
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSteric hindrance-induced selective growth of rhodium on gold nanobipyramids for plasmon-enhanced nitrogen fixation†
Henglei
Jia‡
 a,
Fan
Li‡
a,
Yuanyuan
Yang
a,
Mengxuan
Zhao
a,
Jingzhao
Li
a and
Chun-yang
Zhang
*ab
a,
Fan
Li‡
a,
Yuanyuan
Yang
a,
Mengxuan
Zhao
a,
Jingzhao
Li
a and
Chun-yang
Zhang
*ab
aCollege of Chemistry, Chemical Engineering and Materials Science, Shandong Normal University, Jinan 250014, China. E-mail: cyzhang@sdnu.edu.cn
bSchool of Chemistry and Chemical Engineering, Southeast University, Nanjing 211189, China. E-mail: zhangcy@seu.edu.cn
First published on 2nd May 2023
Abstract
The construction of an antenna–reactor plasmonic photocatalyst that is composed of a plasmonic and a catalytically active metal holds great promise in driving N2 photofixation, but its photocatalytic performance is highly dependent on the spatial distribution of the two components. Up to now, the fabrication of dumbbell-shaped nanostructures featuring spatially separated architecture has remained challenging. Herein, we develop a facile synthetic strategy for the site-selective growth of a Rh nanocrystal ‘reactor’ on two tips of an Au nanobipyramid (NBP) ‘antenna’ through the precise manipulation of steric hindrance toward Rh overgrowth. The obtained Au NBP/tip-Rh nanodumbbells (Au NBP/tip-Rh NDs) can function as an excellent antenna–reactor plasmonic photocatalyst for N2 photofixation. In this scenario, the Au nanoantenna harvests light and generates hot electrons under plasmon resonance, meanwhile the hot electrons are transferred to the active sites on Rh nanocrystals for N2 reduction. In comparison with that of classical core@shell nanostructures, the spatially separated architecture of the Au NBP/tip-Rh NDs facilitates charge separation, greatly improving the photocatalytic activity. This study sheds new light on the structure–function relationship for N2 photofixation and benefits the design and construction of spatially separated plasmonic photocatalysts.
Introduction
N2 fixation, the conversion of naturally abundant N2 to NH3, is an essential process in modern society because NH3 is a fundamental building block for the synthesis of fertilizers and industrial chemical stock.1–3 NH3 is emerging as an alternative hydrogen carrier due to its properties of high hydrogen capacity (17.6 wt%), low liquefaction pressure (∼8 atm), and ease of storage and transportation.4 Although N2 accounts for about 78% of the Earth's atmosphere, the direct utilization of N2 remains challenging because N2 is extremely stable with a N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond dissociation energy of 941 kJ mol−1 and a large energy gap of 10.82 eV between the HOMO and LOMO.5 NH3 synthesis is industrially accomplished by the energy and carbon-intensive Haber–Bosch process, which results in large energy consumption and CO2 emissions. Therefore, the development of a zero-carbon emission NH3 synthetic strategy is highly desired. Artificial photosynthesis of NH3 by using renewable solar energy and H2O as the energy and proton sources has provoked much attention recently,6–11 but traditional photocatalysts are hampered by their weak interaction with N2 and a large reaction activation barrier. Theoretically, transition metal-based catalysts, such as Ru, Rh, and Re, are the most promising materials for N2 fixation.12 When a N2 molecule interacts with a transition metal catalyst, N2 donates electrons from its bonding orbitals to the metal and accepts electrons into its antibonding orbitals, known as back donation.13 This back donation effect can weaken the NN bond and facilitate the cleavage of N2. However, the poor light-harvesting capability of these transition metal-based catalysts limits their applications in N2 photofixation.
N bond dissociation energy of 941 kJ mol−1 and a large energy gap of 10.82 eV between the HOMO and LOMO.5 NH3 synthesis is industrially accomplished by the energy and carbon-intensive Haber–Bosch process, which results in large energy consumption and CO2 emissions. Therefore, the development of a zero-carbon emission NH3 synthetic strategy is highly desired. Artificial photosynthesis of NH3 by using renewable solar energy and H2O as the energy and proton sources has provoked much attention recently,6–11 but traditional photocatalysts are hampered by their weak interaction with N2 and a large reaction activation barrier. Theoretically, transition metal-based catalysts, such as Ru, Rh, and Re, are the most promising materials for N2 fixation.12 When a N2 molecule interacts with a transition metal catalyst, N2 donates electrons from its bonding orbitals to the metal and accepts electrons into its antibonding orbitals, known as back donation.13 This back donation effect can weaken the NN bond and facilitate the cleavage of N2. However, the poor light-harvesting capability of these transition metal-based catalysts limits their applications in N2 photofixation.
The construction of antenna–reactor photocatalysts through the integration of a catalytically active metal with a strongly light-harvesting plasmonic metal holds great promise for both efficient light harvesting and high catalytic activity.14–17 Plasmonic metal nanocrystals (e.g., Au, Ag, and Cu) possess localized surface plasmon resonance (LSPR) property, which arises from the collective oscillations of free electrons confined in metal nanocrystals under resonant excitation.18–20 LSPR can give rise to a strong electromagnetic field at the nanoscale and thereafter enhance the light-matter interaction of metal nanocrystals. More importantly, plasmonic hot electrons and holes are generated through plasmon excitation decay.18 These energetic charge carriers are readily transferred to adjacent metals or directly utilized for chemical reactions. Among various plasmonic noble metal nanocrystals, Au NBPs have recently received increasing attention owing to their unique architecture and the presence of high-index facets.21–25 Therefore, the integration of Au NBPs with catalytically active metals (e.g., Rh) for N2 reduction holds great promise for driving N2 photofixation under ambient conditions.
Precisely manipulating the architecture of bimetallic nanocrystals is of great importance to their photocatalytic performance.26–28 In comparison with traditional core@shell nanostructures, spatially separated nanoarchitectures are more attractive, because they allow hot charge carriers to take part in reduction and oxidation reactions at different active sites independently.29–31 Among different types of spatially separated architectures, dumbbell-shaped nanostructures offer significant benefits for charge separation in the photocatalytic N2 reduction process.32–38 As revealed by theoretical calculations, the sharp tips of Au NBPs can generate extremely large local electric field enhancement and the deposition of catalytically active metals at the hotspots is beneficial for the photocatalytic reaction.21,39 Despite much promise, selective growth of Rh nanocrystals on two tips of Au NBPs remains challenging due to the lack of a wet-chemistry synthesis method.
In this work, we present a facile method for the selective growth of Rh nanocrystals on two tips of Au NBPs to fabricate Au NBP/tip-Rh NDs. The formation of a large steric hindrance at the middle of Au NBPs is vital for selective growth, which is achieved by employing benzyldimethylhexadecylammonium chloride (16-BAC) molecules as the surfactant. Since Rh nanocrystals tend to aggregate in small sizes, the large benzyl group in the 16-BAC molecules presents a large steric hindrance to block the deposition of Rh on the side surface. The unique architecture endows the obtained Au NBP/tip-Rh NDs with excellent photocatalytic performance toward N2 fixation. Benefiting from the spatially separated architecture, the Au NBP/tip-Rh NDs exhibit a N2 photofixation activity of 114.33 μmol h−1 g−1, which is 8.0 and 2.0-fold compared with those of Rh nanocrystals and core@shell nanostructures.
Results and discussion
The Au NBP/tip-Rh NDs are prepared through the selective growth of Rh nanocrystals on two tips of Au NBPs (Fig. 1). The pregrown Au NBPs are stabilized with the surfactant cetyltrimethylammonium bromide (CTAB) molecules. In the absence of 16-BAC, Rh nanocrystals are prone to nucleation on the whole surface of Au NBPs to obtain the core@shell nanostructures. Since Rh nuclei are generally very small, the small steric hindrance of CTAB molecules can hardly prevent the deposition of Rh nuclei on the side surface, and subsequently core@shell nanostructures are formed. 16-BAC molecules have a similar molecular structure to CTAB. A big difference lies in that 16-BAC molecules possess a large benzyl headgroup but CTAB molecules have a small methyl group. The presence of 16-BAC molecules facilitates the selective growth process. First, the large headgroup of 16-BAC molecules can block the entrance of Rh nuclei into the side surfaces of Au NBPs. At an appropriate concentration, the density of 16-BAC molecules at the two tips is smaller than that on the side surface due to the curvature difference,34 which provides an opportunity for Rh nucleation at the two tips. Second, 16-BAC has a stronger capability of stabilizing the Rh precursor than CTAB due to the larger headgroup. The strong bonding capability reduces the number of nucleations and slows down the growth rate of Rh. Upon the nucleation of Rh nanocrystals at the two tips, further deposition of Rh will occur on the original Rh nuclei because of the smaller interfacial free energy than the Au–Rh interface. In addition, the presence of NaI during the synthesis facilitates the epitaxial overgrowth of Rh on Au NBP.40 First, the complexation of I− ions with Rh3+ ions can decrease the nucleation rate of Rh nanocrystals by decreasing the reduction potential. Second, the I− ions possess strong interaction with Au, which can direct the growth of Rh on Au NBPs. Third, the presence of the I−/I2 red/ox pair makes I− ions a good candidate as a reduction/oxidation agent for the activation of the Au surface. Accordingly, Rh nanodomains are formed at two tips of Au NBPs to obtain the Au NBP/tip-Rh NDs.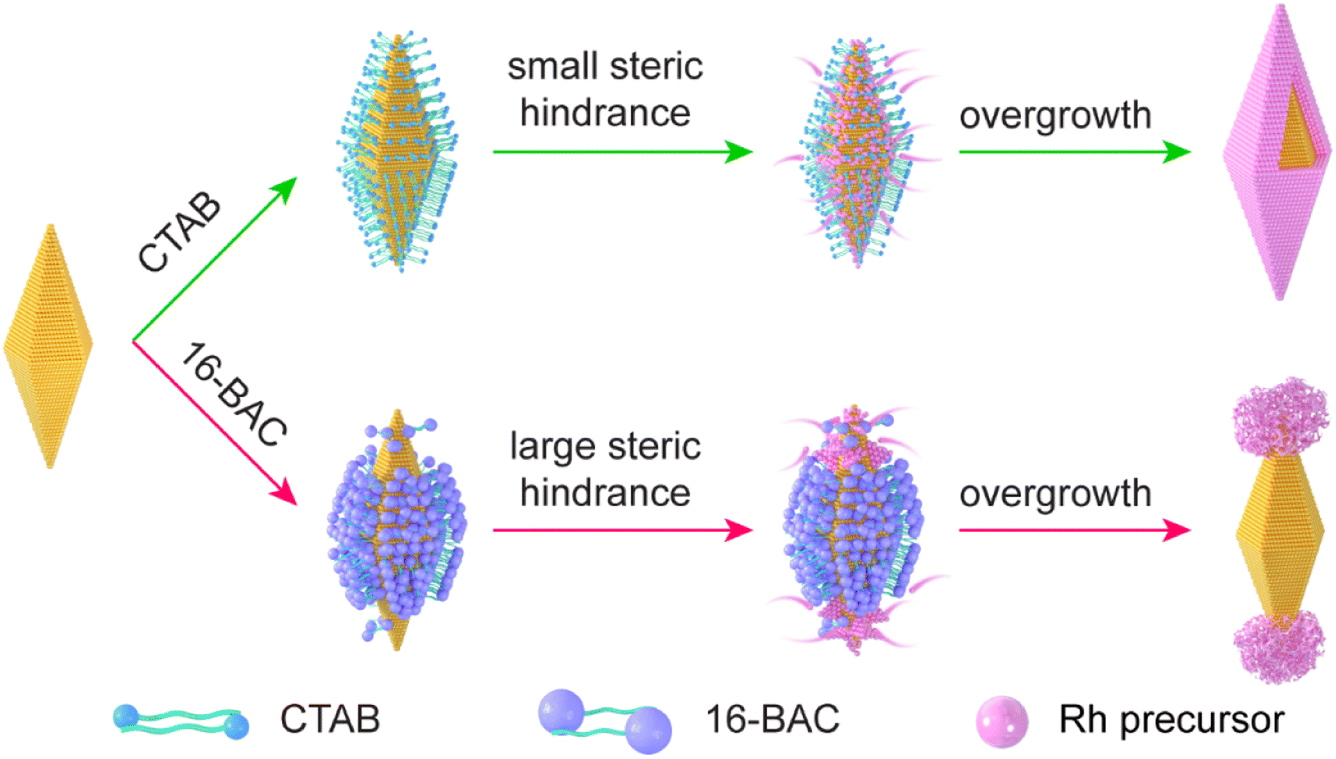 | ||
| Fig. 1 Schematic illustration of the growth behaviors of Rh nanocrystals on Au NBPs in the presence of either CTAB or 16-BAC surfactants. | ||
The starting Au NBPs with a fivefold rotational symmetry structure were prepared using a seeded growth method (Fig. 2a). The as-prepared Au NBPs possess a uniform morphology and a narrow spectral linewidth, with the average length and diameter at the middle being 110.8 ± 2.9 nm and 35.5 ± 1.9 nm, respectively. The excellent monodispersity of Au NBPs provides an ideal platform for Rh selective growth on Au NBPs. The representative transmission electron microscopy (TEM) image of the obtained Au NBP/tip-Rh NDs clearly demonstrates that Rh nanocrystals have been successfully grown on two tips of Au NBPs (Fig. 2b). The Rh nanocrystals exhibit a snowflake morphology with a diameter of 36.9 ± 3.2 nm and are composed of many small dendritic Rh nanostructures (Fig. 2c). After the growth of Rh nanocrystals, a redshift for plasmon resonance wavelength occurs due to the change of the dielectric constant at the two tips (Fig. S1†). To further verify the dumbbell-shaped nanostructures, we carried out high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) imaging and energy-dispersive X-ray (EDX) elemental mapping (Fig. 2d). Evidently, the EDX elemental mapping results reveal the bipyramidal Au core and well-defined Rh nanodendrite-tipped caps, suggesting the formation of dumbbell-shaped nanostructures. In addition, the crystalline properties of Au and Rh are confirmed by aberration-corrected high-resolution TEM (HRTEM) imaging (Fig. 2e). An aberration-corrected HRTEM image clearly demonstrates that the Rh cap consists of many dendritic nanostructures with a diameter of about 1–3 nm. The formation of dendritic structures is attributed to the large lattice mismatch (∼7%) between Au and Rh.40 Dendritic nanostructures with porous channels of Rh nanocrystals are supposed to be ideal architectures for catalysis.
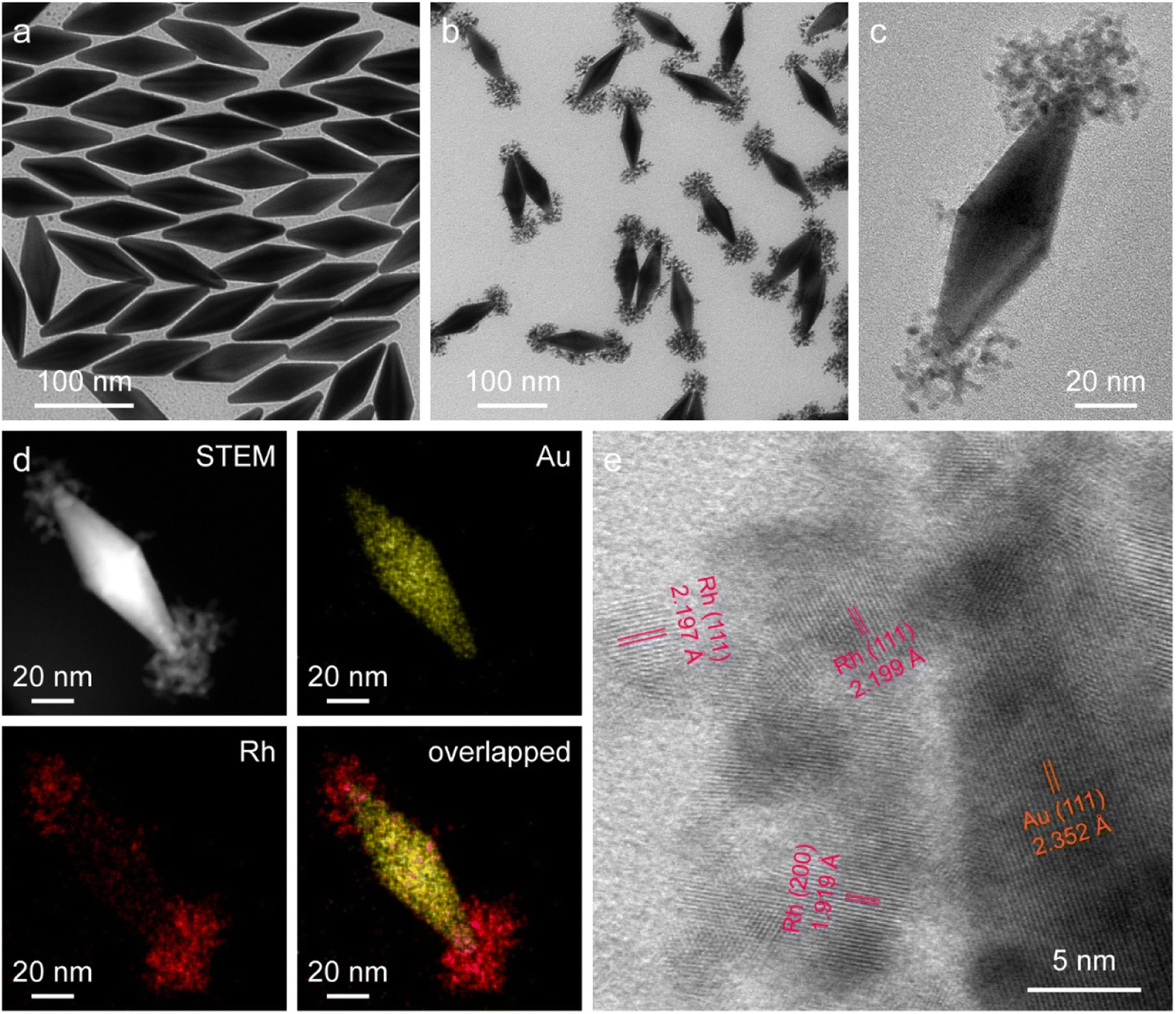 | ||
| Fig. 2 Dumbbell-shaped Au/Rh nanostructures. (a and b) TEM images of the Au NBPs (a) and Au NBP/tip-Rh NDs (b). (c) TEM image of a representative Au NBP/tip-Rh ND. (d) HAADF-STEM image and the corresponding EDX elemental maps of a single Au NBP/tip-Rh ND. (e) Aberration-corrected HRTEM image at the Au–Rh interface. | ||
To examine the structure and composition of the Au NBP/tip-Rh NDs, we performed X-ray diffraction (XRD) and X-ray photoelectron spectroscopy (XPS) (Fig. 3). There are two sets of diffraction patterns in the XRD spectrum, consistent with the combination of standard powder diffraction patterns of Au (JCPDS #4-784) and Rh (JCPDS #5-685), suggesting the highly crystalline nature of the dumbbell-shaped structure (Fig. 3a). Analogously, the survey XPS spectrum further confirms the presence of Au and Rh in the Au NBP/tip-Rh ND sample (Fig. S2†). The high-resolution Au 4f XPS spectrum presents two strong peaks located at 83.8 eV (Au 4f7/2) and 87.5 eV (Au 4f5/2), suggesting the metallic Au0 state (Fig. 3b).24 The Rh 3d XPS spectrum can be fitted with four peaks (Fig. 3c). Two peaks (307.1 eV and 311.9 eV) originate from the Rh0 state, while the other two peaks (308.1 eV and 313.1 eV) are assigned to the Rh3+ state.41,42 The Rh0/Rh3+ ratio is 54![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :46 calculated by integrating the peak areas. The appearance of Rh3+ peaks arises from the surface oxidation of the dendritic Rh nanocrystals,42 because XPS is only sensitive to the surface of materials.10 After the Ar+ ion sputtering, the Rh0/Rh3+ value is 92/8 (Fig. 3d), indicating that the dominant form of Rh is metallic Rh.
:46 calculated by integrating the peak areas. The appearance of Rh3+ peaks arises from the surface oxidation of the dendritic Rh nanocrystals,42 because XPS is only sensitive to the surface of materials.10 After the Ar+ ion sputtering, the Rh0/Rh3+ value is 92/8 (Fig. 3d), indicating that the dominant form of Rh is metallic Rh.
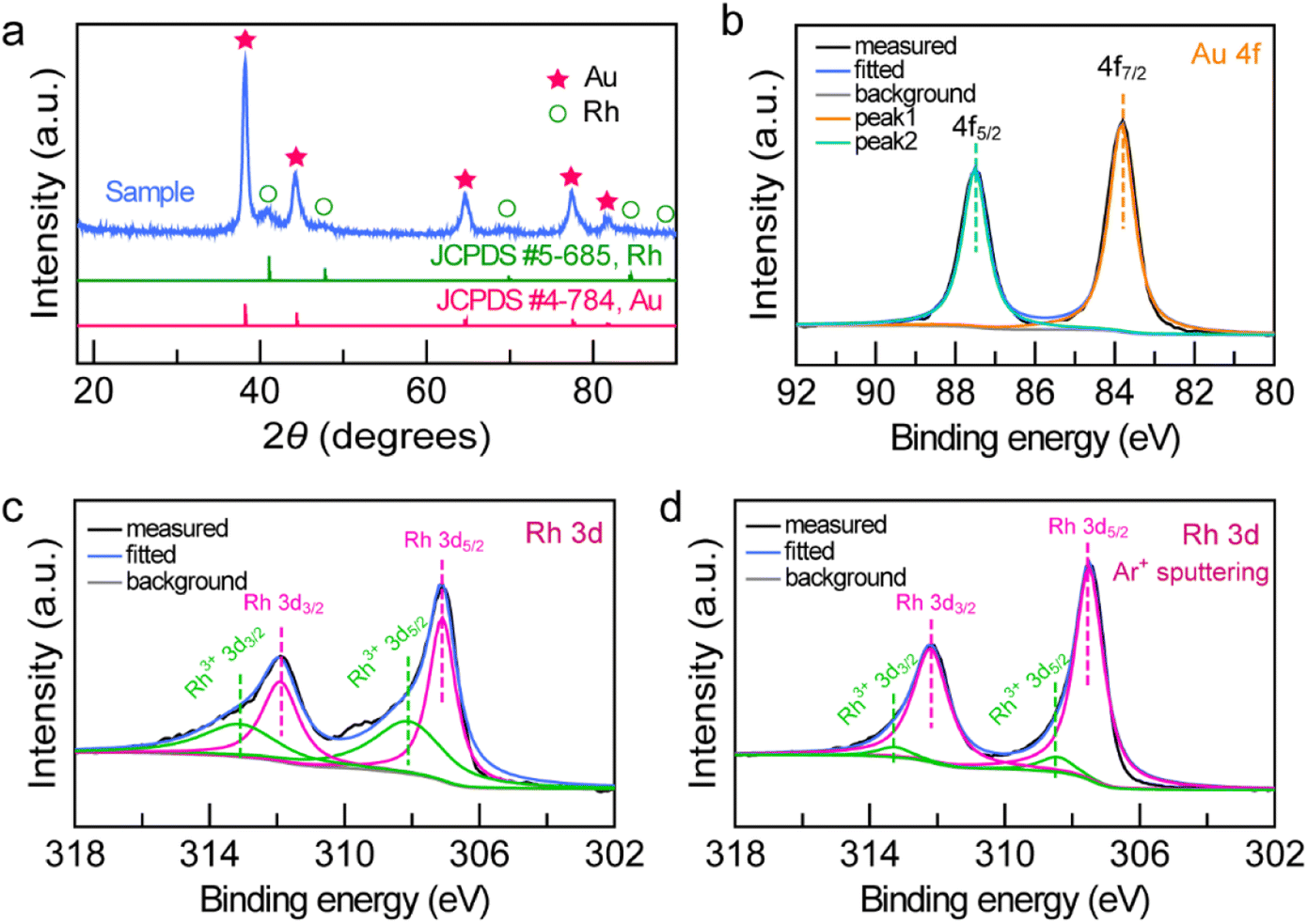 | ||
| Fig. 3 Structure and chemical composition of the Au NBP/tip-Rh NDs. (a) XRD patterns. (b) High-resolution Au 4f XPS spectrum. (c and d) High-resolution Rh 3d XPS spectra before (c) and after (d) the Ar+ ion sputtering experiments. | ||
The unique dumbbell-shaped architecture of the Au NBP/tip-Rh NDs holds great promise for driving N2 photofixation as an antenna–reactor photocatalyst. To evaluate the photocatalytic performance of the dumbbell-shaped nanostructures, we performed N2 fixation experiments under visible-NIR light (λ > 420 nm) illumination (Fig. S3†). To close the photocatalytic cycle, methanol was employed as the hole scavenger. For better comparison, we prepared four other catalysts, including pure Au NBPs, Rh nanocrystals (Fig. S4 and S5†), a mixture of Au NBPs with Rh nanocrystals, and Au NBP@Rh core@shell nanostructures (Fig. S4†). The exact weights of the photocatalysts were determined by inductively coupled plasma optical emission spectroscopy (ICP-OES) (Fig. S6 and Table S1†). Prior to the N2 fixation experiments, a linear calibration relationship was first precalibrated, and the generated NH3 amounts were determined by the indophenol-blue method (Fig. S7†).
The spatially separated design of dumbbell-shaped nanostructures is advantageous for charge separation and may serve as an ideal platform for the study of structure–function relationship. As displayed in Fig. 4a, the generated NH3 amounts on different catalysts gradually increase with the illumination time, suggesting that NH3 originates from the photocatalytic reduction of N2. The Au NBP and Rh nanocrystal samples exhibit very weak photocatalytic activities due to the lack of active sites or poor light-harvesting capability. The improvement of the N2 fixation activity is small by simply mixing the Au NBPs with Rh nanocrystals, suggesting that the antenna–reactor mechanism does not function without direct contact through a sharing interface. Impressively, the Au NBP/tip-Rh NDs exhibit a superior photocatalytic activity toward N2 fixation. The N2 fixation rate of the Au NBP/tip-Rh NDs is 114.33 μmol h−1 g−1, which is 8.0 and 2.0-fold compared with those of Rh nanocrystals and the core@shell nanostructures (Fig. 4b). The improvement in the photocatalytic performance is ascribed to the spatially separated architecture of the dumbbell-shaped nanostructures. To gain more insight, we carried out photocurrent measurements at the open-circuit potential to investigate the charge separation in the dumbbell-shaped and core@shell nanostructures. As displayed in Fig. S8†, the photocurrent generated by the dumbbell-shaped nanostructures is 2.1-fold higher than that generated by the core@shell nanostructures, suggesting that the spatially separated architecture facilitates charge separation in the photocatalytic process. Control experiments confirm that there is no NH3 production under these conditions: (a) without catalysts, (b) without illumination, and (c) replacement of N2 with Ar as the feeding gas, suggesting that the product NH3 originates from the photocatalytic reduction of N2 on the Au NBP/tip-Rh NDs (Fig. S9†). The NH3 generation rate drops to 45.4% (51.9 ± 17.3 μmol h−1 g−1) in the absence of the sacrificial agent, implying the importance of the sacrificial agent in N2 fixation. To further trace the source of NH3, we carried out the isotope labelling experiments by conducting N2 fixation under 14N2 and 15N2 atmospheres, respectively (Fig. 4c). The products in the reaction solutions were analysed using the 1H NMR spectrum. Under a 14N2 atmosphere, triple peaks appear in the 1H NMR spectrum, consistent with the peaks of standard 14NH4Cl solution. When 15N2 is employed as the nitrogen source, only double peaks are detected, which are identical to those of the standard 15NH4Cl solution. The isotope labelling experiments further verify that the generated NH3 is derived from the reduction of N2 rather than contamination or other sources. In addition, the absence of N2H4 in the reaction solution underlines the high selectivity of the N2 photofixation on the Au NBP/tip-Rh NDs (Fig. 4d).
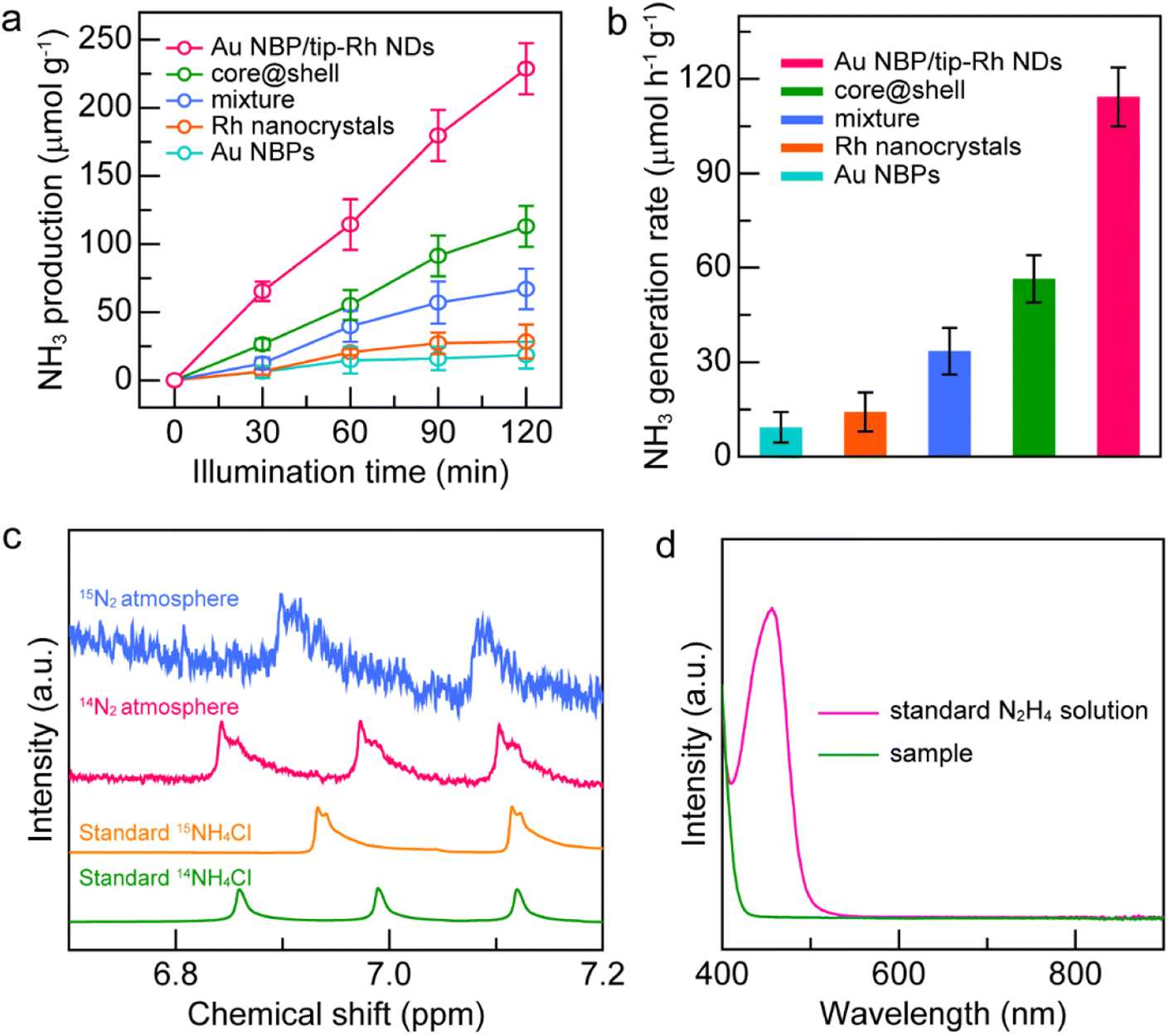 | ||
| Fig. 4 N2 photofixation under visible-NIR light illumination. (a) NH3 productions as a function of time for different catalysts. (b) Comparison of the NH3 production rates on different catalysts. (c) Isotope labelling experiments. (d) Detection of possible N2H4 in the reaction system. The standard N2H4 solution has an absorption peak at 455 nm. | ||
Notably, one of the great challenges in N2 photofixation is the competition between NH3 synthesis and H2 evolution.7 To further determine the selectivity of the Au NBP/tip-Rh ND photocatalyst, we performed the N2 photofixation experiment in a gastight glass reactor under a N2 atmosphere and analyzed the generated H2 using an off-line gas chromatograph (Fig. S10†). The generation rate is 61.2 μmol h−1 g−1 for NH3 and 7.8 μmol h−1 g−1 for H2. Since 2e− and 3e− are required for the generation of one H2 or one NH3 molecule, the selectivity of NH3 was calculated on the basis of the consumed electrons.35 The selectivity of NH3 is 92.2%, suggesting the presence of a competitive H2 evolution side reaction. Even though the reduction of H2O is the main bottleneck for NH3 synthesis, the products are readily separated because NH3 is highly soluble. To confirm the stability of the Au NBP/tip-Rh ND sample in the photocatalytic process, the catalyst was recovered and characterized after a typical photocatalytic reaction (Fig. S11–S13†). No obvious morphological and structural changes are observed, suggesting the excellent stability of the catalyst. Moreover, the photocatalyst exhibits good recyclability, with the photocatalytic activity remaining unchanged after three successive cycles (Fig. S14†). The contact area between the two components in the bicomponent plasmonic nanocrystals may affect the photocatalytic performance, especially in those metal/semiconductor nanostructures with a Schottky junction, because the Schottky barrier only allows hot electrons with sufficient energy to be utilized. It has been reported previously that an appropriate contact area is crucial to photocatalytic activity in the Janus Au–Cu2O nanostructures, striking a balance between the Schottky contact area and charge dissipation.31 In stark contrast, there is an absence of the Schottky barrier at the Au–Rh interface. Direct electron transfer from Au nanoantennas to Rh reactors can easily elevate the transfer efficiency. In addition, the plasmonic energy can transfer to the catalytic metal even if there is no direct contact between the antenna and reactor.43 Moreover, an extremely high electromagnetic field generated at the tips and Rh nanocrystals located at the hotspots of Au NBPs are beneficial for the photocatalytic activity.
The excellent photocatalytic performance of the dumbbell-shaped nanostructures benefits from the antenna–reactor photocatalytic mechanism. To ascertain whether the photocatalytic reaction is driven by the LSPR of Au NBPs, we performed the N2 fixation experiments under the illumination of different monochromatic lights and calculated the apparent quantum efficiencies (AQEs). Fig. 5a displays the action spectrum of the Au NBP/tip-Rh NDs for N2 photofixation. Notably, the AQE values obtained at different wavelengths faithfully follow the absorption spectrum of Au NBPs, confirming the plasmon-driven N2 photofixation process. Specifically, the AQE value is 0.57‰ at 940 nm, which is comparable to that of representative work on N2 photofixation with AuRu core-antenna nanostructures as the photocatalyst.8 The AQE action spectra also suggest that the N2 photofixation is mainly driven by NIR light, different from those catalysts which can only harvest visible light for photocatalysis. Previous studies have shown that plasmonic nanocrystals can accelerate chemical reactions through either plasmonic photocatalysis or the photothermal heating effect.44,45 In the N2 photofixation experiments, the temperature of the reaction solution is kept at 25 °C by using a water-circulation cooling system. To distinguish the contribution of plasmonic photocatalysis and photothermal heating, we carried out N2 fixation experiments in water baths set at different temperatures without illumination (Fig. S15†). In comparison with those under light illumination, the photocatalytic activities in water baths are very low. In addition, the core@shell nanostructures have the same photothermal effect as the dumbbell-shaped nanostructures, but they display lower photocatalytic activity under illumination. Although the photothermal heating effect can hardly be ruled out, these results suggest that plasmon-driven photocatalysis makes a dominant contribution to the excellent activity of the Au NBP/tip-Rh NDs.
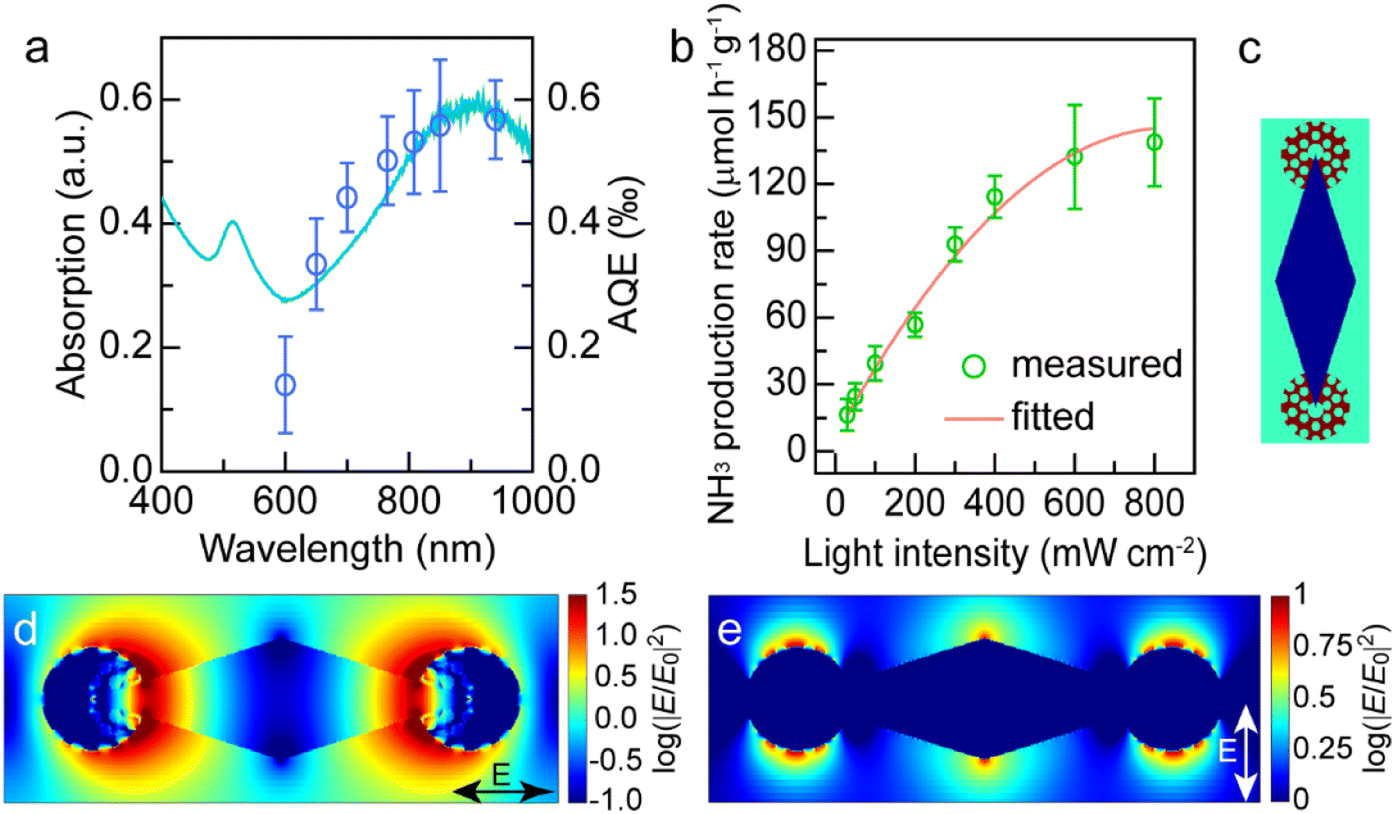 | ||
| Fig. 5 Plasmon-driven N2 photofixation. (a) Extinction (left) and AQE action (right) spectra of the Au NBP/tip-Rh NDs. (b) NH3 generation rates under different light intensities. (c) Schematic model of the Au NBP/tip-Rh ND structure used for FDTD simulations. (d and e) FDTD-simulated electric field intensity enhancement contours under the longitudinal (c) and transverse excitations, respectively. | ||
Plasmon can drive chemical reactions via two classes of mechanisms including the energy transfer mechanism43,46–48 and charge transfer mechanism.44,45,49 We further investigated the underlying mechanism of plasmon-driven N2 photofixation. On the one hand, we studied the dependence of the NH3 production rate upon light intensity to corroborate the contribution of plasmonic hot electrons (Fig. 5b). An approximately linear relationship between the reaction activity and the light intensity substantiates the crucial contribution of plasmonic hot electrons to photocatalytic activity, because the linear dependence is a significant indicator for the electron-driven chemical reaction.50,51 On the other hand, the plasmon-induced resonance energy transfer (PIRET) process can contribute to the plasmonic photocatalysis. To gain a deep insight into the contribution of the PIRET process, we carried out finite-difference time domain (FDTD) simulations to investigate the electromagnetic field enhancement around the dumbbell-shaped nanostructures (Fig. 5c–e). The representative dumbbell-shaped nanostructures were modelled on the basis of the measured geometrical parameters. The electromagnetic field distribution of the Au NBP/tip-Rh NDs was calculated at their plasmon wavelength of 900 nm in two polarization directions. As revealed by the FDTD results, the largest electromagnetic field enhancement is located at the tips of Au NBPs. The excellent photocatalytic activity of the Au NBP/tip-Rh NDs can be ascribed to the strong electromagnetic field enhancement, since the Rh reactor is just located at the hotspot of Au NBPs. The strong local field enhancement can focus light energy onto the Rh catalyst, which efficiently boosts the generation of hot charge carriers and subsequently promotes the photocatalytic activity. Consequently, both hot electron transfer and PIRET processes have significant influence on the plasmonic photocatalysis. In fact, the plasmon-driven chemical reaction is a much more complex process, and it remains elusive to differentiate the contributions of energy transfer and charge transfer to the photocatalytic activity. In future work, we should focus on the study of quantifying charge transfer and energy transfer contributions in plasmon-driven photocatalysis.
To gain a deep insight into the N2 adsorption and activation processes on the Au NBP/tip-Rh NDs, we conducted both experimental and theoretical studies. First, to clarify the interaction of N2 with the photocatalyst, we investigated the active sites for N2 adsorption and activation using density functional theory (DFT) calculations (Fig. 6a–d). The adsorption energies of N2 on Au and Rh are −0.81 eV and −1.06 eV, respectively. The more negative adsorption energy suggests that the active site for N2 fixation is on Rh in Au NBP/Rh NDs. Such a strong interaction of N2 with Rh facilitates the electron transfer from the active sites of Rh to N2 molecules. To gain a deeper understanding of the N2 activation process, we conducted low-temperature Fourier-transform infrared (LT-FTIR) experiments under Ar and N2 atmospheres, respectively (Fig. 6e). Under the Ar atmosphere, only a negative band at 2368 cm−1 that corresponds to the deduction of environmental CO2 can be detected. In contrast, an obvious vibration peak at 2248 cm−1 appears under the N2 atmosphere, suggesting that the N2 molecule chemisorbs on the catalyst via an end-on rather than side-on configuration to form a Rh–N2 complex. The LT-FTIR experiments clearly confirm the activation process of N2 on the Au NBP/Rh NDs. After identifying the Rh active sites, we further investigated the reaction pathways of N2 reduction on the Au NBP/tip-Rh ND catalyst by using DFT calculations. The reduction of N2 to NH3 can go through three classes of pathways including dissociative, distal, and alternating pathways.52 Since the dissociative pathway requires a large amount of energy to break the NN triple bond, the N2 reduction reaction is generally conducted at high temperatures and high pressures (e.g., the traditional Haber–Bosch process). We calculated the Gibbs free energy changes in the alternating and distal pathways on the Au NBP/tip-Rh ND catalyst. As displayed in Fig. 6f, the rate-determining steps in alternating and distal pathways are both the desorption of the first NH3 from the Au NBP/Rh NDs. The uphill energy is 0.67 eV for the alternating pathway and 0.50 eV for the distal pathway, revealing that the distal pathway is the most favourable path for N2 reduction on the Au NBP/tip-Rh NDs. In addition, the calculation results corroborate the fact that there is an absence of byproduct N2H4 in this system. It has been reported that the desorption of *NH2NH2 to obtain byproduct N2H4 is a highly endothermic step.53 As a competitive reaction, the further hydrogenation of *NH2NH2 on the Au NBP/tip-Rh NDs is exothermic, suggesting that NH3 rather than N2H4 is more readily generated in this photocatalytic system.
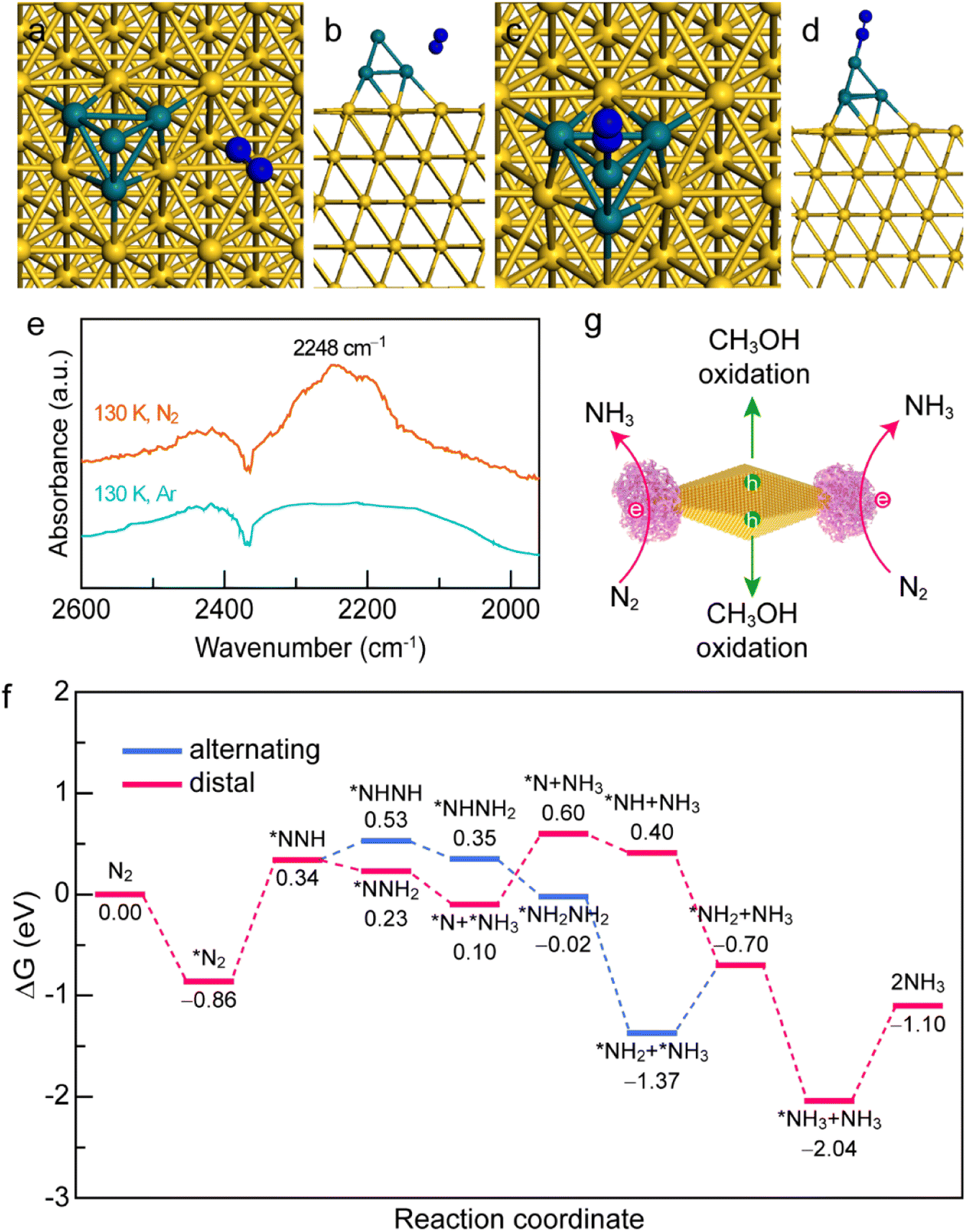 | ||
| Fig. 6 Mechanism for N2 photofixation on Au NBP/tip-NDs. (a and b) N2 adsorption on Au in the Au NBP/tip-Rh NDs (top view (a) and side view (b)). (c and d) N2 adsorption on Rh in the Au NBP/tip-Rh NDs (top view (c) and side view (d)). (e) LT-FTIR spectra of the Au NBP/tip-Rh NDs under Ar and N2 atmospheres, respectively. (f) Gibbs free-energy diagrams of the N2 photofixation on the Au NBP/tip-Rh NDs through alternative and distal pathways. (g) Schematics illustrating the charge carrier separation behaviour in the Au NBP/tip-Rh NDs. | ||
Based on the above experimental and theoretical results, a rational mechanism for the N2 photofixation on the Au NBP/tip-Rh NDs is proposed (Fig. 6g). The N2 molecule chemisorbs on the active sites on Rh via an end-on configuration to form a Rh–N2 complex. The strong interaction of N2 molecules with the Rh catalyst weakens the NN triple bond and activates the N2 molecules. Under plasmon resonance, hot electrons and holes are generated in Au NBPs. Hot electrons are transferred to the Rh catalyst for the reduction of N2 to NH3via the associative distal mechanism, while hot holes are consumed on the side surface of Au NBPs to close the photocatalytic cycle. The antenna–reactor system combined with plasmonic Au NBPs with desired catalytically active Rh nanocrystals efficiently harnesses the light harvesting and facilitates the energy transfer from the Au NBPs to the Rh catalyst for superior N2 photofixation. Moreover, the spatially separated architecture of the dumbbell-shaped nanostructures facilitates charge separation and promotes the catalytic performance.
Conclusions
In summary, we have developed a synthetic strategy to fabricate dumbbell-shaped Au/Rh nanostructures through the site-selective growth of Rh nanocrystals on two tips of Au NBPs. The 16-BAC molecules with a benzyl headgroup can generate a large steric hindrance at the middle of Au NBPs, facilitating the preferential nucleation of Rh at the tips and the formation of dumbbell-shaped architecture. The rational combination of plasmonic Au NBPs with desired catalytically active Rh nanocrystals to form an antenna–reactor system can not only efficiently harness the light harvesting for hot electron generation but also improve the catalytic activity toward N2 photofixation. More importantly, the dumbbell-shaped nanostructures featuring spatially separated architecture allow hot charge carriers to take part in the reduction and oxidation half-reactions at different active sites independently, greatly enhancing the charge separation and promoting the catalytic activity. The N2 photofixation activity of the dumbbell-shaped nanostructures is 8.0 and 2.0-fold higher compared with those of Rh nanocrystals and the core@shell nanostructures. This merit results from the rational design of the antenna–reactor system and the spatially separated architecture of the dumbbell-shaped nanostructures. Since there are also five tips at the middle of Au NBPs due to the fivefold rotational symmetry, the selective deposition of Rh at all seven tips is much more intriguing. Our future work should focus on the combination of hard templates with soft templates to fabricate 7 tip-coated Au NBPs. Above all, our study sheds new light on the precise manipulation of the active site spatial distribution in N2 photofixation and offers instructive guidance for the design and construction of antenna–reactor plasmonic photocatalysts.Data availability
The data that support the finding of this study are available in the main text and the ESI. †Author contributions
H. L. Jia and F. Li contributed equally to this work. H. L. Jia and C.-y. Zhang conceived and supervised the project. F. Li performed the experiments. Y. Y. Yang, M. X. Zhao, and J. Z. Li analysed the data and provided conceptual contributions. H. L. Jia and C.-y. Zhang wrote the paper. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 21735003) and the Natural Science Foundation of Shandong Province (No. ZR2020MB040).Notes and references
- M.-A. Légaré, G. Bélanger-Chabot, R. D. Dewhurst, E. Welz, I. Krummenacher, B. Engels and H. Braunschweig, Science, 2018, 359, 896–900 CrossRef PubMed.
- M. J. Chalkley, M. W. Drover and J. C. Peters, Chem. Rev., 2020, 120, 5582–5636 CrossRef CAS PubMed.
- Y. Ohki, K. Munakata, Y. Matsuoka, R. Hara, M. Kachi, K. Uchida, M. Tada, R. E. Cramer, W. M. C. Sameera, T. Takayama, Y. Sakai, S. Kuriyama, Y. Nishibayashi and K. Tanifuji, Nature, 2022, 607, 86–90 CrossRef CAS PubMed.
- J. P. Guo and P. Chen, Chem, 2017, 3, 709–714 CAS.
- C. Tang and S.-Z. Qiao, Chem. Soc. Rev., 2019, 48, 3166–3180 RSC.
- S.-L. Meng, X.-B. Li, C.-H. Tung and L.-Z. Wu, Chem, 2021, 7, 1431–1450 CAS.
- J. H. Yang, Y. Z. Guo, R. B. Jiang, F. Qin, H. Zhang, W. Z. Lu, J. F. Wang and J. C. Yu, J. Am. Chem. Soc., 2018, 140, 8497–8508 CrossRef CAS PubMed.
- C. Y. Hu, X. Chen, J. B. Jin, Y. Han, S. M. Chen, H. X. Ju, J. Cai, Y. R. Qiu, C. Gao, C. M. Wang, Z. M. Qi, R. Long, L. Song, Z. Liu and Y. J. Xiong, J. Am. Chem. Soc., 2019, 141, 7807–7814 CrossRef CAS PubMed.
- Y. X. Zhao, Y. F. Zhao, R. Shi, B. Wang, G. I. N. Waterhouse, L.-Z. Wu, C.-H. Tung and T. R. Zhang, Adv. Mater., 2019, 31, 1806482 CrossRef PubMed.
- H. Y. Bai, S. H. Lam, J. H. Yang, X. Z. Cheng, S. S. Li, R. B. Jiang, L. Shao and J. F. Wang, Adv. Mater., 2022, 34, 2104226 CrossRef CAS.
- H. L. Jia, M. X. Zhao, A. X. Du, Y. R. Dou and C.-y. Zhang, Chem. Sci., 2022, 13, 13060–13067 RSC.
- S. Z. Andersen, V. Čolić, S. Yang, J. A. Schwalbe, A. C. Nielander, J. M. McEnaney, K. Enemark-Rasmussen, J. G. Baker, A. R. Singh, B. A. Rohr, M. J. Statt, S. J. Blair, S. Mezzavilla, J. Kibsgaard, P. C. K. Vesborg, M. Cargnello, S. F. Bent, T. F. Jaramillo, I. E. L. Stephens, J. K. Nørskov and I. Chorkendorff, Nature, 2019, 570, 504–508 CrossRef CAS.
- H.-P. Jia and E. A. Quadrelli, Chem. Soc. Rev., 2014, 43, 547–564 RSC.
- L. N. Zhou, J. M. P. Martirez, J. Finzel, C. Zhang, D. F. Swearer, S. Tian, H. Robatjazi, M. H. Lou, L. L. Dong, L. Henderson, P. Christopher, E. A. Carter, P. Nordlander and N. J. Halas, Nat. Energy, 2020, 5, 61–70 CrossRef CAS.
- K. Sytwu, M. Vadail, F. Hayee, D. K. Angell, A. Dai, J. Dixon and J. A. Dionne, Science, 2021, 371, 280–283 CrossRef CAS PubMed.
- D. F. Swearer, H. Q. Zhao, L. N. Zhou, C. Zhang, H. Robatjazi, J. M. P. Martirez, C. M. Krauter, S. Yazdi, M. J. McClain, E. Ringe, E. A. Carter, P. Nordlander and N. J. Halas, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 8916–8920 CrossRef CAS PubMed.
- H. L. Jia, Y. Y. Yang, Y. R. Dou, F. Li, M. X. Zhao and C.-y. Zhang, Chem. Commun., 2022, 58, 1013–1016 RSC.
- S. Linic, P. Christopher and D. B. Ingram, Nat. Mater., 2011, 10, 911–921 CrossRef CAS.
- S. S. Li, H. Huang, L. Shao and J. F. Wang, ACS Nano, 2021, 15, 10759–10768 CrossRef CAS PubMed.
- S. Q. Luo, X. H. Ren, H. W. Lin, H. Song and J. H. Ye, Chem. Sci., 2021, 12, 5701–5719 RSC.
- T. H. Chow, N. N. Li, X. P. Bai, X. L. Zhuo, L. Shao and J. F. Wang, Acc. Chem. Res., 2019, 52, 2136–2146 CrossRef CAS PubMed.
- A. Sánchez-Iglesias, N. Winckelmans, T. Altantzis, S. Bals, M. Grzelczak and L. M. Liz-Marzán, J. Am. Chem. Soc., 2017, 139, 107–110 CrossRef.
- X. Z. Zhu, H. K. Yip, X. L. Zhuo, R. B. Jiang, J. L. Chen, X.-M. Zhu, Z. Yang and J. F. Wang, J. Am. Chem. Soc., 2017, 139, 13837–13846 CrossRef CAS PubMed.
- H. L. Jia, Y. Y. Yang, T. H. Chow, H. Zhang, X. Y. Liu, J. F. Wang and C.-y. Zhang, Adv. Funct. Mater., 2021, 31, 2101255 CrossRef CAS.
- X. Z. Zhu, J. Xu, H. Zhang, X. M. Cui, Y. Z. Guo, S. Cheng, C. X. Kan and J. F. Wang, Chem. Sci., 2020, 11, 3198–3207 RSC.
- K. D. Gilroy, A. Ruditskiy, H.-C. Peng, D. Qin and Y. N. Xia, Chem. Rev., 2016, 116, 10414–10472 CrossRef CAS PubMed.
- X. Z. Zhu, H. L. Jia, X.-M. Zhu, S. Cheng, X. L. Zhuo, F. Qin, Z. Yang and J. F. Wang, Adv. Funct. Mater., 2017, 27, 1700016 CrossRef.
- J. Guo, Y. Zhang, L. Shi, Y. F. Zhu, M. F. Mideksa, K. Hou, W. S. Zhao, D. W. Wang, M. T. Zhao, X. F. Zhang, J. W. Lv, J. Q. Zhang, X. L. Wang and Z. Y. Tang, J. Am. Chem. Soc., 2017, 139, 17964–17972 CrossRef CAS PubMed.
- S.-i. Naya, T. Kume, R. Akashi, M. Fujishima and H. Tada, J. Am. Chem. Soc., 2018, 140, 1251–1254 CrossRef CAS PubMed.
- X. Q. Yang, Y. Lu, Y. Liu, J. Wang, L. Shao and J. F. Wang, Small Struct., 2021, 2, 2100101 CrossRef CAS.
- W. J. Xu, J. Jia, T. Wang, C. Li, B. W. He, J. P. Zong, Y. W. Wang, H. J. Fan, H. X. Xu, Y. H. Feng and H. Y. Chen, Angew. Chem., Int. Ed., 2020, 59, 22246–22251 CrossRef CAS PubMed.
- S. Mubeen, J. Lee, N. Singh, S. Krämer, G. D. Stucky and M. Moskovits, Nat. Nanotechnol., 2013, 8, 247–251 CrossRef CAS.
- B. H. Wu, D. Y. Liu, S. Mubeen, T. T. Chuong, M. Moskovits and G. D. Stucky, J. Am. Chem. Soc., 2016, 138, 1114–1117 CrossRef CAS PubMed.
- H. L. Jia, A. X. Du, H. Zhang, J. H. Yang, R. B. Jiang, J. F. Wang and C.-y. Zhang, J. Am. Chem. Soc., 2019, 141, 5083–5086 CrossRef CAS.
- H. L. Jia, F. Li, T. H. Chow, X. Y. Liu, H. Zhang, Y. Lu, J. F. Wang and C.-y. Zhang, Nano Lett., 2022, 22, 7268–7274 CrossRef CAS PubMed.
- X. Q. Yang, Y. Liu, S. H. Lam, J. Wang, S. Z. Wen, C. Yam, L. Shao and J. F. Wang, Nano Lett., 2021, 21, 8205–8212 CrossRef CAS PubMed.
- M. T. Chen, Z. J. Ye, L. Wei, J. Yuan and L. H. Xiao, J. Am. Chem. Soc., 2022, 144, 12842–12849 CrossRef CAS.
- G. L. He, Y. H. Lai, Y. Z. Guo, H. Yin, B. B. Chang, M. Liu, S. R. Zhang, B. C. Yang and J. F. Wang, ACS Appl. Mater. Interfaces, 2022, 14, 53724–53735 CrossRef CAS.
- H. Zhang, S. H. Lam, Y. Z. Guo, J. H. Yang, Y. Lu, L. Shao, B. C. Yang, L. H. Xiao and J. F. Wang, ACS Appl. Mater. Interfaces, 2021, 13, 51855–51866 CrossRef CAS PubMed.
- B. T. Sneed, C.-H. Kuo, C. N. Brodsky and C.-K. Tsung, J. Am. Chem. Soc., 2012, 134, 18417–18426 CrossRef CAS PubMed.
- C. Y. Yang, B. L. Huang, S. X. Bai, Y. G. Feng, Q. Shao and X. Q. Huang, Adv. Mater., 2020, 32, 2001267 CrossRef CAS PubMed.
- Y.-Q. Kang, Q. Xue, Y. Zhao, X.-F. Li, P.-J. Jin and Y. Chen, Small, 2018, 14, 1801239 CrossRef PubMed.
- C. Zhang, H. Q. Zhao, L. N. Zhou, A. E. Schlather, L. L. Dong, M. J. McClain, D. F. Swearer, P. Nordlander and N. J. Halas, Nano Lett., 2016, 16, 6677–6682 CrossRef CAS.
- F. Wang, C. H. Li, H. J. Chen, R. B. Jiang, L.-D. Sun, Q. Li, J. F. Wang, J. C. Yu and C.-H. Yan, J. Am. Chem. Soc., 2013, 135, 5588–5601 CrossRef CAS PubMed.
- L. N. Zhou, D. F. Swearer, C. Zhang, H. Robatjazi, H. Q. Zhao, L. Henderson, L. L. Dong, P. Christopher, E. A. Carter, P. Nordlander and N. J. Halas, Science, 2018, 362, 69–72 CrossRef CAS PubMed.
- S. K. Cushing, J. T. Li, F. K. Meng, T. R. Senty, S. Suri, M. J. Zhi, M. Li, A. D. Bristow and N. Q. Wu, J. Am. Chem. Soc., 2012, 134, 15033–15041 CrossRef CAS PubMed.
- J. T. Li, S. K. Cushing, F. K. Meng, T. R. Senty, A. D. Bristow and N. Q. Wu, Nat. Photonics, 2015, 9, 601–607 CrossRef CAS.
- H. Robatjazi, H. Q. Zhao, D. F. Swearer, N. J. Hogan, L. N. Zhou, A. Alabastri, M. J. McClain, P. Nordlander and N. J. Halas, Nat. Commun., 2016, 8, 27 CrossRef.
- P. Christopher, H. L. Xin, A. Marimuthu and S. Linic, Nat. Mater., 2012, 11, 1044–1050 CrossRef CAS PubMed.
- P. Christopher, H. L. Xin and S. Linic, Nat. Chem., 2011, 3, 467–472 CrossRef CAS PubMed.
- S. Mukherjee, F. Libisch, N. Large, O. Neumann, L. V. Brown, J. Cheng, J. B. Lassiter, E. A. Carter, P. Nordlander and N. J. Halas, Nano Lett., 2013, 13, 240–247 CrossRef CAS PubMed.
- J. H. Yang, Y. Z. Guo, W. Z. Lu, R. B. Jiang and J. F. Wang, Adv. Mater., 2018, 30, 1802227 CrossRef PubMed.
- H. Yin, J. W. Hu, C. H. Fang, Y. Y. Wang, L. X. Ma, N. Zhang, S. R. Zhang, R. B. Jiang and J. F. Wang, Nano Res., 2023, 16, 360–370 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc00081h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |