
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of spirooxindoles via formal acetylene insertion into a common palladacycle intermediate†
Xavier
Abel-Snape
 ,
Colton E.
Johnson
,
Bianca
Imbriaco
and
Mark
Lautens
*
,
Colton E.
Johnson
,
Bianca
Imbriaco
and
Mark
Lautens
*
Davenport Laboratories, Department of Chemistry, University of Toronto, 80 St. George St., Toronto, Ontario M5S 3H6, Canada. E-mail: mark.lautens@utoronto.ca
First published on 10th April 2023
Abstract
A palladium-catalyzed spirocyclization reaction is reported, which is proposed to arise via insertion of an oxabicycle into a palladacycle, formed from carbocyclization and a C–H functionalization sequence. Mechanistic studies suggest the insertion is diastereoselective and a post-catalytic retro-Diels–Alder step furnishes an alkene, wherein the oxibicycle has served as an acetylene surrogate. Aryl iodides and carbamoyl chlorides were compatible as starting materials under the same reaction conditions, enabling the convergent and complementary synthesis of spirooxindoles, as well as other azacycles. These spirooxindoles allowed further transformations that were previously unaccessible.
Introduction
Combining carbopalladation and C–H functionalization has proven to be an attractive entry into palladacyclic species.1 These reactive intermediates have been shown to undergo a variety of transformations, leading to carbo- and heterocyclic compounds including spirooxindoles, spiroindolines and spirodihydrobenzofurans (Scheme 1I).2–4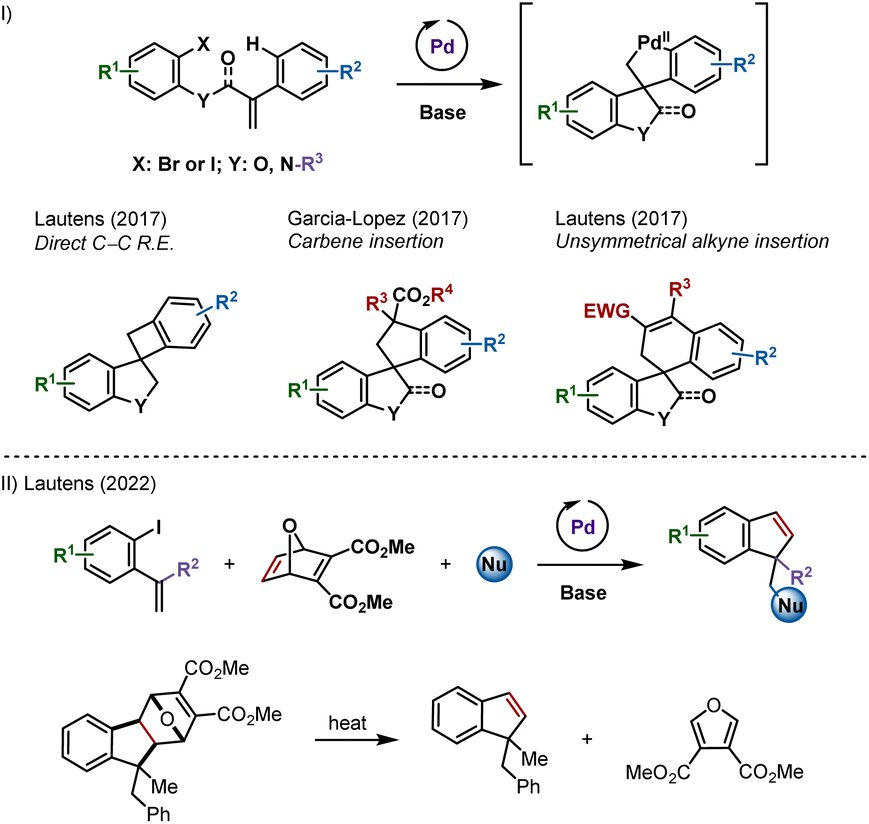 | ||
| Scheme 1 (I) Palladium-catalyzed spirocyclizations via remote C–H activation. (II) Palladium-catalyzed synthesis of indenes and benzofulvenes using an oxabicycle as an acetylene surrogate. | ||
We reported that unsymmetrical polarized alkynes insert into these palladacyclic species (Scheme 1I, right example).4f However, internal symmetrical alkynes failed to react, likely due to a higher activation energy for the requisite insertion step.5 Acetylene, the simplest symmetrical alkyne, was not studied as a potential partner out of concerns for side reactions.
Recently, we described a palladium-catalyzed multicomponent synthesis of indenes and benzofulvenes, using an oxabicycle as an acetylene surrogate, via a post-catalytic retro-Diels–Alder reaction (Scheme 1II).6 Acetylene itself is not well-suited for metal-catalyzed domino processes due to difficulty in handling and has reactive C–H groups that can be favoured over addition to the π-bond.7 Herein, we report the synthesis of spirocycles resulting from formal acetylene insertion using this strategy.
Results and discussion
Reaction optimization
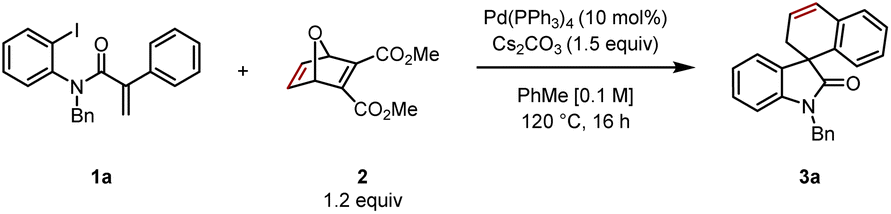
Reacting aryl iodide 1 with oxabicycle 2, catalytic Pd(PPh3)4, base (Cs2CO3) in toluene at 120 °C for 16 h gave the desired product 3a in 55% yield (Table 1, entry 1). Reaction at higher concentration (0.2 M) was detrimental to the transformation, giving 3a in 47% yield (entry 2). Conversely, diluting the mixture to 0.05 M resulted in an improved yield of 69% (entry 3). Conducting the reaction at 130 °C led to a dramatic increase in product formation, and 3a was isolated in 75% yield (entry 4). The combination of higher temperature and lower concentration proved to be best, leading to the product in 85% yield (entry 5). Further reducing the catalyst loading from 10 to 7.5 mol% furnished the desired spirooxindole in an optimized isolated yield of 88% (entry 6).|
|
||
|---|---|---|
| Entry | Variation | Yielda (%) |
| a Reactions were performed on a 0.2 mmol scale; yields were determined by 1H NMR spectroscopy analysis of the crude reaction mixture using 1,3,5-trimethoxybenzene as an internal standard. b Isolated yield. | ||
| 1 | None | 59 (55)b |
| 2 | PhMe [0.2 M] | 47 |
| 3 | PhMe [0.05 M] | 69 |
| 4 | 130 °C | 75 |
| 5 | PhMe [0.05 M], 130 °C | 85 |
| 6 | Pd(PPh3)4 (7.5 mol%), PhMe [0.05 M], 130 °C | 92 (88)b |
Substrate scope
Following optimization, an exploration of the substrate scope was undertaken by varying the substituents on the iodoarene ring (Scheme 2). A variety of 5-substituted spirooxindoles were formed following this protocol: 3b (methoxycarbonyl), 3c (trifluoromethyl), 3d (fluoro), 3e (methyl) and 3f (methoxy) were synthesized in yields ranging from 68 to 78%. A chloro substituent on the 6-position resulted in 73% yield (3g). A more challenging substrate containing a pyridine moiety resulted in the corresponding spirooxazaindole (3h) in only 24% yield. Modifying substituents on the nitrogen atom was also explored. Less steric bulk seemed to disfavour the desired reactivity, as the relatively small methyl and cyanomethyl groups gave the products in 55% (3i) and 32% (3j) yields respectively. In contrast, a yield of 82% was obtained with the tert-butyl-ester derivative 3k. A substrate bearing a nitro group reacted, generating 3l in 85% yield. Thiophene-derived compounds 3m and 3n were both generated in good yields of 81% and 60%, the latter resulting from C–H activation at the 3-position of the corresponding substrate's tethered thiophene ring. An ortho-chloro group on the aryl ring undergoing C–H activation gave rise to 3o in 78% yield.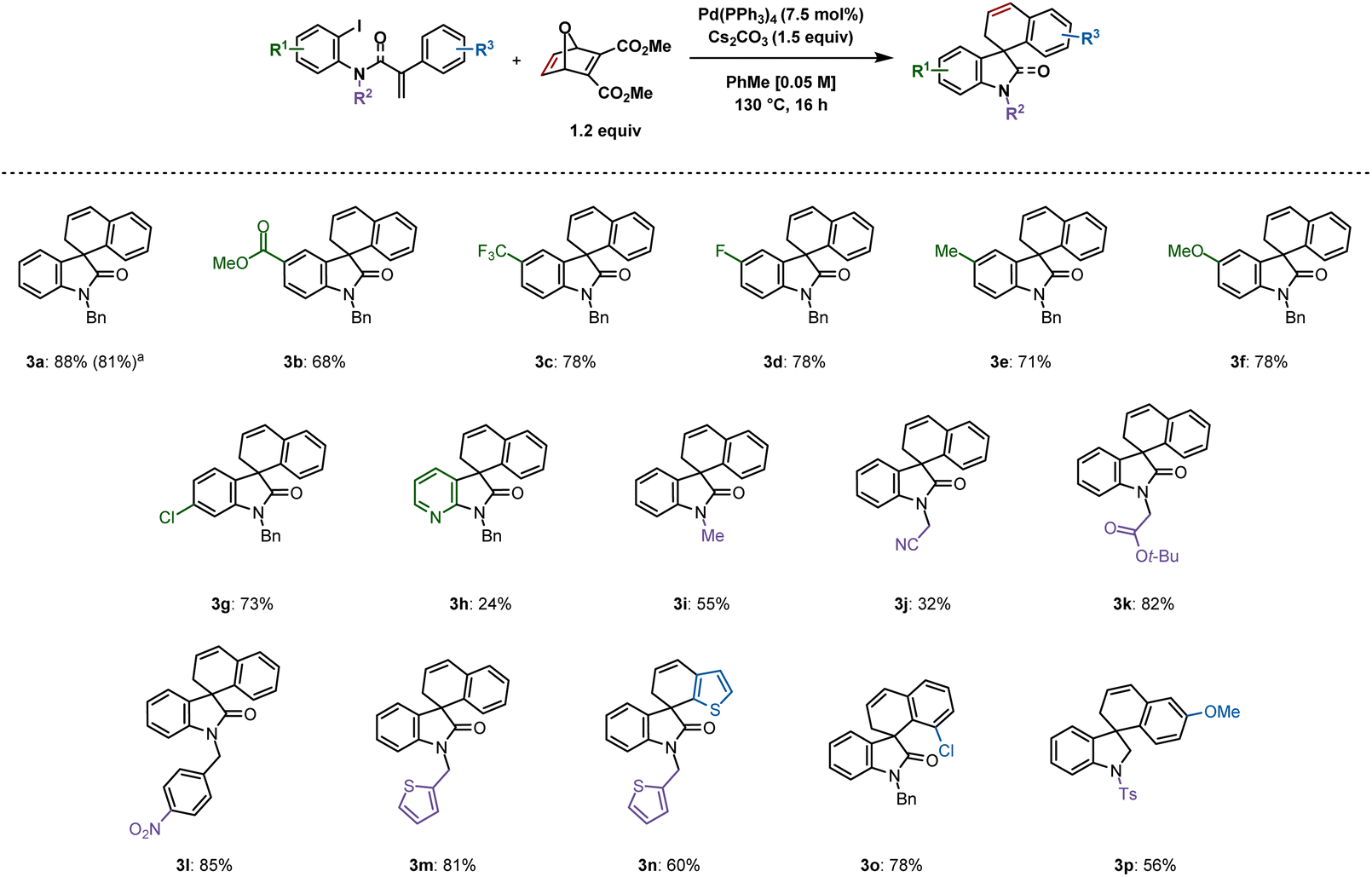 | ||
| Scheme 2 Spirooxindoles synthesized from aryl iodides. Reactions were performed on a 0.2 mmol scale; isolated yields are shown. aReaction was performed on a 1 mmol scale. | ||
A 1 mmol scale reaction of 1a was performed, which provided 3a in 81% yield. We were also able to synthesize spiroindoline 3p in 56% yield from the corresponding sulfonamide. Unfortunately, spirodihydrobenzofurans could not be accessed using this methodology (see ESI† for unsuccessful substrates).
Based on our studies using carbamoyl chlorides as reactive precursors,8 and those of Qu and Chen, wherein carbamoyl chlorides were used to access spirooxindoles following insertion of benzyne or an unsymmetrical alkyne,9 we attempted to synthesize 3a using the previously optimized reaction conditions (Scheme 3). The product was isolated in 78% yield, which was comparable to that reported from the aryl iodide 1. A methyl group at the 6-position generated 3q in 74% yield. The smaller the substituent on the nitrogen, the lower the yield, as demonstrated by isolation of 3i in 51% yield, whereas 3-thiophene derivative 3r was produced in 67% yield. Fluoro and methoxy substituents at the para position of the aryl ring undergoing C–H activation gave similar yields of 3s (60%) and 3t (62%) respectively. Interestingly, a benzodioxol-derived substrate provided 3u in 69% yield in >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 rr, the major regioisomer resulting from C–H activation at the benzodioxole moiety's 6-position. This result is in contrast to a related example in our previous report on benzyne insertion,4c where the major spirodihydrobenzofuran resulted from C–H activation at the 4-position (6.7:1 rr).
:1 rr, the major regioisomer resulting from C–H activation at the benzodioxole moiety's 6-position. This result is in contrast to a related example in our previous report on benzyne insertion,4c where the major spirodihydrobenzofuran resulted from C–H activation at the 4-position (6.7:1 rr).
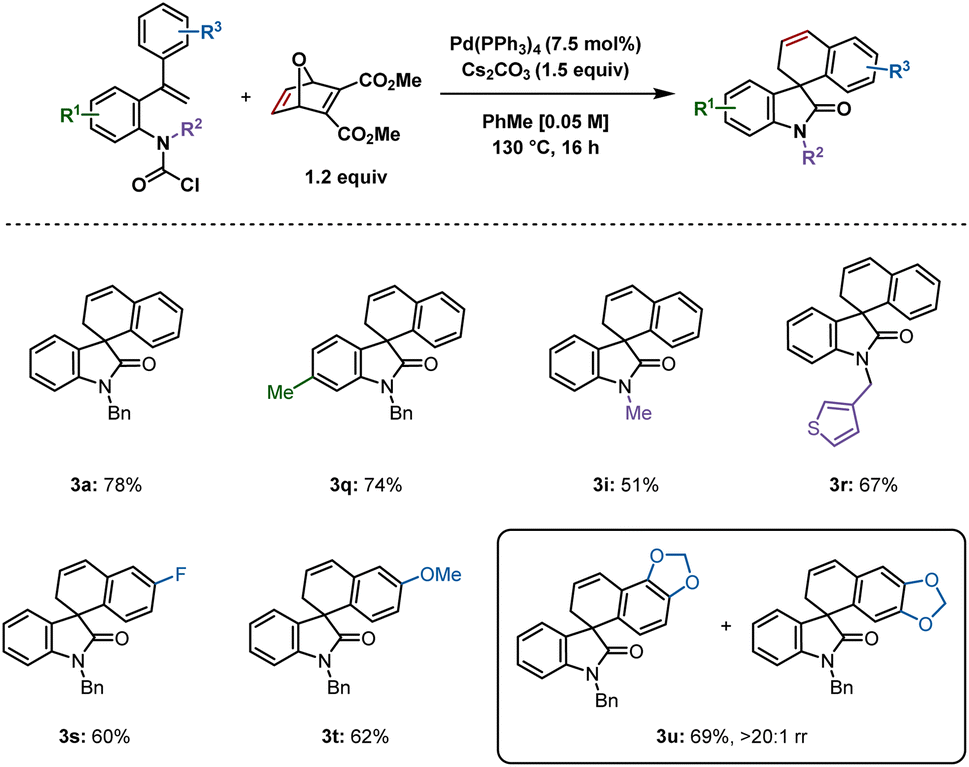 | ||
| Scheme 3 Spirooxindoles synthesized from carbamoyl chlorides. Reactions were performed on a 0.2 mmol scale; isolated yields are shown. | ||
Access to different types of scaffolds via insertion of oxabicycle 2 was also of interest. The spirocyclic pyrroline 5 was formed, though only in 14% yield, from γ,δ-unsaturated oxime ester 4 through a domino Narasaka–Heck/C–H activation reaction (Scheme 4I).10 Spirocycle synthesis is only feasible via remote C(sp2)–H activation, that is, when an aryl ring is bound to the alkene on the starting material, allowing for formation of a spiropalladacycle. In the absence of the aryl group on the alkene, the only C(sp2)–H bond available is located at the ortho position of the aryl iodide, providing a fused palladacycle and ultimately, fused rings following insertion of an unsaturated system.11
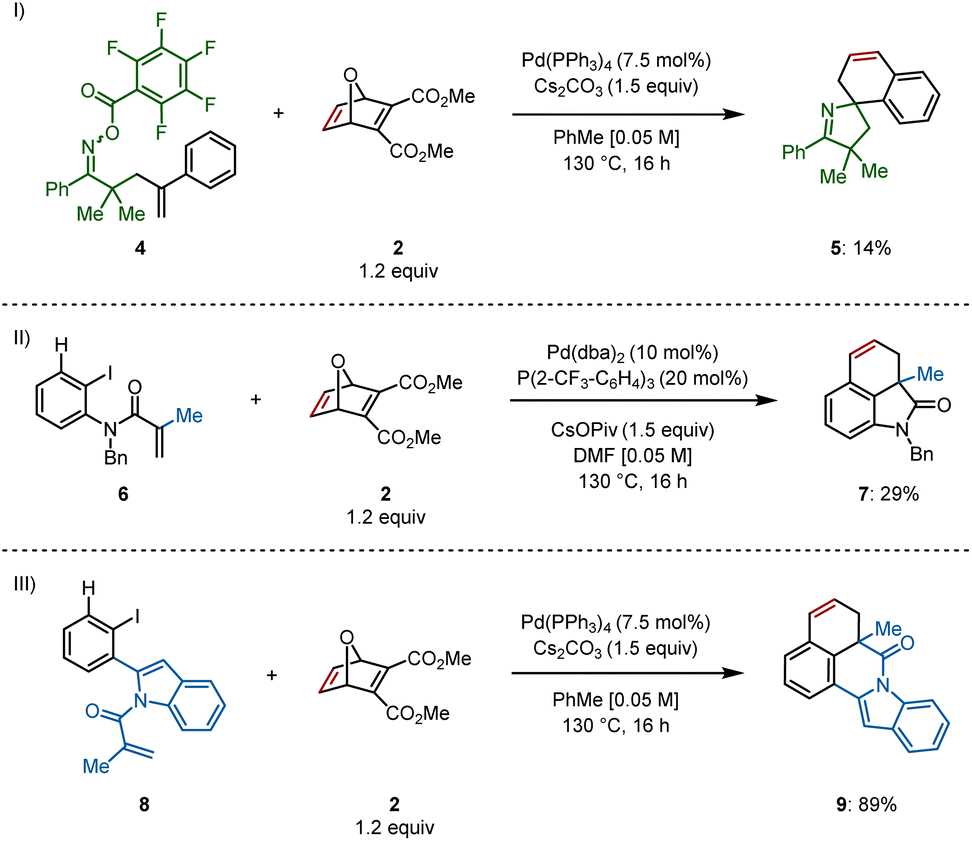 | ||
| Scheme 4 Synthesis of analogous azacycles. (I) Spirocyclic pyrroline. (II) Dihydrobenzoindolone. (III) Indolo[2,1-a]isoquinolinone. Reactions were performed on a 0.2 mmol scale; isolated yields are shown. | ||
For example, methyl-derived 6 provided dihydrobenzoindolone 7 in 29% yield (Scheme 4II, see ESI† for optimization) and indole-derived substrate 8 delivered indolo[2,1-a]isoquinolinone 9 in 89% yield using the standard reaction conditions (Scheme 4III).12 This final example demonstrates successful formation of a six-membered N-heterocycle prior to C–H activation and insertion into the π system of the oxabicycle.
Mechanistic studies
Based on our previous work on the synthesis of indenes,6 we set out to isolate the pre-retro-Diels–Alder intermediate, by stopping the reaction after less time elapsed. In a parallel experiment, the reaction was run at a lower temperature (Scheme 5I). Stopping the reaction after 2 h formed the final product 3a and intermediate cycloadduct 3a* in 50% and 48% yields respectively. Running the reaction at 100 °C led to incomplete conversion of 1a, resulting in the formation of 3a in 8%, as judged by 1H NMR, whereas 3a* was formed in 47% yield. We dissolved cycloadduct 3a* in toluene followed by heating at 130 °C for 16 h, which led to full conversion and provided 3a in 97% yield (Scheme 5II). This experiment also suggests that heat is soley responsible for the retro-Diels–Alder step (see ESI† for control experiments). We also heated 3a* at 130 °C for only 2 h, and separately, at 100 °C for 16 h, which resulted in 48% and 35% consumption of 3a*, providing 3a in quantitative yield (based on recovered starting material).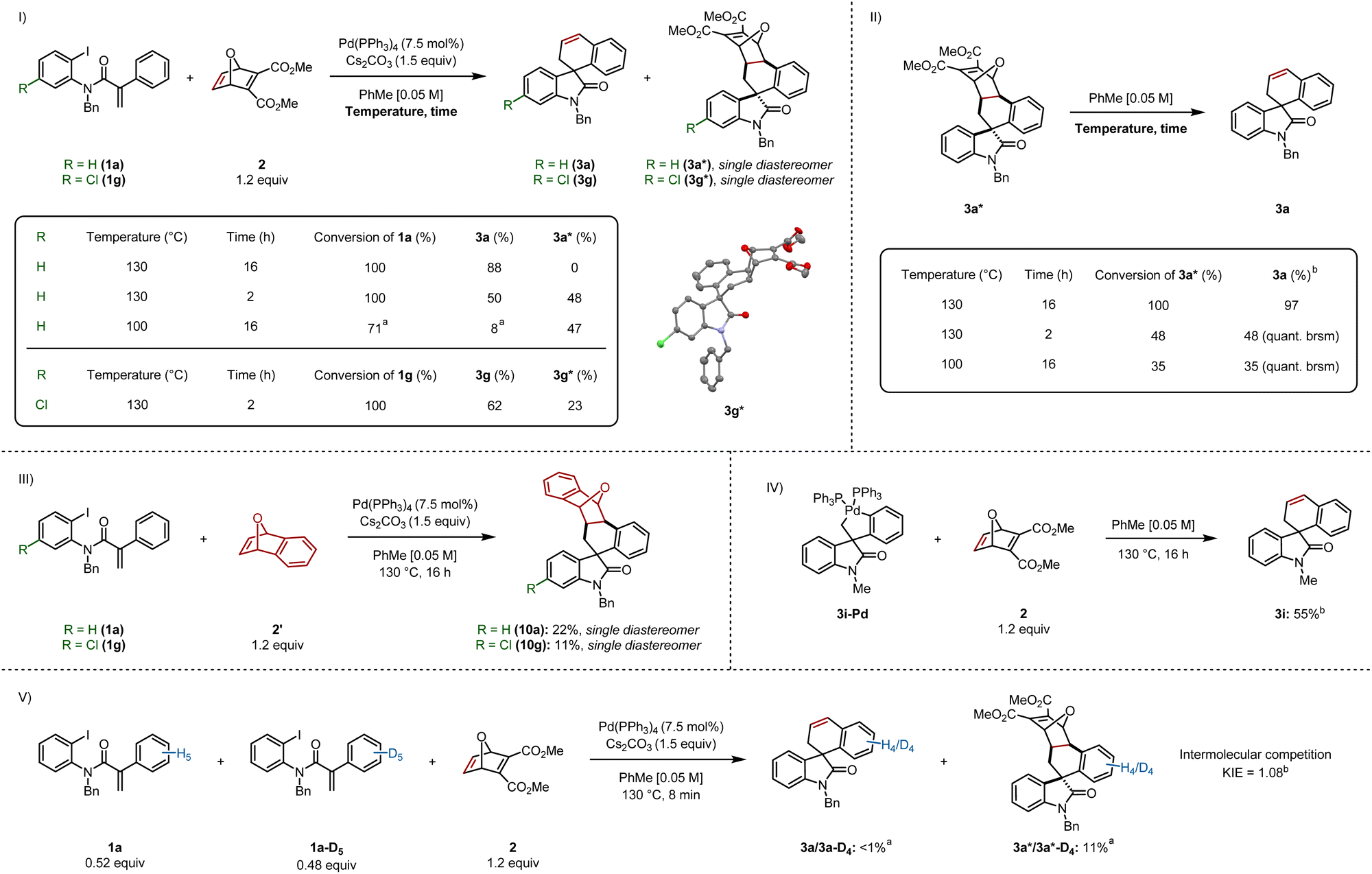 | ||
| Scheme 5 Mechanistic studies. (I) Isolation of pre-retro-Diels–Alder intermediates 3a* and 3g*. (II) Confirmation of 3a* as a precursor to 3a. (III) Insertion of an oxabicycle that cannot undergo a retro-Diels–Alder step. (IV) Palladacycle 3i-Pd as a competent intermediate in the catalytic cycle. (V) KIE experiment. Reactions were performed on a 0.2 mmol scale; isolated yields are shown. aYield was determined by 1H NMR spectroscopic analysis of the crude reaction mixture using 1,3,5-trimethoxybenzene as an internal standard. bReaction was performed on a 0.1 mmol scale. | ||
Interestingly, 3a* was isolated as a single diastereomer. The relative configuration between the spirocenter and the four stereocenters on the oxabicyclic moiety was elucidated via single crystal X-ray diffraction of an analogous chlorinated compound 3g* (Scheme 5I), which revealed the aryl backbone is on the same side of the bridging oxygen.
We sought to generate an adduct that was unable to undergo the retro-Diels–Alder step (Scheme 5III).13 Spirooxindoles 10a and 10g were formed by reacting 1a and 1g respectively with benzooxabicycle 2′, which were generated as a single diastereomer.
We independently synthesized palladacycle 3i-Pd and reacted it with 2, providing 3i in 55% yield, confirming that the five-membered palladacycle is a competent intermediate in the catalytic cycle (Scheme 5IV). Based on this experiment, and other reports,4c,4f,5 we suggest that C–H activation precedes oxabicycle insertion.
An intermolecular competition experiment was carried out between 1a and 1a-D5 with a reaction time of 8 minutes (Scheme 5V). A KIE value of 1.08 was observed, demonstrating C–H activation does not constitute the turnover-limiting step when using an aryl iodide as the substrate (see ESI† for the analogous carbamoyl chloride KIE experiment), which is consistent with previous findings by our group involving benzyne insertion.4c
Based on the mechanistic data presented in Scheme 5, and previous work from our group and others,4c,4f,5 a plausible mechanism is shown in Scheme 6. Aryl iodide 1a or carbamoyl chloride 1a′ would undergo oxidative addition to provide respectively arylpalladium(II) I or acylpalladium(II) I′. Both of these species converge to the same neopentylpalladium(II) intermediate II following intramolecular carbopalladation. Subsequently, a Cs2CO3-assisted concerted metalation–deprotonation (CMD) step gives rise to the key five-membered palladacycle III. Insertion of the strained alkene in 2 occurs not only in exo fashion, but also preferentially into the Csp2–Pd bond, leading to seven-membered palladacycle IV with the palladium atom being bound to the C2 of the oxabicycle. We cannot at this stage exclude the formation of the exo-diastereomer IV′ with the palladium atom being bound to the C1 of the oxabicycle resulting from its opposite regioselective insertion into III.14 Reductive elimination closes the catalytic cycle, thereby regenerating Pd(0) and releasing 3a* and potentially its diastereomer 3a*′. Thereafter, a retro-Diels–Alder process delivers product 3a and the furan 11. It is possible that the rate of the retro-Diels–Alder reaction of 3a*′ is significantly faster than its analogue 3a*, which might explain why it was never isolated. However, the experiment at 100 °C/16 h would suggest if 3a*′ is formed, it would clearly be the minor diastereomer (Scheme 5I).
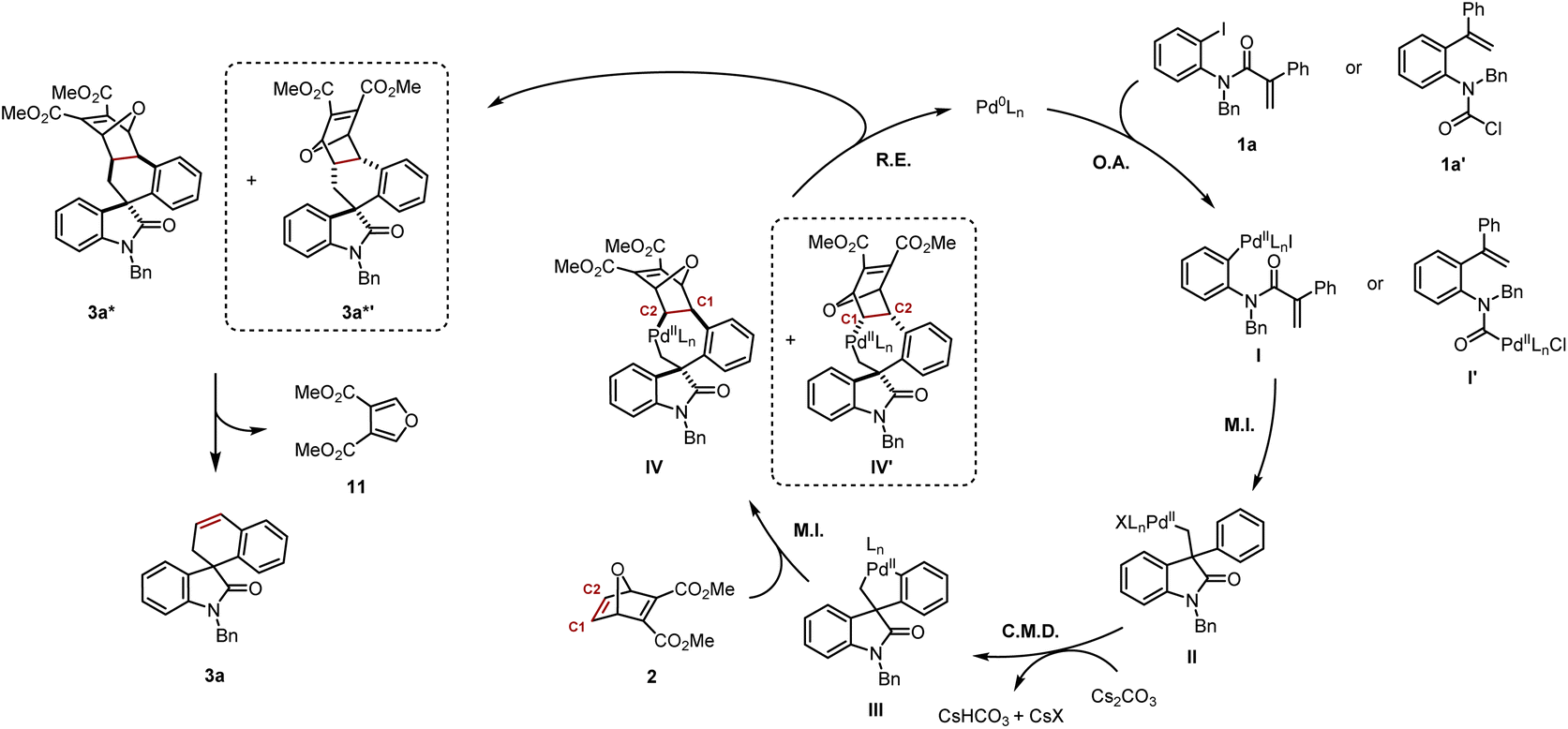 | ||
| Scheme 6 Proposed mechanism. | ||
The resulting cyclic alkene offers multiple opportunities to further diversify the products. A [2 + 2] cycloaddition between 3a and in situ-formed dichloroketene followed by dehalogenation in the presence of zinc and acetic acid was highly regioselective but gave cyclobutanones 12a and 12b in 19% and 8% yields with minimal diastereoselectivity over two steps (Scheme 7I).15 Epoxidation of 3a using in situ-formed methyl(trifluoromethyl)dioxirane gave the diastereomeric epoxides 13a and 13b in 37% and 31% yields respectively, corresponding to a 97% combined yield (based on recovered starting material) (Scheme 7II).16 A regioselective ring opening of 13a with sodium azide gave the 1,2-azido alcohol 14 in 70% yield, resulting in a formal difunctionalization of the olefin in 3a (Scheme 7III). Direct hydrogenation converted 3a to its saturated spirocyclic analogue 14 in 79% yield (Scheme 7IV). Finally, 3a was subjected to an iron-catalyzed Wacker oxidation to provide 16 in 58% yield (Scheme 7V).17
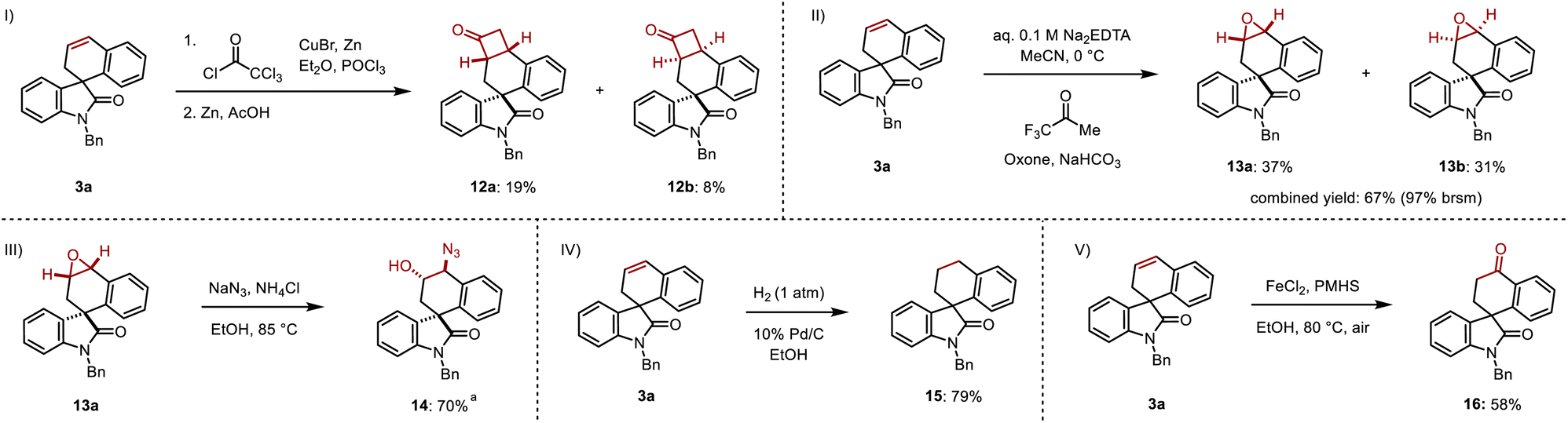 | ||
| Scheme 7 Derivatizations. (I) Ketene [2 + 2] cycloaddition then dehalogenation. (II) Epoxidation. (III) Epoxide ring opening with sodium azide. (IV) Hydrogenation. (V) Iron-catalyzed Wacker oxidation. Reactions were performed on a 0.15 mmol scale; isolated yields are shown. aReaction was performed on a 0.029 mmol scale. | ||
Conclusions
In summary, we have successfully utilized a surrogate to formally insert acetylene in the synthesis of spirooxindoles. Two access points were identified; one using aryl iodides, and the other using carbamoyl chlorides under identical reaction conditions. We determined the oxabicycle acted as a C2-donor by undergoing a thermal post-catalytic retro-Diels–Alder reaction from the corresponding cycloadduct. Various derivatizations were carried out on the resulting alkene, demonstrating its use as an efficient synthetic handle.Data availability
All experimental procedures, characterization, and computational data for this study can be found in the ESI.†Author contributions
X. A.-S., and M. L. conceived and led the project. X. A.-S., C. E. J. and B. I. performed the experiments. X. A.-S., C. E. J. and M. L. analyzed the data and discussed the results. X. A.-S. prepared the ESI† and a first draft of the manuscript, which was edited by all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the University of Toronto, the Natural Science and Engineering Research Council (NSERC), and Kennarshore Inc., for financial support. X. A.-S. thanks NSERC for a graduate scholarship (CGS-D) and the Province of Ontario (OGS) for funding. C. E. J. thanks the Province of Ontario (OGS) for funding. B. I. thanks NSERC for an undergraduate summer research award (USRA). Bijan Mirabi, Dr Austin Marchese and Prof. Mark Taylor are thanked for insightful discussions. Justin Ching is thanked for proofreading. Dr Alan Lough (University of Toronto) is thanked for X-ray crystallographic analysis. We thank Dr Darcy Burns and Dr Jack Sheng (University of Toronto) for their assistance in NMR experiments. We thank Dr Andrei Kutateladze (University of Denver) for assigning the correct regioisomer of cyclobutanones 12a and 12b, using methods developed in his laboratory.Notes and references
- For select reviews on transition-metal catalyzed C–H functionalization methodologies see: (a) D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174 CrossRef CAS PubMed; (b) P. Thansandote and M. Lautens, Chem. - Eur. J., 2009, 15, 5874 CrossRef CAS PubMed; (c) L. McMurray, F. O'Hara and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885 RSC; (d) O. Baudoin, Chem. Soc. Rev., 2011, 40, 4902 RSC; (e) C. Liu, J. Yuan, M. Gao, S. Tang, W. Li, R. Shi and A. Lei, Chem. Rev., 2015, 115, 12138 CrossRef CAS PubMed; (f) J. F. Hartwig, J. Am. Chem. Soc., 2016, 138, 2 CrossRef CAS PubMed; (g) X.-S. Xue, P. Ji, B. Zhou and J.-P. Cheng, Chem. Rev., 2017, 117, 8622 CrossRef CAS PubMed; (h) M. Font, M. Gulías and J. L. Mascareñas, Angew. Chem., Int. Ed., 2022, 61, e202112848 CrossRef CAS PubMed; (i) S. Dutta, S. Chatterjee, S. A. Al-Thabaiti, S. Bawaked, M. Mokhtar and D. Maiti, Chem Catal., 2022, 2, 1046 CrossRef CAS; (j) M.-Z. Lu, J. Goh, M. Maraswami, Z. Jia, J.-S. Tian and T.-P. Loh, Chem. Rev., 2022, 122, 17479 Search PubMed.
- For select reviews on palladium-catalyzed C–H activation methodologies see: (a) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094 CrossRef CAS PubMed; (b) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (c) T. M. Shaikkh and F.-E. Hong, J. Organomet. Chem., 2016, 801, 139 CrossRef; (d) J. Wang and G. Dong, Chem. Rev., 2019, 119, 7478 CrossRef CAS PubMed.
- For reports of Pd catalyzed domino-Heck syntheses of spirocycles resulting from direct C–C reductive elimination see: (a) J. Ye, Z. Shi, T. Sperger, Y. Yasukawa, C. Kingston, F. Schoenebeck and M. Lautens, Nat. Chem., 2017, 9, 361 CrossRef CAS PubMed; (b) A. D. Marchese, A. G. Durant and M. Lautens, Org. Lett., 2022, 24, 95 CrossRef CAS PubMed; (c) B. Xu, D. Ji, L. Wu, L. Zhou, Y. Liu, Z.-M. Zhang and J. Zhang, Chem, 2022, 8, 836 CrossRef CAS.
- For reports of Pd catalyzed domino-Heck syntheses of spirocycles resulting from insertion of an external reagent into a palladacycle intermediate see: (a) H. Zheng, Y. Zhu and Y. Shi, Angew. Chem., Int. Ed., 2014, 53, 11280 CrossRef CAS PubMed; (b) M. Pérez-Gómez and J.-A. García-López, Angew. Chem., Int. Ed., 2016, 55, 14389 CrossRef PubMed; (c) H. Yoon, A. Lossouarn, F. Landau and M. Lautens, Org. Lett., 2016, 18, 6324 CrossRef CAS PubMed; (d) M. Pérez-Gómez, S. Hernández-Ponte, D. Bautista and J.-A. García-López, Chem. Commun., 2017, 53, 2842 RSC; (e) C. Shao, Z. Wu, X. Ji, B. Zhou and Y. Zhang, Chem. Commun., 2017, 53, 10429 RSC; (f) H. Yoon, M. Rölz, F. Landau and M. Lautens, Angew. Chem., Int. Ed., 2017, 56, 10920 CrossRef CAS PubMed; (g) G. Xiao, L. Chen, G. Deng, J. Liu and Y. Liang, Tetrahedron Lett., 2018, 59, 1836 CrossRef CAS; (h) F. Ye, Y. Ge, A. Spannenberg, H. Neumann and M. Beller, Nat. Commun., 2020, 11, 5383 CrossRef CAS PubMed; (i) J. Ma, T.-X. Liu, P. Zhang, C. Zhang and G. Zhang, Chem. Commun., 2021, 57, 49 RSC; (j) Y. Xu, W. Xu, X. Chen, X. Luo, H. Lu, M. Zhang, X. Yang, G. Deng, Y. Liang and Y. Yang, Chem. Sci., 2021, 12, 11756 RSC.
- I. Franzoni, H. Yoon, J.-A. García-López, A. I. Poblador-Bahamonde and M. Lautens, Chem. Sci., 2018, 9, 1496 RSC.
- (a) X. Abel-Snape, G. Wycich and M. Lautens, ACS Catal., 2022, 12, 3291 CrossRef CAS . For the original report on using oxabicycle 2 as an acetylene surrogate, see: ; (b) W. Lv, S. Wen, J. Yu and G. Cheng, Org. Lett., 2018, 20, 4984 CrossRef CAS PubMed.
- (a) I.-T. Trotus, T. Zimmermann and F. Schüth, Chem. Rev., 2014, 114, 1761 CrossRef CAS PubMed; (b) V. V. Voronin, M. S. Ledovskaya, A. S. Bogachenkov, K. S. Rodygin and V. P. Anikov, Molecules, 2018, 23, 2442 CrossRef PubMed.
- For recent reports by our group using carbamoyl chlorides as reactive precursors see: (a) E. M. Larin, A. Torelli, J. Loup and M. Lautens, Org. Lett., 2021, 23, 2720 CrossRef CAS PubMed; (b) J. F. Rodriguez, A. Zhang, J. Bajohr, A. Whyte, B. Mirabi and M. Lautens, Angew. Chem., Int. Ed., 2021, 60, 18478 CrossRef CAS PubMed; (c) R. Arora, J. F. Rodriguez, A. Whyte and M. Lautens, Angew. Chem., Int. Ed., 2021, 61, e202112288 Search PubMed.
- C. Wang, W. Zhao, X. Wu, J. Qu and Y. Chen, Adv. Synth. Catal., 2020, 362, 4996 CrossRef CAS.
- (a) W.-X. Wei, Y. Li, Y.-T. Wen, M. Li, X.-S. Li, C.-T. Wang, H.-C. Liu, Y. Xia, B.-S. Zhang, R.-Q. Jiao and Y.-M. Liang, J. Am. Chem. Soc., 2021, 143, 7868 CrossRef CAS PubMed; (b) W. X. Wei, X. Kong, R. Q. Jiao, X. S. Li, C. T. Wang, Y. Li and Y. M. Liang, Chem. Sci., 2022, 13, 6348 RSC.
- (a) M. Sickert, H. Weinstabl, B. Peters, X. Hou and M. Lautens, Angew. Chem., Int. Ed., 2014, 53, 5147 CrossRef CAS PubMed; (b) T. Yao and D. He, Org. Lett., 2017, 19, 842 CrossRef CAS PubMed; (c) J. F. Rodríguez, A. D. Marchese and M. Lautens, Org. Lett., 2018, 20, 4367 CrossRef PubMed; (d) X. Luo, L. Zhou, H. Lu, G. Deng, Y. Liang, C. Yang and Y. Yang, Org. Lett., 2019, 21, 9960 CrossRef CAS PubMed.
- (a) X. Yang, H. Lu, X. Zhu, L. Zhou, G. Deng, Y. Yang and Y. Liang, Org. Lett., 2019, 21, 7284 CrossRef CAS PubMed; (b) H. Lu, X. Yang, L. Zhou, W. Li, G. Deng, Y. Yang and Y. Liang, Org. Chem. Front., 2020, 7, 2016 RSC; (c) L. Zhou, S. Qiao, F. Zhou, X. Xuchen, G. Deng, Y. Yang and Y. Liang, Org. Lett., 2021, 23, 2878 CrossRef CAS PubMed; (d) M. Zhang, F. Zhou, X. Xuchen, L. Zhou, G. Deng, Y. Liang and Y. Yang, Org. Chem. Front., 2021, 8, 5687 RSC; (e) H. Qi, D. Chi and S. Chen, Org. Lett., 2022, 24, 2910 CrossRef CAS PubMed.
- We were curious as to whether this reaction might only appear to be highly diastereoselective, as it is possible the other diastereomer might be forming but undergoes the retro-Diels-Alder step at a considerably faster rate than its analogue, rendering it unobservable.
- In the case of the enantiomers of IV and IV′, palladium would end up bonded to the oxabicycle’s C1 and C2 atoms respectively.
- K. Surendra, G. Rajendar and E. J. Corey, J. Am. Chem. Soc., 2014, 136, 642 CrossRef CAS PubMed.
- D. Yang, M.-K. Wong and Y.-C. Yip, J. Org. Chem., 1995, 60, 3887 CrossRef CAS.
- B. Liu, F. Jin, T. Wang, X. Yuan and W. Han, Angew. Chem., Int. Ed., 2017, 56, 12712 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2244743 and 2244744. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc01072d |
| This journal is © The Royal Society of Chemistry 2023 |