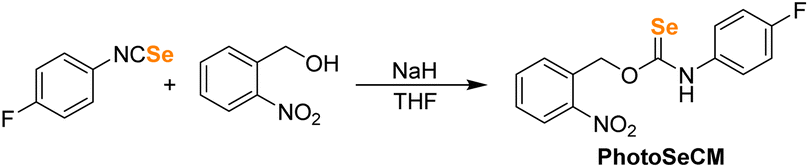 | ||
| Scheme 1 Synthesis of o-nitrobenzyl selenocarbamates. | ||
With PhotoSeCM in hand, we next investigated the product formation after photoactivation. To simplify the overall reaction conditions, we investigated the reaction in THF with the goal of limiting hydrolysis of potential reaction products. Moreover, monitoring the 19F NMR spectrum of the reaction allows for identification of major p-fluoroaniline based reaction products. Upon irradiation at 365 nm, we expected that the primary products would be COSe, p-fluoroaniline, and 2-nitrosobenzaldehyde, however this was not the case. When monitoring the 19F NMR spectrum of the reaction, we observed consumption of the PhotoSeCM signal at −117 ppm but failed to observe the expected p-fluoroaniline product at −130 ppm (Fig. 3). Rather, we observed a new product peak at −123 ppm corresponding to formation of p-fluoroisocyanate, which was confirmed by comparison to an authentic sample. Formation of the isocyanate product suggests that COSe release does not occur upon irradiation, but rather that H2Se/HSe− is released directly from the selenocarbamate through a pathway that is fundamentally different than that of the analogous thiocarbamates (vide infra).
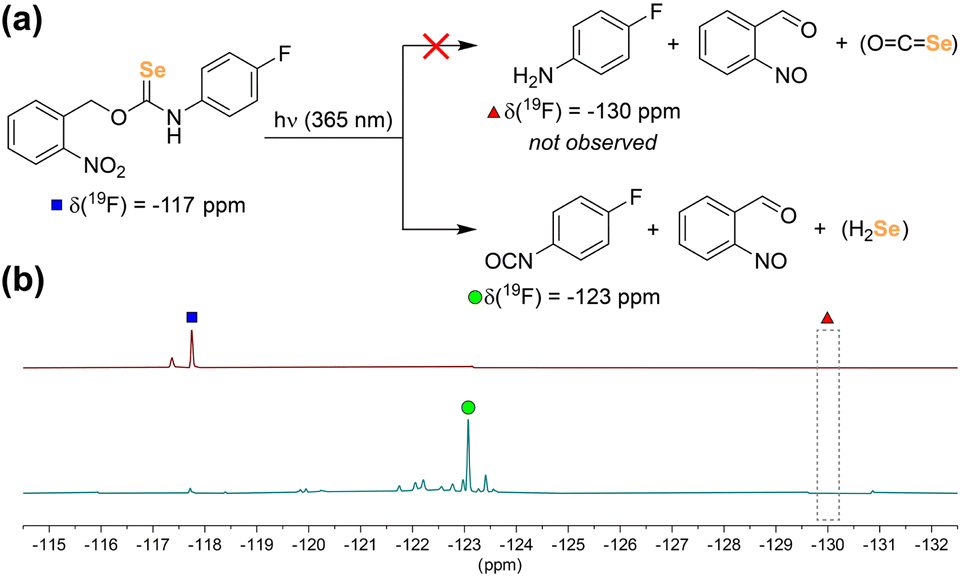 | ||
| Fig. 3 (a) Chemical reactivity of the PhotoSeCM after irradiation. (b) Stacked 19F NMR spectra of PhotoSeCM before (top) and after (bottom) irradiation. The small peak at −117.4 ppm corresponds to a PhotoSeCM rotamer, as is commonly seen for aryl thio- or selenocarbamates. | ||
Building from the PhotoSeCM chemistry, we next wanted to investigate potential COSe/H2Se release from a system that did not require photoactivation. To simplify the activation mechanism, we turned to prior work from our lab on hydrolysis-activated γ-ketothiocarbamates. These systems undergo deprotonation at the β-position to initiate a self-immolative cascade to release COS/H2S and an amine-based payload with reaction rates increasing with increasing pH of solution (Fig. 4a).35 Moreover, inclusion of the p-nitroaniline payload provides a colorimetric response upon donor activation due to the yellow absorbance at 381 nm. To prepare the γ-ketoselenocarbamates, we treated 4-nitrophenyl isoselenocyanate with the desired 4-hydroxybutanone derivative in THF using DBU as a base. We found that the parent compound derived from 4-hydroxybutanone was unstable and could not be isolated, but we were able to prepare the 3-methyl (1) and 3,3-dimethyl (2) derivates, albeit in low isolated yields (Fig. 4b). The γ-ketoselenocarbamates were isolated as the O-alkyl isomers after SiO2 purification, with only minimal Se-alkyl formation that could be removed by purification.
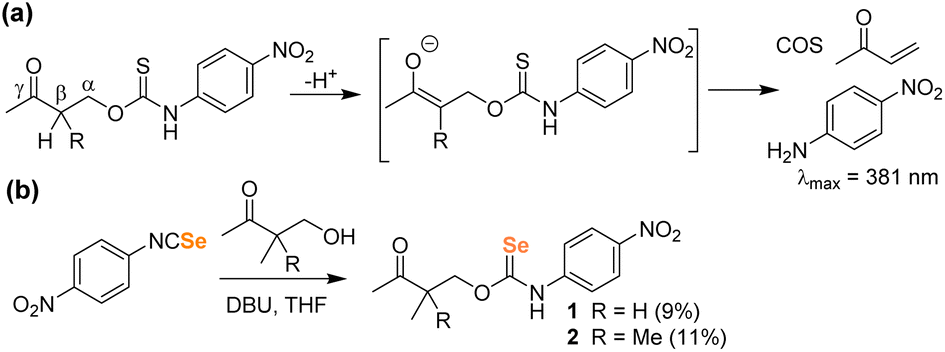 | ||
| Fig. 4 (a) COS/H2S release mechanism from γ-ketothiocarbamates after deprotonation at the β-position. (b) Synthesis of γ-ketoselenocarbamates. | ||
With the γ-ketoselenocarbamate compounds in hand, we next investigated the activation of these donors at different pH values by monitoring the UV-vis spectrum of the released p-nitroaniline product. As expected, compound 2, which lacks deprotonatable hydrogens in the β-position, did not release p-nitroaniline, which also supports the general stability of the selenocarbamate motif in solution. Compound 1, however, did release p-nitroaniline as expected, and the rate of release increased at more basic pH values, which is consistent with the expected β-deprotonation mechanism (Fig. 5). Generation of p-nitroaniline is consistent with either COSe release followed by rapid hydrolysis or direct release of H2Se, formation of the p-nitrophenyl isocyanate, and subsequent hydrolysis. We did not observe the p-nitrophenyl isocyanate product directly, but based on the significantly electron withdrawing NO2 group we expect the isocyanate would be rapidly hydrolyzed to p-nitroaniline and CO2, especially under basic conditions. In addition to the formation of p-nitroaniline, we also observed the precipitation of red Se0, which is consistent with HSe− delivery followed by subsequent autooxidation as is common in aqueous solution. We also compared the second order rate constants for donor activation between 1 and the analogous thiocarbamate (γ-KetoTCM-2).35 We measured k values of 0.38(1) M−1 s−1 and 0.164(4) M−1 s−1 for 1 and γ-KetoTCM-2, respectively, which shows a slightly faster rate of release from the Se than the S analogues.
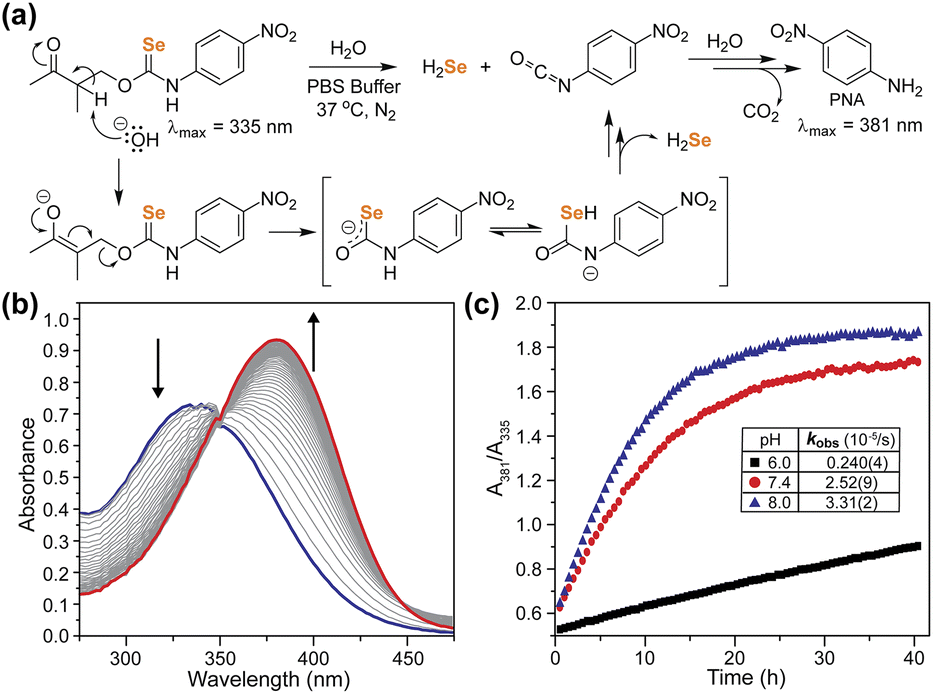 | ||
| Fig. 5 (a) Proposed reaction mechanism to generate H2Se and PNA, (b) representative UV-vis traces of 1 showing PNA formation in pH 7.4, 10 mM PBS, 37 °C under N2, and (c) plotted absorbance ratio corresponding to PNA formation at different pH values. | ||
To investigate H2Se release directly, we first attempted a two-step H2Se volatilization experiment using BnBr as an electrophilic trap under anhydrous and anaerobic conditions as was performed previously for acid-activated H2Se donors.24 Starting with a solution of 1 in DMF, the addition of NaOH resulted in an immediate color change from yellow to red brown, which we attribute to formation of the selenocarbamate intermediate. Upon acidification, we expected to observe H2Se release but instead observed immediate precipitation of elemental Se, which was confirmed by treating the resulting reaction product with PPh3 to form SePPh3 (Fig. S18†). After removal of DMF solvent, the resultant 1H NMR spectrum did not show the p-nitroaniline product, but rather a new product corresponding to protonated p-phenylenediamine, which was confirmed by comparison with the authentic product (Fig. S19†). Based on this observed reactivity, we surmised that the p-nitroaniline product was reduced by the released HSe− in DMF (Fig. 6). Similar chemical reactivities of H2Se toward nitroarenes have been previously reported.42 These results confirm that even in the absence of H2O, HSe− is released directly, rather than being generated indirectly by the hydrolysis of COSe. To validate the hypothesis that HSe− can reduce nitroaromatics under these conditions, we treated p-nitroaniline with [NBu4][HSe] directly in MeCN, which led to an immediate color change to deep red-brown with precipitation of a red brown solid (Fig. 6). The 1H NMR spectrum of the product showed clean formation of p-phenylenediamine (Fig. S20†), and the 77Se NMR spectrum showed consumption of [NBu4][SeH], indicative of oxidation of HSe− to solid Se0 (Fig. S21†). In contrast, treatment of p-nitroaniline with [NBu4][SeH] in aqueous buffer only results in minimal nitro group reduction, which is consistent with the data from Fig. 5. Although Se0 formation could potentially be accounted for by the autooxidation of COSe to form CO and Se0, the experimental observation of p-phenylenediamine formation under the above conditions is most consistent with initial H2Se release followed by subsequent nitro group reduction with concomitant H2Se oxidation to Se0. Taken together, the above data strongly supports the direct release of H2Se from selenocarbamates.
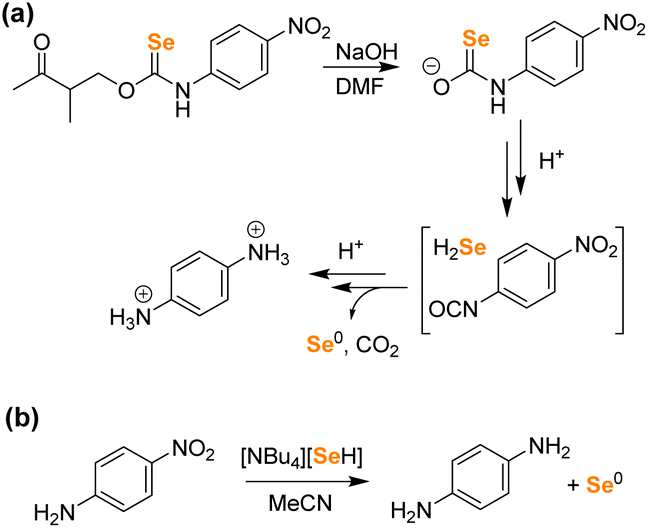 | ||
| Fig. 6 (a) Observed reactivity of 1 after deprotonation in DMF. Both the p-phenylenediamine and Se0 products were observed by NMR spectroscopy. (b) Treatment of p-nitroaniline with HSe− results in nitro group reduction in organic solution, whereas the reduction in water is inefficient. | ||
Computational investigations
To probe the competing decomposition pathways of selenocarbamates, we performed Density Functional Theory (DFT) calculations using the ωB97X-D functional, the aug-cc-pVDZ basis set for geometry optimizations and frequency calculations, and the SMD implicit solvation model for water. Single-point energy refinements were then obtained using the same functional and solvation model but using the triple-zeta aug-cc-pV(T+d)Z basis set. Full computational details can be found in the ESI.†Because little is known about the structure of selenocarbamate anions, we first investigated the preferred tautomer of the anionic phenylselenocarbamate 3 as a computational model (Fig. 7). This selenocarbamate intermediate would be the direct structure from which either COSe or HSe− is generated. We find that the NH tautomer (3a) is the lowest in energy, and the SeH (3b) and OH (3c) tautomers are 17.5 and 14.4 kcal mol−1 higher in free energy, respectively. Tautomer 3a benefits from the stabilization of the negative charge by the most electronegative (O) and most polarizable (Se) atoms, in addition to a flat structure with the phenyl ring trans to selenium. In contrast, the most stable conformers of 3b and 3c have the Ph group cis to selenium, rotated away from the plane of the selenocarbamate. This structure is likely preferred due to strong hyperconjugative interactions between the sp2 lone pair of nitrogen and the C–Se σ* acceptor orbital that are in an antiperiplanar arrangement. As expected from the particularly weak H–Se bond (BDE = 66.0 kcal mol−1, lower than H–I = 70.5 kcal mol−1)43 there is a larger free energy difference between 3b and 3a (17.5 kcal mol−1) than what we computed previously for the NH and SH tautomers in dithiocarbamates (9.7 kcal mol−1).29
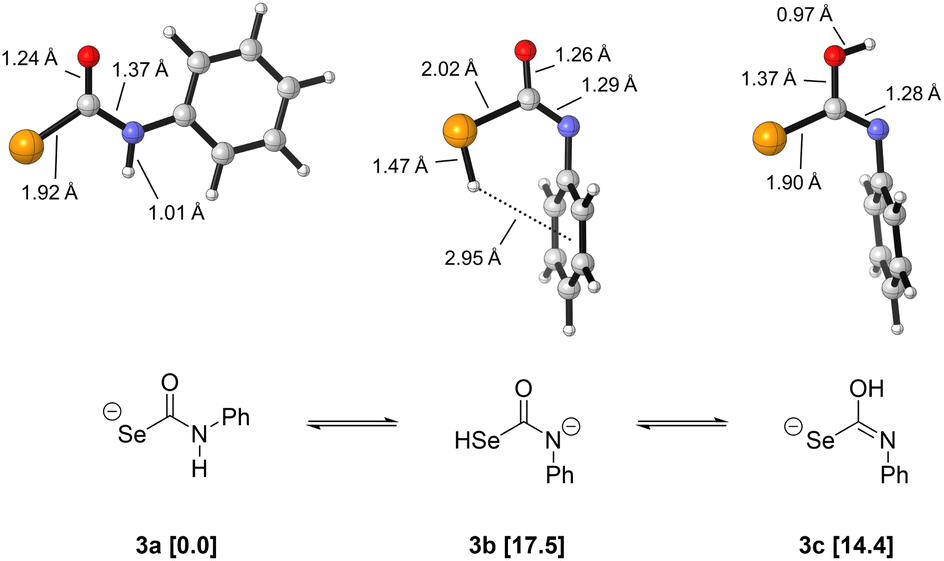 | ||
| Fig. 7 Tautomeric forms and respective free energies (kcal mol−1) of selenocarbamate 3. Calculations at the ωB97X-D/aug-cc-pVDZ/SMD(water)//ωB97X-D/aug-cc-pV(T+d)Z/SMD(water) level of theory. | ||
Our experimental evidence indicates that selenocarbamates like 3 form HSe− directly, but we also investigated the different decomposition pathways of 3 that could result in either COSe or HSe− release to better understand the energetic landscape accessible to selenocarbamates. Starting from 3a, C–N cleavage could occur directly (TS1, Fig. 8), forming COSe and the anilide anion. This pathway has an insurmountable 47.1 kcal mol−1 free energy barrier and is also endergonic by 36.1 kcal mol−1. Despite the expected basicity of anilide in organic solvents, the pKa of anilide in water is not known. Using DFT, we estimate that protonation of anilide by an isolated water molecule (forming hydroxide and aniline) is only favored by 5.1 kcal mol−1. This small value may be due to the difficulty of modeling proton transfer by DFT and the inaccuracy of treating hydroxide and water using only implicit solvation models. Previous computational work from our group showed that the C–N cleavage pathway of thiocarbamates could be accelerated by the presence of an external acid such as water or dihydrogen phosphate (H2PO4−).29 Although there is a slight enthalpic benefit to having an explicit water molecule as an acid mediator to facilitate proton transfer, our calculations for selenocarbamate 3 indicate that there is no free energy benefit and the barrier for TS1 remains too high to represent a viable reaction pathway (see ESI†). Another scenario is the protonation of 3a to form a neutral or zwitterionic selenocarbamic acid, from which decomposition to release COSe could occur. The pKa of selenocarbamic acid has, to the best of our knowledge, never been measured, so we modeled its protonation by water (see ESI†). We find that the barrier for COSe release from a protonated 3a is at least 22.0 kcal mol−1 and would be higher for more acidic selenocarbamates, such as the p-nitro derivative 1. Such a zwitterionic pathway, however, would be counter to the observed increased rate of product formation at basic pH. Alternatively, elimination of PhNCSe and hydroxide from 3c has an even larger barrier (53.0 kcal mol−1, see ESI†) than direct COSe extrusion through TS1, suggesting that neither of these pathways are operable under normal experimental conditions.
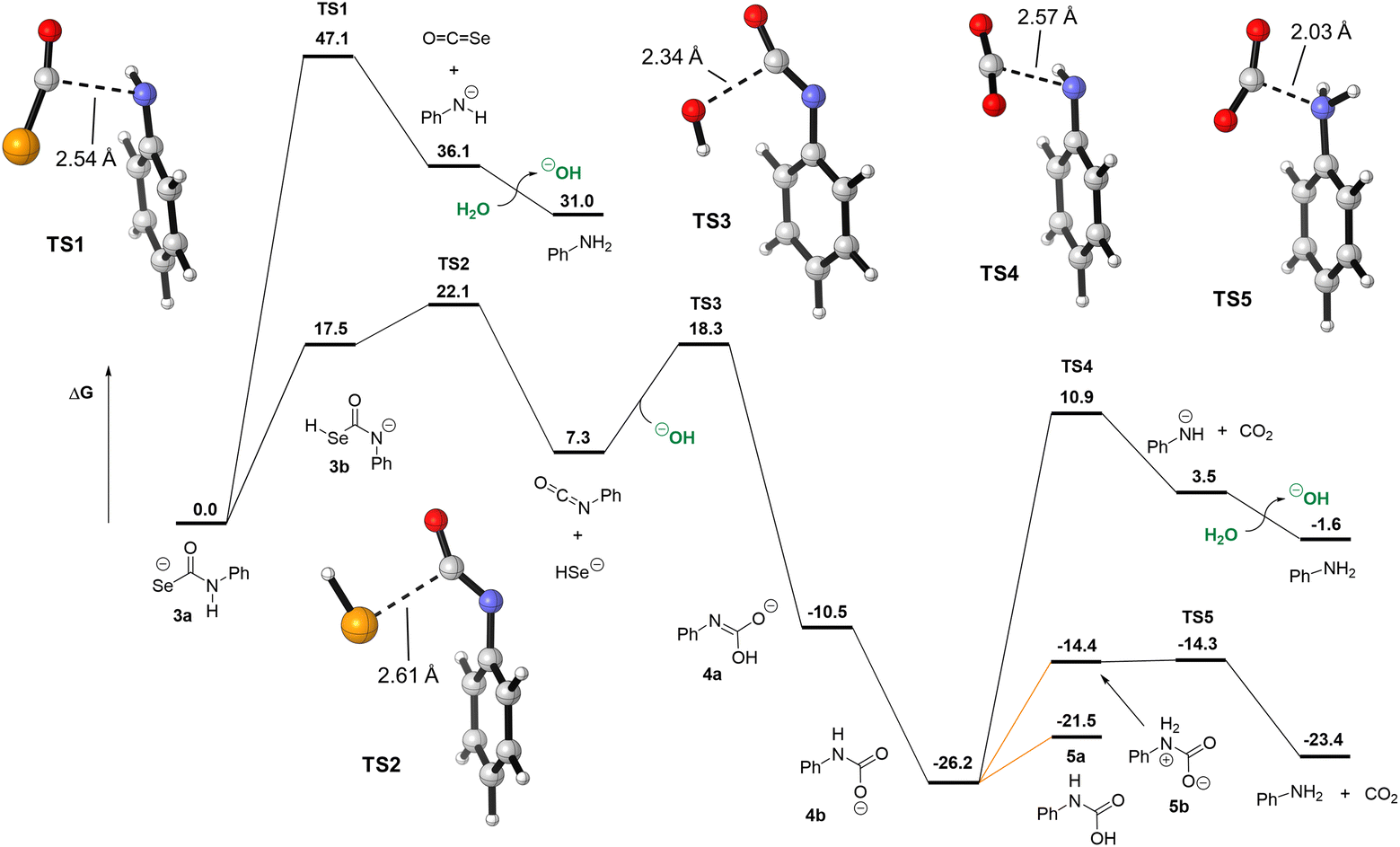 | ||
| Fig. 8 Decomposition pathways of selenocarbamate 3 comparing COSe and HSe− extrusion pathways. Direct release of HSe− is more energetically favorable than COSe release. Free energies (in kcal mol−1) obtained at the ωB97X-D/aug-cc-pVDZ/SMD(water)//ωB97X-D/aug-cc-pV(T+d)Z/SMD(water) level of theory. Orange lines represent protonation free energies computed from the known pKa of phenylcarbamic acid in water and assuming a reaction pH of 8 (see ESI†). | ||
We also investigated the reaction pathway for direct HSe− release from 3. Although 3b is the highest-energy tautomer of the selenocarbamate, this tautomer can readily cleave to form HSe− and PhNCO (TS2). From 3b, this TS only requires an additional 4.6 kcal mol−1 and represents a 22.1 kcal mol−1 barrier from the lowest-energy tautomer 3a. Importantly, TS2 only requires tautomerization from 3a to 3b and so is not affected by the pKa of selenocarbamate 3a in the same way that would be required for elimination of COSe and aniline. This conclusion matches the fact that HSe− generation is the preferred pathway experimentally. Isocyanates, especially those with electron-withdrawing groups, are known to hydrolyze quickly in acidic, basic, or neutral water.44,45 Using hydroxide as nucleophile, we located TS3 that has a 11.0 kcal mol−1 barrier from the phenylisocyanate, forming carbamate anion 4a and its tautomer 4b, which are 10.5 and 26.2 kcal mol−1 more stable than 3a, respectively. The activation energy for TS3 is consistent with the kinetics of hydroxide attack on phenylisocyanate,45 but might be underestimated for a reaction at pH 8. The mechanism of decarboxylation of carbamates such as 4b have been studied, and their rates are known to be pH-dependent, with proton transfer to form the key zwitterionic intermediate 5b being rate-determining.46 Indeed, direct decarboxylation from the anionic 4b requires 37.1 kcal mol−1viaTS4, which is inconsistent with the known reactivity of carbamates. Based on the reported pKa of 4b in water (4.62)46 and a reaction pH of 8, we compute that forming 5a only requires 4.6 kcal mol−1 and that its zwitterionic tautomer 5b is 7.2 kcal mol−1 higher in free energy (see ESI for details†). From 5b, decarboxylation is essentially barrierless, forming the final products aniline and CO2. For the p-nitrophenylcarbamic acid, an intermediate that would be formed in the reaction of 1, this protonation free energy increases to 9.3 kcal mol−1 by virtue of its lower pKa of 1.2. Although the decarboxylation would be slower, the hydrolysis of the isocyanate (TS3) would in contrast benefit from a higher rate. Similarly, we estimate the pKa of the p-nitrophenyl selenocarbamate to be −1.0 (see ESI†), which would preclude its protonation under the basic conditions used in this study and as such make the COSe release mechanism even more energetically difficult.
Discussion
The proposed mechanism for direct H2Se release from selenocarbamates, which is supported by both experimental and computational investigations, differs from the observed prior chemistry with thiocarbamate-based compounds that release COS directly (Fig. 9a). In such systems, direct H2S release is not observed, but rather COS is the primary product. By contrast, similar selenocarbamate compounds do not release COSe, but rather H2Se directly (Fig. 9b). These pathway differences may be in part due to inherent bond strength differences in the O–H (carbamate), S–H (thiocarbamate), and Se–H (selenocarbamate) intermediates that significantly impact the tautomerization thermodynamics. This tautomerization may also be reflected in changes in the acidity, which is consistent with the observation that thioamides and thioureas are ∼5–6 pKa units more acidic than their oxygen congeners, which also suggests that Se incorporation may further acidify the chalcogenocarbamate motifs.47 These factors suggest that the selenocarbamate is less basic than the thiocarbamate, which means that protonation to form the zwitterionic intermediate needed for COSe elimination is less accessible than in the thiocarbamate case, leading to direct HSe− rather than COSe release. In addition, the significant differences in CO, CS, and CSe bond enthalpies mean that the formation is COSe is less favorable than COS or CO2. Moreover, this observed reactivity is similar to prior work on self-immolative aryl dithiocarbamates, which release H2S and Ar-NCS directly rather than extruding CS2 (Fig. 9c).29 In prior work with dithiocarbamates, inclusion of a para-nitro group on the aryl payload acidified the dithiocarbamate and increased the efficiency of direct H2S and isothiocyanate release, which is also consistent with the observed chemistry for the selenocarbamate motifs.
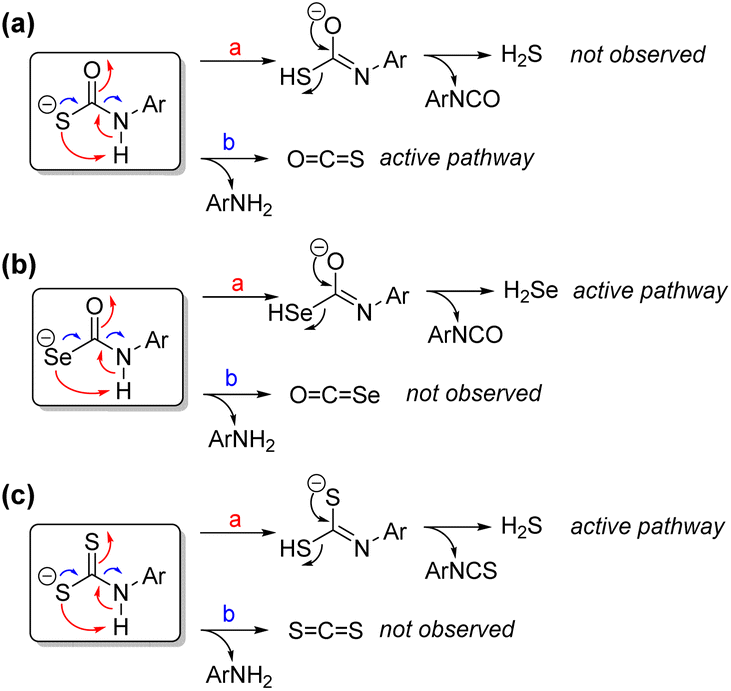 | ||
| Fig. 9 Possible reaction pathways leading to H2S/H2Se and COS/COSe generation from (a) thiocarbamates, (b) selenocarbamates, and (c) dithiocarbamates. | ||
Conclusion
In summary, we show here key differences between the chemistry of thiocarbamates and selenocarbamates with respect to H2S and H2Se release. Prior work has established that thiocarbamates readily release COS, which can be quickly hydrolyzed to H2S by carbonic anhydrases. In contrast, the selenocarbamates investigated in this work release H2Se directly rather than proceeding through the intermediate release of COSe. This observed reactivity is supported by both experimental and computational investigations. More broadly, the direct release of H2Se from activatable selenocarbamates may have a higher utility than if COSe were released as an intermediate because the activity of carbonic anhydrases toward COSe as well as the water stability/reactivity of COSe remains unclear. More broadly, this work further highlights that although sulfur and selenium are often described as two of the most similar elements on the periodic table, their fundamental reaction chemistry is significantly different. Understanding these differences are key for both the development of new chemical tools for investigation bioorganic selenium chemistry and also for understanding the chemical underpinnings of why nature has differentiated between these two otherwise similar elements.Materials and methods
Reagents were purchased from Sigma-Aldrich, Alfa Aesar, or TCI Chemicals and were used directly as received. Deuterated solvents were purchased from Cambridge Isotope Laboratories and used directly as received. [NBu4][SeH] was prepared following literature procedures.481H, 13C{1H}, 19F, 31P and 77Se NMR spectra were recorded on Bruker 500 and 600 MHz instruments. Chemical shifts are reported relative to residual protic solvent resonances for 1H and 13C{1H} spectra. All air-free manipulations were performed in an inert atmosphere using standard Schlenk techniques or an Innovative Technologies N2-filled glove box. Note: Triphosgene and selenium containing species are toxic and should be handled in a fume hood with appropriate personal protective equipment.Synthesis
:1 hexanes/EtOAc; product Rf = 0.55). The reaction mixture was then removed from the oil bath, allowed to cool to room temperature, filtered through a Celite pad, and concentrated to dryness under vacuum. The resulting crude product was purified by SiO2 column chromatography (gradient of hexanes to 1:1 EtOAc/hexanes) to afford a yellow solid (0.571 g, 70%). 1H NMR (600 MHz, CDCl3) δ: 8.26 (d, 2H, J = 8.4 Hz), 8.42 (d, 2H, J = 8.4 Hz). 13C{1H} NMR (151 MHz, CDCl3) δ: 146.2, 135.9, 135.7, 126.9, 125.3. 77Se NMR (115 MHz, CDCl3) δ: −255.9 (s). HRMS m/z [M]− calcd for [C7H4N2O2Se]−: 227.9438; found 227.9434.
:1 EtOAc/hexanes) was used to monitor the reaction over the course of an hour to monitor formation of major O-alkyl product (Rf = 0.50) and minor undesired Se-alkyl isomer (Rf = 0.30). Once any formation of Se-alkyl isomer was observed, the reaction mixture was quenched in brine, extracted in EtOAc (3 × 15 mL), dried over MgSO4, filtered, and concentrated to dryness under vacuum to yield the crude product as an orange solid, further recrystallization by layering hexanes over saturated solution of the crude product in DCM afforded desired product as a white precipitate, which was collected via filtration (181 mg, 51%). There are two rotamers of this compound due to slow rotation around the C–N bond, which is also commonly observed for thiocarbamates. Where resolved the two chemical shifts are separated by a “/” for the two rotamer peaks. 1H NMR (600 MHz, DMSO-d6) δ: 11.96/11.86 (s, 1H), 8.18/8.13 (d, 1H, J = 8.4 Hz), 7.87–7.53 (m, 4H), 7.41/7.39 (d, 1H, J = 8.4 Hz), 7.24/7.19 (t, 2H, J = 8.4 Hz), 6.01/5.92 (s, 2H). 13C{1H} NMR (151 MHz, DMSO-d6) δ: 188.9, 160.0 (d, J1C–F = 251 Hz), 147.3, 134.2, 133.7, 130.9, 129.5, 129.3, 126.6, 124.7 (d, J3C–F = 8.2 Hz), 115.7 (d, J2C–F = 22 Hz), 72.1. 19F NMR (564 MHz, DMSO-d6) δ: −116.1 (m). 77Se NMR (115 MHz, DMSO-d6) δ: 278.3 (s). HRMS m/z [M − H]− calcd for [C14H10FN2O3Se]−: 352.9841; found 352.9842.
:1 hexanes/EtOAc) for 6 hours and then quenched with brine (25 mL). The resulting mixture was extracted with EtOAc (3 × 20 mL), dried over MgSO4, filtered, and concentrated to dryness under vacuum. The resulting crude material was purified via SiO2 column chromatography (gradient of hexanes to 1:1 hexanes/EtOAc) to afford a yellow-orange solid (37.9 mg, 9%). 1H NMR (600 MHz, DMSO-d6) δ: 12.16 (s, 1H), 8.21 (d, 2H, J = 9.0 Hz), 7.64 (br m, 2H), 4.70 (m, 2H), 3.17 (m, 1H), 2.19 (s, 3H), 1.14 (d, 3H, J = 7.2 Hz). 13C{1H} NMR (151 MHz, DMSO-d6) δ: 209.2, 190.9, 143.6, 125.3, 124.7, 121.8, 76.0, 45.0, 28.4, 13.0. 77Se NMR (115 MHz, DMSO-d6) δ: 350.9 (br s). HRMS m/z [M − H]− calcd for [C12H13N2O4Se]−: 329.0041; found 329.0038.
H2Se release experiments
Computational studies
Gaussian 16 was used to perform Density Functional Theory (DFT) calculations, where geometry optimizations were conducted with the ωB97X-D method using the aug-cc-pVDZ basis set. Following this, single-point energy refinements were performed using the ωB97X-D functional and the aug-cc-pV(T+d)Z basis set. The SMD implicit solvation model for water was used at all stages to consider solvation effects. The final free energies were computed by adding the free energy corrections obtained from the frequency analysis to the single-point electronic energies. Full computational details are provided in the ESI.†Data availability
Experimental and computational data are included in the ESI.†Author contributions
MDP and PAC supervised this work. TDN and KL synthesized and characterized all compounds and made experimental measurements. JS and PAC carried out computational investigations and analysis. TDN, KL, JS, PAC, and MDP wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the NSF (CHE-2004150 to MDP, DGE-2022168 to KL) for support of this research. The support of the ACS Petroleum Research Fund donors (61891-DNI4 to PAC) is gratefully acknowledged. Calculations were performed on the Lochness cluster at NJIT.Notes and references
- M. R. Filipovic, J. Zivanovic, B. Alvarez and R. Banerjee, Chem. Rev., 2018, 118, 377–461 CrossRef PubMed.
- L. Liaudet, F. G. Soriano and C. Szabo, Crit. Care Med., 2000, 28, N37–N52 CrossRef CAS PubMed.
- S. W. Ryter and L. E. Otterbein, BioEssays, 2004, 26, 270–280 CrossRef CAS PubMed.
- R. Wang, Physiol. Rev., 2012, 92, 791–896 CrossRef CAS PubMed.
- D. A. Wink and J. B. Mitchell, Free Radical Biol. Med., 1998, 25, 434–456 CrossRef CAS PubMed.
- L. Y. Wu and R. Wang, Pharmacol. Rev., 2005, 57, 585–630 CrossRef CAS PubMed.
- X. Q. Chen, X. Z. Tian, I. Shin and J. Yoon, Chem. Soc. Rev., 2011, 40, 4783–4804 RSC.
- V. S. Lin and C. J. Chang, Curr. Opin. Chem. Biol., 2012, 16, 595–601 CrossRef CAS PubMed.
- C. R. Powell, K. M. Dillon and J. B. Matson, Biochem. Pharmacol., 2018, 149, 110–123 CrossRef CAS PubMed.
- C. M. Levinn, M. M. Cerda and M. D. Pluth, Antioxid. Redox Signaling, 2020, 32, 96–109 CrossRef CAS PubMed.
- P. G. Wang, M. Xian, X. P. Tang, X. J. Wu, Z. Wen, T. W. Cai and A. J. Janczuk, Chem. Rev., 2002, 102, 1091–1134 CrossRef CAS PubMed.
- T. Nagano and T. Yoshimura, Chem. Rev., 2002, 102, 1235–1269 CrossRef CAS PubMed.
- X. Y. Ji, K. Damera, Y. Q. Zheng, B. C. Yu, L. E. Otterbein and B. H. Wang, J. Pharm. Sci., 2016, 105, 406–416 CrossRef CAS PubMed.
- C. C. Romao, W. A. Blattler, J. D. Seixas and G. J. L. Bernardes, Chem. Soc. Rev., 2012, 41, 3571–3583 RSC.
- K. A. Cupp-Sutton and M. T. Ashby, Antioxidants, 2016, 5, 42 CrossRef PubMed.
- M. Roman, P. Jitaru and C. Barbante, Metallomics, 2014, 6, 25–54 CrossRef CAS PubMed.
- C. Hartmann, B. Nussbaum, E. Calzia, P. Radermacher and M. Wepler, Front. Physiol., 2017, 8, 691 CrossRef PubMed.
- M. Kuganesan, K. Samra, E. Evans, M. Singer and A. Dyson, Intensive Care Med. Exp., 2019, 7, 71 CrossRef PubMed.
- X. Y. Kang, H. J. Huang, C. Y. Jiang, L. H. Cheng, Y. Q. Sang, X. K. Cai, Y. L. Dong, L. Sun, X. Wen, Z. Xi and L. Yi, J. Am. Chem. Soc., 2022, 144, 3957–3967 CrossRef CAS PubMed.
- D. Kang, J. Lee, C. Wu, X. Guo, B. J. Lee, J. S. Chun and J. H. Kim, Exp. Mol. Med., 2020, 52, 1198–1208 CrossRef CAS PubMed.
- C. Szabo and A. Papapetropoulos, Pharmacol. Rev., 2017, 69, 497–564 CrossRef CAS PubMed.
- L. Li, M. Whiteman, Y. Y. Guan, K. L. Neo, Y. Cheng, S. W. Lee, Y. Zhao, R. Baskar, C. H. Tan and P. K. Moore, Circulation, 2008, 117, 2351–2360 CrossRef CAS PubMed.
- W. Feng, X. Y. Teo, W. Novera, P. M. Ramanujulu, D. Liang, D. J. Huang, P. K. Moore, L. W. Deng and B. W. Dymock, J. Med. Chem., 2015, 58, 6456–6480 CrossRef CAS PubMed.
- T. D. Newton and M. D. Pluth, Chem. Sci., 2019, 10, 10723–10727 RSC.
- T. D. Newton, S. G. Bolton, A. C. Garcia, J. E. Chouinard, S. L. Golledge, L. N. Zakharov and M. D. Pluth, J. Am. Chem. Soc., 2021, 143, 19542–19550 CrossRef CAS PubMed.
- R. A. Hankins, M. E. Carter, C. L. Zhu, C. Chen and J. C. Lukesh, Chem. Sci., 2022, 13, 13094–13099 RSC.
- A. K. Steiger, Y. Zhao and M. D. Pluth, Antioxid. Redox Signaling, 2018, 28, 1516–1532 CrossRef CAS PubMed.
- Y. Zhao and M. D. Pluth, Angew. Chem., Int. Ed., 2016, 55, 14638–14642 CrossRef CAS PubMed.
- Y. Zhao, H. A. Henthorn and M. D. Pluth, J. Am. Chem. Soc., 2017, 139, 16365–16376 CrossRef CAS PubMed.
- Y. M. Hu, X. Y. Li, Y. Fang, W. Shi, X. H. Li, W. Chen, M. Xian and H. M. Ma, Chem. Sci., 2019, 10, 7690–7694 RSC.
- C. L. Zhu, S. I. Suarez and J. C. Lukesh, Tetrahedron Lett., 2021, 69, 152944 CrossRef CAS.
- Y. Zhao, S. G. Bolton and M. D. Pluth, Org. Lett., 2017, 19, 2278–2281 CrossRef CAS PubMed.
- A. K. Sharma, M. Nair, P. Chauhan, K. Gupta, D. K. Saini and H. Chakrapani, Org. Lett., 2017, 19, 4822–4825 CrossRef CAS PubMed.
- P. Stacko, L. Muchova, L. Vitek and P. Klan, Org. Lett., 2018, 20, 4907–4911 CrossRef CAS PubMed.
- Y. Zhao, A. K. Steiger and M. D. Pluth, Angew. Chem., Int. Ed., 2018, 57, 13101–13105 CrossRef CAS PubMed.
- A. K. Gilbert, Y. Zhao, C. E. Otteson and M. D. Pluth, J. Org. Chem., 2019, 84, 14469–14475 CrossRef CAS PubMed.
- P. Chauhan, P. Bora, G. Ravikumar, S. Jos and H. Chakrapani, Org. Lett., 2017, 19, 62–65 CrossRef CAS PubMed.
- A. K. Steiger, M. Marcatti, C. Szabo, B. Szczesny and M. D. Pluth, ACS Chem. Biol., 2017, 12, 2117–2123 CrossRef CAS PubMed.
- C. Y. Zhang, Q. Z. Zhang, K. Zhang, L. Y. Li, M. D. Pluth, L. Yi and Z. Xi, Chem. Sci., 2019, 10, 1945–1952 RSC.
- K. Eriksen, A. Ulfkjaer, T. I. Solling and M. Pittelkow, J. Org. Chem., 2018, 83, 10786–10797 CrossRef CAS PubMed.
- A. Sorensen, B. Rasmussen, S. Agarwal, M. Schau-Magnussen, T. I. Solling and M. Pittelkow, Angew. Chem., Int. Ed., 2013, 52, 12346–12349 CrossRef CAS PubMed.
- T. Miyata, K. Kondo, S. Murai, T. Hirashima and N. Sonoda, Angew. Chem., Int. Ed. Engl., 1980, 19, 1008 CrossRef.
- Lange's Handbook of Chemistry, McGraw-Hill Education, New York, 17th edn, 2017 Search PubMed.
- A. F. Hegarty, C. N. Hegarty and F. L. Scott, J. Chem. Soc., Perkin Trans. 2, 1975, 1166–1171 RSC.
- E. A. Castro, R. B. Moodie and P. J. Sansom, J. Chem. Soc., Perkin Trans. 2, 1985, 737–742 RSC.
- S. L. Johnson and D. L. Morrison, J. Am. Chem. Soc., 1972, 94, 1323–1334 CrossRef CAS PubMed.
- F. G. Bordwell, Acc. Chem. Res., 1988, 21, 456–463 CrossRef CAS.
- H. A. Fargher, N. Lau, L. N. Zakharov, M. M. Haley, D. W. Johnson and M. D. Pluth, Chem. Sci., 2019, 10, 67–72 RSC.
- D. H. R. Barton, S. I. Parekh, M. Tajbakhsh, E. A. Theodorakis and C. L. Tse, Tetrahedron, 1994, 50, 639–654 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR spectra and full computational details. See DOI: https://doi.org/10.1039/d3sc01936e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |