
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceScalable synthesis of soluble crystalline ionic-graphdiyne by controlled ion expansion†
Lingling
Wang
a,
Lu
Qi
a,
Qinglei
Zhang
 b,
Binghui
Xue
c,
Zhiqiang
Zheng
a,
Panchao
Yin
c,
Yurui
Xue
a,
Wenlong
Yang
*a and
Yuliang
Li
*ade
b,
Binghui
Xue
c,
Zhiqiang
Zheng
a,
Panchao
Yin
c,
Yurui
Xue
a,
Wenlong
Yang
*a and
Yuliang
Li
*ade
aShandong Provincial Key Laboratory for Science of Material Creation and Energy Conversion, Science Center for Material Creation and Energy Conversion, Institute of Frontier and Interdisciplinary Science, Shandong University, Qingdao, 266237, P. R. China. E-mail: wlyang@sdu.edu.cn; ylli@iccas.ac.cn
bState Key Laboratory of Luminescent Materials and Devices, Institute of Polymer Optoelectronic Materials and Devices, South China University of Technology, Guangzhou, 510640, P. R. China
cSouth China Advanced Institute for Soft Matter Science and Technology, South China University of Technology, Guangzhou, 510640, P. R. China
dInstitute of Chemistry, Chinese Academy of Sciences, Beijing, 100190, P. R. China
eUniversity of Chinese Academy of Sciences, Beijing, 100049, P. R. China
First published on 11th April 2023
Abstract
Graphdiyne (GDY) is a promising material possessing extensive electronic tunability, high π conjugacy, and ordered porosity at a molecular level for the sp/sp2-hybridized periodic structures. Despite these advantages, the preparation of soluble and crystalline graphdiyne is limited by the relatively compact stacking interactions, mostly existing in thick-layer and insoluble solids. Herein, we proposed a strategy of “framework charge-induced intercalation (FCII)” for the synthesis of a soluble (4.3 mg ml−1) and yet interlayer-expanded (∼0.6 Å) crystalline ionic graphdiyne, named as N+-GDY, through regulating the interlayer interactions. The skeleton of such a sample is positively charged, and then the negative ions migrate to the interlayer to expand the space, endowing the N+-GDY with solution processability. The crystal structure of N+-GDY is proved through analysis of HR-TEM images under different axes of observation and theoretical simulations. The resulting N+-GDY possesses high dispersity in organic solvents to produce a pure-solution phase which is conducive to the formation of oriented N+-GDY films, accompanied by exfoliation–nanosheet restacking. The film exhibits a conductivity of 0.014 S m−1, enabling its applications in electronic devices.
Introduction
Graphdiyne (GDY) has emerged as a functional two-dimensional (2D) carbon material that is constructed via the integration of molecular building blocks, recently attracting broad interest in versatile application areas,1 such as catalysts,2 solar cells,3 rechargeable batteries,4 electronic devices,5 detectors,6 biomedicine and therapy,7 and water purification.8 So far, researchers have made considerable efforts to synthesize well-defined GDY, whereas there are still existing numerous challenges, including the preparation of mono- or few-layer crystalline nanosheets and GDY solutions. For example, the strong inter-layer π–π stacking as well as the relatively possible restacking result in ill-defined cleave nanolayers, which renders it highly challenging to partition 2D GDY to harvest large-size, mono-layer, and multi-layer sheets. The highly crystalline few-layer nanosheets provide the profound fundamental condition for the preparation of GDY solutions to further synthesize highly crystalline GDY films. Significant attempts have been made in the preparation of few-layer highly crosslinked polymer (CP) nanosheets, finally forming crystalline films for expanding the applications.9 In recent years, few layered GDY nanosheets have been synthesized by conventional delamination methods, including interface-confined growth,10 mechanical,11 and chemical12 exfoliation methods; nevertheless, these methods are difficult to scale up and it is difficult to form high-quality nanosheets using these methods. So far, surface-assisted methods13 have been recommended as the most successful example to form highly crystalline mono- or multi-layer nanosheets of CPs. The search is to weaken the interlayer binding force in CPs to allow simple exfoliation into mono-layer sheets, hopefully with high crystallinity. Through decoupling the interlayer stacking, turbostratic GDY nanosheets, displaying random aggregation, can be recognized, which should weaken the interlayer interaction, facilitate the exfoliation, and finally achieve relative CP solutions. GDY is an emerging 2D carbon material, and it is urgent to develop a solution-treatable GDY synthesis method to promote the further development of this field.The essence of the formation of soluble GDY is to weaken the interlayer stacking forces and enhance framework–solvent interactions of nanosheets to allow self-exfoliation via electrostatic repulsion to obtain high solubility in certain solvents with retaining the GDY basic skeleton.14 Interlayer engineering has been key in preparing expanded interlayer and monolayer nanosheets in expansion–exfoliation, simultaneously tuning properties and improving performances.15 This method can readily intercalate or trap substantial charge species into the inter-layer skeletons to reduce the two adjacent-layer interactions. Therefore, this strategy is achievable for soluble GDY triggered by interlayer expansion to regulate the dispersibility of GDY in various solvents. Herein, we have incorporated charges into the GDY scaffold for the first time to synthesize an interlamellar expansion few-layer crystalline graphite diacetylene compound by the “framework charge-induced intercalation (FCII)” method under facile conditions to endow the nanosheets with uniform restacking thickness, and its solubility can be enhanced (Fig. 1). The unique features of our design include three key processes: concise quaternization of the pyridine-N via methylation (Fig. 1(a)), exfoliation of stacked nanosheets to achieve multilayers (Fig. 1(b)), and the restacking of delaminated nanosheets (Fig. 1(c)). To the best of our knowledge, such a two-dimensional GDY design incorporating building blocks that bear out-of-plane negative ions embedded between layers has not been reported previously. The charge is encoded into the skeleton to regulate the interlayer π–π stacking, so that the resulting pyridine+-based graphdiyne (N+-GDY) features an obvious expanded-interlayer space with a layer enlargement of 0.6 Å. Such a well-crystallized material can be dissolved in organic solvents for feasible film processing. The prepared GDY nanostructured films with interlayer-expanded GDY-based nanostructures and solubility showed well-defined ionic frameworks and a conductivity of 0.014 S m−1.
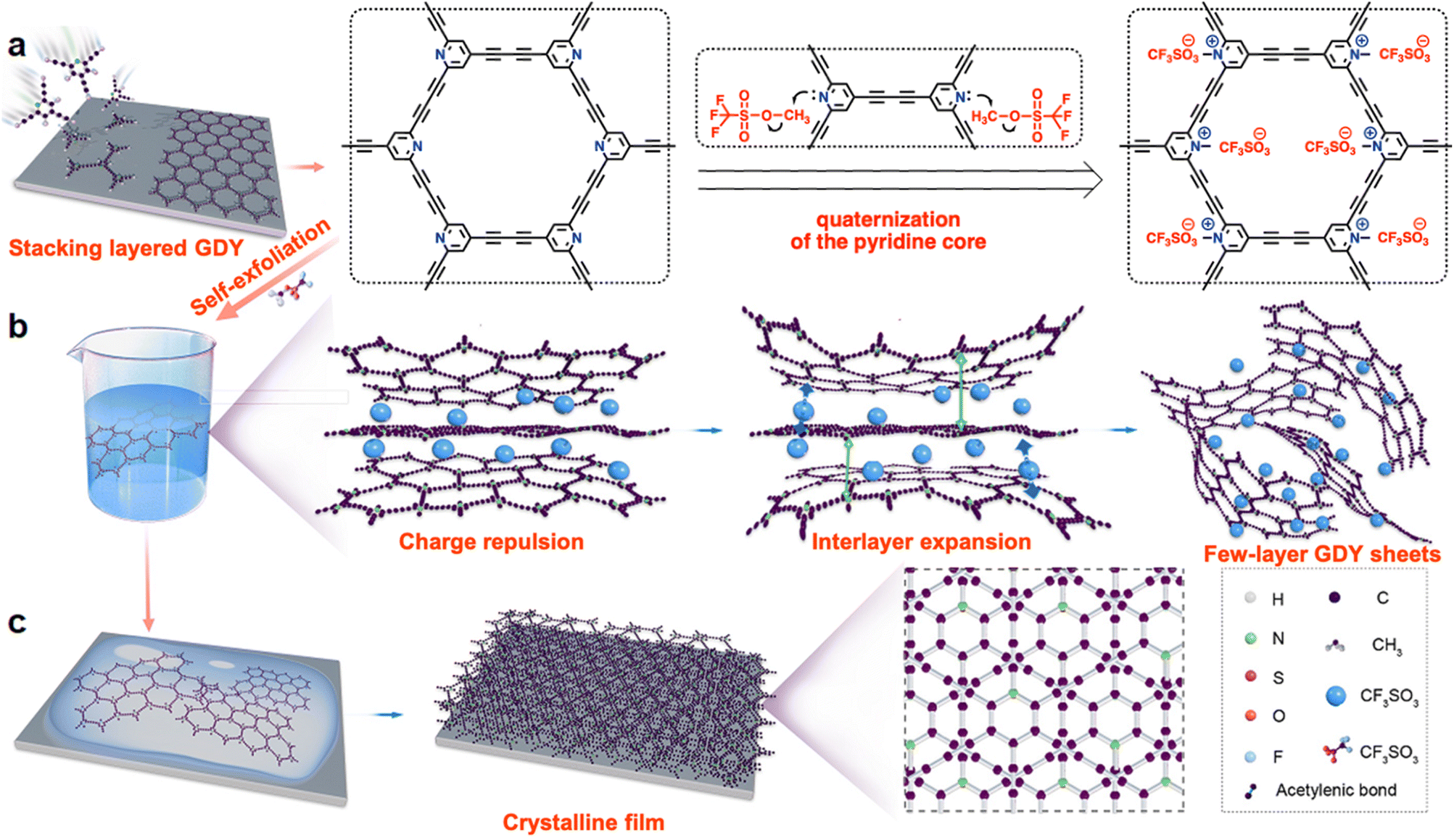 | ||
| Fig. 1 Schematic representation of (a) the synthesis, (b) solubility mechanism, and (c) restacking processes of the N+-GDY. | ||
Results and discussion
We chose 2,4,6-trialkynylpyridine (TAP) as the precursor to prepare pyridine-based graphdiyne (N-GDY), in which the pyridine units are easily converted into 2,4,6-trialkynylpyridinium derivatives through simple one-step quaternization.16 By integrating the pyridinium salts into the skeletons, the positive charges of N+-GDY provide electrostatic repulsion and expand the interlayer space of nanosheets. Interlayer expansion is a preliminary condition for preparing GDY nanosheets in a typical two-step expansion–exfoliation process (Fig. 1). This process facilitates the graphdiyne skeletons to disperse well in organic solvents and form uniform films in a solution-processed manner. The optimal reaction conditions for the synthesis of N-GDY were acetone/pyridine (3/1 v/v) at 60 °C for 3 days.17 Subsequently, the achieved N-GDY powder was added into dichloromethane (DCM), and then methyl trifluoromethanesulfonate (MeOTf) was added dropwise in the mixture at room temperature for 2 hours to obtain N+-GDY powder. By simple manual shaking, the powder could readily dissolve in dimethyl sulfoxide (DMSO) to attain N+-GDY dispersion. In the abovementioned system, the collected powder product was modified with MeOTf, performing as the strong methylating reagent, and eventually the desired N+-GDY solution was obtained. By encoding high-density electrostatic repulsion into the N+-GDY skeleton to withstand the inter-layer π–π interaction, the stacking nanosheets could self-exfoliate into multilayers. The charged N+-GDY was unambiguously characterized by various analytical methods.The chemical skeletons and functional groups of N+-GDY were unambiguously determined by a variety of characterization methods (Fig. 2, S1–S5, Tables S1 and S2†). Fourier transform infrared (FT-IR) spectra of N+-GDY nanosheets showed that the new absorption peak attributed to the C–N+ bond at 1619 cm−1 appeared after methylation, verifying that the pyridine-N was positively charged successfully (Fig. 2(a)). Specifically, the peak at 2115 cm−1 belonging to the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C stretching vibration remained unchanged, assigned to the formation of an asymmetric diacetylene moiety (R–CC–CC–R′), indicative of the N+-GDY maintaining the structural integrity and no decomposition in the ionization process. The obvious characteristic peaks at 1190, 1050, and 630 cm−1 illustrated the successful incorporation of CF3SO3− into the nanosheets. Additionally, the N 1s XPS spectrum of N+-GDY exhibited two obvious peaks at 399.5 and 402.1 eV, corresponding to pyridine N and pyridinium N+-CH3 (Fig. 2(b)); simultaneously it can be calculated that the ratio of pyridine N to pyridinium N+-CH3 is ∼1
C stretching vibration remained unchanged, assigned to the formation of an asymmetric diacetylene moiety (R–CC–CC–R′), indicative of the N+-GDY maintaining the structural integrity and no decomposition in the ionization process. The obvious characteristic peaks at 1190, 1050, and 630 cm−1 illustrated the successful incorporation of CF3SO3− into the nanosheets. Additionally, the N 1s XPS spectrum of N+-GDY exhibited two obvious peaks at 399.5 and 402.1 eV, corresponding to pyridine N and pyridinium N+-CH3 (Fig. 2(b)); simultaneously it can be calculated that the ratio of pyridine N to pyridinium N+-CH3 is ∼1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5, revealing that five-sixths of pyridine N were methylated and one-sixth kept the neutral state. These above-mentioned results clearly proved that the ionized skeletons were synthesized and the anions were incorporated successfully.
:5, revealing that five-sixths of pyridine N were methylated and one-sixth kept the neutral state. These above-mentioned results clearly proved that the ionized skeletons were synthesized and the anions were incorporated successfully.
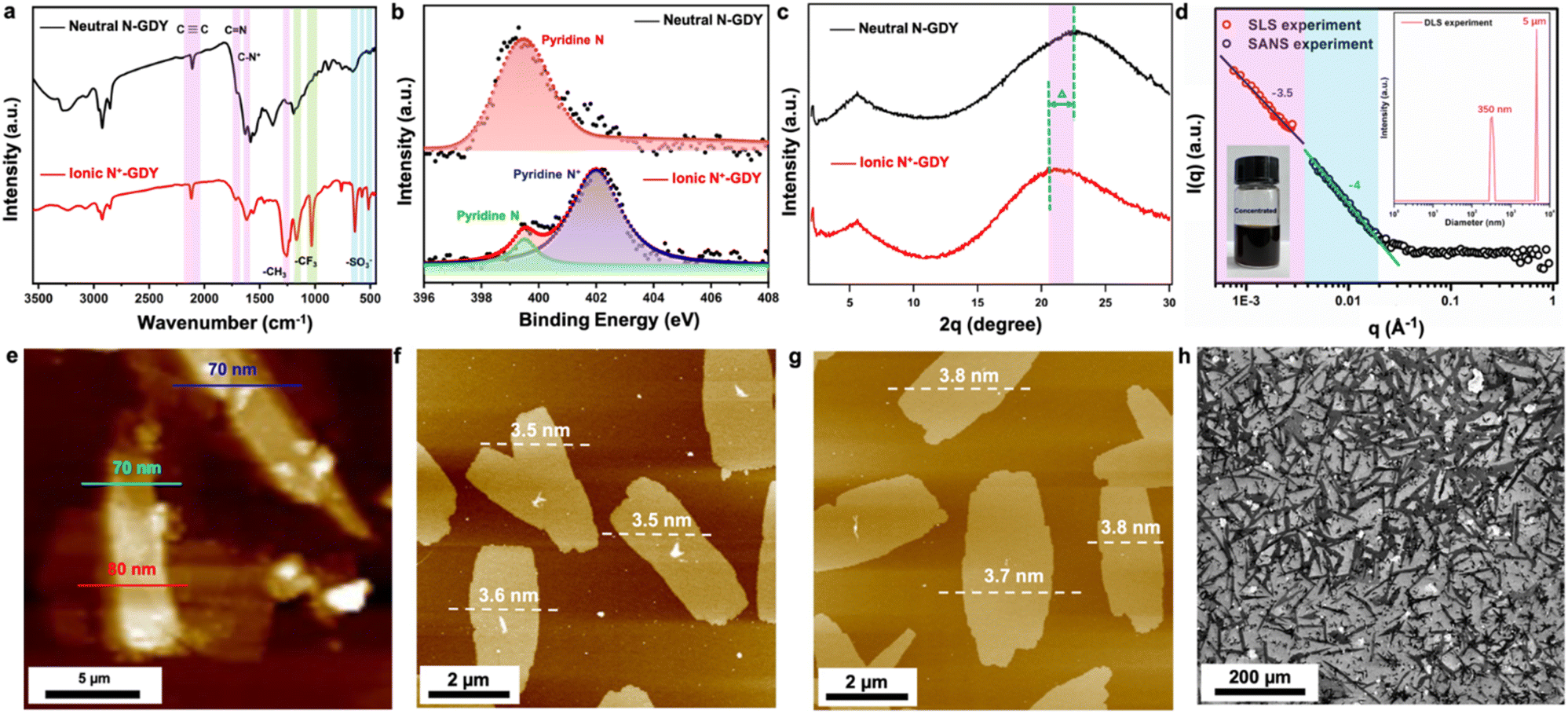 | ||
| Fig. 2 Structural characterization. (a) FTIR, (b) high-resolution N 1s, and (c) PXRD of N-GDY and N+-GDY. (d) SLS, DLS, and SANS measurements of N+-GDY. (e) AFM and thickness of N-GDY stacking nanosheets, and (f) and (g) AFM and thickness, and (h) SEM images of N+-GDY nanosheets. | ||
The crystallinity of N-GDY and N+-GDY powders was determined by using powder X-ray diffraction (PXRD) measurements (Fig. 2(c)). Notably, PXRD of N-GDY and N+-GDY exhibited obviously distinguished π–π stacking peaks, showing a distinctly expanded interlayer space. As mentioned above, the PXRD pattern of neutral N-GDY revealed the most intense peak at 23° which could be attributed to the (001) Bragg's reflection,1g corresponding to a π–π stacking distance of approximately 0.38 nm. However, the ionic N+-GDY showed a peak at 20.5° with a stacking distance of 0.44 nm, indicative of an obvious negative shift of ∼0.6 Å with an interlayer spacing expansion of 15.8%. The obvious discrepancy proves that the CF3SO3− has successfully intercalated into GDY layers, finally expanding the π–π stacking distance. Remarkably, unlike the N-GDY, which is insoluble and forms thick aggregates, the N+-GDY nanosheets readily dissolve in DMSO with a solubility of 4.3 mg ml−1 (inset in Fig. 2(d)), which could exfoliate nanosheets into multilayers that were well dispersed in solutions, because of its positively charged pyridinium ions. Atomic force microscopy (AFM) studies of N+-GDY reveal the 2D sheet-like morphology with a uniform thickness of 3.5–3.8 nm, approximately 8–9 layers (Fig. 2(f), (g), S7(b) and (c)†), in sharp contrast to stacking N-GDY nanosheets yielding a higher thickness of around 70 nm (Fig. 2(e) and S7(a)†), which directly demonstrated that the N+-GDY was successfully delaminated into few-layer formation. Moreover, the thickness of exfoliated N+-GDY flakes is uniform, validating that tailoring the structure of the charges interlocking with the GDY provides a handle to control the thickness of the exfoliated flakes. To gain insight into the solubility mechanism, we calculated the inter-layer and skeleton–solvent interactions of N+-GDY and N-GDY. The resulting inter-layer interactions of N+-GDY were −2.1 eV, whereas they were around −1.0 eV for the N-GDY which is relatively smaller than that of the N+-GDY in terms of absolute value; similarly, the ionic skeleton of skeleton–solvent interactions was as high as −2.5 eV compared to the neutral of −0.3 eV in terms of absolute value, enabling high dispersity of N+-GDY in DMSO (Table S3†). The results clearly validated that the essence of solubility could be attributed to the remarkable skeleton–solvent interactions and relatively weak inter-layer interactions. It can be concluded that the N+-GDY could dissolve in DMSO and form true solutions. As shown in the scanning electron microscopy images (Fig. 2(h)), N+-GDY maintained the 2D skeleton and formed delaminated structures. The N+-GDY could exfoliate into monolayers or multilayers that were well dispersed in the solution. To further explore the size dimension and aggregation behavior of the N+-GDY in solution, we conducted dynamic/static light scattering (DLS/SLS) and small-angle neutron scattering (SANS) measurements of a concentrated solution in DMSO-d6 (Fig. 2(d)). The DLS data showed two types of specifically narrow size distributions of 350 nm and 5 μm, representing single nanosheets and their aggregates, respectively. Subsequently, on account of the large areas, the results of SLS and SANS were well fitted with the Porod law with characteristic slopes as −4 in the low Q range, indicating the smooth feature of local structures of N+-GDY in solutions. Moreover, SLS data in the ultra-low Q range show a similar slope (−3.5), suggesting the surface-wrinkled and the so-resulting collapsed structure of N+-GDY nanosheets. This is actually consistent with transmission electron microscopy (TEM) observations. The sheet dimension is large enough to endow the flexible character of the 2D sheet, leading to its collapsed structure.
The morphology and crystallinity of stacking N-GDY and exfoliated N+-GDY nanosheets were analyzed by SEM and TEM. The initial N-GDY powder exhibited a tightly stacked skeleton (Fig. 3(a) and (b)), whereas the N+-GDY exhibited large-area two-dimensional thin-layer nanosheets without aggregation (Fig. 3(e) and (f)), which obviously illustrated that the incorporation of charges facilitates the easy exfoliation of ultrathick stacking GDY into few layers via ionic repulsion. Interestingly, the high-resolution TEM (HR-TEM) images of N-GDY and N+-GDY nanosheets exhibited an apparently distinctive inter-layer distance. As shown in Fig. 3(c), (d), (g) and (h), the resulting N-GDY nanolayers exhibited 0.38 nm d-spacing; however, as for N+-GDY, the lattice fringes with a d-spacing of 0.44 nm were observed, thus pointing to the good inter-layer crystallinity and an obvious interlayer-expanded space of ∼0.6 Å. The distinct π–π stacking spaces of N-GDY and N+-GDY nanosheets are consistent with the calculated results of PXRD performance, confirming that the anions were encoded into the skeletons thus increasing the inter-layer space. Additionally, energy-dispersive X-ray spectroscopy (EDX) (Fig. 3(i)–(l) and S7†) revealed that the elements are uniformly distributed throughout the nanosheets, displaying the homogeneity of flexible N+-GDY nanosheets. These results certified that the FCII strategy facilitated the exfoliation and restacking of N+-GDY nanosheets with the trapping of CF3SO3− during the post-modification process.
 | ||
| Fig. 3 The morphology and microscopy characterization of N-GDY and N+-GDY. (a) SEM, (b) TEM images of N-GDY stacking nanosheets at low magnification. (e) SEM, (f) TEM images of N+-GDY nanosheets. HRTEM images of (c) and (d) N-GDY and (g) and (h) N+-GDY nanosheets. (i)–(l) Elemental distribution mapping of overlapping, C, N, and S of the N+-GDY. | ||
Although recording HR-TEM images under different axes of observation has been proven to be an efficient way to determine the sample structure, so far only limited resolution has been achieved to explicitly analyze the structure of GDY-based materials. The good crystallinity and stability of the N+-GDY allow us to study its structure in detail by TEM. As shown in Fig. 4, a series of HRTEM images of N+-GDY with distinct structures were obtained under different angles of tilt (corresponding to different axes of observation), revealing overall retention of the crystallinity; likewise, the corresponding selected area electron diffraction (SAED) patterns were detected, in which the positions, d-values, and intensities of several diffraction spots were extracted. From the construction of the reciprocal lattice, several uniquely unit cell parameters were determined, and the lattice fringes are depicted in Fig. 4, revealing ordered alignments of hexagonal, rhombic, and square lattices, respectively. To further determine the optimal stacking mode and crystal structure, structural simulations were conducted by using the first-principles density functional theory (DFT) method to optimize the mono-layer and multi-layer framework with AA, AB, ABC, and staggered-ABC stacking modes. The achieved N+-GDY exhibited the most stable state of staggered-ABC stacking mode showing the lowest energy among the different stacking models. As shown in Fig. 4(a) and (b), the ordered hexagonal nanopore with a uniform diameter of ∼0.45 nm was visible along the [001] zone axis consisting of the simulated pattern generated by the Fourier transform (inset in Fig. 4(b)), directly unveiling the intactness of the skeletons upon delamination and also showing the presence of the continuous one-dimensional channels when viewing along [hk0]. Notably, the SAED pattern displayed straight porous periodic channels that extend through entire crystal domains (Fig. 4(c)), which matches with the Fourier transform pattern (inset in Fig. 4(b)), indicating its single-crystalline nature. Additionally, Fig. 4(e)–(g) exhibit a reticular rhombic lattice fringe of the [010] facet with spacings of 0.85 nm, and the SAED displayed a rhombic diffraction pattern with the nearest reflection. Square lattice fringes with an interplanar distance of 1.2 nm are observed in Fig. 4(i)–(k), and the SAED pattern was acquired along the [100] axis and showed diffraction spots corresponding to the (0kl) planes. Simultaneously, rectangular and second-rhombic diffraction patterns were observed (Fig. S8 and S9†) along the [504] and [581] facets with spacings of 1.2 and 0.32 nm, respectively, which further proved the successful synthesis of N+-GDY. On the basis of the structural simulations, the experimental diffraction patterns matched almost exactly that is expected for the proposed staggered-ABC stacking model (Fig. 4(d), (h) and (l)); the calculated interlayer distance was around 0.44 nm in intrinsic configuration (staggered-ABC stack) that was in good agreement with the experimental profile, which confirmed that the N+-GDY adopted a staggered-ABC stacking arrangement and 0.44 nm inter-layer distance. Furthermore, the N-GDY adopted a dominant AA stacking configuration,18 displaying a totally different stacking mode from the ionic N+-GDY with the staggered-ABC stack. These results verified that the N+-GDY nanosheets underwent restacking and self-assembling processes from AA to staggered-ABC stacking modes during dissolution. Therefore, combined with the corresponding tilt angles of different SAED patterns and structural simulations, we acquired the clear structural assignment of N+-GDY.
 | ||
| Fig. 4 TEM images and electronic diffraction of N+-GDY collected from a crystal of 3.0 × 2.0 μm2 (inset). (a), (e), and (i) TEM images; (b), (f), and (j) HRTEM images showing the hexagonal, rhombic, square lattices (insets: simulated SAED patterns generated by the Fourier transform); and (c), (g), and (k) SAED patterns, and (d), (h), and (l) calculation of the stacking model of a single-crystalline domain along the [001], [010], and [100] directions, respectively, shows 9-fold interpenetration of a diamond net. | ||
Our FCII strategy provides the high soluble N+-GDY combined with interlayer-expanded N+-GDY nanosheets, facilitating us to prepare GDY-based optoelectronic devices with diverse functions. High-quality films could be directly prepared by drop-casting or spin-casting the dispersed solutions onto conductive substrates, followed by thermal evaporation (Fig. 5(a) and (b)). Meanwhile, the thickness of the smooth morphology N+-GDY films was around 4 μm. The surface morphology revealed that the N+-GDY nanosheets were dropped on ITO electrodes, thereby showing the uniform distribution of C, N, S, F, and O elements, suggesting homogeneity of the films (Fig. 5(b) and S11†). To further gain insight into the crystallinity of the N+-GDY films, we measured the grazing incidence small angle X-ray scattering (GISAXS) of the exfoliated N+-GDY flakes on the silica substrate (Fig. 5(c) and (d)). It exhibited a typical “face-on” orientation of the GDY skeletons parallel to the substrate, where the diffraction peaks were at 0.23, 0.28, 0.81, 0.86 nm−1, clearly revealing a 2D layered diffraction mode of the N+-GDY film. Compared with the original powder, the restacking film exhibited oriented crystallinity, further illustrating that the delamination–restacking facilitated the formation of the corresponding crystal. Based on the oriented N+-GDY film, the conductivity was performed. However, the observed conductivity of N-GDY is a weighted average of the electrical conductivity in each crystallographic direction. Therefore, to see the differences, we measured the intra-layer (horizontal direction) and inter-layer (vertical direction) conductivity in two types of device structures of insulating glass and conductive ITO substrates, respectively (Fig. 5(e)). Both films exhibited a linear current–voltage (I–V) profile, indicative of ohmic conduction, and we could calculate the conductivities from the slope of the curves. The ionic N+-GDY films in the as-synthesized states displayed obvious anisotropic conduction with an inherently high vertical conductivity of 0.014 S m−1 and a negligible horizontal conductivity of 3.3 × 10−5 S m−1 (Fig. 5(f)), which exhibits anisotropic conductive properties. The poor horizontal conductivity of N+-GDY may be restrained because of the charge combination in the interlaced networks, while the high vertical conduction may originate from the electrons or holes hopping from a single interlayer to another. All the results indicated that N+-GDY possessed the nature of electrical anisotropy.
 | ||
| Fig. 5 (a) Preparation of the N+-GDY film by solution processing. (b) The surface morphology (inset graph: cross-sectional SEM images), (c) GISAXS and (d) the out-of-plane diffraction patterns of drop-cast N+-GDY films. (e) Device structures of horizontal and vertical directions. (f) I–V curves of the 4 μm-thick films measured in horizontal (red line) and vertical (black line) directions of N+-GDY films. | ||
Conclusions
In summary, we proposed a new “framework charge-induced intercalation” strategy (FCII) to synthesize soluble and interlayer-expanded few-layer N+-GDY crystalline sheets, the clear ionic structure of which was confirmed by TEM, FT-IR, XPS, and theoretical calculations. By integrating charges into their backbone functionalities and inserting ions into interlayers, the N+-GDY nanosheets were formed following the sequence of exfoliation and restacking with the trapping of ions. The FCII strategy is highly beneficial for the scalable production of few-layer GDY with controllable well-defined thickness. The prepared N+-GDY nanostructured films showed well-defined ionic frameworks and anisotropic conductivity, enabling their applications in electronic devices. We anticipate that by incorporating pyridine-N into graphdiyne, such a structure screening strategy could be utilized to synthesize other kinds of knots, such as pyrimidine-, pyridazine-, pyrazine-, and triazine-linked graphite diacetylenes, thus being developed into a general method for synthesizing exfoliated and highly soluble crystalline GDYs.Data availability
The authors declare that additional experimental details and data are available in the ESI.†Author contributions
L. L. Wang conceived and designed the project, carried out experiments, analyzed the materials by various characterization methods, and wrote the original draft. Z. Q. Zheng carried out GISAXS studies. Q. L. Zhang investigated the electronic conductivity of devices. L. Qi performed TEM. B. H. Xue and P. C. Yin helped in setting up and carrying out SANS measurements. Y. R. Xue provided guidance. W. L. Yang, and Y. L. Li helped in funding acquisition, review and editing, supervision, and project administration. All authors contributed to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (22071136, 21790050, and 21790051), the Taishan Scholar Project of Shandong Province of China (62460082161012), Natural Science Foundation of Shandong Province (ZR2022QB189), and the National Key Research and Development Project of China (2018YFA0703501). We would like to thank Xiaoju Li and Haiyan Sui from Shandong University Core Facilities for Life and Environmental Sciences for their help with TEM.Notes and references
-
(a) G. X. Li, Y. L. Li, H. B. Liu, Y. B. Guo, Y. J. Li and D. B. Zhu, Chem. Commun., 2010, 46, 3256–3258 RSC
; (b) C. S. Huang, Y. J. Li, N. Wang, Y. R. Xue, Z. C. Zuo, H. B. Liu and Y. L. Li, Chem. Rev., 2018, 118, 7744–7803 CrossRef CAS PubMed
-
(a) Y. Gao, Y. R. Xue, L. Qi, C. Y. Xing, X. C. Zheng, F. He and Y. L. Li, Nat. Commun., 2022, 13, 5227 CrossRef CAS PubMed
-
(a) Z. Jin, M. Yuan, H. Li, H. Yang, Q. Zhou, H. Liu, X. Lan, M. Liu, J. Wang, E. H. Sargent and Y. Li, Adv. Funct. Mater., 2016, 26, 5284–5289 CrossRef CAS
-
(a) J. J. He, N. Wang, Z. L. Cui, H. P. Du, L. Fu, C. S. Huang, Z. Yang, X. Y. Shen, Y. P. Yi, Z. Y. Tu and Y. L. Li, Nat. Commun., 2017, 8, 1172 CrossRef PubMed
-
(a) C. Lu, Y. Yang, J. Wang, R. P. Fu, X. X. Zhao, L. Zhao, Y. Ming, Y. Hu, H. Z. Lin, X. M. Tao, Y. L. Li and W. Chen, Nat. Commun., 2018, 9, 752 CrossRef PubMed
-
(a) H. L. Yan, S. Y. Guo, F. Wu, P. Yu, H. B. Liu, Y. L. Li and L. Q. Mao, Angew. Chem., Int. Ed., 2018, 57, 3922–3926 CrossRef CAS PubMed
-
(a) N. Parvin, Q. Jin, Y. Z. Wei, R. B. Yu, B. Zheng, L. Huang, Y. Zhang, L. H. Wang, H. Zhang, M. Y. Gao, H. J. Zhao, W. P. Hu, Y. L. Li and D. Wang, Adv. Mater., 2017, 29, 1606755 CrossRef PubMed
-
(a) S. C. Lin and M. J. Buehler, Nanoscale, 2013, 5, 11801–11807 RSC
-
(a) J. Li, X. Jing, Q. Li, S. Li, X. Gao, X. Feng and B. Wang, Chem. Soc. Rev., 2020, 49, 3565–3604 RSC
-
(a) J. W. Colson, A. R. Woll, A. Mukherjee, M. P. Levendorf, E. L. Spitler, V. B. Shields, M. G. Spencer, J. Park and W. R. Dichtel, Science, 2011, 332, 228–231 CrossRef CAS PubMed
-
(a) D. N. Bunck and W. R. Dichtel, J. Am. Chem. Soc., 2013, 135, 14952–14955 CrossRef CAS PubMed
- M. A. Khayum, S. Kandambeth, S. Mitra, S. B. Nair, A. Das, S. S. Nagane, R. Mukherjee and R. Banerjee, Angew. Chem., Int. Ed., 2016, 55, 15604–15608 CrossRef CAS PubMed
-
(a) J. F. Dienstmaier, A. M. Gigler, A. J. Goetz, P. Knochel, T. Bein, A. Lyapin, S. Reichlmaier, W. M. Heckl and M. Lackinger, ACS Nano, 2011, 5, 9737–9745 CrossRef CAS PubMed
-
(a) L. L. Wang, C. Zeng, H. Xu, P. C. Yin, D. C. Chen, J. Deng, M. Li, N. Zheng, C. Gu and Y. G. Ma, Chem. Sci., 2019, 10, 1023–1028 RSC
-
(a) K. Kobayashi and J. Yamauchi, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 17085 CrossRef CAS PubMed
- B. Gui, X. F. Liu, Y. P. Chen, Y. Zhang, P. H. Chen, M. H. He, J. L. Sun and C. Wang, Angew. Chem., Int. Ed., 2021, 60, 274–280 CrossRef PubMed
- Z. Yang, X. Y. Shen, N. Wang, J. J. He, X. D. Li, X. Wang, Z. F. Hou, K. Wang, J. Gao, T. G. Jiu and C. S. Huang, ACS Appl. Mater. Interfaces, 2018, 11, 2608–2617 CrossRef PubMed
-
(a) F. Tuinstra and J. L. Koenig, J. Chem. Phys., 1970, 53, 1126 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc01393f |
| This journal is © The Royal Society of Chemistry 2023 |