
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTrimethylaluminum-mediated one-pot peptide elongation†
Tomohiro
Hattori
 * and
Hisashi
Yamamoto
*
* and
Hisashi
Yamamoto
*
Peptide Research Center, Chubu University, 1200 Matsumoto-cho, Kasugai, Aichi 487-8501, Japan. E-mail: thattori@isc.chubu.ac.jp; hyamamoto@isc.chubu.ac.jp
First published on 27th April 2023
Abstract
Efficient and straightforward peptide bond formation of N-, and C-terminal unprotected amino acids was successfully achieved by using trimethylaluminum. The coupling reaction was accomplished by pre-reaction of N-, and C-terminal unprotected amino acids and trimethylaluminum to form a five-membered ring that smoothly reacted with nucleophilic amino acid esters. This simple and highly efficient reaction system allows one-pot tripeptide synthesis without the need for expensive coupling reagents. Furthermore, peptide bond formation can be effectively achieved even for amino acids with bulky substituents at the side chain to afford the corresponding tripeptides in high yields in a one-pot manner. In addition, the reaction can be applied for further peptide elongation by the subsequent addition of amino acids and trimethylaluminum. We anticipate that this cost-effective, straightforward, and efficient protocol will be useful for the synthesis of a wide variety of peptides.
Introduction
Peptides, the hetero-polymers of various amino acid residues, are essential biomolecules with enormous applications in various fields. In particular, there appears a growing interest in therapeutic peptides,1 and hundreds of peptides have been employed in preclinical and clinical developments.2 Therefore, the development of an efficient and cost-effective methodology for peptide synthesis is pivotal for organic chemists. Despite this goal, protection and deprotection of amino acid groups are indispensable steps in peptide elongation. Peptide bond formation has heavily relied on the protection system for the N-and/or C-terminal of amino acids. This is important for the prevention of unexpected byproducts, such as homo-coupled3 and cyclized4 products, that arise in the absence of protective measures. It is therefore accurate to state that the success of peptide bond formation has been dramatically influenced by the use of protective groups. On the other hand, the use of protecting groups is a bottleneck in rapid and economical peptide synthesis such as one-pot synthesis because protection/deprotection and purification at each step are necessary.An elegant and straightforward strategy for peptide bond formation using unprotected amino acids is the temporary formation of five-membered rings from unprotected amino acids using silicon dichloride,5 boron,6 phosphorous7 or hexafluoroacetone8 (Scheme 1A). Cyclic amino acids not only have higher solubility in certain organic solvents such as DCM and MeCN, but also prevent side reactions such as homo-coupling reactions by the virtue of steric hindrance. Very recently, we identified silacyclic amino acids/peptides that enabled efficient peptide bond formation (Scheme 1B).9 Although the use of these silacyclic dipeptides is a promising strategy for elongation at both the terminals, silacyclic dipeptides need to be isolated when they are used as substrates for further elongation. Furthermore, expensive or complex reagents such as tantalum catalysts and bis(imidazolyl)silane were required for effective amide bond formation. Even when such reagents were used, the reactivity of amino acids bearing bulky substituents remains low, because it was not easy for nucleophilic amino acids to react with five-membered intermediates composed of bulky amino acids such as valine, leucine, and isoleucine. To achieve economical, readily available, and general peptide bond formation, completely new systems are immensely desired.
 | ||
| Scheme 1 Backgrounds of peptide bond formation utilizing five-membered cyclic amino acids. | ||
Trimethylaluminum (AlMe3) is commonly used as a Lewis acid in various organic reactions.10 Some studies have shown that AlMe3 facilitates the effective amidation of carboxylic acids even in the absence of coupling reagents,11 indicating that AlMe3 is an exceptionally attractive and useful reagent for peptide bond formation. In fact, one of the studies demonstrated the possibility of constructing peptide bonds from unprotected amino acids and AlMe3.12 However, because of the low nucleophilicity of the intermediate Al-five-membered ring, this method exhibits low reactivity and has limited substrate scope. Actually, the intermediate five-membered ring did not well function as a nucleophile, and only moderate dipeptides were obtained (31–60% yields). Herein, we have discovered the formation of a five-membered ring composed of Al and unprotected amino acids. The ring behaved as an electrophile to form peptide bonds with nucleophilic amino acid esters. The resulting dipeptide reacted with electrophilic amino acids to afford tripeptides in a one-pot manner. Furthermore, this one-pot system could be applied to further longer peptide synthesis such as tetra-/pentapeptide by sequentially adding the prepared five-membered cyclic amino acids (Scheme 1C).
Results and discussion
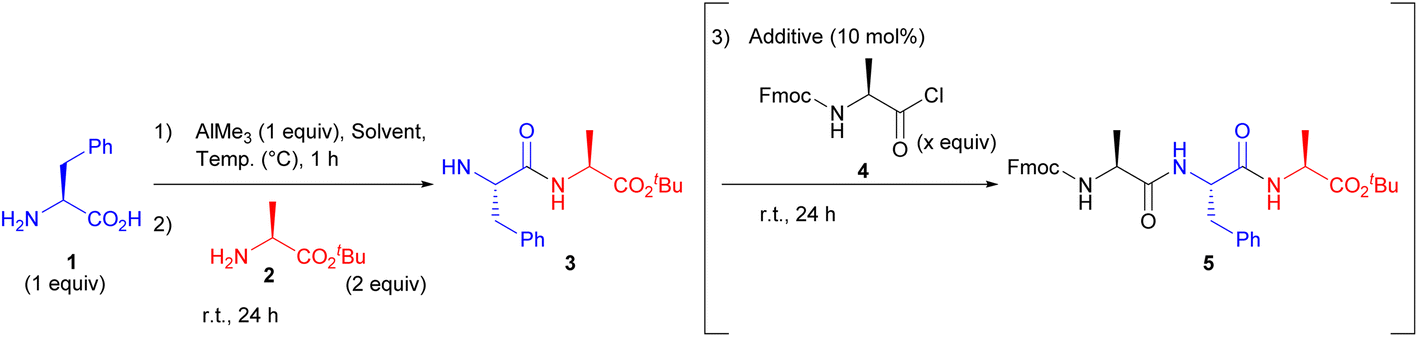
We first investigated the compatibility of AlMe3 for peptide bond formation. After pre-mixing the unprotected H-L-Phe-OH (1) and 1 equiv. AlMe3 in various organic solvents, H-L-Ala-OtBu (2) was added. As a result, the peptide bond formation hardly proceeded in THF, CPME, toluene, DMF, and DMSO (Table 1, entries 1–3, 5 and 6), while dipeptide (3) was obtained using MeCN, CHCl3, or DCM as a solvent (entries 4, 7, and 8). Reducing the temperature from room temperature to 0 °C during the pre-stirring step slightly improved the reaction efficiency, presumably because the five-membered ring consisting Al was relatively unstable at room temperature (entries 8 vs. 9 and 10 vs. 11). Furthermore, the use of 2 equiv. of AlMe3 enhanced the reaction efficiency (entries 8 vs. 10 and 9 vs. 11). Additionally, five-membered intermediates were barely formed using other common metal reagents, and unprotected amino acid was recovered in most cases (see ESI†). Interestingly, isolated yields increased upon addition of Fmoc-L-Ala-Cl (4) to form the corresponding tripeptide (entry 12). This is probably because treatment with 4 cleaved the N–Al bond, which was generated from the first coupling reaction, and the resulting product (5) could be fully recovered by column chromatography.13 Moreover, the addition of a catalytic amount of TMS-OTf and PivOH in the final step enhanced the reaction efficiency, and the addition of PivOH was especially effective (entry 17). It is likely that the acid catalyst has dual roles. It acts as a ligand for Al, and as a Lewis-acid to facilitate the peptide bond formation between the acid chloride and dipeptide. In addition, the reaction efficiency was improved to a satisfactory level by further increasing the amount of Fmoc-L-Ala-Cl to 3 equiv. (entry 18). Although the excess substrate results in the generation of undesired products such as Fmoc-L-Ala-L-Ala-OtBu, peptides of different lengths could be easily isolated, similar to the separation of unreacted substrates.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | Temp. (°C) | Additive | Fmoc-Ala-Cl (x equiv.) | Yield of 3a (%) | Yield of 5a (%) |
| a Isolated yields. b 2 equiv. of AlMe3 was used. | ||||||
| 1 | THF | r.t. | — | — | 0 | — |
| 2 | CPME | r.t. | — | — | 0 | — |
| 3 | Toluene | r.t. | — | — | 0 | — |
| 4 | MeCN | r.t. | — | — | 39 | — |
| 5 | DMF | r.t. | — | — | 0 | — |
| 6 | DMSO | r.t. | — | — | 0 | — |
| 7 | CHCl3 | r.t. | — | — | 55 | — |
| 8 | DCM | r.t. | — | — | 61 | — |
| 9 | DCM | 0 | — | — | 66 | — |
| 10b | DCM | r.t. | — | — | 67 | — |
| 11b | DCM | 0 | — | — | 77 | — |
| 12b | DCM | 0 | — | 2 | — | 85 |
| 13b | DCM | 0 | TMS-OTf | 2 | — | 89 |
| 14b | DCM | 0 | TfOH | 2 | — | Trace |
| 15b | DCM | 0 | AcOH | 2 | — | 84 |
| 16b | DCM | 0 | TFA | 2 | — | Trace |
| 17b | DCM | 0 | PivOH | 2 | — | 91 |
| 18b | DCM | 0 | PivOH | 3 | — | 95 |
With the optimised conditions in hand, we then investigated the one-pot synthesis of various tripeptides (Scheme 2). Generally, the peptide bond formation of sterically hindered amino acids, such as valine, leucine, and isoleucine, has rarely been accomplished. To clarify the effects of steric hindrance on the reaction outcome, we examined the substrate scope using valine as the nucleophile or electrophile. Consequently, various natural and unnatural amino acids were applicable for this one-pot system to produce the corresponding tripeptides. Under the optimal reaction conditions, substituents in the side chain had a negligible impact on the reaction yields (5a–5aq). Specifically, proline, which is an amino acid having secondary amine, could also be used as the nucleophile in the coupling reaction (5r). Furthermore, unnatural amino acids with quaternary carbons in the α-position could be used as both nucleophiles and electrophiles (5j, 5k, 5p). And various functional groups such as S, NH, ether, and ester were all well tolerated in this one-pot reaction (5ae–5aq). Fortunately, we did not face the issue of loss of enantiopurity presumably because of the rapid reaction and steric fixation of the five-membered intermediate. In fact, racemisation of the amino acids barely occurred even when Phg was used, which is extremely prone to epimerisation/racemisation14 (5v and 5ab). Although Boc-protected lysine afforded a low yield owing to the partial decomposition of the Boc-protected group in the side chain under the standard reaction conditions, the issue was resolved using an Fmoc-group in the side chain (5aq). The relatively low yield of 5aq is explained by the poor solubility of 5aq in DCM. Thus, our study presents a new strategy for synthesising various tripeptides, regardless of steric hindrances.
 | ||
| Scheme 2 One-pot tripeptide synthesis (1 + 1 + 1). | ||
Based on the impressive results of the one-pot tripeptide synthesis, we attempted to expand this strategy for synthesising longer peptides. Because the product of the first step is an N-unprotected dipeptide, that can be used as a nucleophile in the next step, we speculated that this protocol had enormous potential for further peptide elongation. To enhance the utility of this system, we investigated the one-pot tetrapeptide synthesis (Scheme 3). After performing the dipeptide synthesis under the standard reaction conditions, another unprotected amino acid (1′) and AlMe3 were added into the dipeptide solution and stirred at room temperature for further 24 h. The corresponding tetrapeptides were obtained in moderate yields upon treatment with amino acid chlorides (4) even when an amino acid with bulky substituents was used as the substrate (Scheme 3, 6a–6d).
 | ||
| Scheme 3 One-pot tetrapeptide synthesis (1 + 1 + 1 + 1). | ||
Furthermore, the protocol was amenable to one-pot pentapeptide synthesis (Scheme 4). First, we investigated the one-pot pentapeptide synthesis from a tripeptide. The condensation of H-Aib-OH, amino acid chloride (4), and tripeptide tert-butyl ester (5e′), which was prepared from 5evia the deprotection of the Fmoc-group, smoothly afforded the corresponding pentapeptide in 34% yield (Scheme 4A, 7a). Finally, the one-pot pentapeptide synthesis was achieved by adding the amino acids one by one at a time. After preparing the tripeptide according to Scheme 3, unprotected amino acid (1′′) and AlMe3 were added. After stirring for 24 h at 50 °C, Fmoc-amino acid chloride (4) was added. The corresponding pentapeptide was obtained in a moderate yield (Scheme 4B, 7b–7e). These results suggest that peptides can be elongated as long as the resulting peptide is soluble in the reaction medium. Thus, we developed the first step for the viable one-pot synthesis of long peptides.
 | ||
| Scheme 4 Pentapeptide synthesis. | ||
During the first pre-mixing process of the reagents, a gas bubble formation was observed. Although the generated gas was not characterised, it is well known that methane gas is evolved during the reaction of AlMe3 with protic nucleophiles.15 Based on this fact, additional experiments were performed to gain insights into the reaction mechanism, especially for the formation of the five-membered rings by gathering the generated gas via water displacement.16 First, we investigated the amount of gas generated using simple substrates bearing carboxylic acid or amine moieties by stirring with AlMe3 (Fig. 1). AlMe3 was slowly added to a two-necked flask charged with a DCM solution of the substrate at room temperature. The flask was connected to an inverted cylinder filled with water, and the volume of the generated gas was measured. As a result, the gas was quickly detected, and its amount was quantitatively estimated after the treatment of 3-phenylpropionic acid with AlMe3, while this gas was barely detected in the case of benzylamine. Notably, when AlMe3 was added in the presence of H-L-Phe-OH in DCM, gas was smoothly produced. And further gradual gas generation led to the accumulation of almost two equivalents of gas in 1 h. This indicates that the carboxylic acid moiety of H-L-Phe-OH reacted with AlMe3 first, and a subsequent reaction with the proximate amino group afforded the stable five-membered ring.
 | ||
| Fig. 1 Time course of gas generation. Generated gas was measured via a water displacement technique. For yield of gas calculations, methane was treated as an ideal gas. | ||
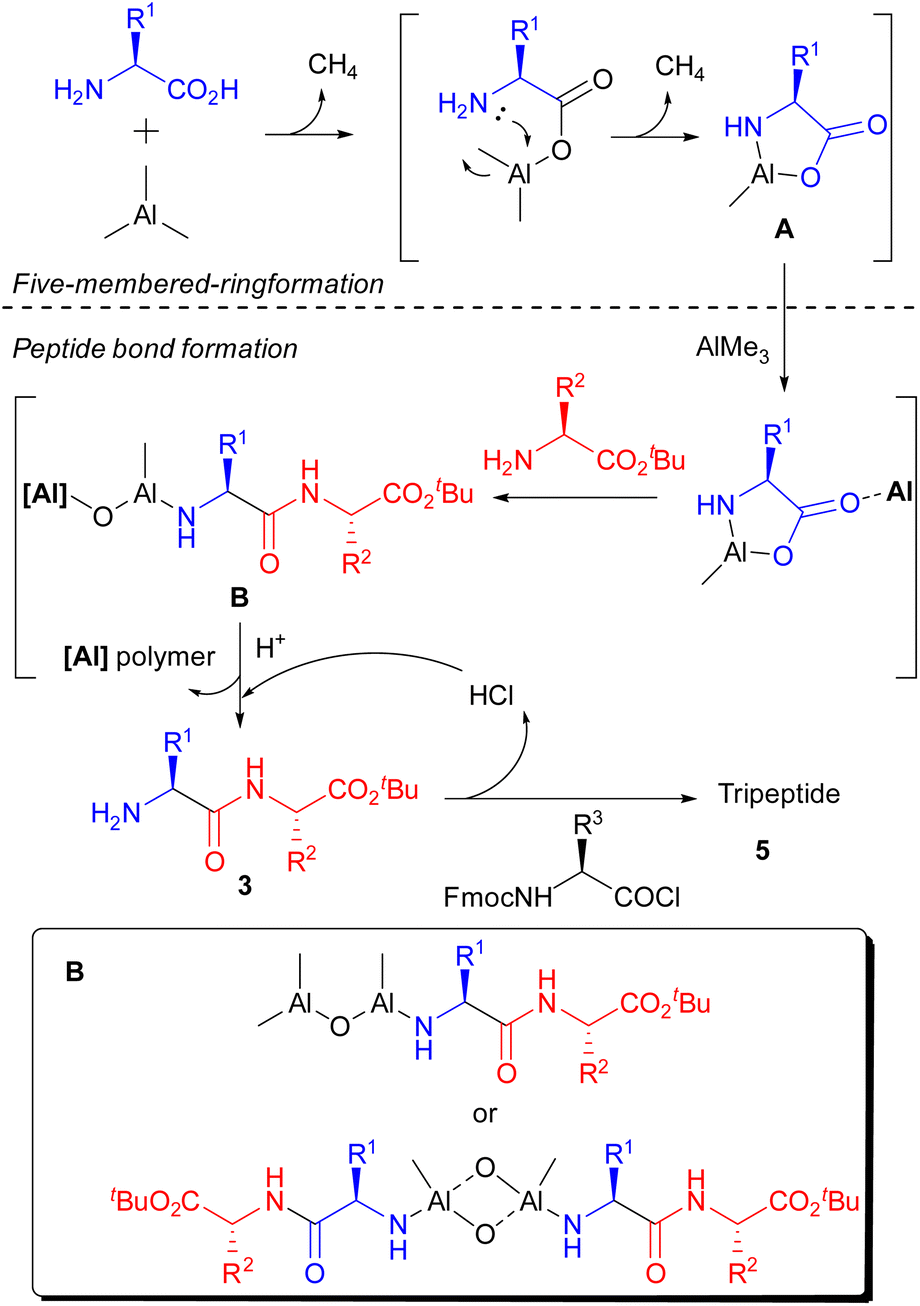
According to the result of this study on gas generation and literature, a proposed mechanism is illustrated in Scheme 5. Initially, AlMe3 reacted with the C-terminus of the amino acid, quickly releasing an equivalent amount of gas. And the N–Al bond, which could not be formed using benzylamine, was gradually constructed to afford a stable five-membered ring in 1 h (A). Then, peptide bonds were formed smoothly in the presence of nucleophilic amino acid ester via opening of the five-membered ring by the help of unreacted AlMe3 as a Lewis catalyst. We speculate that Al–N and Al–O bonds at the N-terminal of the produced dipeptide are retained under these reaction conditions, and the Al–O- bond reacted with unreacted AlMe3 or the constructed dimer to prevent further condensation with A (B).17 The generation of B prevented the recovery of produced peptides even though the key reaction proceeded sufficiently. After deprotection of the N-terminus by an acid to form 3, the condensation reaction with an Fmoc-protected amino acid chloride was carried out to afford the corresponding tripeptide (5).
 | ||
| Scheme 5 Plausible mechanism. | ||
Conclusions
AlMe3-mediated peptide bond formation of N-, and C-terminal free amino acids, followed by one-pot peptide elongation was accomplished. Peptide bonds were smoothly formed even with amino acids bearing bulky side chains, because the reactions proceeded via a less sterically hindered five-membered ring compared to linear amino acids. Furthermore, the system could be used for synthesising longer peptides like tetra- and pentapeptides by sequentially adding N-, and C-terminal unprotected amino acids and AlMe3. Cost-effectiveness, functional group tolerance, and the ability to undergo further elongation are important for the wide spread practical application of peptide synthesis. We expect that this novel and simple technique for the elongation of amino acids/peptides will represent a major breakthrough in the field of peptide synthesis in the near future.Data availability
All the data including experimental procedures, NMR, IR, and HRMS, are recorded in the ESI.†Author contributions
Funding acquisition and supervision were performed or provided by H. Yamamoto. Conception of the experiments, funding acquisition, investigation, analysis, and preparation of the manuscript were performed by T. Hattori.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Yuki Matsunaga for assistance with measurement of HPLC data. This work was partly supported by Grant-in-Aid for Scientific Research from the New Energy and Industrial Technology Development Organization (NEDO) project (JPNP14004), Shiono Wellness Foundation, The Naito Foundation, and Sumitomo Foundation.Notes and references
- (a) C. Morrison, Nat. Rev. Drug Discovery, 2018, 17, 531–533 CrossRef CAS PubMed; (b) A. Henninot, J. C. Collins and J. M. Nuss, J. Med. Chem., 2018, 61, 1382–1414 CrossRef CAS PubMed; (c) K. Fosgerau and T. Hoffmann, Drug Discovery Today, 2015, 20, 122–128 CrossRef CAS PubMed; (d) A. A. Kaspar and J. M. Reichert, Drug Discovery Today, 2013, 18, 807–817 CrossRef CAS PubMed; (e) D. J. Craik, D. P. Fairlie, S. Liras and D. Price, Chem. Biol. Drug Des., 2013, 81, 136–147 CrossRef CAS PubMed; (f) F. Albericio and H. G. Kruger, Future Med. Chem., 2012, 4, 1527–1531 CrossRef CAS PubMed; (g) J. J. Nestor Jr, Curr. Med. Chem., 2009, 16, 4399–4418 CrossRef CAS PubMed; (h) E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS PubMed.
- F. Hamdan, F. Tahoori and S. Balalaie, RSC Adv., 2018, 8, 33893–33926 RSC.
- (a) G. W. Anderson and A. C. McGregor, J. Am. Chem. Soc., 1957, 79, 6180–6183 CrossRef CAS; (b) F. C. McKay and N. F. Albertson, J. Am. Chem. Soc., 1957, 79, 4686–4690 CrossRef CAS; (c) L. A. Carpino and G. Y. Han, J. Org. Chem., 1972, 37, 3404–3409 CrossRef CAS; (d) A. Ishidro-Llobet, M. Álvarez and F. Albericio, Chem. Rev., 2009, 109, 2455–2504 CrossRef PubMed.
- (a) D. T. W. Chu, C. W. Nordeen, D. J. Hardy, R. N. Swanson, W. J. Giardina, A. G. Pernet and J. J. Plattner, J. Med. Chem., 1991, 34, 168–174 CrossRef CAS PubMed; (b) C. Prasad, Peptides, 1995, 15, 151–164 CrossRef PubMed; (c) J. Bujdák and B. M. Rode, Catal. Lett., 2003, 91, 149–154 CrossRef; (d) J. L. Sorrells and F. M. Menger, J. Am. Chem. Soc., 2008, 130, 10072–10073 CrossRef CAS PubMed; (e) F. de Azambuja and T. N. Parac-Vogt, ACS Catal., 2019, 9, 10245–10252 CrossRef CAS; (f) J. G. Kettle, S. K. Bagal, S. Bickerton, M. S. Bodnarchuk, J. Breed, R. J. Carbajo, D. J. Cassar, A. Chakraborty, S. Cosulich, I. Cumming, M. Davies, A. Eatherton, L. Evans, L. Feron, S. Fillery, E. S. Gleave, F. W. Goldberg, S. Harlfinger, L. Hanson, M. Howard, R. Howells, A. Jackson, P. Kemmitt, J. K. Kingston, S. Lamont, H. J. Lewis, S. Li, L. Liu, D. Ogg, C. Philips, R. Polanski, G. Robb, D. Robinson, S. Ross, J. M. Smith, M. Tonge, R. Whiteley, J. Yang, L. Zhang and X. Zhao, J. Med. Chem., 2020, 63, 4468–4483 CrossRef CAS PubMed; (g) F. de Azambuja, A. Loosen, D. Conic, M. van den Besselaar, J. N. Harvey and T. N. Parac-Vogt, ACS Catal., 2021, 11, 7647–7658 CrossRef CAS; (h) C. Simms, F. de Azambuja and T. N. Parac-Vogt, Chem.–Eur. J., 2021, 27, 17230–17239 CrossRef CAS PubMed.
- (a) S. H. van Leeuwen, P. J. L. M. Quaedflieg, Q. B. Broxterman and R. M. J. Liskamp, Tetrahedron Lett., 2002, 43, 9203–9207 CrossRef CAS; (b) A. N. Balaev, V. N. Osipov, K. A. Okhmanovich and V. E. Fedorov, Pharm. Chem. J., 2015, 49, 334–339 CrossRef CAS.
- (a) R. M. Lanigan, V. Karaluka, M. T. Sabatini, P. Starkov, M. Badland, L. Boulton and T. D. Sheppard, Chem. Commun., 2016, 52, 8846–8849 RSC; (b) M. T. Sabatini, V. Karaluka, R. M. Lanigan, L. T. Boulton, M. Badland and T. D. Sheppard, Chem.–Eur. J., 2018, 24, 7033–7043 CrossRef CAS PubMed.
- Z. Li, H. Fu, H. Gong and Y. Zhao, Bioorg. Chem., 2004, 32, 170–177 CrossRef CAS PubMed.
- J. Spengler, C. Böttcher, F. Albericio and K. Burger, Chem. Rev., 2006, 106, 4728–4746 CrossRef PubMed.
- T. Hattori and H. Yamamoto, J. Am. Chem. Soc., 2022, 144, 1758–1765 CrossRef CAS PubMed.
- (a) K. Tomooka, K. Matsuzawa, K. Suzuki and G. Tsuchihashi, Tetrahedron Lett., 1987, 28, 6339–6342 CrossRef CAS; (b) P. Deelertpaiboon, V. Reutrakul, S. Jarussophon, P. Tuchinda, C. Kuhakarn and M. Pohmakotr, Tetrahedron Lett., 2009, 50, 6233–6235 CrossRef CAS; (c) C. H. Fotsch and A. R. Chamberlin, J. Org. Chem., 1991, 56, 4141–4147 CrossRef CAS; (d) G. C. Crawley and M. T. Briggs, J. Org. Chem., 1995, 60, 4264–4267 CrossRef CAS; (e) K. Suzuki, T. Nagasawa and S. Saito, e-EROS Encyclopedia of Reagents for Organic Synthesis, John Wiley & Sons, Ltd., 2001 Search PubMed; (f) S. Drissi-Amraoui, Synlett, 2015, 26, 1424–1425 CrossRef CAS.
- (a) S. W. Chung, D. P. Uccello, H. Choi, J. I. Montgomery and J. Chen, Synlett, 2011, 14, 2072–2074 Search PubMed; (b) J. Li, K. Subramaniam, D. Smith, J. X. Qiao, J. J. Li, J. Qian-Cutrone, J. F. Kadow, G. D. Vite and B.-C. Chen, Org. Lett., 2012, 14, 214–217 CrossRef PubMed; (c) R. M. Lanigan and T. D. Sheppard, Eur. J. Org. Chem., 2013, 7453–7465 CrossRef CAS.
- S. F. Martin, M. P. Dwyer and C. L. Lynch, Tetrahedron Lett., 1998, 39, 1517–1520 CrossRef CAS.
- This was partially supported from the results that the yield of N-unprotected dipeptide was slightly improved by addition of potassium sodium tartrate or ammonium chloride in the extraction step.
- (a) G. G. Smith and T. Sivakua, J. Org. Chem., 1983, 48, 627–634 CrossRef CAS; (b) S. Popovic, H. Bieräugel, R. J. Detz, A. M. Kluwer, J. A. A. Koole, D. E. Streefkerk, H. Hiemstra and J. H. van Maarseveen, Chem.–Eur. J., 2013, 19, 16934–16937 CrossRef CAS PubMed; (c) M. A. Elsawy, C. Hewage and B. Walker, J. Pept. Sci., 2012, 18, 302–311 CrossRef CAS PubMed; (d) S. Fuse, Y. Mifune, H. Nakamura and H. Tanaka, Nat. Commun., 2016, 7, 13491 CrossRef CAS PubMed; (e) C. Liang, M. A. M. Behnam, T. R. Sundermann and C. D. Klein, Tetrahedron Lett., 2017, 58, 2325–2329 CrossRef CAS; (f) Y. Yeboue, M. Jean, G. Subra, J. Martinez, F. Lamaty and T.-X. Métro, Org. Lett., 2021, 23, 631–635 CrossRef CAS PubMed.
- (a) S. J. Young, B. Kellenberger, J. H. Reibenspies, S. E. Himmel, M. Manning, O. P. Anderson and J. K. Stille, J. Am. Chem. Soc., 1988, 110, 5744–5753 CrossRef CAS; (b) C. J. Linnen, D. E. Macks and R. D. Coombe, J. Phys. Chem. B, 1997, 101, 1602–1608 CrossRef CAS; (c) R. Liu, S. Yang, Y. Ding and D. Xia, J. Organomet. Chem., 2022, 958, 122192 CrossRef CAS.
- The water displacement method was used because methane gas has almost no solubility in water at room temperature under ambient pressure as shown below; (a) S. Yamamoto, J. B. Alcauskas and T. E. Crozier, J. Chem. Eng. Data, 1976, 21, 78–80 CrossRef CAS; (b) E. Wilhelm, R. Battino and R. J. Wilcock, Chem. Rev., 1977, 77, 219–262 CrossRef CAS; (c) Z. Duan, N. Møller, J. Greenberg and J. H. Weare, Geochim. Cosmochim. Acta, 1992, 56, 1451–1460 CrossRef CAS.
- (a) T. N. P. Luhtanen, M. Linnolahti and T. A. Pakkanen, J. Organomet. Chem., 2002, 648, 49–54 CrossRef CAS; (b) R. Glaser and X. Sun, J. Am. Chem. Soc., 2011, 133, 13323–13336 CrossRef CAS PubMed; (c) M. Linnolahti, A. Laine and T. A. Pakkanen, Chem.–Eur. J., 2013, 19, 7133–7142 CrossRef CAS PubMed; (d) K. Yang and R. Glaser, Catalysts, 2022, 12, 312 CrossRef CAS; (e) M. Zhang, Y. Feng, L. Lou, H. Zhang, J. Wang and Y. Yang, Org. Process Res. Dev., 2022, 26, 1506–1513 CrossRef CAS; (f) C. Wang, X. Wu, H. Li, J. Qu and Y. Chen, Angew. Chem., Int. Ed., 2022, 61, e202210484 CAS; (g) J. Liu, D. Liu, W. Nie, H. Yu and J. Shi, Org. Chem. Front., 2022, 9, 163–172 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc00208j |
| This journal is © The Royal Society of Chemistry 2023 |