
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChiral aldehyde catalysis enables direct asymmetric α-substitution reaction of N-unprotected amino acids with halohydrocarbons†
Hao-Ran
Shen‡
,
Chao-Xing
Li‡
,
Xin
Jiang
,
Yao
Lin
,
Jian-Hua
Liu
,
Fang
Zhu
,
Zhu-Lian
Wu
 ,
Tian
Cai
,
Wei
Wen
*,
Rong-Xing
He
and
Qi-Xiang
Guo
*
,
Tian
Cai
,
Wei
Wen
*,
Rong-Xing
He
and
Qi-Xiang
Guo
*
Key Laboratory of Applied Chemistry of Chongqing Municipality, Chongqing Key Laboratory of Soft-Matter Material Chemistry and Function Manufacturing, School of Chemistry and Chemical Engineering, Southwest University, Chongqing, 400715, China. E-mail: wenwei1989@swu.edu.cn; qxguo@swu.edu.cn
First published on 3rd May 2023
Abstract
The direct catalytic α-hydrocarbylation of readily available amino acids with halohydrocarbons is one of the most straightforward methods leading to α,α-disubstituted non-proteinogenic α-amino acid compounds. However, all the reported methodologies depend on N-protected amino acids as starting materials. Herein, we report on three highly efficient aldehyde-catalyzed direct α-hydrocarbylations of N-unprotected amino acid esters with aryl-, allyl-, and benzyl halides. By promoting a simple chiral BINOL-aldehyde catalyst or combining catalysts of a chiral aldehyde and Lewis acid ZnCl2, the asymmetric α-arylation, α-allylation, and α-benzylation of amino acid esters with the corresponding halohydrocarbons proceed smoothly, producing α,α-disubstituted α-amino acids in moderate-to-high yields and good-to-excellent enantioselectivities. The asymmetric α-arylation reaction can be applied in the formal synthesis of the clinical candidate compound (+)-AG-041R. Based on the results given by control experiments, three reaction models are proposed to illustrate the stereoselective-control outcomes.
Introduction
The halohydrocarbon-involved substitution reaction is one of the most classic transformations in organic chemistry for the formation of carbon–carbon and carbon-heteroatom bonds.1 With the utilization of halohydrocarbons such as aryl, alkenyl, and alkyl halides as reactants, a large number of optically active molecules have been constructed via SNAr,2 SN1, or SN2 substitutions,3 or via couplings.4 Among these reactions, the catalytic asymmetric α-substitution reactions of readily available amino acid derivatives with halohydrocarbons provide a highly efficient pathway for the preparation of optically active unnatural α,α-disubstituted α-amino acids. Especially, chiral phase-transfer catalysis has enabled important advances, benefiting from highly efficient catalytic ability, broad application scope and lack of chemoselectivity issues. Conversely, protection and deprotection steps are unavoidable (Fig. 1, path a).5 In contrast, an ideal catalytic strategy leading to such products could directly promote the α-hydrocarbylation of N-unprotected amino acid esters with high efficiency (Fig. 1, path b); however, such a valuable methodology has not been disclosed until now. As an alternative strategy, the chiral aldehyde/transition-metal dual catalysis allows for the one-step synthesis of NH2-unprotected α,α-disubstituted α-amino acids by utilizing electrophiles generated in situ. (path c).6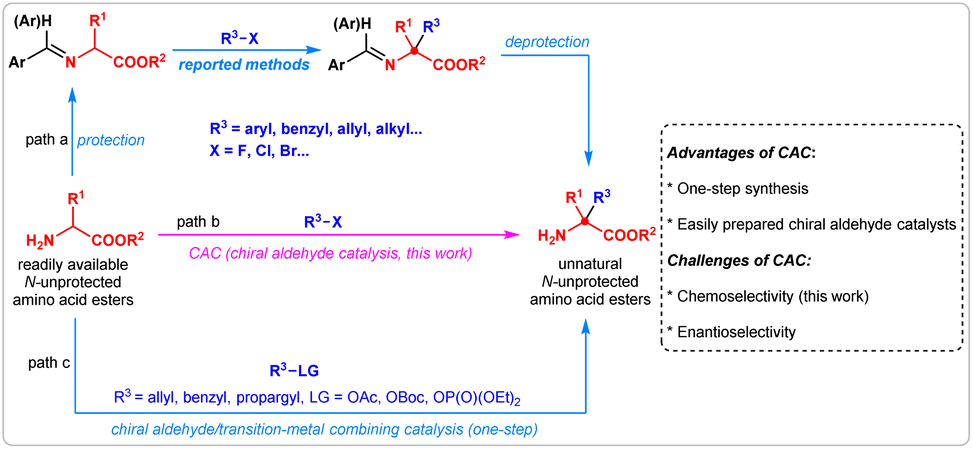 | ||
| Fig. 1 Catalytic asymmetric α-hydrocarbylation reaction of amino acid esters. | ||
A unique property of chiral aldehyde catalysis,6 directly promoting the asymmetric α-functionalization of N-unprotected amino acid esters by increasing the acidity through catalytic formation of a chiral imine, has been demonstrated in alkylation,7 Michael addition,8 Mannich,9 aldol,10 allylation,11 benzylation,12 and propargylation13 reactions. We envisioned that this strategy may be promising for the direct asymmetric α-hydrocarbylation of amino acid esters with halohydrocarbons. However, it was challenging to control the chemoselectivity of C/N-functionalization in this transformation. In the previously reported reactions employing chiral aldehyde catalysis, either the electrophiles were gradually generated in situ during the reaction process (for alkylation, allylation, benzylation, and propargylation reactions), or the N-functionalization side products were unstable and reversely decomposed to starting material (for Michael, Mannich, and aldol reactions). Therefore, the side reaction of N-functionalization in those reactions was efficiently suppressed. In our proposed reaction, chiral aldehyde-catalyzed α-hydrocarbylation of N-unprotected amino acid esters with halohydrocarbons, the equivalent amounts of halohydrocarbon electrophiles and the possible formation of stable N-hydrocarbylation byproducts made chemoselectivity more difficult to control. Thus, it was necessary to develop an aldehyde-catalyzed direct α-hydrocarbylation reaction of amino acid esters with improved chemoselectivity and enantioselectivity. Here, we report three chiral aldehyde-catalyzed asymmetric α-substitution reactions of N-unprotected amino acid esters with aryl halides,14 allyl chlorides, and benzyl chlorides.15 Although the N-functionalization byproducts are not suppressed completely, the desired C-functionalization products can be generated in moderate-to-high yields with good-to-excellent enantioselectivities for all three transformations. Furthermore, the α-arylation product is used for the formal total synthesis of the clinical candidate compound (+)-AG-041R, and three reaction models are proposed based on the results of control experiments.
Results and discussion
Initial studies
We initially investigated the possibility of our proposal in the asymmetric reactions of amino acid ester 1a with 2-nitro fluorobenzene 2a, allyl chloride 5a, and benzyl chloride 8a. The simple chiral aldehyde CA-1 was chosen as a catalyst, and the base Cs2CO3, or tetramethylguanidine (TMG), was added to accelerate the deprotonation process. We found that the arylation reaction of tert-butyl alaninate 1a and 2-nitro fluorobenzene 2a took place smoothly, providing the desired product 3a in 25% yield with >99% enantioselective excess (ee), although the side product 4a was obtained in 16% yield (Fig. 2a). For the allylation reaction of 1a with cinnamyl chloride 5a, the desired product 7a was obtained in 23% yield with 8% ee. However, no product 9a was observed in the benzylation of 1a with 8a. To suppress the N-functionalization side reaction, the Lewis acid ZnCl2 was added. As expected, the yield of the allylation product 6a was enhanced to 89%, and the benzylation product 9a was obtained in 15% yield (Fig. 2b and c).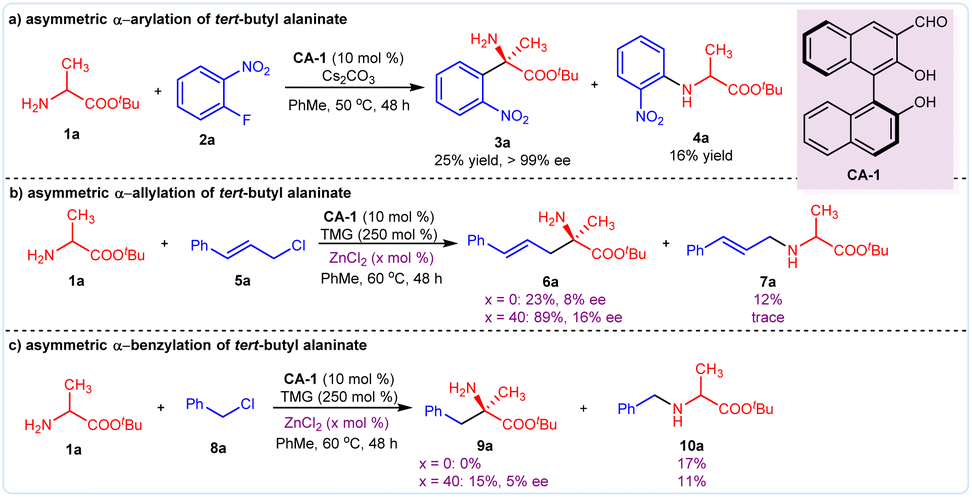 | ||
| Fig. 2 Initial investigation of the catalytic asymmetric α-hydrocarbylation of amino acid esters. (a) Asymmetric arylation; (b) asymmetric allylation; (c) asymmetric benzylation. | ||
Asymmetric α-arylation reaction
Encouraged by the initial results, we systematically studied the chiral aldehyde-catalyzed arylation reaction. Because of the excellent enantioselectivity of 3a obtained in Fig. 2a, chiral aldehyde CA-1 was chosen for optimization of the reaction conditions. Base screening indicated that inorganic bases were suitable for this reaction, and K3PO4 provided the greatest yield of 3a (Fig. 3a). The choice of solvent also affected the yield of 3a. When this reaction was conducted in Et2O, product 3a was obtained in 86% yield with >99% ee (Fig. 3b). Then, the base equivalent and the reaction concentration were tuned. The results indicated that using 5 equivalents of K3PO4 and 0.2 M 2a (y = 1) produced the best yield (Fig. 3c and d).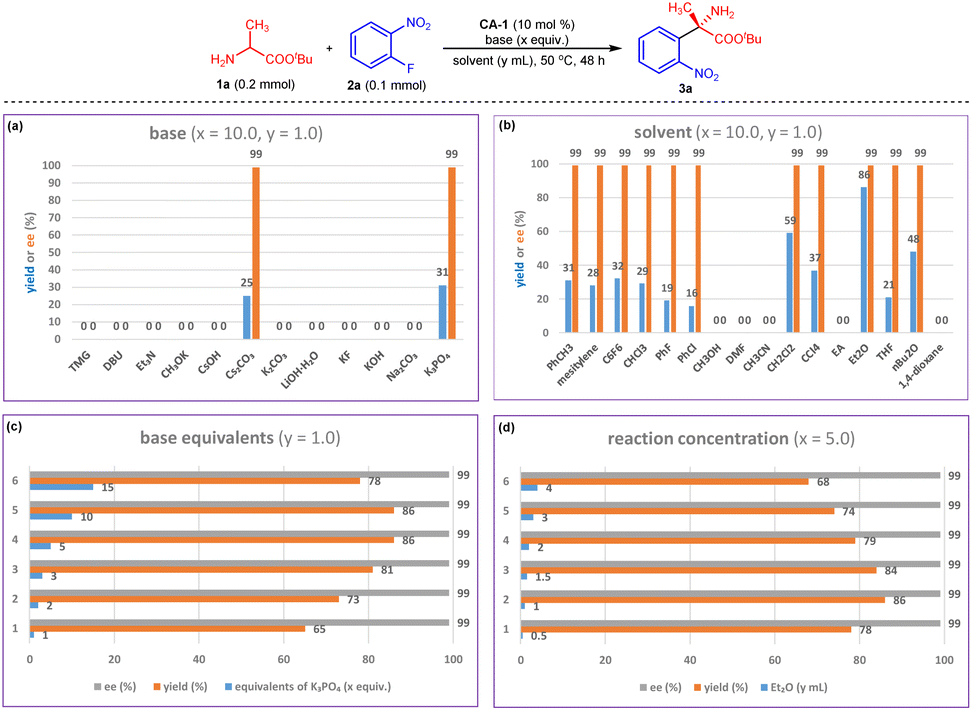 | ||
| Fig. 3 Reaction condition optimization for arylation of 1a with 2a. (a) Bases screening; (b) solvents screening; (c) base equivalents screening; (d) reaction concentration optimization. | ||
After determining the optimal reaction conditions, we examined the substrate scopes for this reaction. First, various substituted amino acid esters were employed as reaction partners with 2-nitro fluorobenzene 2a (Fig. 4a). The variation of the alkoxyl group on reactant 1 affected the yields but had little influence on the enantioselectivity (Fig. 4a, 3a–3e). Generally, the benzyl and methyl alaninate produced corresponding products 3c and 3e in moderate yields. The decrease in yields may be caused by the amine-ester exchange reaction of amino acid esters containing less bulky alkoxyl groups. Amino acid esters bearing saturated linear alkyls were effective reaction partners for 2a, producing 3f–3i in good-to-high yields with excellent enantioselectivities. Similar results were observed when the saturated branched alkyl- or unsaturated linear alkyl-substituted amino acid esters participated in this reaction (Fig. 4a, 3j–3m). Other functional groups, such as ester, sulfoxide, and indolyl, that are contained in the amino acid esters were well tolerated under the optimal reaction conditions; products 3n–3q were obtained in moderate-to-good yields with excellent enantioselectivities. The model reaction was readily expanded to the gram scale. With 10 mmol of 2-nitro fluorobenzene, compound 3a was produced in 74% yield (1.97 g) with >99% ee.
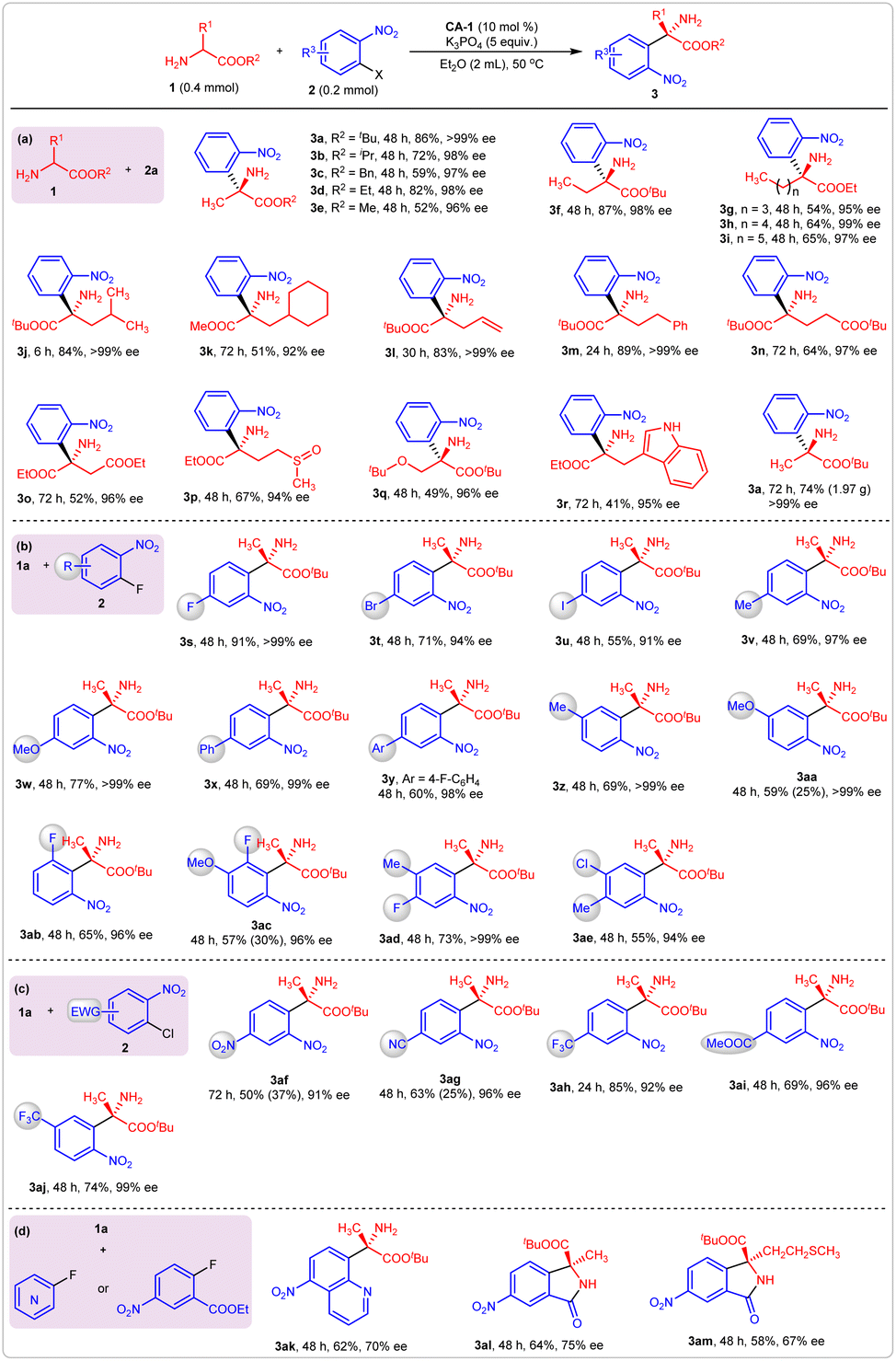 | ||
| Fig. 4 Substrate scopes of the asymmetric arylation reaction. (a) For amino acid esters; (b) for substituted 2-nitro fluorobenzenes; (c) for substituted 2-nitro chlorobenzenes; (d) for other substituted fluorobenzenes. | ||
Then, various substituted 2-nitro fluorobenzenes 2 were examined (Fig. 4b). The introduction of a third substituent did not affect the enantioselectivity, but the yield was affected by its electronic effects. For example, the 1,4-difluoro-2-nitrobenzene gave product 3s in 91% yield with >99% ee. When a third substituent with less electronegativity than the fluoro moiety was installed at the para-position of the benzene ring, the yields slightly decreased (Fig. 4b, 3t–3yvs.3s). Besides the para-substituted 2-nitro fluorobenzenes, the meta- and ortho-substituted ones also gave the desired products in good yields with excellent enantioselectivities (Fig. 4b, 3z–3ab). When two substituents were introduced simultaneously on the benzene ring, the combination of an electron-donating and electron-withdrawing group was necessary to obtain satisfactory yields (Fig. 4b, 3ac–3ae) because the increase of strong electron-withdrawing groups accelerated the generation of N-arylation side products. Particularly, when 2,4-dinitro fluorobenzene was introduced as an arylation reagent, only the N-arylation byproduct was observed. For most substrates, the generation of N-functionalization side products was unavoidable. For example, accompanying the formation of C-functionalization products 3aa and 3ac, corresponding N-arylation side products were obtained in 25% and 30% yield, respectively.
To overcome the drawback that only N-arylation byproducts were generated in reactions using a strong electron-withdrawing group-substituted 2-nitro fluorobenzene, corresponding 2-nitro chlorobenzenes were employed as reactants. As expected, the desired C-arylation became dominant, and only a small amount of N-arylation was observed. For example, nitro, cyano, trifluoromethyl, and ester-substituted 2-nitro chlorobenzenes gave products 3af–3aj in good-to-high yields with excellent enantioselectivities (Fig. 4c). 8-Fluoro-5-nitroquinoline was also a suitable reaction partner with amino acid ester 1a, which gave products 3ak in 62% yield with 70% ee. Furthermore, when ethyl 2-fluoro-5-nitrobenzoate was used as an arylation reagent, optically active chiral isoindolinones were produced via a tandem arylation/cyclization process (Fig. 4d, 3al–3am). The absolute configuration of product 3a was assigned as S by comparing its optical rotation value with the literature data (see ESI†), and the stereochemistries of the other products in Fig. 4 were assigned accordingly.
Asymmetric α-allylation and benzylation reactions
Allyl and benzyl chlorides are two other types of commonly used alkylation reagents for carbon–carbon and carbon-heteroatom bonds formations. However, the N-functionalization byproducts can form spontaneously because of their high reactivity. Thus, the chemoselectivity of the chiral aldehyde-catalyzed α-functionalization of N-unprotected amino acid esters with allyl or benzyl chlorides becomes extremely difficult to control. Nevertheless, our initial experimental results revealing that the target α-allylation product could be obtained in 89% yield with 16% ee under the promotion of a chiral aldehyde-ZnCl2 system encouraged us to pursue high efficiency using allyl or benzyl chlorides. Therefore, we systematically investigated the reaction conditions of the catalytic asymmetric allylation of tert-butyl alaninate 1a with cinnamyl chloride 5a. Chiral aldehyde catalyst screening indicated that most of the 3-formyl BINOL aldehydes could produce 6a in good-to-excellent yields; however, the enantioselectivities were not satisfactory. The 2-formyl BINOL-aldehyde CA-15 gave excellent enantioselectivity (94% ee), but the yield was moderate (61%) (Fig. 5a, i). We chose CA-15 as the chiral aldehyde catalyst for further condition optimization. Various solvents were evaluated, and mesitylene was optimal in terms of yield and enantioselectivity (Fig. 5a, ii). When this reaction was conducted at 50 °C, product 6a was obtained in 65% yield with 94% ee (Fig. 5a, iii). We found that the reaction concentration affected the yield to some degree: the usage of 0.4 mL mesitylene at 0.2 mmol scales of reactant 1a could produce compound 6a in 73% yield with 94% ee (Fig. 5a, iv). Based on these results, the optimal reaction conditions for the asymmetric α-allylation reaction were determined.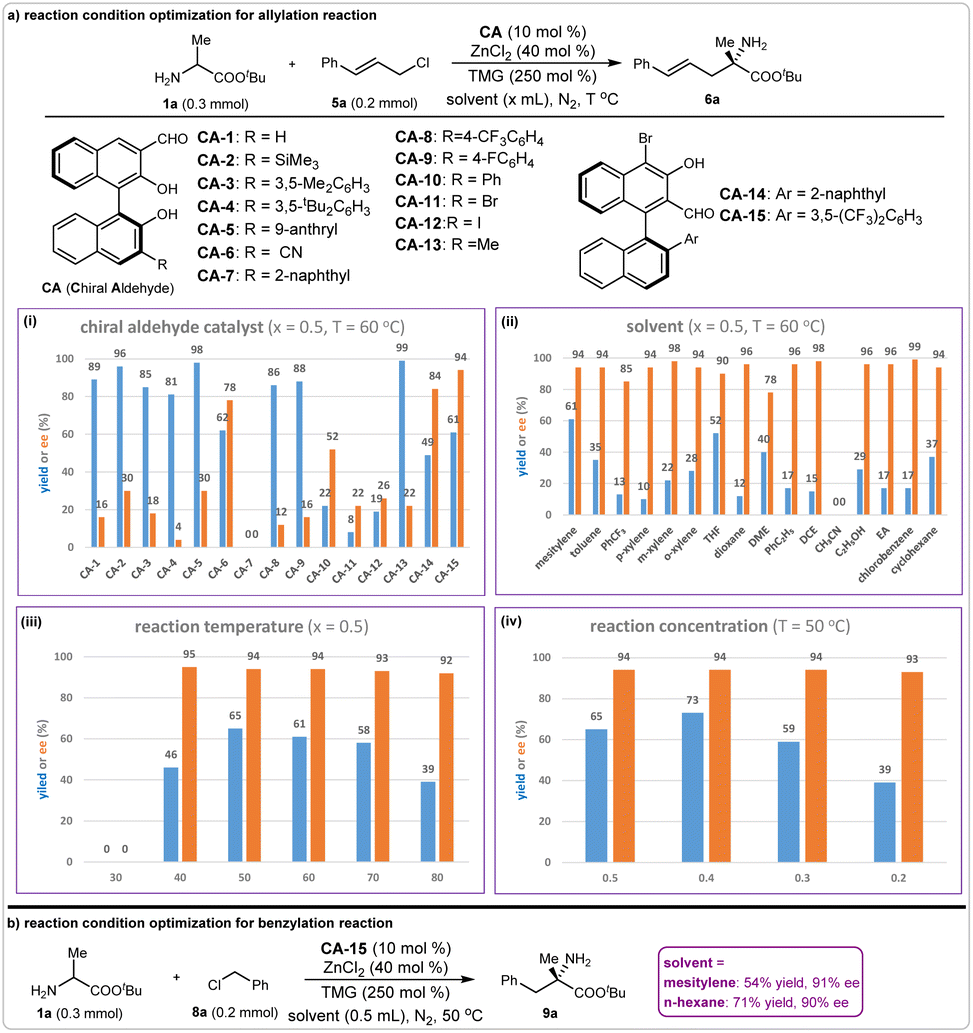 | ||
| Fig. 5 Condition optimizations for asymmetric allylation and benzylation reactions. (a) Catalysts screening; (b) solvents screening; (c) reaction temperature optimization; (d) reaction concentration optimization. | ||
Due to the similar reaction activities of benzyl chlorides with allyl chlorides, we directly applied the above optimal reaction conditions in the asymmetric α-benzylation reaction. As expected, this reaction took place smoothly, giving the desired product 9a in 54% yield with 90% ee. Then, optimization of the reaction conditions was performed, including the screening of chiral aldehyde catalysts, bases, solvents, and reaction temperature (see ESI†). Finally, we found that product 9a could be obtained in 71% yield with 91% ee when n-hexane was used as the reaction solvent (Fig. 5b). Thus, the optimal reaction conditions for this asymmetric benzylation were determined.
Under the optimal reaction conditions, we examined the corresponding substrate scopes of these asymmetric allylation and benzylation reactions. First, various substituted cinnamyl chlorides were assessed. The results indicated that cinnamyl chlorides bearing single or double substituents on benzene ring were effective reaction partners with amino acid ester 1a, providing compounds 6b–6k in moderate-to-good yields with excellent enantioselectivities. The experimental outcomes were not affected by the steric and electronic properties of the substituent. Concerning the substrates of amino acid esters, the yields of corresponding products were affected by the steric influence of their α-substituents. For all eight amino acid esters we employed in this reaction, products 6l–6s were obtained in moderate yields with excellent enantioselectivities.
Similar results were observed in the asymmetric benzylation reaction. Various substituted benzyl chlorides exhibited good activities in this reaction and gave the corresponding products in moderate-to-high yields with good enantioselectivities (Fig. 6, 9b–9j). When the methyl 2-(chloromethyl)benzoate 8b was used as an acceptor, the product 9k was obtained in 52% yield with 92% ee via a tandem benzylation/lactamization process. Because of the steric influence of their α-substituents, amino acid esters other than 1a gave products with moderate yields. In addition, all the enantioselectivities for these products were maintained at a high level (Fig. 6, 9l–9r). The absolute configurations of products 6b (S) and 9a (S) were determined by comparing their optical rotation values with the literature data (see ESI†), and the stereochemistries of the other products in Fig. 6 were assigned accordingly.
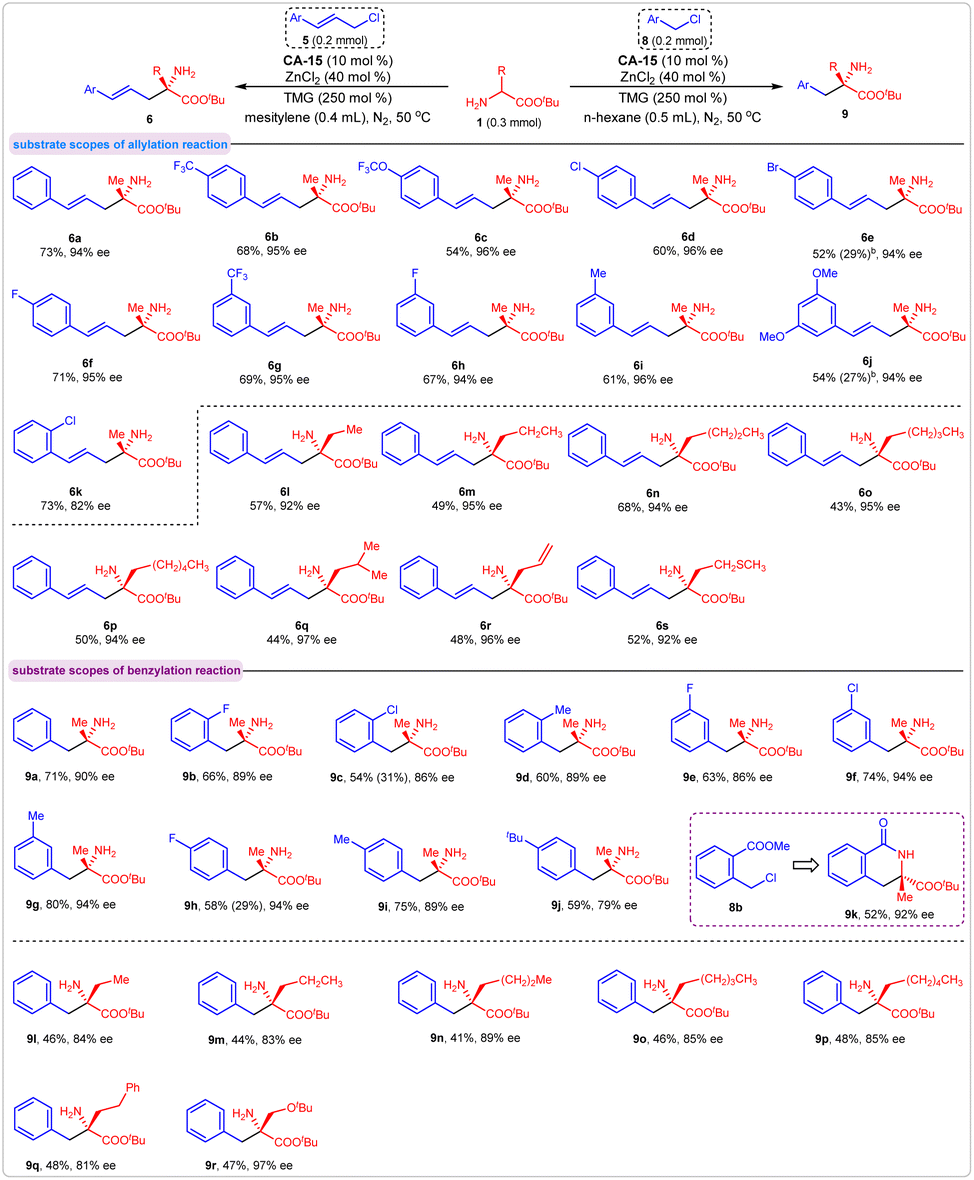 | ||
| Fig. 6 Substrate scopes of the asymmetric allylation and benzylation reactions. | ||
Synthetic application
The clinical candidate compound (+)-AG-041R is a gastrin/CCK-B receptor antagonist with an IC50 of 1.1 nmol.16 Few catalytic asymmetric methodologies have been developed for preparing this compound,17 but we envisioned that its core oxindole unit can be constructed from our arylation product by a reduction of the nitro group and an in situ intramolecular amidation reaction, and a new catalytic asymmetric synthetic route for this clinical candidate was anticipated. As we discussed in the section on substrate scope, the diethyl aspartate 1b reacted smoothly with 2-nitro fluorobenzene 2a under the optimal reaction conditions, and product 3o was obtained in 52% yield with >99% ee. Then, we reduced the nitro group of 3o to an amine. An intramolecular amidation took place in situ to give 3-amino oxindole, product 11. Using the reported methods, urea moiety-contained oxindole 12 and Iwabuchi intermediate 13 (ref. 17a) were prepared, successively (Scheme 1). The spectra data and optical rotation of compound 13 agree with the literature (see ESI†). Thus, based on the chiral aldehyde-catalyzed arylation of amino acid esters, a new synthetic route for the formal synthesis of AG-041R was achieved.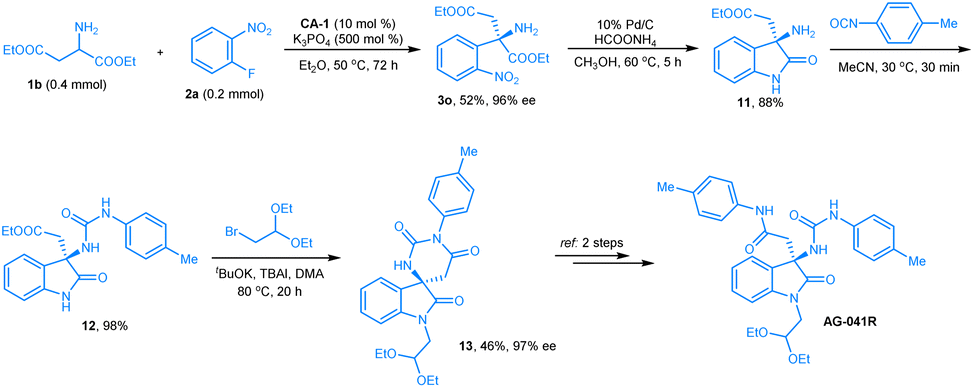 | ||
| Scheme 1 The formal synthesis of AG-041R. | ||
Reaction mechanism investigation
The possible reaction models for these three reactions were investigated. For the asymmetric arylation of amino acid ester 1a with 2a, once the reactant 2-nitro fluorobenzene 2a was replaced by 3-nitro fluorobenzene or 4-nitro fluorobenzene, the reaction efficiency was greatly decreased. Protecting the 2′ hydroxyl of the chiral aldehyde catalyst CA-1 also prevented the formation of product 3a with high efficiency (Fig. 7a). These results indicated that hydrogen bond interactions may have occurred between the 2′ hydroxyl of CA-1 and the nitro group of 2-nitro fluorobenzene 2a. Thus, a possible transition state I (TS I) was proposed for the production of (S)-3a (Fig. 7c).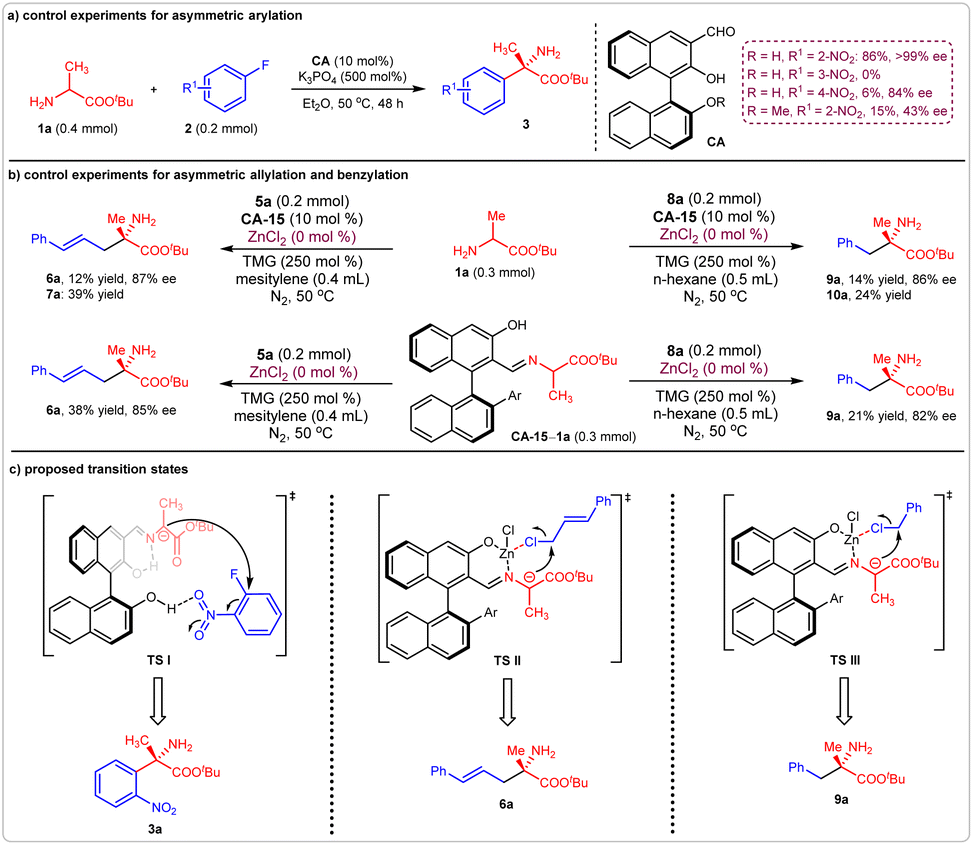 | ||
| Fig. 7 Control experiments and possible transition states. (a) Control experiments for asymmetric arylation reaction; (b) control experiments for asymmetric allylation and benzylation reactions; (c) proposed transition states. | ||
For the asymmetric allylation and benzylation reactions, all of the yields of the corresponding products decreased greatly in the absence of Lewis acid ZnCl2, no matter whether the amino acid ester 1a or the formed Schiff base CA-15-1a was employed as donor (Fig. 7b), indicating that the Lewis acid can accelerate the processes of Schiff base formation and deprotonation. Furthermore, the slight decreases in enantioselectivities showed that the Lewis acid can enhance the stereoselective-control ability of the corresponding transition states. It was reasonable to deduce that these reactions took place between the Zn-Schiff base complexes and halohydrocarbons. Therefore, two transition states, TS II and TS III, were proposed. Thus, products (S)-6a and (S)-9a were generated respectively (Fig. 7c).
Conclusions
In conclusion, we demonstrated highly efficient chiral aldehyde-catalyzed α-substitution reactions of α-amino acid esters with aryl, allyl, and benzyl halides. In the promotion of the simple chiral aldehyde CA-1, the α-arylation reaction of N- unprotected amino acid esters with aryl halides takes place smoothly, giving α-aryl α,α-disubstituted amino acid derivatives in moderate-to-high yields with good-to-excellent enantioselectivities. The combinational catalytic system comprising chiral aldehyde CA-15 and Lewis acid ZnCl2 efficiently promotes the reactions of asymmetric allylation and benzylation, giving α-allyl and α-benzyl α,α-disubstituted amino acid derivatives in moderate-to-good yields with good-to-excellent enantioselectivities. The α-arylation product 3o is used for the formal total synthesis of AG-041R, and three transition states are proposed to illustrate the stereoselective-control results.Data availability
All data supporting the findings of this study are available within the article and its ESI† file.Author contributions
W. W. and G. Q. X. conceived this project. S. H. R., L. C. X., J. X., L. Y., L. J. H. and Z. F. carried out the experiments. W. Z. L. and C. T. performed the HRMS analysis. H. R. X. and J. X. discussed the transition states. G. Q. X. wrote the manuscript. All authors discussed the results.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from NSFC (22071199, 22201235), the Innovation Research 2035 Pilot Plan of Southwest University (SWU-XDZD22011), and the Chongqing Science Technology Commission (cstccxljrc201701, cstc2018jcyjAX0548).Notes and references
- (a) J. McMurry, Organic Chemistry, 9th edn; Cengage Learning, 2015 Search PubMed; (b) F. Carey and R. Giuliano, Organic Chemistry, 8th edn; McGraw-Hill, 2021 Search PubMed; (c) L. G. Wade, Organic Chemistry, 8th edn; Pearson Education India, 2012 Search PubMed; (d) J. Clayden, N. Greeves, S. Warren and P. Wothers, Organic Chemistry, 2nd edn; Oxford University Press, Oxford, 2012 Search PubMed.
- O. F. Terrier, Modern Nucleophilic Aromatic Substitution, Wiley-VCH, Weinheim, Germany, 2013 Search PubMed.
- (a) S. R. Hartshorn, Aliphatic Nucleophilic Substitution, Cambridge University Press, London, 1973 Search PubMed; (b) E. V. Anslyn and D. A. Dougherty, Modern Physical Organic Chemistry, University Science Books, Sausalito, 2006 Search PubMed.
- (a) J. Choi and G. C. Fu, Science, 2017, 356, eaaf7230 CrossRef PubMed; (b) A. de Meijere, S. Bräse and M. Oestreich, Metal-Catalyzed Cross-Coupling Reactions and More, Wiley-VCH, Weinheim, 2014 CrossRef; (c) T. J. Colacot, New Trends in Cross-Coupling: Theory and Applications, Royal Society of Chemistry, Cambridge, 2015 Search PubMed; (d) Y.-J. Hao, X.-S. Hu, Y. Zhou, J. Zhou and J.-S. Yu, ACS Catal., 2020, 10, 955–993 CrossRef CAS; (e) G. C. Fu, ACS Cent. Sci., 2017, 3, 692–700 CrossRef CAS PubMed; (f) M. Orlandi, M. Escudero-Casao and G. Licini, Synthesis, 2021, 53, 4559–4566 CrossRef.
- (a) C. Nájera and J. M. Sansano, Chem. Rev., 2007, 107, 4584–4671 CrossRef PubMed; (b) A. Perdih and M. S. Dolenc, Curr. Org. Chem., 2007, 11, 801–832 CrossRef CAS; (c) C. Cativiela and M. D. Diaz-De-Villegas, Tetrahedron: Asymmetry, 2007, 18, 569–623 CrossRef CAS; (d) K. Maruoka and T. Ooi, Chem. Rev., 2003, 103, 3013–3028 CrossRef CAS PubMed; (e) B. Lygo and B. I. Andrews, Acc. Chem. Res., 2004, 37, 518–525 CrossRef CAS PubMed; (f) M. J. O'Donnell, Acc. Chem. Res., 2004, 37, 506–517 CrossRef PubMed; (g) M. J. O'Donnell, Aldrichimica Acta, 2001, 34, 3–15 Search PubMed.
- (a) W. Wen and Q.-X. Guo, Synthesis, 2023, 55, 719–732 CrossRef CAS; (b) K.-J. Lin, A. Shi, C.-H. Shi, J.-B. Lin and H.-G. Lin, Front. Chem., 2021, 9, 687–817 Search PubMed; (c) Z.-Q. Yuan, J. Liao, H. Jiang, P. Cao and Y. Li, RSC Adv., 2020, 10, 35433–35448 RSC; (d) Q. Wang, Q. Gu and S.-L. You, Angew. Chem., Int. Ed., 2019, 58, 6818–6825 CrossRef CAS PubMed.
- B. Xu, L.-L. Shi, Y.-Z. Zhang, Z.-J. Wu, L.-N. Fu, C.-Q. Luo, L.-X. Zhang, Y.-G. Peng and Q.-X. Guo, Chem. Sci., 2014, 5, 1988–1991 RSC.
- (a) W. Wen, L. Chen, M.-J. Luo, Y. Zhang, Y.-C. Chen, Q. Ouyang and Q.-X. Guo, J. Am. Chem. Soc., 2018, 140, 9774–9780 CrossRef CAS PubMed; (b) W.-Z. Wang, H.-R. Shen, J. Liao, W. Wen and Q.-X. Guo, Org. Chem. Front., 2022, 9, 1422–1426 RSC; (c) J.-G. Ma, Q.-H. Zhou, G.-S. Song, Y.-C. Song, G.-Q. Zhao, K.-L. Ding and B.-G. Zhao, Angew. Chem., Int. Ed., 2021, 60, 10588–10592 CrossRef CAS PubMed.
- (a) J.-F. Chen, X. Gong, J.-Y. Li, Y.-K. Li, J.-G. Ma, C.-K. Hou, G.-Q. Zhao, W.-C. Yuan and B.-G. Zhao, Science, 2018, 360, 1438–1442 CrossRef CAS PubMed; (b) W. Wen, M.-J. Luo, Y. Yuan, J.-H. Liu, Z.-L. Wu, T. Cai, Z.-W. Wu, Q. Ouyang and Q.-X. Guo, Nat. Commun., 2020, 11, 5372 CrossRef CAS PubMed.
- (a) A. Cheng, L. Zhang, Q. Zhou, T. Liu, J. Cao, G. Zhao, K. Zhang, G. Song and B. Zhao, Angew. Chem., Int. Ed., 2021, 60, 20166–20172 CrossRef CAS PubMed; (b) P. Ji, X. Liu, J. Xu, X. Zhang, J. Guo, W.-W. Chen and B. Zhao, Angew. Chem., Int. Ed., 2022, 61, e202206111 CAS; (c) C. Hou, B. Peng, S. Ye, Z. Yin, J. Cao, X. Xiao and B. Zhao, Nat. Catal., 2022, 5, 1061–1068 CrossRef CAS.
- (a) L. Chen, M.-J. Luo, F. Zhu, W. Wen and Q.-X. Guo, J. Am. Chem. Soc., 2019, 141, 5159–5163 CrossRef CAS PubMed; (b) F. Zhu, Q.-W. Shen, W.-Z. Wang, Z.-L. Wu, T. Cai, W. Wen and Q.-X. Guo, Org. Lett., 2021, 23, 1463–1467 CrossRef CAS PubMed; (c) J. Ma, B. Gao, G. Song, R. Zhang, Q. Wang, Z. Ye, W.-W. Chen and B. Zhao, Angew. Chem., Int. Ed., 2022, 61, e202200850 CAS.
- J.-H. Liu, W. Wen, J. Liao, Q.-W. Shen, Y. Lin, Z.-L. Wu, T. Cai and Q.-X. Guo, Nat. Commun., 2022, 13, 2509 CrossRef CAS PubMed.
- F. Zhu, C.-X. Li, Z.-L. Wu, T. Cai, W. Wen and Q.-X. Guo, Nat. Commun., 2022, 13, 7290 CrossRef CAS PubMed.
- (a) M. Chaari, A. Jenhi, J. P. Lavergne and P. Viallefont, Tetrahedron, 1991, 47, 4619–4630 CrossRef CAS; (b) S. Shirakawa, K. Yamamoto and K. Maruoka, Angew. Chem., Int. Ed., 2015, 54, 838–840 CrossRef CAS PubMed; (c) S. Shirakawa, K. Yamamoto, T. Tokuda and K. Maruoka, Asian J. Org. Chem., 2014, 3, 433–436 CrossRef CAS; (d) K. Tomohara, T. Yoshimura, R. Hyakutake, P. Yang and T. Kawabata, J. Am. Chem. Soc., 2013, 135, 13294–13297 CrossRef CAS PubMed; (e) Y. Li, H. Pan, W.-Y. Li, X. Feng and X. Liu, Synlett, 2021, 32, 587–592 CrossRef CAS; (f) F. Foschi, A. Tagliabue, V. Mihali, T. Pilati, I. Pecnikaj and M. Penso, Org. Lett., 2013, 15, 3686–3689 CrossRef CAS PubMed.
- (a) J.-S. Yang, K. Lu, C.-X. Li, Z.-H. Zhao, X.-M. Zhang, F.-M. Zhang and Y.-Q. Tu, Angew. Chem., Int. Ed., 2022, 61, e202114129 CAS; (b) Y. Kubota, S. Shirakawa, T. Inoue and K. Maruoka, Tetrahedron Lett., 2012, 53, 3739–3741 CrossRef CAS; (c) Y. P. Park, S. Kang, Y. J. Lee, T. S. Kim, B. S. Jeong, H. Park and S. Jew, Org. Lett., 2009, 11, 3738–3741 CrossRef CAS PubMed; (d) T. S. Kim, Y. J. Lee, K. Lee, B. S. Jeong, H. Park and S. Jew, Synlett, 2009, 4, 671–674 Search PubMed; (e) S. Jew, B. S. Jeong, J. H. Lee, M. S. Yoo, Y. J. Lee, B. Park, M. G. Kim and H. Park, J. Org. Chem., 2003, 68, 4514–4516 CrossRef CAS PubMed; (f) S. Woo, Y. G. Kim, B. Lim, J. Ho, Y. Lee, H. Gwon and K. Nahm, RSC Adv., 2018, 8, 2157–2160 RSC; (g) T. Ooi, T. Miki and K. Maruoka, Org. Lett., 2005, 7, 191–193 CrossRef CAS PubMed; (h) M. Kitamura, S. Shirakawa and K. Maruoka, Angew. Chem., Int. Ed., 2005, 44, 1549–1551 CrossRef CAS PubMed; (i) T. Achard, Y. N. Belokon, J. A. Fuentes, M. North and T. Parsons, Tetrahedron, 2004, 60, 5919–5930 CrossRef CAS; (j) Y. N. Belokon, D. Bhave, D. D'Addario, E. Groaz, M. North and V. Tagliazucca, Tetrahedron, 2004, 60, 1849–1861 CrossRef CAS; (k) B. Lygo, J. Crosby and J. A. Peterson, Tetrahedron Lett., 1999, 40, 8671–8674 CrossRef CAS; (l) T. Ooi, M. Takeuchi, M. Kameda and K. Maruoka, J. Am. Chem. Soc., 2000, 122, 5228–5229 CrossRef CAS; (m) M. J. O'Donnell and S. Wu, Tetrahedron: Asymmetry, 1992, 3, 591–594 CrossRef; (n) U.-H. Dolling, P. Davis and E. J. J. Grabowski, J. Am. Chem. Soc., 1984, 106, 446–447 CrossRef CAS; (o) M. J. O'Donnell, W. D. Bennett and S. Wu, J. Am. Chem. Soc., 1989, 111, 2353–2355 CrossRef.
- (a) T. Chiba, Y. Kinoshita, M. Sawada, K. Kishi, A. Baba and E. Hoshino, Yale J. Biol. Med., 1998, 71, 247–255 CAS; (b) E. Lindstrcm, M. Bjçrkqvist and R. Hakanson, Br. J. Pharmacol., 1999, 127, 530–536 CrossRef PubMed; (c) X.-Q. Ding, E. Lindstrom and R. Hakanson, Clin. Pharmacol., 1997, 81, 232–237 CAS; (d) M. Ochi, K. Kawasaki, H. Kataoka, Y. Uchio and H. Nishi, Biochem. Biophys. Res. Commun., 2001, 283, 1118–1123 CrossRef CAS PubMed; (e) H. Kitamura and M. Okazaki, Osteoarthr. Cartilage, 2005, 13, 287–296 CrossRef PubMed; (f) W.-H. Sun, F. Zhu, G.-S. Chen, H. Su, C. Luo, Q.-S. Zhao, Y. Zhang, J. Shao, S.-M. Sun, G.-X. Zhou, Y.-L. Ding and Y.-L. Cheng, Cancer Lett., 2008, 263, 302–311 CrossRef CAS PubMed.
- (a) T. Emura, T. Esaki, K. Tachibana and M. Shimizu, J. Org. Chem., 2006, 71, 8559–8564 CrossRef CAS PubMed; (b) S. Sato, M. Shibuya, N. Kanoh and Y. Iwabuchi, J. Org. Chem., 2009, 74, 7522–7524 CrossRef CAS PubMed; (c) S. Sato, M. Shibuya, N. Kanoh and Y. Iwabuchi, Chem. Commun., 2009, 41, 6264–6266 RSC; (d) S. Hajra, S. Laskar and B. Jana, Chem.–Eur. J., 2019, 25, 14688–14693 CrossRef CAS PubMed; (e) X.-J. Xia, Q.-Q. Zhu, J. Wang, J. Chen, W.-G. Cao, B. Zhu and X.-Y. Wu, J. Org. Chem., 2018, 83, 14617–14625 CrossRef CAS PubMed; (f) M. Sawa, S. Miyazaki, R. Yonesaki, H. Morimoto and T. Ohshima, Org. Lett., 2018, 20, 5393–5397 CrossRef CAS PubMed; (g) J. Dai, D. Xiong, T.-R. Yuan, J. Liu, T. Chen and Z.-H. Shao, Angew. Chem., Int. Ed., 2017, 56, 12697–12701 CrossRef CAS PubMed; (h) O. D. Engl, S. P. Fritz and H. Wennemers, Angew. Chem., Int. Ed., 2015, 54, 8193–8197 CrossRef CAS PubMed; (i) J. Wang, Q.-X. Zhang, B.-Y. Zhou, C. Yang, X. Li and J.-P. Cheng, Iscience, 2019, 16, 511–533 CrossRef CAS PubMed; (j) K. Zhao, T. Shu, J.-Q. Jia, G. Raabe and D. Enders, Chem.–Eur. J., 2015, 21, 3933–3936 CrossRef CAS PubMed; (k) N. Hara, S. Nakamura, M. Sano, R. Tamura, Y. Funahashi and N. Shibata, Chem.–Eur. J., 2012, 18, 9276–9280 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc01294h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |