
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSingle-atom copper catalyst for the S-arylation reaction to produce diaryl disulfides†
Yiming
Zhao
 ,
Yan
Zhou
,
Shanshan
Lv
,
Han
Li
,
Qikang
Wu
,
Shaohuan
Liu
,
Wanying
Li
,
Taiyu
Chen
and
Zheng
Chen
*
,
Yan
Zhou
,
Shanshan
Lv
,
Han
Li
,
Qikang
Wu
,
Shaohuan
Liu
,
Wanying
Li
,
Taiyu
Chen
and
Zheng
Chen
*
Key Laboratory of Functional, Ministry of Education, College of Chemistry and Materials Science, Anhui Normal University, Wuhu 241000, China. E-mail: chenzh07@mail.ahnu.edu.cn
First published on 29th March 2023
Abstract
Single-atom Cu supported on CeOx nanorod catalysts (Cu1/CeOx) have been synthesized through the anchoring of copper by terminal hydroxyl groups on the CeOx surface. The oxygen defect characteristics of the CeOx nanorods promote electron transfer between Cu and CeOx through a Ce–O–Cu interface, which realizes flexible electronic regulation of the Cu sites. Single-atom Cu species with an oxidation state of between +1 and +2 were formed, which was confirmed by X-ray photoelectron spectroscopy, X-ray fine structure spectroscopy, and electron paramagnetic resonance spectroscopy. Cu1/CeOx emerged as a catalyst with advanced catalytic performance for elemental sulfur in S-arylation using aryl iodides, achieving 97.1% iodobenzene conversion and 94.8% selectivity toward diphenyl disulfide. The substituted iodobenzene with different electronic or steric groups successfully realized S-arylation and produced the corresponding diaryl disulfides with high selectivity. The fully exposed single-atom Cu with flexible electronic characteristics successively realized oxidative addition or coordination of multiple substrates, making it possible to obtain diaryl disulfide with high selectivity.
Introduction
Transition-metal oxides (TMOs) have been widely used as supports for the preparation of single-atom metal catalysts, which show unique catalytic properties in heterogeneous catalysis.1–4 Single metal atoms are usually anchored in the oxygen vacancies5–8 or metal vacancies9–12 generated by TMOs, or other coordination unsaturated sites, such as edges, steps and terraces.13–16 Through metal–support interactions (MSI), not only can a single metal atom is stabilized, but also the electronic structure of the metal can be adjusted, especially on reducible TMOs.17–21 Although the metal utilization of single-atom catalysts can be maximized compared with nanoparticles, the steric hindrance of the metal center in the above anchoring types is large because metal atoms are embedded in the lattice structure. This not only has a great impact on the simple reaction of small molecule activation, it also hinders the reaction of multiple complex substrates on a single metal site. Cerium oxide (CeOx) easily produces oxygen vacancies due to the coexistence of +3 and +4 valence states, which are used as support to anchor single metal atoms in automobile exhaust elimination and industrial catalytic processes.22,23 The anchoring effect of hydroxyl groups on a CeOx surface to metal species during the preparation of catalysts is usually ignored and rarely considered as sites for anchoring single atoms, although many examples of CeOx-anchored single-atom Cu have been reported. Although the surface hydroxyl groups of CeOx will be removed after heat treatment, it is inevitable that it will come into contact with oxygen and water in the atmospheric environment or in aqueous solutions of metal salts. In this way, the hydroxyl groups on the surface of CeOx will still exist and play an important role in the anchoring of metal species.1,24 MSI still exist in this type of single-atom metal catalyst after heat treatment and electron transfer can be realized through a metal-O-Ce interface.25,26 Compared with vacancy-anchored single-atom metal species, the anchoring of surface hydroxyl groups is conducive to the full exposure of single metal atoms, which is more favorable for application in the transformation of complex organic molecules, especially the activation of multiple substrates.Diaryl disulfide compounds are valuable reagents in organic synthesis due to their unique S–S bonds, which exhibit high activity in radical tandem reactions or transition metal catalyzed pathways to synthesis functional sulfur-containing organic compounds.27–29 With the special properties of organic disulfides and the continuous development of their application in synthetic chemistry and biochemistry,30,31 the synthesis of disulfides has also been greatly developed. Using elemental sulfur as a sulfur source is an advanced alternative to constructing C–S bonds in the synthesis of functional materials and biological compounds, which has the advantages of being odorless, tasteless, stable, and cheap, and having no need for pre-functionalization.32,33 Through the transition metal catalyzed S-arylation reaction, the coupling of aryl halides and elemental sulfur can result in the formation of aryl disulfides or aryl sulfides.34,35 However, the preferentially formed disulfide products continue to react with the substrate aryl iodides to form diarylsulfides or undergo reductive cracking to form thiophenols, reducing the selectivity of the reaction.36,37 To develop heterogeneous catalysts, especially single-atom catalysts with uniform active sites, it is conducive to realize high selectivity towards diaryldisulfides.
Here, single-atom Cu anchored on CeOx nanorods is developed through the anchoring and dispersing of copper ions by terminal hydroxyl groups on the CeOx surface. The hydroxyl-rich environment on the surface of CeOx effectively hinders the aggregation or sintering of Cu atoms, and also realizes charge transfer through Cu–O–Ce units. The grafted single-atom Cu sites have a special valence state of between +1 and +2, and are more fully exposed to substrate molecules than embedded metal sites. Therefore, the coordination of S22− and the oxidative addition of Ph-I can be realized at a single Cu atom, and then diphenyl disulfide can be produced efficiently through reductive elimination. Under the optimized reaction conditions, 97.1% of iodobenzene conversion and 94.8% selectivity toward diphenyl disulfide could be achieved using Cu1/CeOx as catalyst. Therefore, optimizing the exposure space of single-atom metal sites with flexible regulation of the electronics by the support will be conducive to multicomponent reactions to produce complex organic molecules.
Results and discussion
Synthesis and characterization of Cu1/CeOx nanorods
CeOx nanorods containing defects were prepared via a reported method with a few modifications38 and used as the support to fabricate the single-atom Cu catalyst (Fig. S1†). The Cu1/CeOx catalyst was obtained by wet impregnation using copper(II) chloride dihydrate (CuCl2·2H2O) as the copper source and a subsequent reducing treatment. Inductively coupled plasma optical emission spectrometry (ICP-OES) revealed a Cu loading of 0.69 weight percent (wt%) in the single-atom Cu catalyst. The Cu1/CeOx sample was characterized by transmission electron microscopy (TEM) and scanning electron microscopy (SEM). The overall morphology was nanorods, as shown in Fig. 1a and S2,† no nanoparticles were observed in the CeOx nanorods, and the defects of the nanorods can also be clearly observed. The energy-dispersive X-ray (EDX) elemental mapping shows that Cu and Ce are uniformly distributed on the Cu1/CeOx sample (Fig. 1b). Aberration corrected high-angle annular dark-field scanning transmission electron microscopy (AC HAADF-STEM) was unable to show the atomic dispersion of the Cu as the Ce atom is heavier than the Cu atom (Fig. 1c). The fast Fourier transform (FFT) results show that the included angle between each diffraction spot is 60°, which corresponds to a face-centered cubic (fcc) CeO2 crystal (see the inset in Fig. 1c). The diffraction spot should correspond to the {220} crystal plane, which indicates that the [110] crystal plane is clearly exposed. The X-ray diffraction (XRD) pattern of Cu1/CeOx is the same as that of the CeOx nanorods, without peaks attributed to copper and copper oxide nanoparticles, suggesting that no changes to the crystal structure and no aggregation of Cu species occurred in the final catalysts (Fig. 1d). The Cu/CeOx samples with higher loading (1 wt% and 2 wt%) also showed no obvious Cu nanoparticles (Fig. S3 and S4†), nor did the characteristic peaks involving Cu elements and oxides appear in the XRD data (Fig. S5†).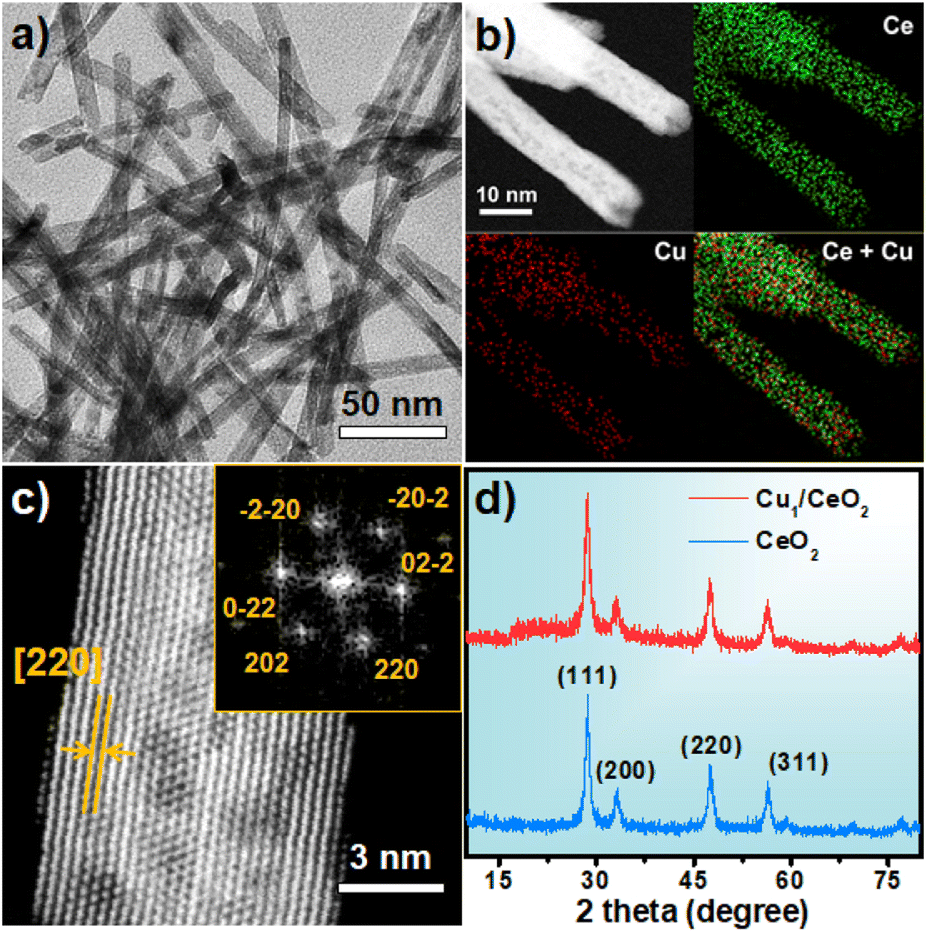 | ||
| Fig. 1 (a) Representative TEM image of the Cu1/CeOx nanorods; (b) energy-dispersive X-ray spectroscopy for elemental mapping of the Cu1/CeOx nanorods; (c) representative HAADF-STEM image of a Cu1/CeOx nanorod; (d) XRD patterns of the Cu1/CeOx and CeOx nanorods. | ||
Compared with the X-ray absorption near edge structure spectra (XANES) of the Cu2O and CuO standard samples, the valence state of Cu in Cu1/CeOx should be between +1 and +2.39,40 Moreover, we found that the pre-edge absorption peak reflecting the 1s–4p transitions did not appear in the data of the Cu1/CeOx sample, while Cu2O and CuO were located at 8982.6 eV and 8986.1 eV, respectively. This result shows that the local geometric structure of Cu in Cu1/CeOx is completely different from the linear O–Cu–O coordination of Cu2O or the square coordination of oxygen atoms in CuO.41,42 The Fourier-transform extended-edge X-ray absorption fine structure (FT EXAFS) analysis shows that the Cu K-edge spectrum of the Cu1/CeOx sample is different from those of Cu foil, Cu2O and CuO, in which the Cu species are coordinated to O atoms and present atomic dispersion (Fig. 2b). The XANES absorption edge energy of the Cu/CeOx catalyst with 1 wt% and 2 wt% loading is slightly higher than that of the Cu1/CeOx catalyst (Fig. S6a†), and the overall coordinative structure is still based on Cu–O bonds (Fig. S6b†). We determined the dispersion of Cu in the three catalysts through N2O titration, among which the dispersion of the single-atom Cu was close to 100% (Table S1†). The dispersion of the Cu/CeOx catalyst with 1 wt% and 2 wt% decreases gradually, which proves that there were unexposed Cu sites. The EXAFS fitting results show that Cu is more inclined to coordinate with two O atoms, and that the associated bond length is 1.8 Å (Fig. 2c and S7, Table S2†). Since the length of the Ce–O bond is generally >2.3 Å, the Cu atom cannot be in any vacancy of CeOx according to the Cu–O bond length from the EXAFS results. Therefore, the most likely structural model is single-atom Cu anchored by the oxygen atom of the hydroxyl group on the CeOx surface, which is illustrated in the inset of Fig. 2c. We also attempted to place Cu atoms in the Ce vacancy or oxygen vacancy of CeOx, which did not lead to ideal fitting results through comparison between the experimental Cu K-edge XANES spectra of Cu1/CeOx and the theoretical spectra of four different structures (Fig. S8†). The wavelet transform (WT) contour plot of Cu1/CeOx contains just one obvious intensity of Cu–O near 6.5 Å−1 (Fig. 2d), while the intensity of Cu–Cu of Cu foil appears at 8.0 Å−1 (Fig. 2e). The Cu–O coordination of the second shell in the Cu2O and the Cu–Cu coordination of the second shell in CuO do not appear in Cu1/CeOx (Fig. 2f and g), which also proves the uniqueness of the Cu–O coordination structure in Cu1/CeOx.
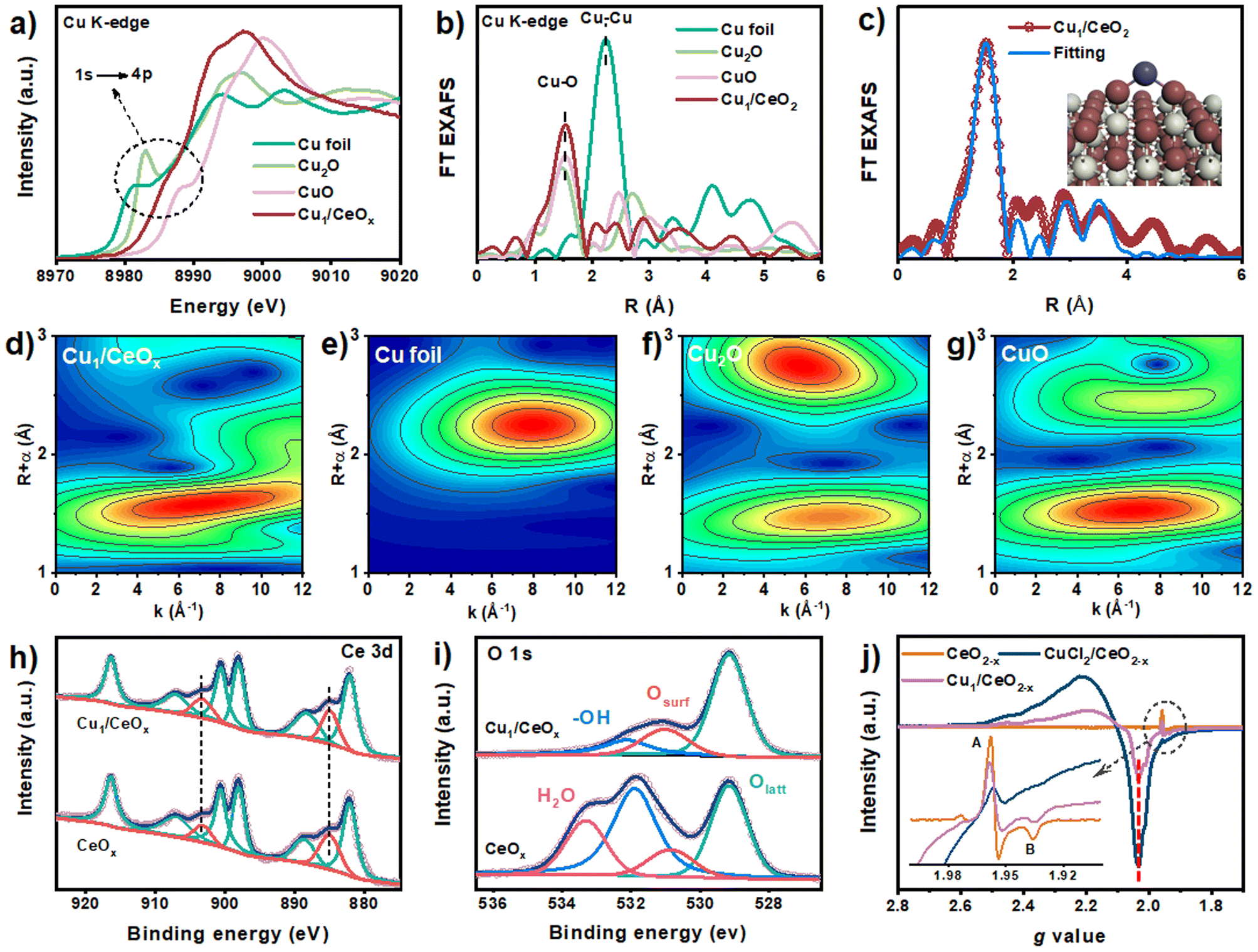 | ||
| Fig. 2 (a) XANES spectra at the Cu K-edge of Cu1/CeOx with CuO, Cu2O, and Cu foil; (b) FT EXAFS spectra of Cu1/CeOx with CuO, Cu2O, and Cu foil; (c) the fitting curve of Cu1/CeOx and the experimental data, the inset shows the fitted structure; high-resolution spectra of (d) Ce 3d and (e) O 1s in the Cu1/CeOx and CeOx samples; (f) EPR spectra of the CeOx, CuCl2/CeOx and Cu1/CeOx samples; (g–j) WT-EXAFS curves of the Cu1/CeOx, Cu2O, CuO and Cu foil samples. | ||
X-ray photoelectron spectroscopy (XPS) data show that there is no obvious difference in the Ce 3d spectra between Cu1/CeOx and CeOx, with obvious characteristic peaks of Ce3+ located at 885.1 eV and 903.4 eV (Fig. 2h).43 We analyzed the O 1s spectra of CeOx and found that CeOx is prone to adsorbing water (H2O, located at 533.3 eV) from the environment and formed a large number of surface hydroxyl groups (–OH, located at 532.0 eV), although the samples were all characterized after heat treatment (Fig. 2i).44 We also obtained consistent information from the infrared spectra, in which the characteristic peak located at ∼3320 cm−1 revealed the existence of abundant surface –OH groups on the CeOx support (Fig. S9†). The obtained Cu1/CeOx sample showed an obvious signal in its XPS data for surface O species (Osurf, located at 530.9 eV), and the lattice O (Olatt, located at 529.1 eV) peak basically unchanged.45 In the Cu 2p region, the binding energy position of Cu 2p3/2 belonging to the +1 oxidation state of Cu can be clearly observed, located at 932.5 eV (Fig. S10†).46 There is a weak shake-up satellite peak near 940.0 eV, indicating the existence of a Cu2+ state.47,48 No significant peaks were observed for Cl 2p, indicating that there is no Cl present in Cu1/CeOx (Fig. S11†). Through electron paramagnetic resonance (EPR) spectroscopy, as shown in Fig. 2j, the data show a Cu2+ signal (g value = 2.03) for the Cu1/CeOx nanorods, but this was less than for the unreduced CuCl2/CeOx sample. There is no hyperfine splitting assigned to isolated Cu2+, which proves the specific nature of single-atom Cu species in the Cu1/CeOx sample. Signals A and B can be attributed to Ce3+ species, which show that Ce3+ is the most likely oxidation state in the CeOx support.49 The loading of CuCl2 reduces the signal intensity of Ce3+, indicating that there is electron transfer from Ce3+ to Cu2+ species. This electron transfer occurs via a Cu–O–Ce interface, which proves the occurrence of MSI. After reduction treatment, an increase in Ce3+ components was observed in Cu1/CeOx, which is due to the electron transfer from Cu to the CeO2 support. The Raman spectra of both CeOx and Cu1/CeOx exhibit an obvious strong peak at ∼457 cm−1, which can be attributed to the F2g fluorite structure of CeO2 (Fig. S12†).45 The unchanged position of this peak indicates that the introduction of Cu species does not change the overall crystal structure of the CeOx support. The weak broad peak at ∼608 cm−1 attributed to the defect induced (D) mode indicates that there is little difference in the oxygen vacancy concentration between the two samples.50 The weak peak of the Cu sample at ∼262 cm−1 can be attributed to the vibration of Cu–O bond.51
Catalytic performance of the Cu1/CeOx nanorods in the S-arylation reaction of iodobenzene
Iodobenzene and elemental sulfur were used as the initial exploration substrates for the S-arylation reaction (Fig. 3 and Table S3†). Several solvents were selected to test the catalytic performance of Cu1/CeOx on a dithiolation reaction at 100 °C for 12 h (Table S3†). The amount of H2O in optimized solvent DMF had a great influence on the catalytic performance, which significantly improved the selectivity for diphenyl disulfide (2a). This may be due to the fact that H2O promotes the reaction between elemental S and alkali to produce S22−, but too much H2O also led to a decline in catalytic activity (Table S3†). No obvious base-controlled selectivity was observed for the Cu1/CeOx catalyzed S-arylation reaction,34 and 97.4% selectivity towards 2a was attained with 82.6% conversion when NaOH was used as a strong base (Table S4†). When the reaction temperature was reduced, the conversion decreased significantly with a slight decrease in selectivity (Table S5,† entries 1 and 2). When the temperature rose to 110 °C, the conversion significantly increased to 97.7%, and the selectivity only slightly reduced (Table S5,† entry 4). Further increasing the reaction temperature led to a significant decrease in selectivity (Table S5,† entries 5 and 6). When the reaction time was prolonged to 14 h at 110 °C, the target product 2a decomposed to produce phenylthiophenol (4a) and reduced the selectivity to 87.1% (Table S5,† entry 7). If the reaction time was shortened to 10 h, Cu1/CeOx achieved 97.1% conversion and 94.8% selectivity (Table S5,† entry 9). Cu/CeOx samples synthesized with higher loading prepared and then reduced by H2/Ar at 150 °C without washing with ultrapure water also showed poor catalytic activity (Table S5,† entries 18–21). The pure CeOx nanorods showed no catalytic activity, where the catalyst was obtained through a synthesis method and then reduced by H2/Ar at 150 °C. The Cu NPs showed only 34.2% transformation and 75.9% selectivity for 2a. Therefore, CeOx promotes the smooth S-arylation reaction by constructing single-atom Cu of a unique coordination environment with O atoms and charge characteristics. The Cu1/CeOx catalyst was stable in the S-arylation reaction, and the recycled catalyst retained its activity for at least four cycles (Fig. 3d, Table S6†). The ICP-OES analysis revealed a Cu loading of 0.638 wt% in the recycled Cu1/CeOx, which shows no difference compared to the original catalyst in the XRD patterns (Fig. S13†). Hot filtration experiments showed that almost no conversion of iodobenzene to diphenyldisulfide products was detected in the absence of the Cu catalyst, and <0.01 ppm of the Cu species was leached (Fig. S14†). These results prove that the catalysis of the S-arylation reaction by Cu1/CeOx is heterogeneous in essence, and also show that the MSI through the Cu–O–Ce interface endow the single-atom Cu with superior activity, selectivity and stability.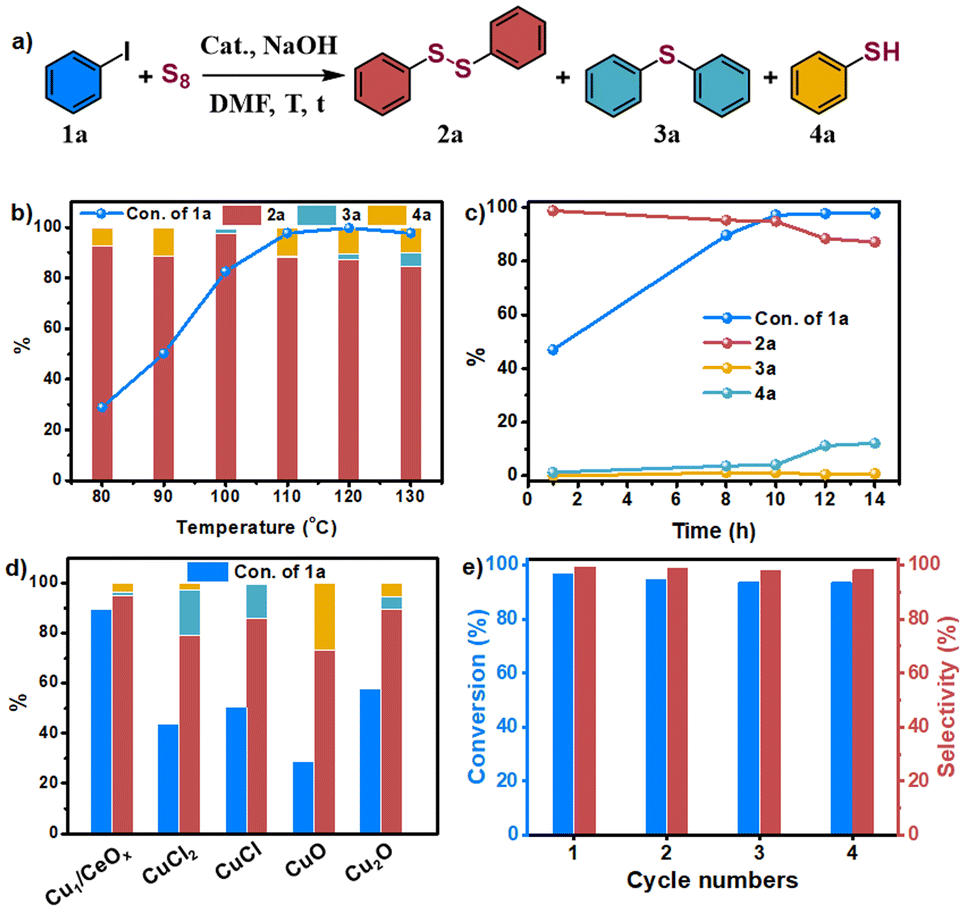 | ||
| Fig. 3 (a) The reaction equation of the S-arylation using iodobenzene; (b) Cu1/CeOx catalyzed S-arylation using iodobenzene at different temperatures; (c) time-course plot of the conversion and different products at 110 °C; (d) catalyst comparison for the S-arylation reaction using iodobenzene at 110 °C for 10 h; (e) the recycling performance of Cu1/CeOx. | ||
The Cu1/CeOx catalyst performed well in S-arylation with a wide substrate scope of aryl iodides under the optimized conditions. As summarized in Scheme 1, the S-arylation of iodobenzene with electron-donating groups led to higher yields being obtained. p-Methyl iodobenzene achieved complete conversion of iodobenzene and almost 100% selectivity of 4,4′-dimethyl-diphenyl disulfide (2b). The tolerance of electron-donating groups such as –Me, –OMe, –Et, –iPr and –tBu at different substitution positions enabled the synthesis of structurally diverse diaryl disulfide products (2c–2i) in moderate to good conversions and selectivity. While iodobenzene with electron-withdrawing groups underwent the S-arylation reaction like –F, –Cl, –Br, –CF3 and –OCF3 at para-position or meta-position, the yields of corresponding diaryl disulfide generally decreased slightly (2j–2p). Several aryl iodides multi-substituted at different positions underwent the S-arylation reaction smoothly to deliver the corresponding product in up to 86% yield (2r–2t). Even 2,4,6-trimethyliodosbenzene with steric hindrance can produce the corresponding disulfide product in 73.8% yield (2u). These results indicate that most of the substituted aryl iodides with various electronic and steric effects can be used to realize the S-arylation reaction with high efficiency and produce the corresponding diaryl disulfide products.
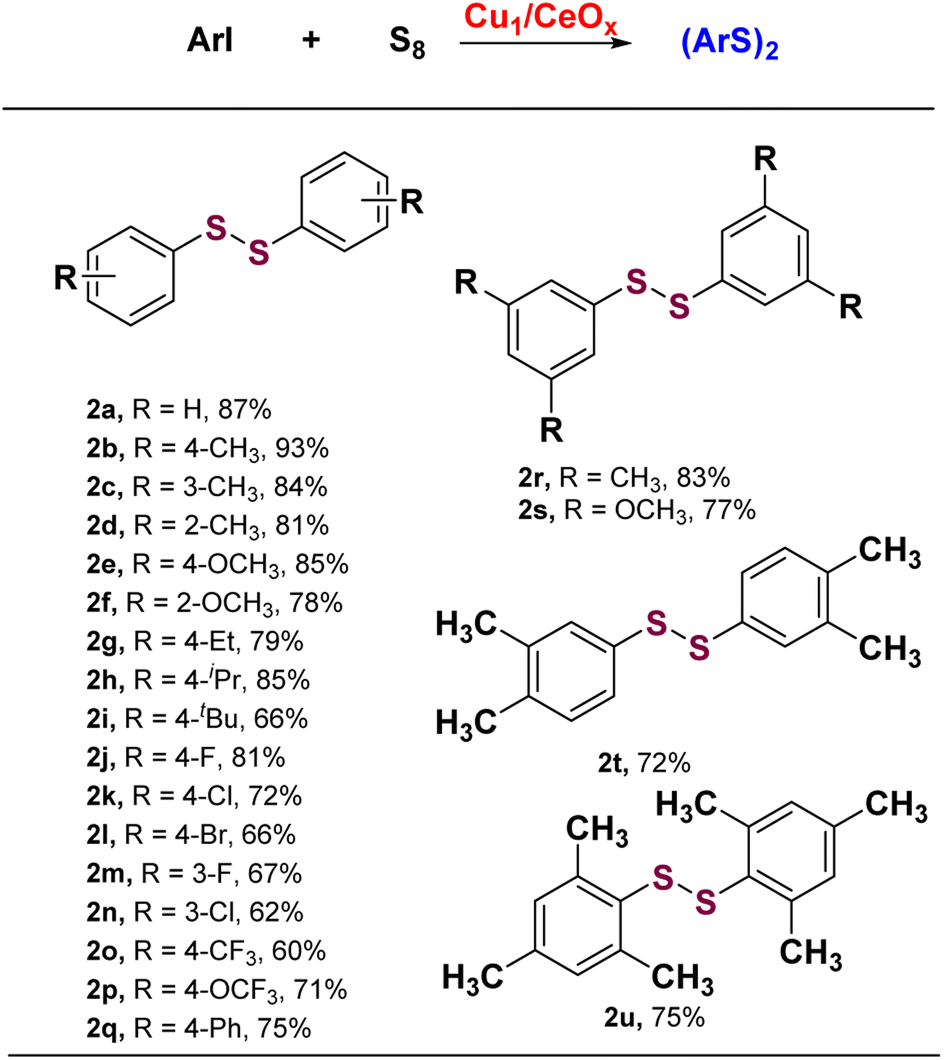 | ||
| Scheme 1 Substrate scope of iodoarylene. Reaction conditions: 1 (0.5 mmol), S8 (0.75 mmol), Cu1/CeOx (20 mg), NaOH (2.0 mmol), DMF (2 mL) and H2O (67 μL), Ar, 110 °C, 10 h, isolated yield. aNa2CO3 was used as the base. | ||
Control experiments and mechanism of the Cu1/CeOx catalyzed S-arylation reaction of iodobenzene
Control experiments were performed to gain insight into the mechanism of the Cu1/CeOx catalyzed S-arylation. When 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) or butylated hydroxytoluene (BHT) were added to the standard conditions as radical trapping reagents, almost no change in conversion and selectivity was observed, providing evidence that the Cu1/CeOx catalyzed S-arylation reaction is not a radical process (Scheme 2a and b). No products were detected when the base was removed from the standard conditions, which shows that base plays an important role in the S-arylation process (Scheme 2c). We also used Na2S or Na2S2 to replace elemental sulfur to react under standard conditions, and almost no desired product was observed using Na2S (Scheme 2d) but 78% conversion was observed using Na2S2 (Scheme 2e). NaOH reacts with elemental sulfur to form sodium thiosulfate (Na2S2), and the addition of H2O in the solvent promotes the process. For single-atom Cu, the oxidative addition of iodobenzene must occur to form a Cu–I bond firstly before it reacts with Na2S2 to release NaI.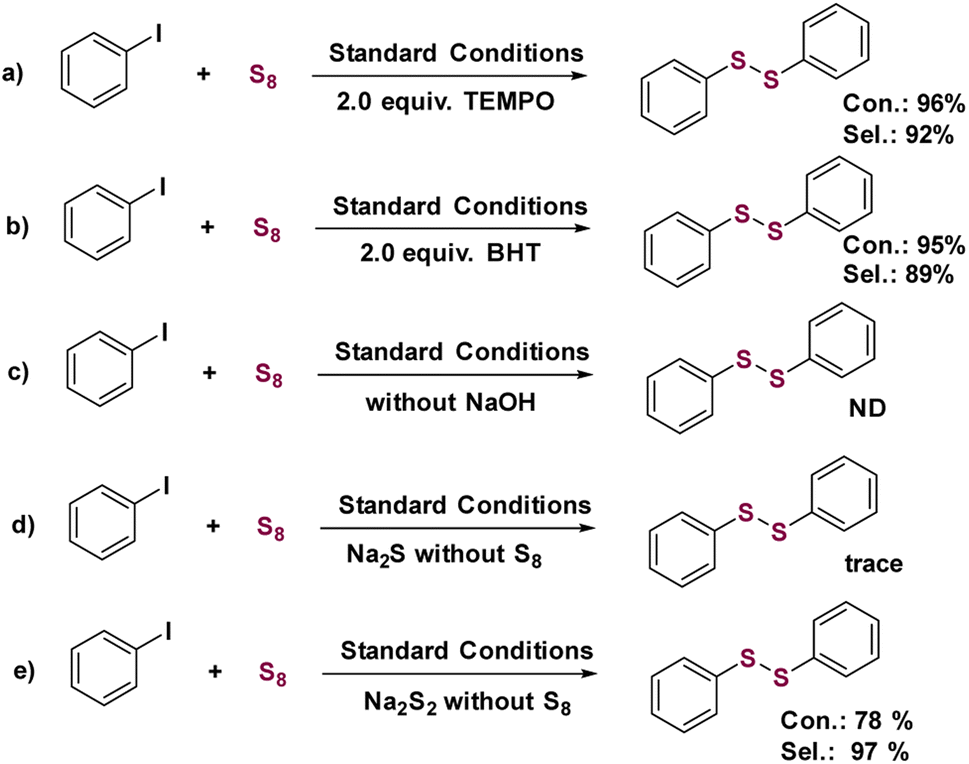 | ||
| Scheme 2 Control experiments for the S-arylation reaction of iodobenzene with the free radical trapping reagents (a) TEMPO and (b) BHT. (c) Control experiments for the S-arylation reaction of iodobenzene without NaOH. (d) Control experiments for the S-arylation reaction of Na2S without S8. (e) Control experiments for the S-arylation reaction of Na2S2 without S8. | ||
The copper halide catalysts involve the coordination of two Cu sites to S22− firstly, and this cyclic intermediate structure is then conducive to the subsequent rupturing of S–S bonds.34,35 The catalyst comparison results in Fig. 3d show that Cu–Cl coordination obviously produces more diphenyl sulfide than Cu–O coordination. Even if the S-arylation reaction is carried out without Ar protection, Cu1/CeOx still produces more diphenyldisulfide, while CuCl2 mainly produces diphenylsulfide (Table 1). The coordination mode of single-atom Cu with S22− is different from that of copper halide, and it is difficult to form two single-atom Cu coordinated with S22−.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Cat | Con. (%) of 1a | Sel. (%) | ||
| 2a | 3a | 4a | |||
| 1 | Cu1/CeOx | 83.3 | 88.1 | 5.3 | 6.6 |
| 2 | CuCl2 | 61.5 | 38.4 | 59.7 | 1.9 |
Based on the control experiment and previous studies, the mechanism of dithiolation for aryl iodides was predicted (Scheme 3). It is clear that the elemental sulfur reacts with NaOH to generate Na2S2,34,35 which then reacts with the intermediate Ph–CuIII–I (I) formed via the oxidative addition of Ph–I on single-atom CuI.52,53 We speculate that the formed intermediate II forms a feedback bond structure, making the valence state of Cu in the intermediate II return to the +1 oxidation state. The CuI in intermediate II may continue to coordinate with Ph–I to provide the π-complex III.54–56
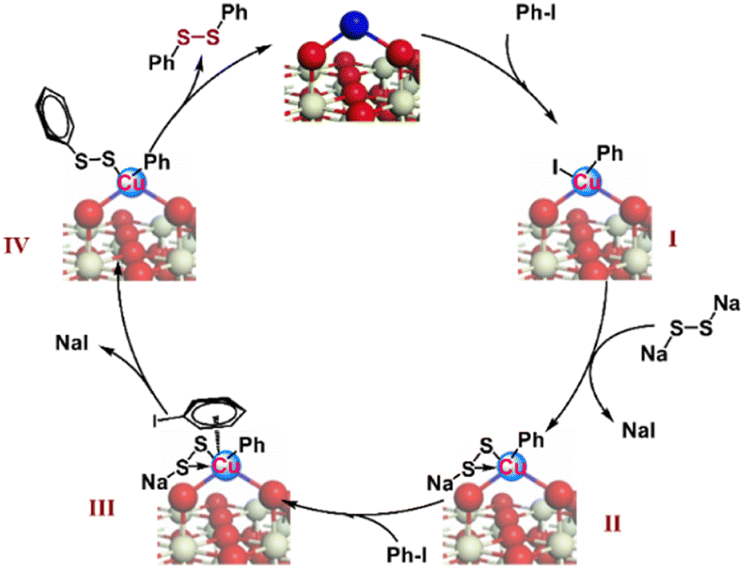 | ||
| Scheme 3 The mechanism of the S-arylation reaction of iodobenzene with the Cu1/CeOx catalyst. | ||
Evidence of the η6-coordination mode of π-complex III can also be deduced from the substituent effect of the substrate scope, in which electro-donating substituents exhibit better reactivity than electron-withdrawing substituents. Next, intramolecular nucleophilic substitution occurs at the aromatic ring to give the intermediate IV, and diphenyl disulfide is finally formed via reductive elimination. Based on the complexity of this Ullmann-type reaction and the uncertainty of the mechanism,57 we can only speculate its possible mechanism according to existing comparative experiments. However, the change in the oxidation state of single-atom Cu caused by the redox cycle will not result in leaching of the metal species for the MSI between CeOx and Cu, which highlights the advantages of the single-atom Cu catalyst in the transformation of complex organic molecules. Anchoring outside the CeOx surface fully exposes the space of single-atom Cu sites, which is conducive to the coordination and reaction of multiple substrates.
Conclusions
In summary, partially positively charged single atom Cu coordinated with two oxygen atoms was obtained via hydroxyl anchoring on the surface of CeOx. This Cu1/CeOx catalyst realizes electron transfer through a Cu–O–Ce interface, and fully exposes the space of single-atom Cu. In the S-arylation reaction, the coordination and addition of multiple substrate molecules can be realized, and thus shows superior activity and specific selectivity for disulfides. Iodobenzene with substituents that exhibit different electronic properties and steric hindrance can realize the S-arylation reaction smoothly. Therefore, the oxide support with redox functionality is similar to organic ligands with flexible electronic regulation and more optimized spatial configuration, which makes anchored single-atom metal catalysts show promising application value in heterogeneous catalysis.Data availability
All experimental data is available in the ESI.†Author contributions
Yiming Zhao contributed to the investigation and formal analysis; Yan Zhou and Shanshan Lv contributed to data curation; Han Li and Qikang Wu contributed to the methodology; Shaohuan Liu, Wanying Li and Taiyu Chen contributed to resources; Zheng Chen contributed to writing the original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Key Research and Development Project of Anhui Province (2022a05020048) and the Open Project of Anhui Province Key Laboratory for Degradation and Monitoring of Pollution of the Environment (FSKFKT007D). We would like to express our gratitude to Prof. Guofeng Zhao and Mr Fengyang Jiang at East China Normal University for help with the N2O titration experiment. We thank Prof. Kun Liu at Shandong First Medical University for help with the CO adsorption.Notes and references
- R. Lang, X. Du, Y. Huang, X. Jiang, Q. Zhang, Y. Guo, K. Liu, B. Qiao, A. Wang and T. Zhang, Chem. Rev., 2020, 120, 11986–12043 CAS.
- J. Park, S. Lee, H. E. Kim, A. Cho, S. Kim, Y. Ye, J. W. Han, H. Lee, J. H. Jang and J. Lee, Angew. Chem., Int. Ed., 2019, 58, 16038–16042 CAS.
- R. Lang, T. Li, D. Matsumura, S. Miao, Y. Ren, Y.-T. Cui, Y. Tan, B. Qiao, L. Li, A. Wang, X. Wang and T. Zhang, Angew. Chem., Int. Ed., 2016, 55, 16054–16058 CAS.
- R. Zhang, Y. Chen, M. Ding and J. Zhao, Nano Res., 2022, 15, 2810–2833 Search PubMed.
- Y. Wang, Z. Chen, P. Han, Y. Du, Z. Gu, X. Xu and G. Zheng, ACS Catal., 2018, 8, 7113–7119 CAS.
- Z. Hu, C. Yang, K. Lv, X. Li, Q. Li and J. Fan, Chem. Commun., 2020, 56, 1745–1748 CAS.
- S. Hejazi, S. Mohajernia, B. Osuagwu, G. Zoppellaro, P. Andryskova, O. Tomanec, S. Kment, R. Zbořil and P. Schmuki, Adv. Mater., 2020, 32, 1908505 CAS.
- X. Li, H. Rong, J. Zhang, D. Wang and Y. Li, Nano Res., 2020, 13, 1842–1855 CAS.
- S. Duan, R. Wang and J. Liu, Nanotechnology, 2018, 29, 204002 Search PubMed.
- W. Qu, X. Liu, J. Chen, Y. Dong, X. Tang and Y. Chen, Nat. Commun., 2020, 11, 1532 CAS.
- S. Hoang, Y. Guo, A. J. Binder, W. Tang, S. Wang, J. Liu, H. Tran, X. Lu, Y. Wang, Y. Ding, E. A. Kyriakidou, J. Yang, T. J. Toops, T. R. Pauly, R. Ramprasad and P.-X. Gao, Nat. Commun., 2020, 11, 1062 CAS.
- B.-H. Lee, S. Park, M. Kim, A. K. Sinha, S. C. Lee, E. Jung, W. J. Chang, K.-S. Lee, J. H. Kim, S.-P. Cho, H. Kim, K. T. Nam and T. Hyeon, Nat. Mater., 2019, 18, 620–626 CAS.
- D. Kunwar, S. Zhou, A. DeLaRiva, E. J. Peterson, H. Xiong, X. I. Pereira-Hernández, S. C. Purdy, R. ter Veen, H. H. Brongersma, J. T. Miller, H. Hashiguchi, L. Kovarik, S. Lin, H. Guo, Y. Wang and A. K. Datye, ACS Catal., 2019, 9, 3978–3990 CAS.
- L. F. Allard, A. Borisevich, W. Deng, R. Si, M. Flytzani-Stephanopoulos and S. H. Overbury, J. Electron. Microsc., 2009, 58, 199–212 CAS.
- L. DeRita, J. Resasco, S. Dai, A. Boubnov, H. V. Thang, A. S. Hoffman, I. Ro, G. W. Graham, S. R. Bare, G. Pacchioni, X. Pan and P. Christopher, Nat. Mater., 2019, 18, 746–751 CAS.
- W. Chen, H. Jin, F. He, P. Cui, C. Cao and W. Song, Nano Res., 2022, 15, 3017–3025 CAS.
- X. Sui, L. Zhang, J. Li, K. Doyle-Davis, R. Li, Z. Wang and X. Sun, Adv. Energy Mater., 2022, 12, 2102556 CAS.
- W. Zhang, H. Ma, H. Wang, J. Jiang, Z. Sui, Y.-A. Zhu, D. Chen and X. Zhou, Catal. Sci. Technol., 2021, 11, 7840–7843 CAS.
- W.-H. Li, J. Yang, H. Jing, J. Zhang, Y. Wang, J. Li, J. Zhao, D. Wang and Y. Li, J. Am. Chem. Soc., 2021, 143, 15453–15461 CAS.
- J. Yang, W.-H. Li, S. Tan, K. Xu, Y. Wang, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 19085–19091 CAS.
- J. Zhang, C. Zheng, M. Zhang, Y. Qiu, Q. Xu, W.-C. Cheong, W. Chen, L. Zheng, L. Gu, Z. Hu, D. Wang and Y. Li, Nano Res., 2020, 13, 3082–3087 Search PubMed.
- L. Chen, Z. Qi, X. Peng, J.-L. Chen, C.-W. Pao, X. Zhang, C. Dun, M. Young, D. Prendergast, J. J. Urban, J. Guo, G. A. Somorjai and J. Su, J. Am. Chem. Soc., 2021, 143, 12074–12081 CAS.
- B. B. Sarma, J. Kim, J. Amsler, G. Agostini, C. Weidenthaler, N. Pfänder, R. Arenal, P. Concepción, P. Plessow, F. Studt and G. Prieto, Angew. Chem., Int. Ed., 2020, 59, 5806–5815 CAS.
- S. Zhao, F. Chen, S. Duan, B. Shao, T. Li, H. Tang, Q. Lin, J. Zhang, L. Li, J. Huang, N. Bion, W. Liu, H. Sun, A.-Q. Wang, M. Haruta, B. Qiao, J. Li, J. Liu and T. Zhang, Nat. Commun., 2019, 10, 3824 Search PubMed.
- F. Wang, J. Ma, S. Xin, Q. Wang, J. Xu, C. Zhang, H. He and X. Cheng Zeng, Nat. Commun., 2020, 11, 529 CAS.
- H. V. Thang, G. Pacchioni, L. DeRita and P. Christopher, J. Catal., 2018, 367, 104–114 CAS.
- C. Liu, Y. Fang, S.-Y. Wang and S.-J. Ji, ACS Catal., 2019, 9, 8910–8915 CAS.
- C. R. Reddy, R. Ranjan and S. K. Prajapti, Org. Lett., 2019, 21, 623–626 CAS.
- X. S. Zhang, J. Y. Jiao, X. H. Zhang, B. L. Hu and X. G. Zhang, J. Org. Chem., 2016, 81, 5710–5716 CAS.
- G. Liang, M. Liu, J. Chen, J. Ding, W. Gao and H. Wu, Chin. J. Chem., 2012, 30, 1611–1616 CAS.
- G. Saito, J. A. Swanson and K.-D. Lee, Adv. Drug Deliver. Rev., 2003, 55, 199–215 CAS.
- F. Wei, X.-Q. Shen, X.-H. Zhang and X.-G. Zhang, Adv. Synth. Catal., 2018, 360, 3911–3915 CAS.
- R. Semwal, C. Ravi, S. Saxena and S. Adimurthy, J. Org. Chem., 2019, 84, 14151–14160 CAS.
- H.-Y. Chen, W.-T. Peng, Y.-H. Lee, Y.-L. Chang, Y.-J. Chen, Y.-C. Lai, N.-Y. Jheng and H.-Y. Chen, Organometallics, 2013, 32, 5514–5522 CAS.
- C. Ravi, N. N. Reddy, V. Pappula, S. Samanta and S. Adimurthy, J. Org. Chem., 2016, 81, 9964–9972 CAS.
- M. Martinek, M. Korf and J. Srogl, Chem. Commun., 2010, 46, 4387–4389 CAS.
- Y. Li, C. Nie, H. Wang, X. Li, F. Verpoort and C. Duan, Eur. J. Org. Chem., 2011, 2011, 7331–7338 CAS.
- Z. Chen, F.-X. Cao, W. Gao, Q.-C. Dong and Y.-Q. Qu, Rare Met., 2019, 39, 806–814 Search PubMed.
- D. Chen, L. H. Zhang, J. Du, H. Wang, J. Guo, J. Zhan, F. Li and F. Yu, Angew. Chem., Int. Ed., 2021, 60, 24022–24027 CAS.
- F. Wang, J. Tian, M. Li, W. Li, L. Chen, X. Liu, J. Li, A. Muhetaer, Q. Li, Y. Wang, L. Gu, D. Ma and D. Xu, Angew. Chem., Int. Ed., 2020, 59, 8203–8209 CAS.
- X. Hu, J. Chen, S. Li, Y. Chen, W. Qu, Z. Ma and X. Tang, J. Phys. Chem. C, 2020, 124, 701–708 CAS.
- C. Maurizio, F. d'Acapito, M. Benfatto, S. Mobilio, E. Cattaruzza and F. Gonella, Eur. Phys. J. B, 2000, 14, 211–216 CAS.
- C. Anandan and P. Bera, Appl. Surf. Sci., 2013, 283, 297–303 CAS.
- J. Guo, X. Feng, S. Wang, Q. Wu, S. Lv, Y. Zhou, H. Li, Z. Chen and Y. Zhang, Dalton Trans., 2021, 50, 18061–18068 CAS.
- J. Zhang, Z. Wang, W. Chen, Y. Xiong, W.-C. Cheong, L. Zheng, W. Yan, L. Gu, C. Chen, Q. Peng, P. Hu, D. Wang and Y. Li, Chem, 2020, 6, 725–737 CAS.
- Y. Xiong, S. Wang, W. Chen, J. Zhang, Q. Li, H. S. Hu, L. Zheng, W. Yan, L. Gu, D. Wang and Y. Li, Small, 2021, 17, e2006834 Search PubMed.
- M. Yin, C.-K. Wu, Y. Lou, C. Burda, J. T. Koberstein, Y. Zhu and S. O'Brien, J. Am. Chem. Soc., 2005, 127, 9506–9511 CAS.
- W. Wang, D. I. Sharapa, A. Chandresh, A. Nefedov, S. Heißler, L. Heinke, F. Studt, Y. Wang and C. Wöll, Angew. Chem., Int. Ed., 2020, 59, 10514–10518 CAS.
- C. Oliva, L. Forni, A. M. Ezerets, I. E. Mukovozov and A. V. Vishniakov, J. Chem. Soc. Faraday. Trans., 1998, 94, 587–592 CAS.
- S. Zhao, D. Kang, Y. Liu, Y. Wen, X. Xie, H. Yi and X. Tang, ACS Catal., 2020, 10, 11739–11750 CAS.
- R. Kydd, W. Y. Teoh, K. Wong, Y. Wang, J. Scott, Q.-H. Zeng, A.-B. Yu, J. Zou and R. Amal, Adv. Funct. Mater., 2009, 19, 369–377 CAS.
- D. Liu, H.-X. Ma, P. Fang and T.-S. Mei, Angew. Chem., Int. Ed., 2019, 58, 5033–5037 CAS.
- Y. Jiang, Y. Qin, S. Xie, X. Zhang, J. Dong and D. Ma, Org. Lett., 2009, 11, 5250–5253 CAS.
- D. Ma, Y. Zhang, J. Yao, S. Wu and F. Tao, J. Am. Chem. Soc., 1998, 120, 12459–12467 CAS.
- D. Ma and Q. Cai, Acc. Chem. Res., 2008, 41, 1450–1460 CAS.
- R. T. Stibrany, C. Zhang, T. J. Emge, H. J. Schugar, J. A. Potenza and S. Knapp, Inorg. Chem., 2006, 45, 9713–9720 CAS.
- E. Sperotto, G. P. van Klink, G. van Koten and J. G. de Vries, Dalton Trans., 2010, 39, 10338–10351 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc06738b |
| This journal is © The Royal Society of Chemistry 2023 |