
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReshaping aromatic frameworks: expansion of aromatic system drives metallabenzenoids to metallapentalenes†
Qian
Li
 a,
Jiawei
Fei
a,
Kaidong
Ruan
a,
Yuhui
Hua
b,
Dafa
Chen
b,
Ming
Luo
*b and
Haiping
Xia
*ab
a,
Jiawei
Fei
a,
Kaidong
Ruan
a,
Yuhui
Hua
b,
Dafa
Chen
b,
Ming
Luo
*b and
Haiping
Xia
*ab
aState Key Laboratory of Physical Chemistry of Solid Surfaces, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, China
bShenzhen Grubbs Institute, Department of Chemistry, Southern University of Science and Technology, Shenzhen 518055, China. E-mail: luom3@sustech.edu.cn; xiahp@sustech.edu.cn
First published on 24th April 2023
Abstract
Reshaping an aromatic framework to generate other skeletons is a challenging issue due to the stabilization energy of aromaticity. Such reconfigurations of aromatics commonly generate non-aromatic products and hardly ever reshape to a different aromatic framework. Herein, we present the transformation of metallaindenols to metallapentalenes and metallaindenes in divergent pathways, converting one aromatic framework to another with an extension of the conjugation framework. The mechanistic study of this transformation shows that phosphorus ligands play different roles in the divergent processes. Further theoretical studies indicate that the expansion of the aromatic system is the driving force promoting this skeletal rearrangement. Our findings offer a new concept and strategy to reshape and construct aromatic compounds.
Introduction
Aromaticity is a core fundamental concept in chemistry which has gained great interest from experimental and theoretical chemists.1 Due to the aromatic stabilization energy, aromatic molecular structures normally exhibit a higher stability than other geometric frameworks.2 In general, reconfiguration of benzene and heterobenzenes results in non-aromatic products. For example, the ring contraction of benzene by ultraviolet light,3 a trinuclear titanium cluster4 or rare-earth metal complexes5 led to the corresponding cyclopentene derivatives, and the products of the photoinduced contraction reactions of heterobenzenes are analogous to the results with benzene.6 On the other hand, reshaping one aromatic framework to another skeleton with maintenance of aromaticity has been rarely reported. A prominent example is the rearrangement between azulene and naphthalene. Azulene can change to the thermodynamically more stable naphthalene at high temperatures.7 Recently, the reverse rearrangement from the naphthalene unit to an azulene moiety during the synthesis of nanographenes was reported.8 Collectively, reshaping aromatic frameworks regularly results in non-aromatic products and seldom affords another aromatic framework.Metallaaromatics represent a unique member of an aromatic family in which a transition metal fragment is involved in an aromatic ring.9 To date, although various metallaaromatics including metallabenzenes,10 metallabenzynes,11 metallapentalenes,12 metallapentalynes,13 dianion metalloles,14 heterometallaaromatics,15 and spiro metalloles16 have been found, the novel structures and reactivities of metallaaromatics are still of interest.17 For recent progress on metallabenzenes, iridabenzenes and rhodabenzenes have been prepared17c,d,f,h and the first structurally characterised Dewar metallabenzenes were isolated17g after spectroscopic observation.10d The ring contraction reactions of metallabenzenes, such as iridabenzene, are similar to the contraction of benzene and heterobenzenes, and give the metallacyclopentadienes with loss of the original aromaticity.18 Inspired by the reshaping of the organic aromatic fused rings that maintain the aromaticity and the ability of the transition metals to possess a number of metal–carbon bonds,19 we hypothesised that the fused metallaaromatics could expand the aromatic system when reshaped into other metallacycles.
Herein, we describe our design and synthesis of the first metallaindenol assembled from one nine-atom unsaturated carbon chain with OsCl2(PR3)3 (R = Ph, p-tolyl). Ring contraction of the metallaindenols gives metallapentalenes. In contrast to the transformations of metallabenzynes to metallabenzenes20 and metallapentalynes to metallapentalenes12,13a,21 both of which retain the same skeletons, this rearrangement represents the first skeletal reshaping from one metallaaromatic framework to another (Fig. 1). With the aid of experimental observations and theoretical calculations, the reaction mechanism, and the aromaticity of these metallaaromatics together with the photophysical properties during the transformation are systematically studied.
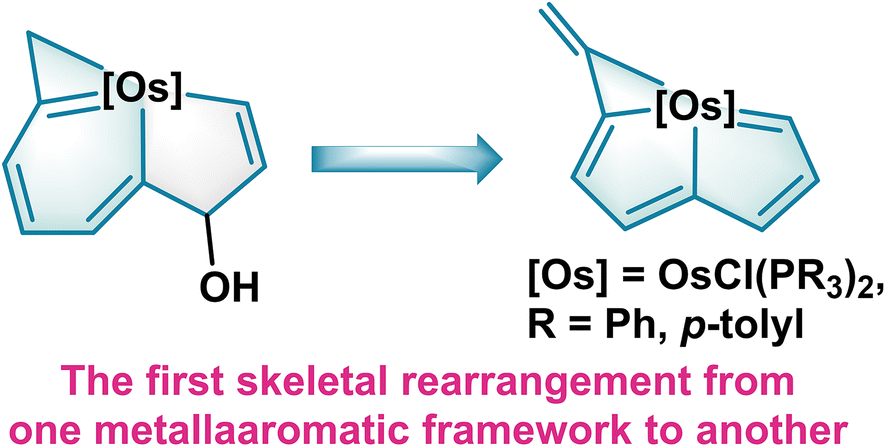 | ||
| Fig. 1 Reshaping of metallabenzenoids to metallapentalenes driven by expansion of the aromatic system. | ||
Results and discussion
Synthesis and characterization of metallaindenols (1 and 2) and their reactions with acids
We have previously developed a simple strategy to construct the metal bridgehead polycyclic framework via an unsaturated carbon chain chelated with a transition metal fragment.13b Encouraged by these unprecedented findings, we next wanted to investigate the assembly behaviours of other novel unsaturated carbon chains with the metal complexes, to construct novel aromatic organometallic frameworks. We designed and synthesized a carbon chain (L) embracing two alkynyl groups and one terminal allene group connected to each other by a secondary alcohol.The reaction of OsCl2(PR3)3 (R = Ph, p-tolyl), the carbon chain L, and HCl·Et2O at room temperature (RT) in dichloromethane (DCM) gave complexes 1 (R = Ph) and 2 (R = p-tolyl) (Fig. 2A). The structure of 1 was determined by single-crystal X-ray diffraction, which shows nine carbon atoms are chelated with the osmium fragment, to give the osmaindenol framework (Fig. 2B). The bond lengths in the six-membered ring (6MR) (C4–C5 1.384 Å, C5–C6 1.380 Å, C6–C7 1.378 Å, C7–C8 1.362 Å, Os1–C4 2.044 Å, and Os1–C8 2.064 Å) are similar to those found in osmabenzenes.22 The five-membered ring (5 MR) has an sp3 hybridized C3 bearing an hydroxyl group which is confirmed by the bond distances of three single bonds (C3–O1 1.460 Å, C2–C3 1.484 Å, and C3–C4 1.552 Å). The bond length of Os1–C9 is 2.209 Å, which is typical of an Os–C single bond, together with C8–C9 (1.338 Å), indicating the three-membered ring (3MR) an osmacyclopropene moiety (Fig. 2C).22b,23
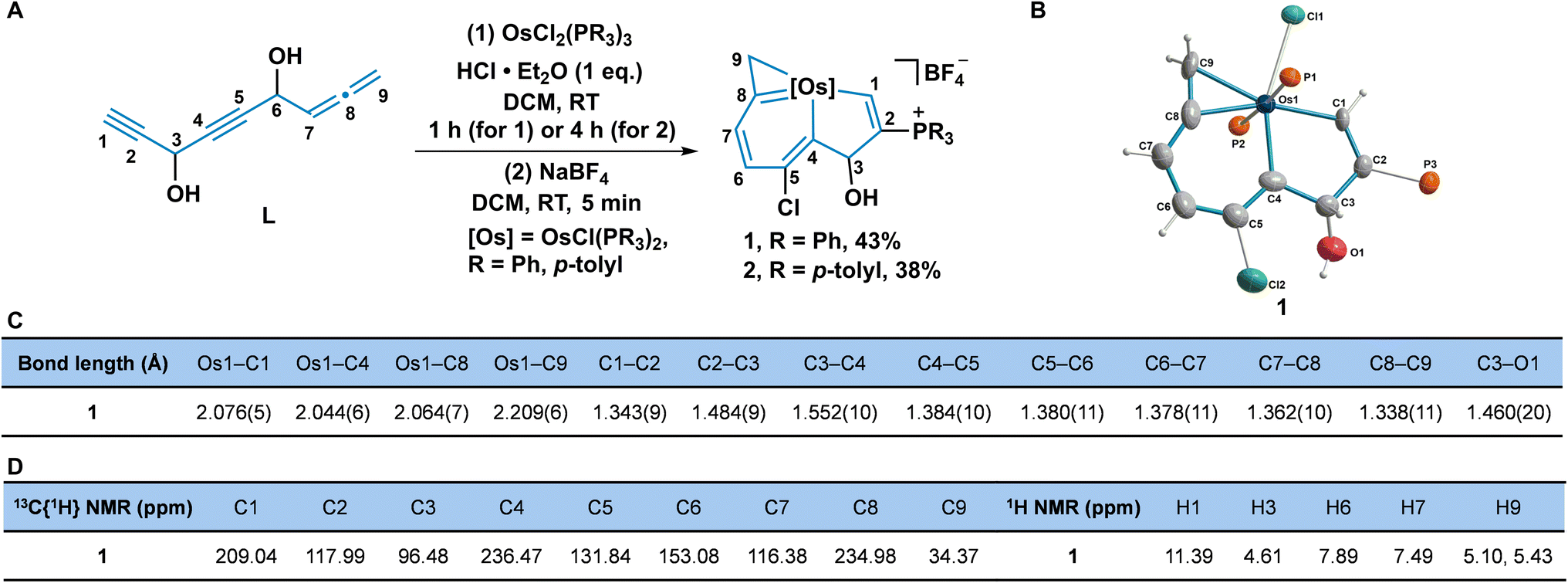 | ||
| Fig. 2 (A) Synthesis of osmaindenols 1 and 2. (B) The solid-state structure for the cation of 1. The phenyl groups in the PPh3 moieties have been removed for clarity. (C) Selected bond lengths (Å) for 1. (D) Selected NMR chemical shifts (ppm) for 1. | ||
The structure of the osmaindenol 1 was also verified by nuclear magnetic resonance (NMR) spectroscopy and high-resolution mass spectrometry (HRMS). In the 31P{1H} NMR spectrum, CPPh3 appears at 13.02 ppm (singlet) and OsPPh3 appears as a doublet at −14.18 and −20.22 ppm as a result of the nonequivalent circumstances caused by the C3-hydroxyl group. The large coupling constant (JP–P = 284.4 Hz) of the trans-phosphine ligands is similar to that in a reported alkynol coordinated osmium complex.22a The protons in the osmabenzene moiety have peaks at 7.89 (H6) and 7.49 (H7) ppm in the aromatic region. The signal of H1 appears as a doublet at 11.39 ppm (d, JP–H = 23.3 Hz) because of its coupling to phosphorus. The chemical shifts of C1 (209.04 ppm), C4 (236.47 ppm), and C8 (234.98 ppm) indicate the covalent bonds between osmium and carbons, and are consistent with the crystal structure. The signals from H9 (5.10 and 5.43 ppm) and C9 (34.37 ppm) agree with those observed in osmacyclopropenes (Fig. 2D).22b,23 The crystal and NMR data suggest that 1 can be described by the resonance structures 1A–1D (Scheme S1, ESI†). Osmaindenol 2 was characterized by NMR and HRMS as well, which indicate it has a similar structure to complex 1. A possible mechanism for the formation of osmaindenol involves the assembly of the fused metallacycle from the carbon chain L and the osmium fragment accompanied by a nucleophilic addition of the chloride and PPh3 (Scheme S2, ESI†).
Up to now, the synthesis and reactivity of the metal-bridged indene derivatives were rarely surveyed.24 Because the addition of acid could remove the hydroxyl group on C3 and produce a conjugated metallacycle, we further investigated the reactivities of osmaindenols (1 and 2) with acid. When excess HCl·Et2O and PPh3 were added into the solution of 1, elimination of the hydroxyl group occurred. Surprisingly, products 3 and 4 were isolated with 52% and 25% yields, respectively (Fig. 3A). The X-ray analysis reveals that 3 has a structural osmapentalene fused with an osmacyclopropenylidene unit. An osmaindenol has thus been reshaped to an osmapentalene by a ring contraction reaction (Fig. 3B). The coplanar structure of Os1 and C1–C7 is supported by their small mean deviation of 0.030 Å from the least-squares plane. The bond lengths of Os1–C1 (2.054 Å), Os1–C4 (2.077 Å), Os1–C7 (2.009 Å), and Os1–C8 (2.156 Å) are comparable to those of the osmapentalene derivative fused with an osmacyclopropenylidene moiety.13b The delocalized fused metallacycles are confirmed by C–C bond lengths of 1.352–1.417 Å which are within the range of C–C single and double bonds (Fig. 3D). The NMR spectra are consistent with the crystal structure. Three phosphorus signals are observed at 17.53 (CPPh3), 12.07 (CPPh3), and −24.17 ppm (OsPPh3). The signals for H1 (14.18 ppm) and H3 (8.81 ppm) are more downfield than those of 1, indicating an aromatic osmapentadiene moiety25 (Fig. 3E). The observed carbon signals in the fused metallacycles agree with those of the similar structural osmapentalenes.13b It is worth mentioning that the transformation of 1 to 3 represents a new approach to the preparation of metallapentalene derivatives.
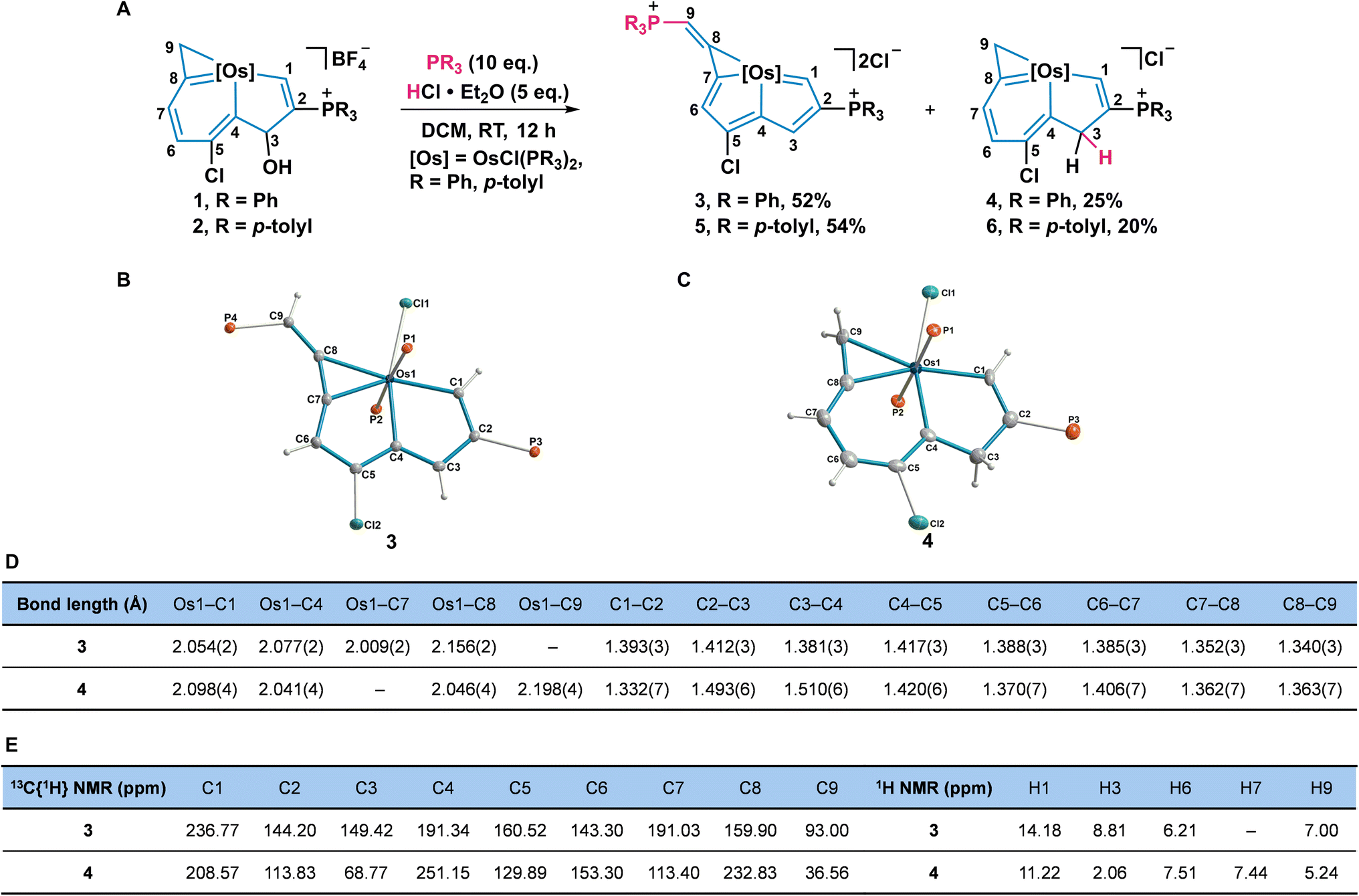 | ||
| Fig. 3 Acid-mediated transformation of metallaindenols to metallapentalenes and metallaindenes. (A) Synthesis of 3 to 6. (B and C). The solid-state structures for cations of 3 (B) and 4 (C). The phenyl groups in the PPh3 moieties have been removed for clarity. (D) Selected bond lengths (Å) for 3 and 4. (E) Selected NMR chemical shifts (ppm) for 3 and 4. | ||
The osmaindene (4) was formed by an electrophilic addition reaction and its structure is similar to that of 1 (Fig. 3C). The bond lengths of Os1–C1 (2.098 Å), Os1–C4 (2.041 Å), Os1–C8 (2.046 Å), Os1–C9 (2.198 Å), and the C–C bonds in the fused rings of 4 are similar to those observed in 1. The C2–C3 (1.493 Å) and C3–C4 (1.510 Å) bonds are typical C–C single bonds, indicating that C3 retains its sp3 hybridization. The signals for CPPh3 and OsPPh3 are assigned to 12.75 and −15.32 ppm, respectively. The chemical shifts observed in the 3MR and 6MR are comparable to those in 1. The signals of H1 (11.22 ppm) and C1 (208.57 ppm) in 4 are analogous to those observed in 1 as well. The upfield signals of H3 (2.06 ppm) and C3 (68.77 ppm) of 4 indicate that C3 maintains its sp3 hybridized state (Fig. 3D and E). Compared with the reported metallaindenes which are comprised of conjugated frameworks,24 complex 4 contains a sp3 hybridized carbon in 5MR and is more closely related to the classical 1H-indene. Furthermore, the transformation of osmaindenol 2 to osmapentalene 5 and osmaindene 6 was realized under the same conditions.
Mechanistic study
To further understand the mechanism of this reaction, density functional theory (DFT) calculations were employed for the formation of 3 (Fig. 4A). The initial protonation of the OH group with acid, and subsequent elimination of the OH group gave the intermediate (A) which has an exergonic energy of 13.8 kcal mol−1. Then excess PPh3 as a nucleophile, attacked at the C9 of A and the ring opening of osmacyclopropene occurred to form the intermediate (B) with an exergonic energy of 16.8 kcal mol−1.26 The elimination of a proton from C9 together with the ring opening of osmacyclohexadiene results in an intermediate (C) which contains a negatively charged metal centre and a coordinated allene group. Next, the reaction of the negatively charged metal centre with acid produces the intermediate (D) with a metal hydride and finally, the metal-hydride complex D gives the product 3 and H2 with a large exothermicity (38.5 kcal mol−1).27 The whole exothermicity for formation of 3 is 37.9 kcal mol−1. The in situ NMR spectra were also employed to monitor the reaction process in which 1 gradually transformed 3 in 12 h (Fig. S1 and S2, ESI†). In addition, gas chromatography (GC) was employed to examine the H2 product in this process in which H2 gas from an in situ reaction was detected at 0.58 min (Fig. 4B).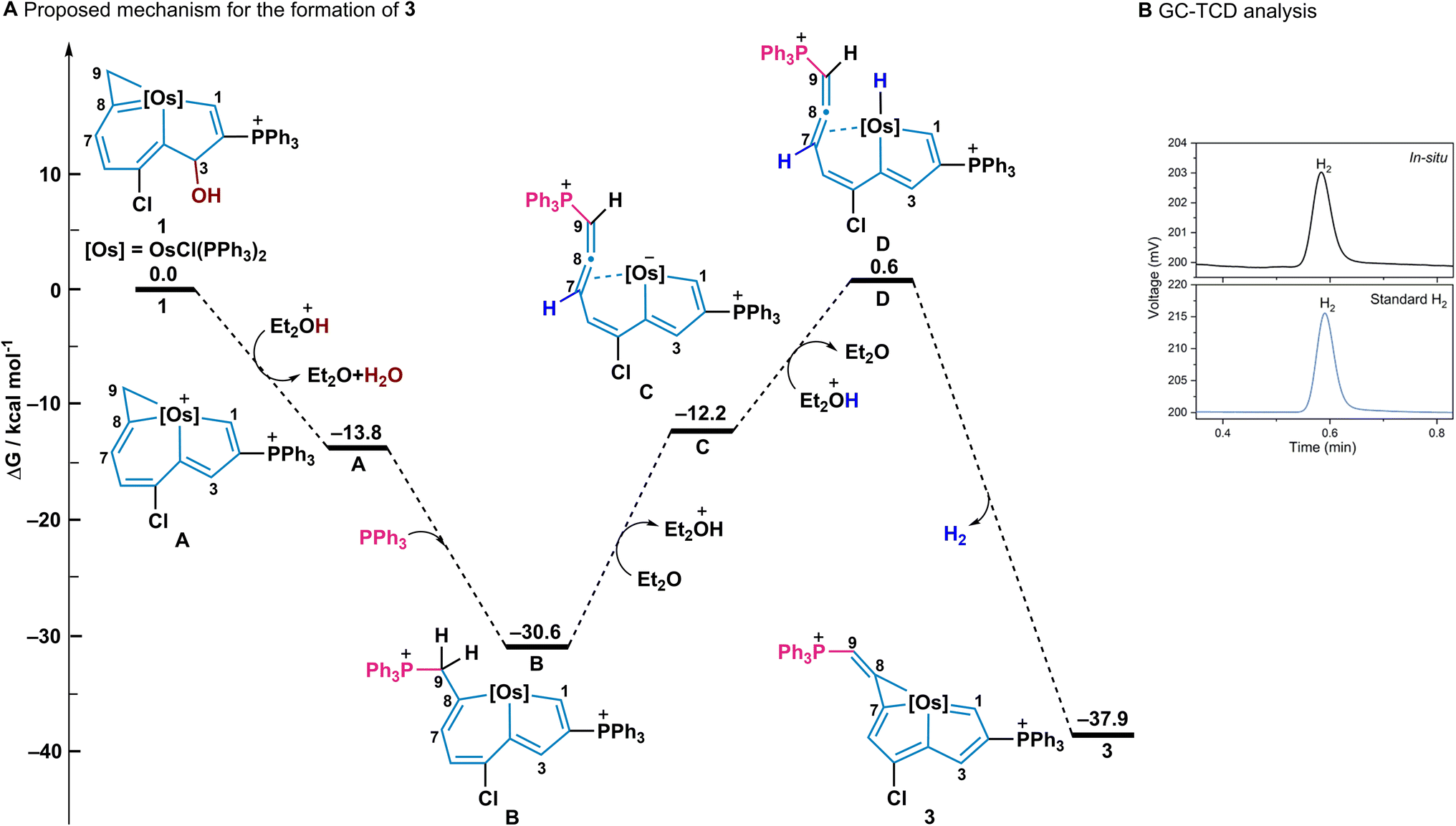 | ||
| Fig. 4 (A) A DFT calculated mechanism for the formation of 3. (B) The GC chromatograms of the in situ atmosphere in the formation of 3, and the standard gas of H2 obtained from thermal conductivity detector (TCD). Carrier gas: nitrogen. The peak of the in situ atmosphere was identified as H2 (0.58 min). | ||
In addition, a different pathway for forming osmaindene 4via an intermediate A was proposed, assisted by the DFT calculations and an isotope labelling experiment (Fig. 5). Excess PPh3 is initially involved as a reducing agent28 and an 18-electron intermediate (E) and triphenylphosphine oxide adduct are produced. The inset box in Fig. 5A illustrates the process in the conversion of A to E reported in a previous paper where oxygenation of PPh3 by a metal complex via electron transfer occurs.28b Electron transfer from A to PPh3 produces E and  and this is followed by the nucleophilic attack of H2O to give
and this is followed by the nucleophilic attack of H2O to give 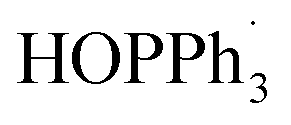 which was then disproportionated to OPPh3, PPh3, and H2O. The electrophilic addition of acid to E generates product 4, which has an exergonic energy of 34.1 kcal mol−1. It should be noted, that the umpolung relay which has been used in organic synthesis29 was discovered when 1 has an electrophilic addition on the C3 position in the formation of 4, which can be attributed to the positively charged osmium centre being reduced to a negatively charged centre by PPh3. To confirm the role of PPh3 in this reaction, we reduced the amount of PPh3 to one equivalent. The in situ NMR results showed that only complex 4 was formed in 2 h, together with a triphenylphosphine oxide adduct whose signal was detected at 38.62 ppm in the 31P{1H} NMR spectrum (Fig. S4, ESI†). This is because the dynamic adduct HCl·PPh3 was first formed when one equivalent of PPh3 was used, and as a result, the nucleophilicity of PPh3 was diminished and the transformation then tends to produce 4 (Fig. 5B, left equation). The amounts of acid in this reaction were also screened, three equivalents of acid promote the complete conversion and five equivalents of acid make the highest yield of 3 (Table S1, ESI†). An isotope labelling experiment was performed to shed light on the mechanism of the formation of 4 (Fig. 5B, right equation). The NMR results showed that deuterium was observed not only on the expected C3 position but also on C9, indicating a proton exchange of the C9H2 entity due to their acidity (Fig. S5 and S7, ESI†).30
which was then disproportionated to OPPh3, PPh3, and H2O. The electrophilic addition of acid to E generates product 4, which has an exergonic energy of 34.1 kcal mol−1. It should be noted, that the umpolung relay which has been used in organic synthesis29 was discovered when 1 has an electrophilic addition on the C3 position in the formation of 4, which can be attributed to the positively charged osmium centre being reduced to a negatively charged centre by PPh3. To confirm the role of PPh3 in this reaction, we reduced the amount of PPh3 to one equivalent. The in situ NMR results showed that only complex 4 was formed in 2 h, together with a triphenylphosphine oxide adduct whose signal was detected at 38.62 ppm in the 31P{1H} NMR spectrum (Fig. S4, ESI†). This is because the dynamic adduct HCl·PPh3 was first formed when one equivalent of PPh3 was used, and as a result, the nucleophilicity of PPh3 was diminished and the transformation then tends to produce 4 (Fig. 5B, left equation). The amounts of acid in this reaction were also screened, three equivalents of acid promote the complete conversion and five equivalents of acid make the highest yield of 3 (Table S1, ESI†). An isotope labelling experiment was performed to shed light on the mechanism of the formation of 4 (Fig. 5B, right equation). The NMR results showed that deuterium was observed not only on the expected C3 position but also on C9, indicating a proton exchange of the C9H2 entity due to their acidity (Fig. S5 and S7, ESI†).30
 | ||
| Fig. 5 (A) A DFT calculated mechanism for the formation of 4 (the inset box illustrates the process in the conversion of A to E according to previous research28b). (B) Reactions of 1 and 2 with one equivalent of PR3 (R = Ph, p-tolyl) and HCl·Et2O/DCl·D2O. | ||
The intermediates A and E apparently exhibit interesting conjugated systems, but attempts to isolate them from this reaction failed. In organic systems, the acid-induced indenol derivatives can generate 8π-electron antiaromatic indenyl cation intermediates, which further produce the corresponding SN1 substitution products.31 In sharp contrast, when metallaindenols react with acid, the metallaindenols are transformed to metallapentalenes driven by the expansion of the aromatic system, and a skeletal rearrangement is observed in the organometallic system.
Theoretical studies of aromaticity
To clarify the alteration of aromaticity in the transformation of 1 to 3 and 4, nucleus independent chemical shift (NICS) values32 were calculated based on simplified model compounds 1′, 3′, and 4′, in which the PPh3 ligands were replaced by PH3 groups (Fig. 6A). In general, the more negative NICS values indicate the more aromatic rings. The negative NICS(1)zz values of the conjugated 6MR of 1′ (−11.1 ppm) and 4′ (−7.8 ppm) imply the aromatic nature of the osmabenzene units in the fused frameworks. In comparison, the two fused 5MRs of 3′ show that NICS(1)zz has more negative values (−15.7 and −16.5 ppm), revealing the aromaticity of 3′. At the same time, the NICS(1)zz grids demonstrate that the inner cavity area of the osmabenzene units in 1′ and 4′, and the osmacyclopentadienes in 3′ is shielded (green to red, negative values). All the 3MRs of 1′, 3′, and 4′ show negative NICS(1)zz values (−24.7 ppm for 1′, −32.5 ppm for 3′, and −25.2 ppm for 4′) and indicate aromaticity.22b,33 The anisotropy of the induced current density (ACID)34 analysis which has proved to be a versatile tool to describe aromaticity also supported the aromaticity changes in these complexes (Fig. 6B). Distinct clockwise circulations were observed in the osmabenzene units of 1′ and 4′, the two fused 5MRs of 3′, and the 3MRs of 1′, 3′, and 4′, suggesting they had aromatic characteristics. Isomerization stabilization energy (ISE) equations were used to evaluate the aromatic stabilization energies of 1-Me, 3-Me, and 4-Me, indicating that they are all aromatic frameworks with the ISE of 3-Me being slightly larger than those of 1-Me and 4-Me (Fig. 6C). Collectively, both NICS(1)zz values and NICS(1)zz grids indicate that ring A in 3′ is more aromatic than the corresponding ring in 1′ or 4′. In particular, ring B in 3′ was converted from a non-aromatic unit to an aromatic ring. In addition, similar ISE results of 2-Me, 5-Me, and 6-Me are found as well, indicating their aromatic nature (Fig. S9, ESI†). Thus, these theoretical studies of aromaticity indicate that the expansion of aromatic systems plays a crucial role for driving the rearrangement of two typical metallaaromatics, i.e., achieving metallapentalenes from metallabenzenoids.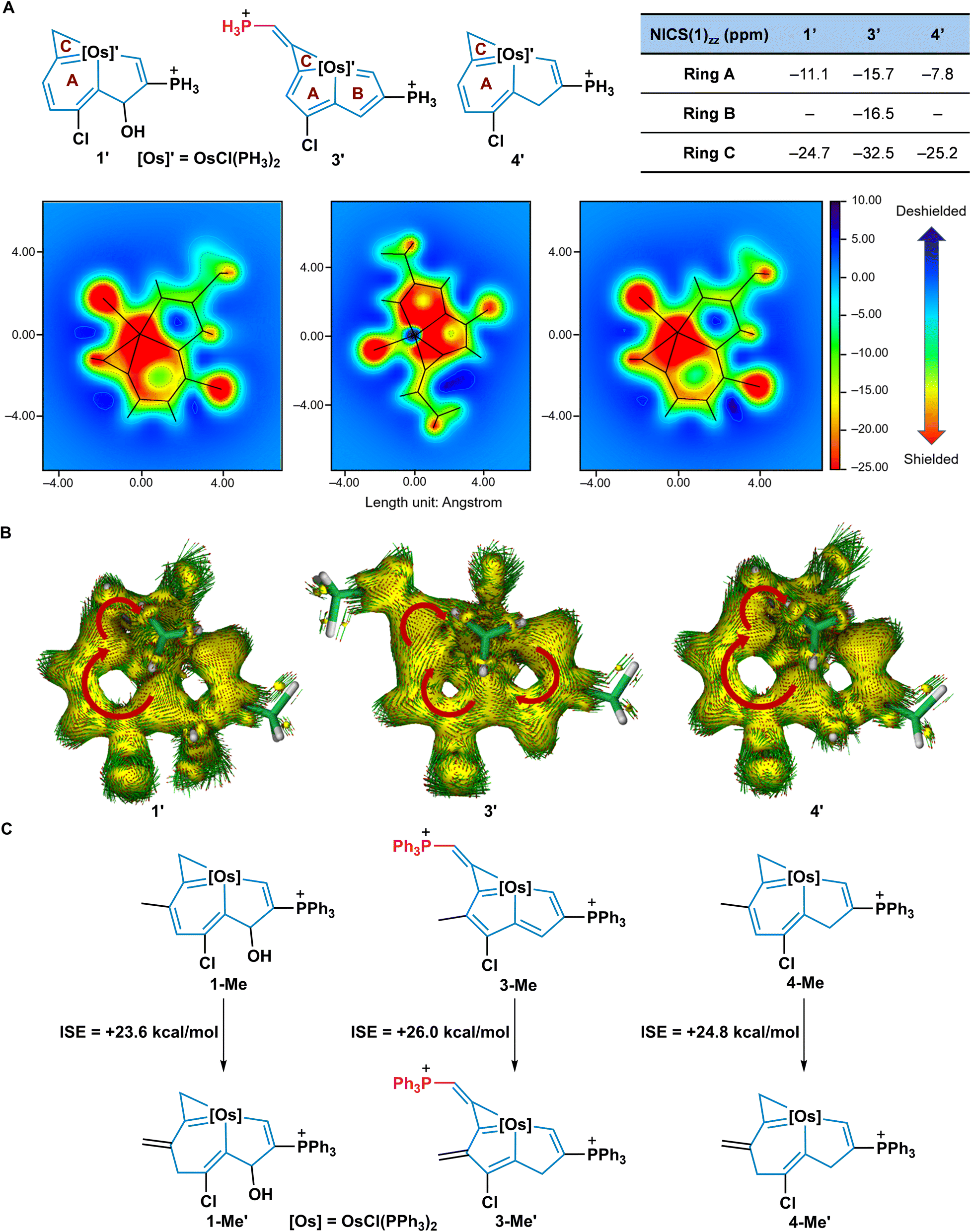 | ||
Fig. 6 Aromaticity evaluation. (A) NICS(1)zz values and NICS(1)zz grids for model complexes 1′, 3′, and 4′, NICS(1)zz were calculated at 1.0 Å above the ring centres. Each grid is equal to the fused ring with a 0.01 Å resolution and 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 points. A fixed colour scale (−25.0 to +10.0 ppm) gives a visualized comparison. Projections of the framework are provided on the maps and linked by lines. (B) The ACID plots of 1′ (left), 3′ (middle), and 4′ (right). The isosurface value of the ACID plots is 0.030 a.u. The magnetic field vector is orthogonal in relation to the ring plane (the red arrows represent the corresponding diatropic ring currents). (C) The isomerization stabilization energy (ISE) for 1-Me, 3-Me, and 4-Me. 000 points. A fixed colour scale (−25.0 to +10.0 ppm) gives a visualized comparison. Projections of the framework are provided on the maps and linked by lines. (B) The ACID plots of 1′ (left), 3′ (middle), and 4′ (right). The isosurface value of the ACID plots is 0.030 a.u. The magnetic field vector is orthogonal in relation to the ring plane (the red arrows represent the corresponding diatropic ring currents). (C) The isomerization stabilization energy (ISE) for 1-Me, 3-Me, and 4-Me. | ||
Photophysical properties of 1, 3, and 4
The electronic absorption spectra converge with the expansion of the aromatic system (Fig. 7). Compared with the absorption spectra of complexes 1 and 4, complex 3 displays two peaks at 511 nm (logε = 3.91, ε: molar extinction coefficient in L mol−1 cm−1) and 408 nm (logε = 3.67) in the visible region because of its extended conjugation framework. Time-dependent density functional theory (TD-DFT) (B3LYP/def2-TZVP) calculations show that the absorption bands for 3 are characterized as HOMO−1 → LUMO and HOMO−2 → LUMO transitions, respectively, (Table S2, ESI†). The absorption spectra together with calculations of complexes 2, 5, and 6 are also investigated, and the results were consistent with the expansion of the aromatic system (Fig. S10 and Table S2, ESI†).
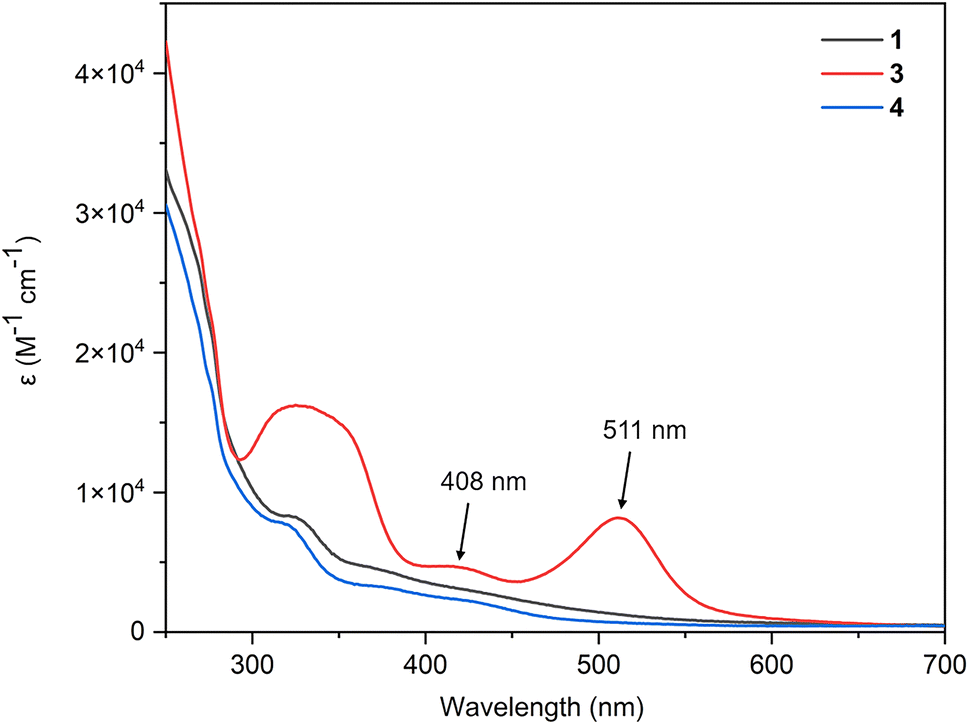 | ||
| Fig. 7 The UV-Vis absorption spectra for complexes 1, 3, and 4 in DCM (1.0 × 10−5 M, RT). | ||
Conclusion
To conclude, this unusual ring contraction reshapes metallabenzenoids to metallapentalenes by the expansion of an aromatic system, and exemplify a skeletal rearrangement from one aromatic framework to another in an organometallic system. Combined with new structures and reaction types, our results will support further advances in metallaaromatics and promote the development of methodology of unsaturated carbon chains with organometallic reagents. We anticipate that these findings could potentially result in a new way for further studying reshaping and construction of other aromatic compounds.Data availability
The validation experiments, experimental procedures, characterization, and computational details of this manuscript can be found in the ESI.† Crystallographic data has been deposited at the Cambridge Crystallographic Data Centre (CCDC) under accession numbers 2190320, 2190323, and 2190338.Author contributions
QL, ML, and HX designed this project. QL performed the experiments. JF solved all the X-ray structures. KR, YH, and ML designed and performed the theoretical calculations. QL, JF, DC, ML, and HX analyzed the data and prepared the paper. All of the authors discussed the results and contributed to the preparation of the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Science Foundation of China (Grant No. 21931002, 22071098, 92156021, and 22101115), the Shenzhen Science and Technology Innovation Committee (Grant No. JCYJ20200109140812302 and JCYJ20210324105013035), the Guangdong Provincial Key Laboratory of Catalysis (Grant No. 2020B121201002), and the Introduction of Major Talent Projects in Guangdong Province (Grant No. 2019CX01C079). We gratefully acknowledge Prof. Liu Leo Liu and Dr Chaopeng Hu at Sustech for their assistance with the mechanistic study, and Prof. Hong Zhang at XMU for his constructive comments.Notes and references
- (a) J. Mortier, Arene Chemistry: Reaction Mechanisms and Methods for Aromatic Compounds, John Wiley & Sons Ltd, New Jersey, 2016 Search PubMed; (b) I. Fernandez, Aromaticity: Modern Computational Methods and Applications, Elsevier, 2021 Search PubMed.
- S. W. Slayden and J. F. Liebman, Chem. Rev., 2001, 101, 1541–1566 CrossRef CAS PubMed.
- (a) L. Kaplan, J. S. Ritscher and K. E. Wilzbach, J. Am. Chem. Soc., 1966, 88, 2881–2882 CrossRef CAS; (b) T. Slanina, R. Ayub, J. Toldo, J. Sundell, W. Rabten, M. Nicaso, I. Alabugin, I. F. Galván, A. K. Gupta, R. Lindh, A. Orthaber, R. J. Lewis, G. Grönberg, J. Bergman and H. Ottosson, J. Am. Chem. Soc., 2020, 142, 10942–10954 CrossRef CAS PubMed.
- (a) S. Hu, T. Shima and Z. Hou, Nature, 2014, 512, 413–415 CrossRef CAS PubMed; (b) X. Kang, G. Luo, L. Luo, S. Hu, Y. Luo and Z. Hou, J. Am. Chem. Soc., 2016, 138, 11550–11559 CrossRef CAS PubMed.
- M. Zhu, Z. Chai, Z.-J. Lv, T. Li, W. Liu, J. Wei and W.-X. Zhang, J. Am. Chem. Soc., 2023, 145, 6633–6638 CrossRef CAS PubMed.
- (a) L. Kaplan, J. W. Pavlik and K. E. Wilzbach, J. Am. Chem. Soc., 1972, 94, 3283–3284 CrossRef CAS; (b) K. Wakita, N. Tokitoh, R. Okazaki, N. Takagi and S. Nagase, J. Am. Chem. Soc., 2000, 122, 5648–5649 CrossRef CAS; (c) M. A. Clark, R. C. Schoenfeld and B. Ganem, J. Am. Chem. Soc., 2001, 123, 10425–10426 CrossRef CAS PubMed.
- (a) R. W. Alder, R. W. Whiteside, G. Whittaker and C. Wilshire, J. Am. Chem. Soc., 1979, 101, 629–632 CrossRef CAS; (b) L. T. Scott, Acc. Chem. Res., 1982, 15, 52–58 CrossRef CAS; (c) R. W. Alder, S. P. East, J. N. Harvey and M. T. Oakley, J. Am. Chem. Soc., 2003, 125, 5375–5387 CrossRef CAS PubMed.
- Y. Han, Z. Xue, G. Li, Y. Gu, Y. Ni, S. Dong and C. Chi, Angew. Chem., Int. Ed., 2020, 59, 9026–9031 CrossRef CAS PubMed.
- D. Chen, Y. Hua and H. Xia, Chem. Rev., 2020, 120, 12994–13086 CrossRef CAS PubMed.
- (a) C. W. Landorf and M. M. Haley, Angew. Chem., Int. Ed., 2006, 45, 3914–3936 CrossRef CAS PubMed; (b) B. J. Frogley and L. J. Wright, Coord. Chem. Rev., 2014, 270–271, 151–166 CrossRef CAS; (c) I. Fernández, G. Frenking and G. Merino, Chem. Soc. Rev., 2015, 44, 6452–6463 RSC; (d) L. J. Wright, Metallabenzenes: An Expert View, John Wiley & Sons Ltd, Hoboken, NJ, 2017 CrossRef; (e) M. Talavera and S. Bolaño, Molecules, 2021, 26, 4655 CrossRef CAS PubMed.
- (a) G. Jia, Acc. Chem. Res., 2004, 37, 479–486 CrossRef CAS PubMed; (b) J. Chen and G. Jia, Coord. Chem. Rev., 2013, 257, 2491–2521 CrossRef CAS; (c) G. Jia, Organometallics, 2013, 32, 6852–6866 CrossRef CAS.
- C. Zhu, M. Luo, Q. Zhu, J. Zhu, P. v. R. Schleyer, J. I.-C. Wu, X. Lu and H. Xia, Nat. Commun., 2014, 5, 3265 CrossRef PubMed.
- (a) C. Zhu, S. Li, M. Luo, X. Zhou, Y. Niu, M. Lin, J. Zhu, Z. Cao, X. Lu, T. Wen, Z. Xie, P. v. R. Schleyer and H. Xia, Nat. Chem., 2013, 5, 698–703 CrossRef CAS PubMed; (b) Q. Zhuo, J. Lin, Y. Hua, X. Zhou, Y. Shao, S. Chen, Z. Chen, J. Zhu, H. Zhang and H. Xia, Nat. Commun., 2017, 8, 1912 CrossRef PubMed.
- J. Wei, W.-X. Zhang and Z. Xi, Chem. Sci., 2018, 9, 560–568 RSC.
- (a) H. Wang, Y.-M. Lin, S. Chen, Y. Ruan and H. Xia, Chin. J. Chem., 2021, 39, 3435–3442 CrossRef CAS; (b) M. Luo, Y. Cai, X. Lin, L. Long, H. Zhang and H. Xia, Chin. J. Chem., 2021, 39, 1558–1564 CrossRef CAS; (c) M. L. Buil, M. A. Esteruelas, E. Oñate and N. R. Picazo, Organometallics, 2021, 40, 4150–4162 CrossRef CAS PubMed; (d) V. Adamovich, M. Benítez, P.-L. Boudreault, M. L. Buil, M. A. Esteruelas, E. Oñate and J.-Y. Tsai, Inorg. Chem., 2022, 61, 9019–9033 CrossRef CAS PubMed; (e) M. Benítez, M. L. Buil, M. A. Esteruelas, S. Izquierdo, E. Oñate and J.-Y. Tsai, Inorg. Chem., 2022, 61, 19597–19611 CrossRef PubMed; (f) M. L. Buil, M. A. Esteruelas, E. Oñate and N. R. Picazo, Organometallics, 2023, 42, 327–338 CrossRef CAS.
- (a) Z. Huang, Y. Zhang, W.-X. Zhang, J. Wei, S. Ye and Z. Xi, Nat. Commun., 2021, 12, 1319 CrossRef CAS PubMed; (b) Y. Zhang, C. Yu, Z. Huang, W.-X. Zhang, S. Ye, J. Wei and Z. Xi, Acc. Chem. Res., 2021, 54, 2323–2333 CrossRef CAS PubMed.
- (a) S. Chen, L. Liu, X. Gao, Y. Hua, L. Peng, Y. Zhang, L. Yang, Y. Tan, F. He and H. Xia, Nat. Commun., 2020, 11, 4651 CrossRef CAS PubMed; (b) Y. Cai, Y. Hua, Z. Lu, Q. Lan, Z. Lin, J. Fei, Z. Chen, H. Zhang and H. Xia, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2102310118 CrossRef CAS PubMed; (c) M. Talavera, K. M. Cid-Seara, Á. Peña-Gallego and S. Bolaño, Dalton Trans., 2021, 50, 11216–11220 RSC; (d) R. Pereira-Cameselle, Á. Peña-Gallego, K. M. Cid-Seara, J. L. Alonso-Gómez, M. Talavera and S. Bolaño, Inorg. Chim. Acta, 2021, 517, 120189 CrossRef CAS; (e) F.-H. Cui, Y. Hua, Y.-M. Lin, J. Fei, L.-H. Gao, X. Zhao and H. Xia, J. Am. Chem. Soc., 2022, 144, 2301–2310 CrossRef CAS PubMed; (f) K. Masada, S. Kusumoto and K. Nozaki, Angew. Chem., Int. Ed., 2022, 61, e202117096 CAS; (g) W. Wei, X. Xu, H. H. Y. Sung, I. D. Williams, Z. Lin and G. Jia, Angew. Chem., Int. Ed., 2022, 61, e202202886 CAS; (h) M. Talavera, R. Pereira-Cameselle, Á. Peña-Gallego, I. Vázquez-Carballo, I. Prieto, J. L. Alonso-Gómez and S. Bolaño, Dalton Trans., 2023, 52, 487–493 RSC.
- (a) J. R. Bleeke and R. Behm, J. Am. Chem. Soc., 1997, 119, 8503–8511 CrossRef CAS; (b) E. Álvarez, M. Paneque, M. L. Poveda and N. Rendón, Angew. Chem., Int. Ed., 2006, 45, 474–477 CrossRef PubMed.
- (a) C. Zhu and H. Xia, Acc. Chem. Res., 2018, 51, 1691–1700 CrossRef CAS PubMed; (b) M. Luo, Y. Hua, K. Zhuo, L. Long, X. Lin, Z. Deng, Z. Lin, H. Zhang, D. Chen and H. Xia, CCS Chem., 2020, 2, 758–763 Search PubMed.
- (a) W. Y. Hung, J. Zhu, T. B. Wen, K. P. Yu, H. H. Y. Sung, I. D. Williams, Z. Lin and G. Jia, J. Am. Chem. Soc., 2006, 128, 13742–13752 CrossRef CAS PubMed; (b) G. He, J. Zhu, W. Y. Hung, T. B. Wen, H. H.-Y. Sung, I. D. Williams, Z. Lin and G. Jia, Angew. Chem., Int. Ed., 2007, 46, 9065–9068 CrossRef CAS PubMed; (c) W. Y. Hung, B. Liu, W. Shou, T. B. Wen, C. Shi, H. H.-Y. Sung, I. D. Williams, Z. Lin and G. Jia, J. Am. Chem. Soc., 2011, 133, 18350–18360 CrossRef CAS PubMed.
- M. Luo, C. Zhu, L. Chen, H. Zhang and H. Xia, Chem. Sci., 2016, 7, 1815–1818 RSC.
- (a) H. Xia, G. He, H. Zhang, T. B. Wen, H. H. Y. Sung, I. D. Williams and G. Jia, J. Am. Chem. Soc., 2004, 126, 6862–6863 CrossRef CAS PubMed; (b) Z. Chu, G. He, X. Cheng, Z. Deng and J. Chen, Angew. Chem., Int. Ed., 2019, 58, 9174–9178 CrossRef CAS PubMed.
- (a) M. Batuecas, R. Castro-Rodrigo, M. A. Esteruelas, C. García-Yebra, A. M. López and E. Oñate, Angew. Chem., Int. Ed., 2016, 55, 13749–13753 CrossRef CAS PubMed; (b) Z. Chu, J. Li, D. Chen, Z. Lu, W. Meng, M. Luo and H. Xia, Chin. J. Chem., 2023 DOI:10.1002/cjoc.202300116.
- (a) M. Luo, L. Long, H. Zhang, Y. Yang, Y. Hua, G. Liu, Z. Lin and H. Xia, J. Am. Chem. Soc., 2017, 139, 1822–1825 CrossRef CAS PubMed; (b) J. Li, H. Kang, K. Zhuo, Q. Zhuo, H. Zhang, Y.-M. Lin and H. Xia, Chin. J. Chem., 2018, 36, 1156–1160 CrossRef CAS; (c) Q. Li, Y. Hua, C. Tang, D. Chen, M. Luo and H. Xia, J. Am. Chem. Soc., 2023, 145, 7580–7591 CrossRef CAS PubMed.
- W. Ma, C. Yu, T. Chen, L. Xu, W.-X. Zhang and Z. Xi, Chem. Soc. Rev., 2017, 46, 1160–1192 RSC.
- (a) T. Beweries, C. Fischer, S. Peitz, V. V. Burlakov, P. Arndt, W. Baumann, A. Spannenberg, D. Heller and U. Rosenthal, J. Am. Chem. Soc., 2009, 131, 4463–4469 CrossRef CAS PubMed; (b) S. Zhang, W.-X. Zhang, J. Zhao and Z. Xi, J. Am. Chem. Soc., 2010, 132, 14042–14045 CrossRef CAS PubMed; (c) Z.-J. Lv, Z. Huang, J. Shen, W.-X. Zhang and Z. Xi, J. Am. Chem. Soc., 2019, 141, 20547–20555 CrossRef CAS PubMed.
- M. S. Chinn and D. M. Heinekey, J. Am. Chem. Soc., 1990, 112, 5166–5175 CrossRef CAS.
- (a) H. Xin, J. Li, R.-Q. Lu, X. Gao and T. M. Swager, J. Am. Chem. Soc., 2020, 142, 13598–13605 CrossRef CAS PubMed; (b) N. Sharma, H.-B. Zou, Y.-M. Lee, S. Fukuzumi and W. Nam, J. Am. Chem. Soc., 2021, 143, 1521–1528 CrossRef CAS PubMed.
- (a) S. Wang, B.-Y. Cheng, M. Sršen and B. König, J. Am. Chem. Soc., 2020, 142, 7524–7531 CrossRef CAS PubMed; (b) X.-R. Chen, Y.-M. Li, X. Li, J. Xuan, H.-P. Zhou, Y.-P. Tian and F. Li, Org. Lett., 2021, 23, 1302–1308 CrossRef CAS PubMed.
- L. Chen, L. Lin, A. R. Nath, Q. Zhu, Z. Chen, J. Wu, H. Wang, Q. Li, W.-F. Lin, J. Zhu and H. Xia, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2215900120 CrossRef CAS PubMed.
- (a) E. C. Friedrich and T. M. Tam, J. Org. Chem., 1982, 47, 315–319 CrossRef CAS; (b) A. D. Allen, M. Fujio, N. Mohammed, T. T. Tidwell and Y. Tsuji, J. Org. Chem., 1997, 62, 246–252 CrossRef CAS PubMed; (c) H. Jiao, P. v. R. Schleyer, Y. Mo, M. A. McAllister and T. T. Tidwell, J. Am. Chem. Soc., 1997, 119, 7075–7083 CrossRef CAS.
- Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. v. R. Schleyer, Chem. Rev., 2005, 105, 3842–3888 CrossRef CAS PubMed.
- K. Zhuo, Y. Liu, K. Ruan, Y. Hua, Y.-M. Lin and H. Xia, Nat. Synth., 2023, 2, 67–75 CrossRef.
- D. Geuenich, K. Hess, F. Köhler and R. Herges, Chem. Rev., 2005, 105, 3758–3772 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2190320, 2190323, and 2190338. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc01491f |
| This journal is © The Royal Society of Chemistry 2023 |