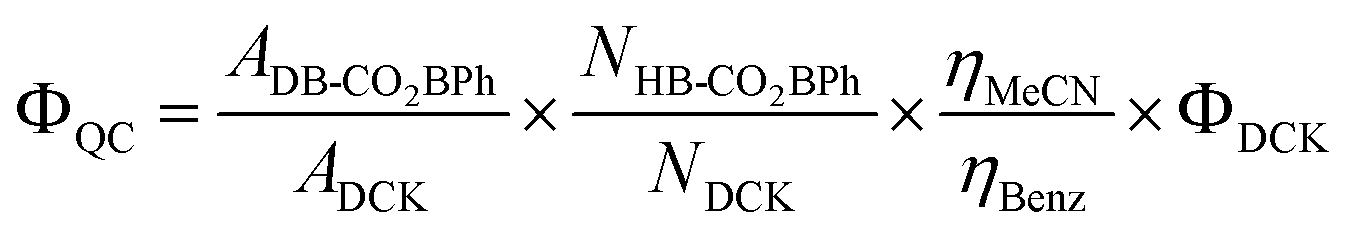DOI:
10.1039/D3SC01074K
(Edge Article)
Chem. Sci., 2023,
14, 5802-5810
Quantum chain amplification in nanocrystalline Dewar benzenes by intramolecular sensitization†
Received
27th February 2023
, Accepted 19th April 2023
First published on 25th April 2023
Abstract
Quantum chain reactions are characterized by the formation of several photoproducts per photon absorbed (ΦQC > 1) and constitute a promising signal amplification mechanism. The triplet-sensitized isomerization of Dewar benzene is known to undergo quantum chain reactions characterized by an adiabatic valence-bond isomerization to the excited state of Hückel benzene, which is able to transfer its triplet energy to a new ground state Dewar benzene that reacts to continue the chain. Given that diffusion-mediated energy transfer is the chain-limiting event in solution, we demonstrate here that reactions in crystals are significantly more efficient by taking advantage of energy transfer by a presumed exciton delocalization mechanism. Using Dewar benzenes with covalently attached, high energy triplet sensitizers we have demonstrated the efficiency of the solid state by the amplification of a quantum yield of ca. ΦQC ≈ 76 in acetonitrile solution to as much as ca. ΦQC ≈ 100–120 in submicron size specimens prepared by the re-precipitation method, and up to ca. ΦQC ≈ 300 with microcrystalline powders suspended in water.
Introduction
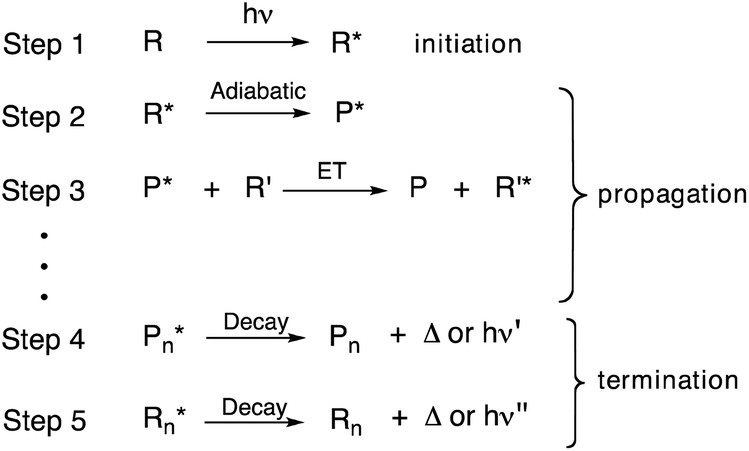
Most organic photochemical processes involve the excitation of a single molecule followed by return to the ground state by releasing heat, emitting a single photon, transferring its energy to another molecule, or undergoing a reaction to give a primary photoproduct.1 Exceptions from this norm include nonlinear photochemical processes involving a non-identical number of photochemical events and photons absorbed. Such systems can be utilized for signal amplification devices, catalysis, and solar cells.2–5 Examples include coherent and stepwise two photon absorption,6,7 fusion of two or more low energy photons to generate one high-energy excited state by triplet–triplet annihilation,8 and the fission of a single excitation generated with a high-energy photon that results in two excited states, each with half the energy of the original.9,10 An equally interesting, but significantly less explored process, occurs when a single photon leads to many chemical events in a “quantum chain reaction”.11 Quantum chain reactions start by formation of an excited state (step 1, Scheme 1) and are followed by a relatively uncommon propagation step involving an adiabatic photochemical reaction where the photoproduct is formed in the excited state (step 2, Scheme 1).12–15 This is followed by energy transfer to a new ground-state reactant that prolongs the chain (step 3, Scheme 1). Chain termination occurs when either the excited-state product (P*) or reactant (R*) undergo thermal or radiative decay (step 4 and/or step 5, Scheme 1).
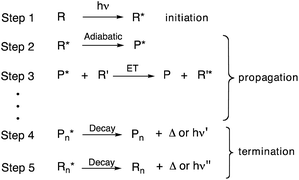 |
| | Scheme 1 Initiation, propagation and termination steps of a quantum chain reaction. | |
Despite the promise offered by chemical systems leading to multiple events per photon absorbed, quantum chain reactions are limited both by the small number of adiabatic reactions known at this time and the lack of convenient strategies to optimize the energy transfer step.16–32 In this regard, singlet state quantum chains are severely limited by the short lifetimes of the excited photoproducts, which make energy transfer by diffusion-mediated mechanisms unfavourable. In fact, adiabatic reactions that take place in the singlet state are commonly established by detecting the fluorescence33,34 or transient absorption of the excited photoproduct, rather than by observation of a quantum yield of product formation that is greater than one, which is the key signature of a quantum chain. We recently proposed that an effective way to circumvent this limitation is by carrying out quantum chain reactions in crystalline solids,35,36 where energy transfer can occur by an ultrafast exciton delocalization mechanism.37–41 We showed that the adiabatic decarbonylation of diphenylcyclopropenone to diphenyl acetylene could not enter a quantum chain process in solution despite having an adiabatic quantum yield ΦAR = 1 because there is no time for diffusion-mediated energy transfer within the ca. 8 ps lifetime of the excited photoproduct.35,36,42 By contrast, quantum chain reactions with quantum yields up to ΦQC = 3.3 could be measured in aqueous nanocrystalline suspensions of diphenylcyclopropenone,35,36 where energy transfer is mediated by a rapid exciton delocalization mechanism.42 Efficient quantum chains (ΦQC ≫ 1) require adiabatic reactions with high quantum efficiencies (ΦAR ≈ 1) and are limited by the number of energy transfer steps, n, as shown in eqn (1)–(3),36
| | | ΦQC = (ΦAR)1 + (ΦAR)2 + (ΦAR)3 + … (ΦAR)n | (1) |
| | | ΦAR = kAR/[kAR + kR-dec] | (2) |
It should be noted that efficient adiabatic reactions (ΦAR ≈ 1) require reaction rates that are much greater than that of the decay of the excited reactant by all other pathways (kAR ≫ kR-dec). The maximum value n in eqn (1) is determined by the relative rates of energy transfer from the excited photoproduct (kET) in relation to its rate of decay (kP-dec).36
Based on the above analysis, one may expect that triplet quantum chain reactions carried out in crystalline solids have the potential of reaching very high ΦQC values by taking advantage of energy transfer by triplet exciton hopping, which is known to approach the picosecond time scale.37–41,43 Assuming ideal adiabatic reactions with ΦAR ≈ 1, the theoretical limit for a triplet quantum chain is given by the value of n in eqn (1), as defined in eqn (3). Based on a general approximation, one may expect n = kET/kP-dec to take values as large as 109 if triplet excitons were to have jumping rates of ca. kET ≈ 1012 s−1 and triplet lifetimes (τT) were to extend into the millisecond time scales (τT = 1/kdec ≈ 10−3 s).
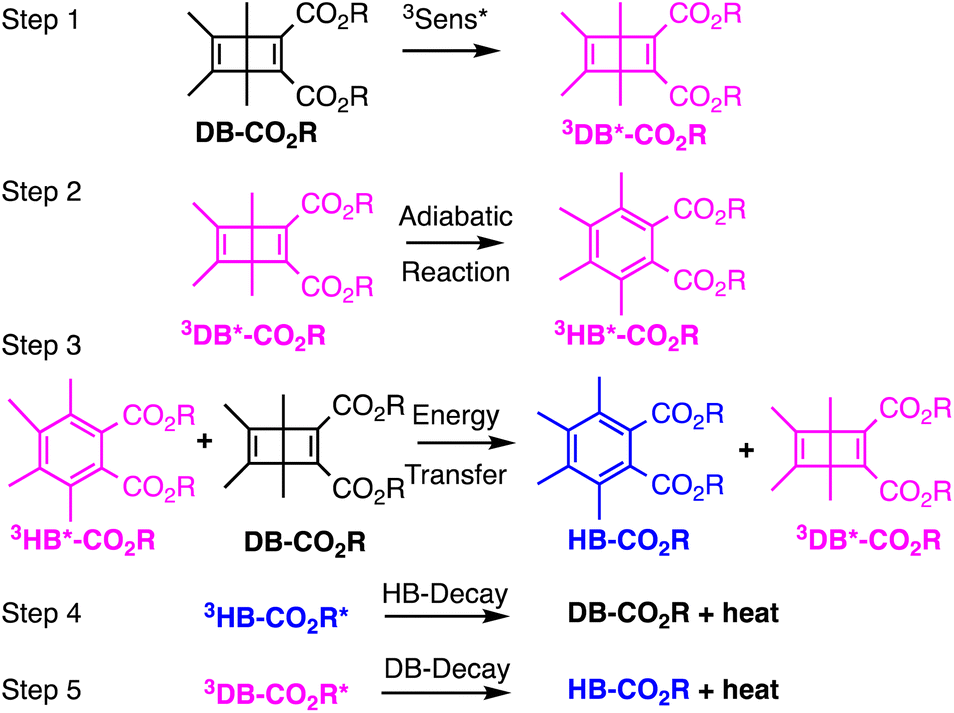
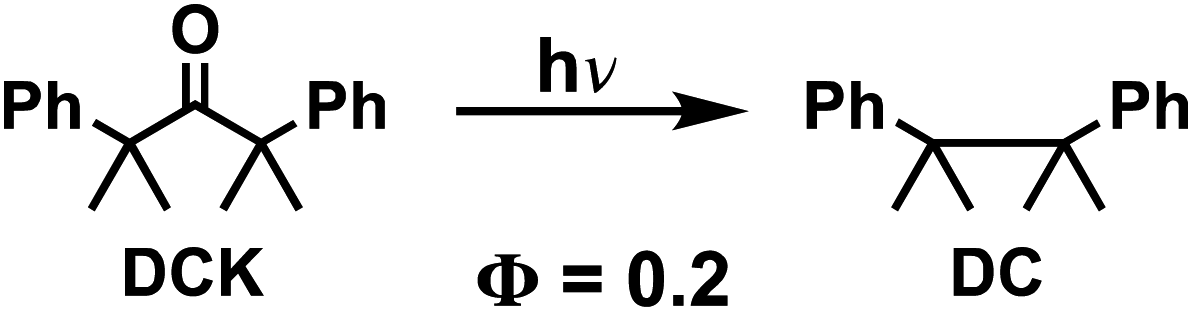
As a first test of this hypothesis, we explore here the quantum chain reaction of crystalline Dewar benzene to Hückel benzene. Early studies by Turro44 established a concentration-dependent quantum chain reaction that reaches a limiting experimental value of ca. ΦQC = 0.5 but extrapolates to ΦQC ≈ 10 at infinite Dewar benzene concentration. It was shown that triplet energy sensitizers with ET > 63 kcal mol−1 are required for efficient energy transfer, indicating an experimental upper limit for the triplet excitation energy of Dewar benzene.44 More recently, using a Dewar benzene 3,4,5,6-tetramethyl-1,2-diester derivative (DB-CO2Me, Scheme 2), Merkel et al. reported quantum yields as high as ΦQC = 120 in ethyl acetate,45 and Ferrar et al. showed the feasibility of quantum chain reactions in polymer matrices upon the addition of external sensitizers and co-sensitizers.46 In the latter example, reactions were carried out with reactant, sensitizer and co-sensitizer randomly dispersed in the polymer matrix, such that a distribution of donor and acceptor distances can lead to a range of energy transfer efficiencies that affect both sensitization and chain propagation steps. By contrast, Dewar benzene molecules in crystals have the close proximity required for efficient energy transfer by an exciton delocalization mechanism, especially if the reactant is equipped with an intramolecular sensitizer. A simple solution to this problem is to substitute the dicarboxymethyl groups in DB-CO2Me for 4-hydroxy-benzophenones in DB-CO2BPh so that triplet sensitization can occur intramolecularly. It is well known that electronic excitation of the benzophenone chromophore leads to intersystem crossing and efficient formation of the triplet n, π* excited state within a few picoseconds. Thus, the original excited state is localized on the benzophenone part of the bichromophoric compound (DB-CO2-3BPh*, not shown in Scheme 2) and is followed by intramolecular energy transfer to the covalently attached Dewar benzene moiety 3DB*-CO2BPh, which initiates the quantum chain (step 1). An adiabatic triplet state reaction forms the triplet excited state of the Hückel benzene photoproduct, 3HB*-CO2BPh step 2, which acts as the quantum chain carrier. In step 3, the triplet photoproduct 3HB*-CO2BPh should be well suited in crystals to transfer energy to a neighboring ground-state Dewar benzene DB-CO2BPh to propagate the chain. While chain termination is expected to occur via steps 4 or 5 when either excited state decays back to the ground state manifold, step 5 is accounted for in the quantum yield of the adiabatic reaction, so that the chain length given by the value of n depends on the relative rates of step 3 and 4. As described below, we were able to demonstrate a concentration-dependent quantum chain in solution with modest values ranging from ca. ΦQC ≈ 2.5–76 for reactant concentrations ranging from 5–50 mM. By contrast, quantum chain reactions carried out with samples prepared by the re-precipitation method led to average quantum chain values of ca. ΦQC ≈ 100, even though the samples were shown to be semi-crystalline. Notably, a qualitative comparison of the latter with microcrystalline powders suspended in water showed an increase in quantum chain values up to ca. ΦQC ≈ 300. Evidence for a fast adiabatic reaction was obtained by nanosecond laser flash photolysis experiments showing that a long-lived transient generated from the starting Dewar benzene DB-CO2BPh is identical to the one generated from the Hückel benzene isomer, which corresponds to the chain carrier 3HB*-CO2BPh.
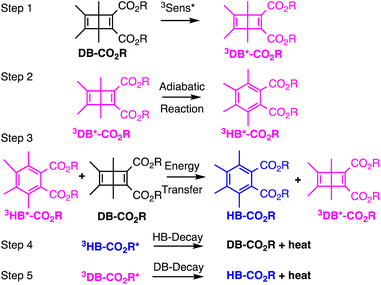 |
| | Scheme 2 Steps for the triplet sensitized quantum chain reaction of Dewar benzenes. | |
Experimental section
Synthesis and characterization of Dewar benzenes
Samples of Dewar benzene derivatives used in this study were prepared by conventional synthetic procedures that are described in the ESI section.†
Laser flash photolysis detection of the quantum chain carrier
Nanosecond laser flash photolysis experiments with DB-CO2BPh and HB-CO2BPh were carried out using a Brilliant B Quantel Nd:YAG laser operating at 355 nm with a pulse width of ca. 8 ns as the excitation source. Samples were introduced to a mounted 1 cm quartz flow cell through a continuous one-pass flow system to ensure that only unreacted material was continuously sampled. Samples were sparged with argon for at least an hour prior to flowing into the flow cell and remained continuously sparged for the entire experiment. The characteristic triplet–triplet absorption of the benzophenone sensitizer was determined by measuring the triplet absorption and lifetime of 4-acetoxybenzopheone (4-AcOBPh).
Quantum chain reaction in solution
The quantum yields of product formation from DB-CO2BPh (ΦHB-OBPh) were determined using the photodecarbonylation of dicumyl ketone (DCK) as a chemical actinometer.49 The selection of DCK was based on its known quantum yield in solution and in the solid state,49 with the latter measured in nanocrystalline suspensions.50 Thus, photochemical excitation of DCK in benzene solution at λ = 312 nm leads to the consumption of starting material with a quantum yield of ΦDCK(soln) = 0.41 ± 0.04. Similarly, photochemical reactions in the crystalline state using optically dense nanocrystalline suspensions proceed with the exclusive formation of dicumene (DC) with a quantum yield of ΦDC(cryst) = 0.20 ± 0.02.49
Results and discussion
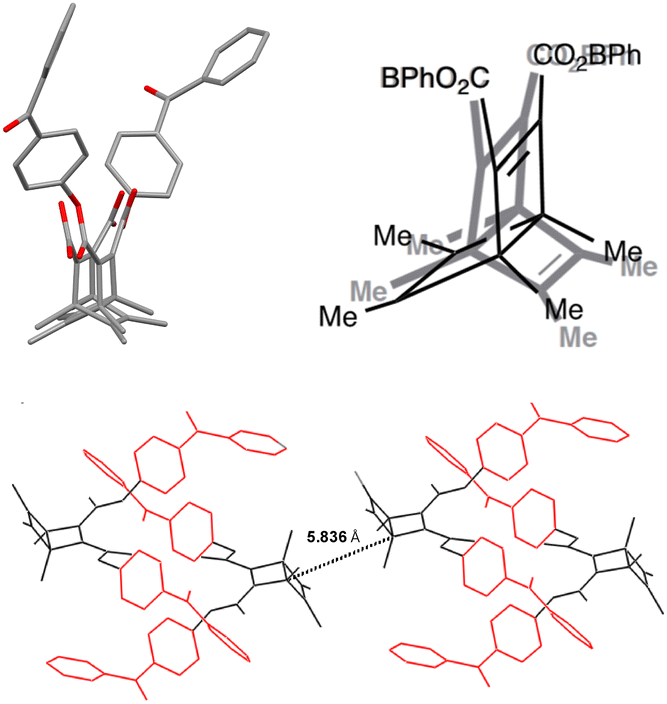
Samples of DB-CO2BPh proved to be crystalline after purification. Melting with concomitant reaction to the valence-bond isomerized product HB-CO2BPh was shown to occur at ca. 109 °C. Diffraction data from cubic prisms obtained by slow evaporation from diethyl ether were solved in the space group P21/n with 4 molecules per unit cell. Molecules of DB-CO2BPh crystallize with the Dewar benzene fragments disordered over two positions, alternating their concave and convex faces with occupancies of 64% and 36%. The disorder extends to the carboxylate groups, which connect to the corresponding benzophenones in a manner that they themselves are not disordered, as illustrated in Fig. 1.
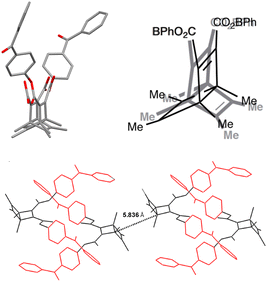 |
| | Fig. 1 (Upper left) Capped stick representation of the X-ray molecular structure of DB-CO2BPh illustrating the disorder of the Dewar benzene group and, (upper right) line structure illustrating its two orientations. (Bottom) Packing view illustrating the interdigitated benzophenone arrangement between pairs of molecules, and the closest distance between neighbouring Dewar benzenes, which may be related to the distance for energy transfer. | |
Nanocrystalline suspensions
Knowing that the determination of an efficient quantum chain must be established by measuring the quantum yield of reaction, which is given by the number of product molecules formed per photon absorbed, we had to verify that samples of DB-CO2BPh are able to form nanocrystalline suspensions. It is well known that spectroscopic methods based on measurements of the transmission of light in bulk solids are extremely challenging due to the high optical density, scattering, birefringence, and dichroism that characterize single crystals and polycrystalline samples. These effects also make it difficult to measure the number of photons absorbed by the sample, which is essential for quantum yield measurements. We have previously shown that crystals with sizes that are equal or smaller than the wavelength of light are able to mitigate these optical challenges,40,41,49,51 making it possible to measure transmission spectra, and to determine quantum yields with particle loadings that are high enough to trap every photon emitted from a calibrated light source. It is now well established that many compounds are able to form aqueous nanocrystalline suspensions of well-defined crystals with average dimensions on the order of 50–500 nm, depending on the specific sample and experimental parameters. In our case, concentrated MeCN solutions of DB-CO2BPh were slowly added to a rapidly stirring solution of cetyl trimethyl ammonium bromide (CTAB) at concentrations that are 1/50th of the critical micelle concentration (CMC) in millipore water. Size determination by dynamic light scattering (DLS, ESI page S14†) and scanning electron microscopy revealed prismatic crystals in the range ca. 200 nm to ca. 1 μm in size (Fig. 2).
 |
| | Fig. 2 SEM images of DB-CO2BPhca. 1 μm size crystals obtained from a dried suspension on a Si surface. Scale bars of (a) 10 micron and (b) 100 nm are shown. | |
Product analysis in solution and in powder samples
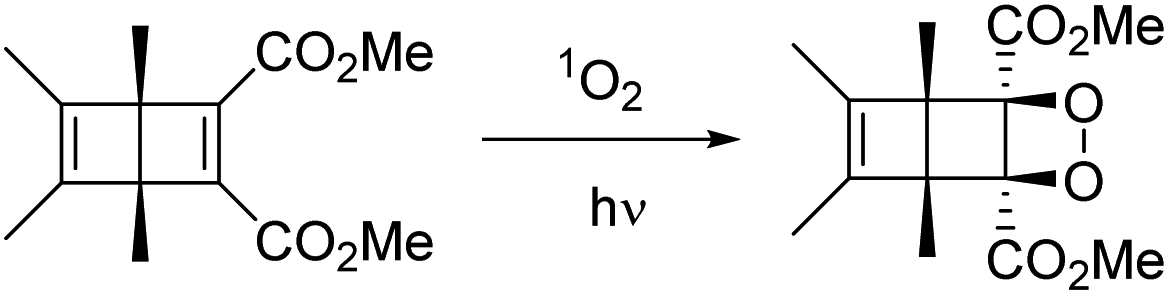
Dilute (ca. 0.1 M) degassed acetonitrile solutions of DB-CO2H and DB-CO2BPh exposed to UV light with λ ≥ 295 nm resulted in full conversion to the corresponding Hückel benzenes HB-CO2H and HB-CO2BPh as the only product. Similar results were observed with dry powders placed between two glass slides, indicating that the reaction can proceed as a solid-to-solid transformation. We noticed that reactions of DB-CO2BPh carried out in solution in the presence of oxygen lead to the formation of a new product consistent with the formation of an endoperoxide by reaction of the Dewar benzene with singlet oxygen (Scheme 3).
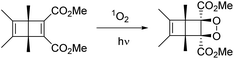 |
| | Scheme 3 Reaction of Dewar benzene with singlet oxygen in solution. | |
As expected, reactions carried out with DB-CO2BPh powders and nanocrystalline suspensions were not affected by the presence of oxygen. Samples of HB-CO2BPh could be crystallized and were shown to melt at 142 °C, higher than the melting point of the DB-CO2BPh reactant (ca. 109 °C). This suggests the possibility of a solid-to-solid reaction. However, powder X-ray diffraction analysis of samples of DB-CO2BPh before and after reaction revealed that the reaction proceeds by amorphization, rather than a potential single-crystal to single-crystal transformation, or a more common solid-to-solid reaction by a reconstructive phase transition. Unfortunately, we were not able to get diffraction-quality single crystals of the Hückel benzene photoproduct HB-CO2BPh.
Spectroscopic characterization
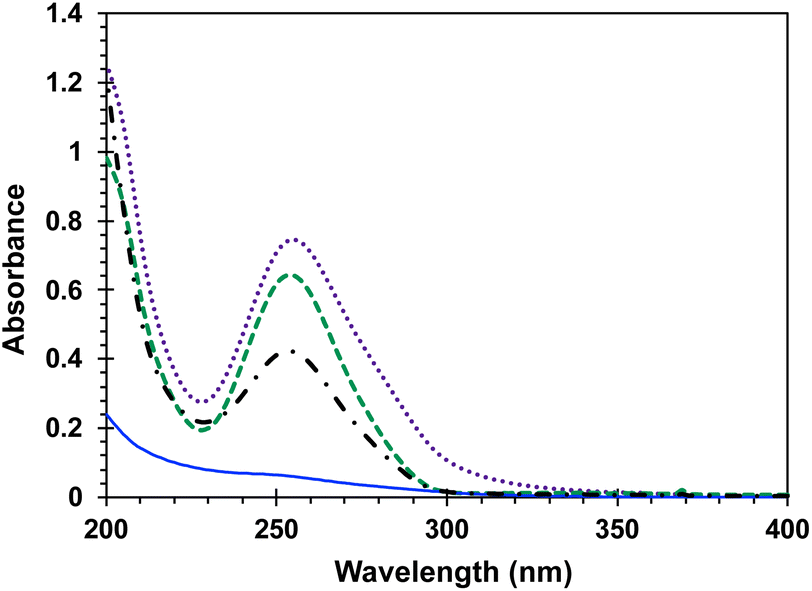
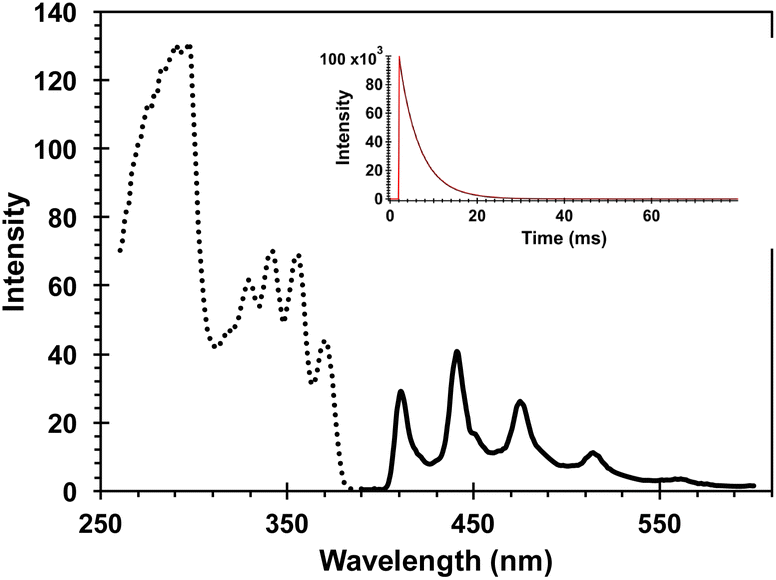
Included in Fig. 3 is a comparison of the steady state UV spectra of the sensitizer-linked Dewar benzene DB-CO2BPh along with those of samples containing equimolar Dewar benzene methyl ester DB-CO2Me, two equivalents of 4-AcOBPh that was used as a model chromophore, and a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 4-AcOBPh and DB-CO2Me. A relatively weak interaction of the two proximal benzophenone esters and the Dewar benzene can be inferred by noting that the UV spectrum of DB-CO2BPh is more intense and slightly broader than the spectrum obtained when the two independent chromophores are mixed in the same molar ratio (Fig. 3). From this absorption spectra a ground-state energy value of ca. 59 kcal mol−1 can be calculated for DB-CO2BPh. We determined the triplet energy of the benzophenone sensitizer by measuring the phosphorescence spectrum of Hückel benzene HB-CO2BPh in dilute 2-methyltetrahydrofuran glass at 77 K (Fig. 4). A characteristic emission with vibrational resolution and a relatively short (4.8 ms) lifetime typical of an n, π* excited state was obtained, along with a triplet energy of ca. 65 kcal mol−1 estimated from the onset of the 0–0 vibrational band.
:1 mixture of 4-AcOBPh and DB-CO2Me. A relatively weak interaction of the two proximal benzophenone esters and the Dewar benzene can be inferred by noting that the UV spectrum of DB-CO2BPh is more intense and slightly broader than the spectrum obtained when the two independent chromophores are mixed in the same molar ratio (Fig. 3). From this absorption spectra a ground-state energy value of ca. 59 kcal mol−1 can be calculated for DB-CO2BPh. We determined the triplet energy of the benzophenone sensitizer by measuring the phosphorescence spectrum of Hückel benzene HB-CO2BPh in dilute 2-methyltetrahydrofuran glass at 77 K (Fig. 4). A characteristic emission with vibrational resolution and a relatively short (4.8 ms) lifetime typical of an n, π* excited state was obtained, along with a triplet energy of ca. 65 kcal mol−1 estimated from the onset of the 0–0 vibrational band.
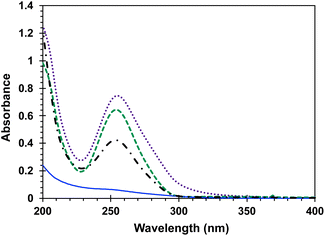 |
| | Fig. 3 Steady state UV absorption spectra in MeCN of 2.08 × 10−5 M DB-CO2Me (blue), 4.16 × 10−5 M 4-AcOBPh (green), 1.65 × 10−5 M DB-CO2BPh (purple), and a solution with 1:2 molar ratio of the DB-CO2Me and 4-AcOBPh (black). | |
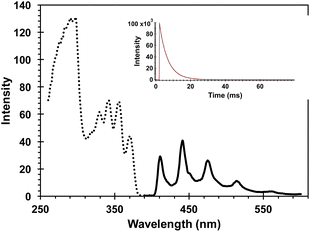 |
| | Fig. 4 Phosphorescence excitation (dashed line) and emission (solid line) spectra of HB-CO2BPh in a 2-methyltetrahydrofuran glass at 77 K. The emission spectrum was detected at 441 nm and the emission obtained by excitation at 343 nm. The phosphorescence decay shown in the inset occurs with a time constant of 4.8 ms. | |
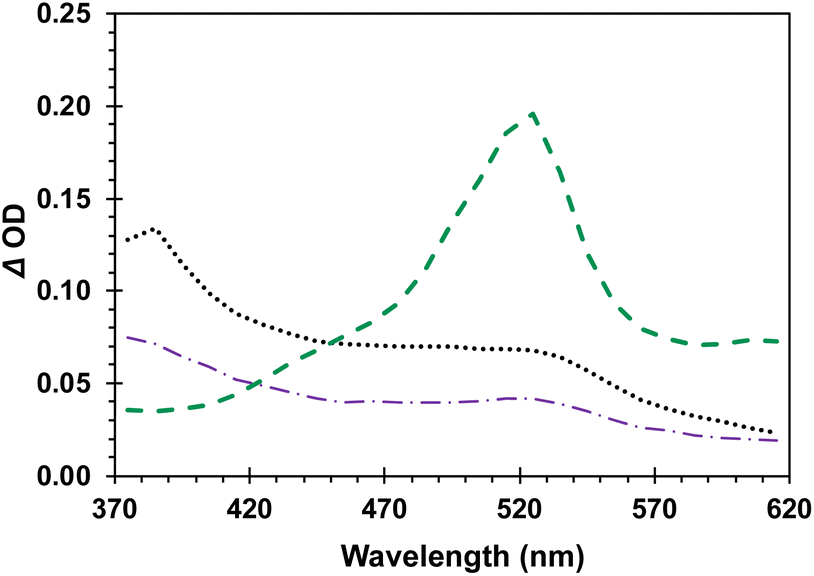
As shown by the green dashed line in Fig. 5, triplet 4-AcOBPh has the characteristic triplet benzophenone transient absorption with a λmax = 520 nm. Freeze-pump-thawed samples revealed a lifetime of 14.6 μs in dilute MeCN solution (ESI, page S19†). The transient spectrum obtained from solutions of DB-CO2BPh was significantly different with broad absorption bands at ca. 480 and 520 nm. The spectrum of DB-CO2BPh can be quenched with oxygen.
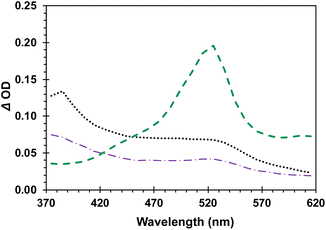 |
| | Fig. 5 Transient absorption spectra (λexc = 355 nm) of 5 mM 4-AcOBPh (green), 0.76 mM Dewar benzene DB-CO2BPh (purple), and 1.5 mM Hückel benzene HB-CO2BPh (black) acquired immediately after the laser pulse in Ar saturated MeCN. | |
Subsequent experiments carried out with Hückel benzene HB-CO2BPh showed that the same transient is formed within the 8 ns pulse, suggesting the spectrum obtained upon irradiation of DB-CO2BPh is indeed the triplet state 3HB*-CO2BPh formed adiabatically from the originally sensitized 3DB*-CO2BPh, in agreement with steps 1 and 2 in Scheme 2.
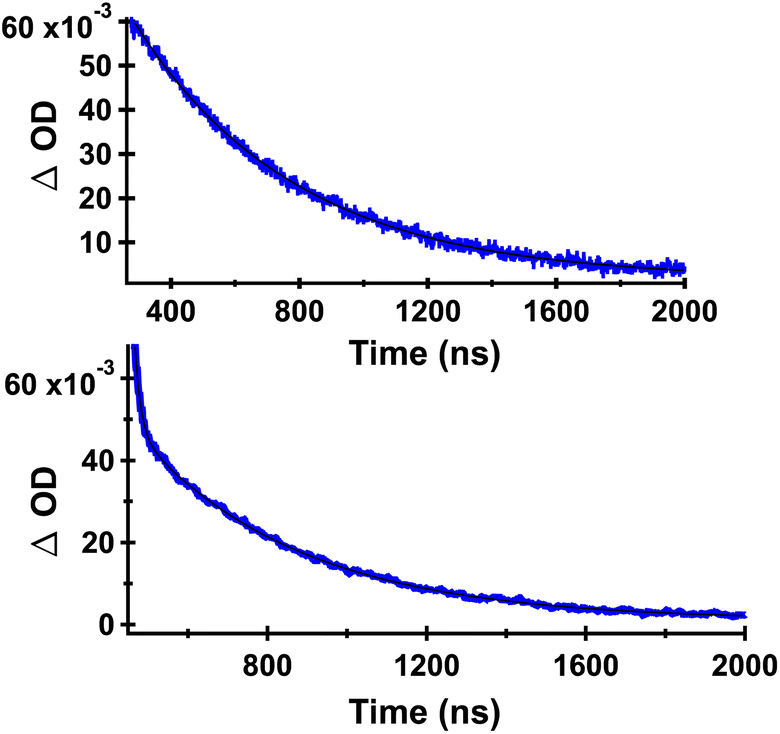
Decays measured at 380–420 nm for DB-CO2BPh showed clean monoexponential decay (top of Fig. 6) with a lifetime of 503 ns (Table 1). Interestingly, absorption decays for the same sample measured at 530 nm revealed a double exponential with a short-lived component of ca. 16 ns that accounts for only 2% of the decay, and a majority (98%) long-lived component of 420 ns. As indicated in Table 1, the decay kinetics obtained from samples of HB-CO2BPh were very similar. We tentatively assign the short-lived component at 530 nm as originating from a residual fraction of the originally excited benzophenone-localized triplet states, in the process of transferring their triplet energy to the corresponding Dewar or Hückel benzene chromophore, with the former undergoing a fast adiabatic reaction to form the observed triplet Hückel benzene. A monoexponential behavior at the shorter wavelength is consistent with having no significant absorption from the benzophenone chromophore at 385 nm, suggesting that it corresponds to the benzophenone-linked triplet benzene.
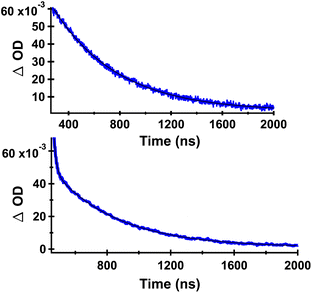 |
| | Fig. 6 Decay curves of DB-CO2BPh in Ar-saturated MeCN detected at 385 nm (top) and at 530 nm (bottom). Data points are shown in blue and the corresponding fit with a black line. The decay data obtained from HB-CO2BPh were essentially identical, indicating that they arise from the same species (Table 1). | |
Table 1 Transient lifetimes and pre-exponential factors of DB-CO2BPh and HB-CO2BPh in Ar-saturated MeCN
| Compound (λdet)a |
A
1
|
τ
1 (%w1)b,c |
A
2
|
τ
2 (%w2)b,c |
χ
2 (104) |
|
Detection wavelength in parentheses.
Percentage weighted contribution of component “i” is given by %wi = 100 [(Aiτi)/(A1τ1 + A2τ2)].
Lifetimes in nanoseconds.
|
|
DB-CO2BPh (530 nm) |
0.024 |
16.2 (2%) |
0.046 |
420 (98%) |
5.7 |
|
DB-CO2BPh (385 nm) |
— |
— |
0.058 |
503 |
2.2 |
|
HB-CO2BPh (530 nm) |
0.022 |
14.5 (2%) |
0.14 |
419 (98%) |
11.5 |
|
HB-CO2BPh (385 nm) |
— |
— |
1 |
465 |
6.5 |
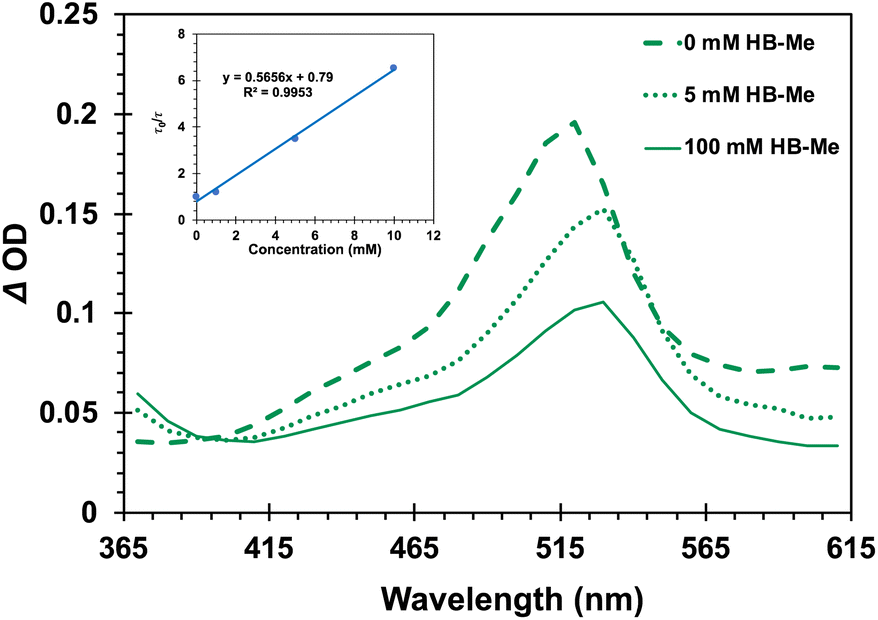
The importance of the intramolecular benzophenone sensitizer for the formation of the triplet state chain carrier was also demonstrated with samples of the Dewar benzene methyl ester DB-CO2Me, which failed to produce an observable triplet transient on its own, as expected for a fast non-adiabatic singlet-state reaction and a low triplet yield that results from inefficient intersystem crossing. However, the spectroscopic signature of the suggested chain carrier was obtained using the ground-state Hückel benzene methyl ester HB-CO2Me as a quencher of triplet 4-AcOBPh.47 As shown in Fig. 7, the spectrum of 34-AcOBPh* (ET ∼ 60 kcal mol−1) evolved into that of 3HB*-CO2Me as the concentration of the ground-state Hückel benzene increased from 0 mM (Fig. 7, dashed green line) to 100 mM (solid green line). The relatively intense signal of 34-AcOBPh* with a maximum at ca. 520 nm was replaced by a red-shifted weaker signal with λmax ≈ 530 nm accompanied by an increase of the absorption intensity at 365 nm. Stern–Volmer analysis using changes in the decay rate as a function of increasing quencher concentration (Fig. 7, inset) revealed a bimolecular quenching rate constant of kq = 6.3 × 107 M−1 s−1, which is about two orders of magnitude slower than diffusion control.48
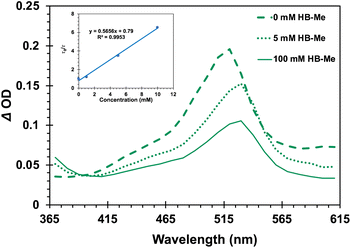 |
| | Fig. 7 Evolution of the transient absorption (λexc = 355 nm) of 4-AcOBPh as a function of added Hückel benzene HB-CO2Me Inset: Stern–Volmer plot built with the lifetime of triplet 4-AcOBPh in the presence of increasing concentrations of HB-CO2Me. A bimolecular rate constant kq = 6.3 × 107 M−1 s−1 can be estimated from this analysis. | |
Quantum chain reaction in solution
Knowing that the efficiency of a quantum chain reaction in solution depends on the efficiency of a concentration-dependent energy transfer step, one should expect the observed quantum yield to increase as a function of reactant concentration. With that in mind, we carried out quantum yield determinations with DB-CO2BPh concentrations varying from ca. 3–50 mM in MeCN. Optically dense, deoxygenated samples of the Dewar benzene and the actinometer DCK (Scheme 4) were irradiated in parallel at 312 nm in 5 mL Pyrex tubes to assure that all photons entering each sample were absorbed and all samples were similarly exposed to the light source. The moles of product (N) formed from the sample and the actinometer were calculated with internal standards using 1H NMR.
 |
| | Scheme 4 Photodecarbonylation of DCK in nanocrystalline suspension. | |
The following equation was utilized to calculate quantum yields,
where
ADB-CO2BPh refers to the absorbance of
DB-CO2BPh,
ADCK is the absorbance of
DCK,
NHB-CO2BPh are the number of moles of isomerized product
HB-CO2BPh,
NDCK is the number of moles of reacted
DCK, and
ηMeCN and
ηbenzene are the refractive indices of MeCN and benzene, respectively. Experiments were run up to five times over different reaction times with conversion values ranging from
ca. 14% to 75%.
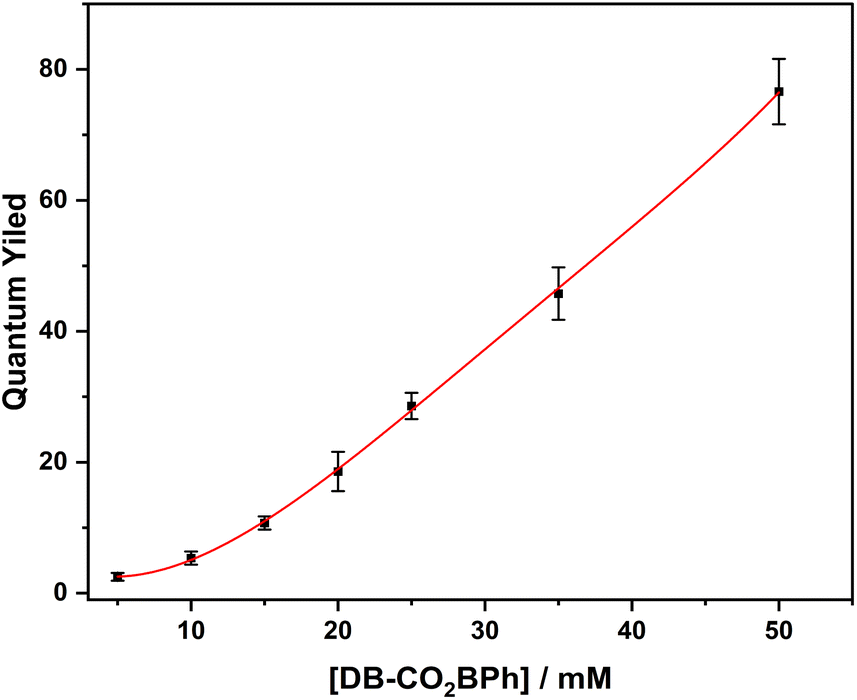
Fig. 8 displays the quantum yield of formation of
HB-CO2BPh photoproduct as a function of the initial concentration of
DB-CO2BPh, varying from 5 mM to 50 mM. An initial value of
ΦHB-CO2BPh = 2.5 ± 0.2 with 5 mM of Dewar benzene increased up to
ΦHB-CO2BPh = 76 ± 3.5 with a reactant concentration of 50 mM.
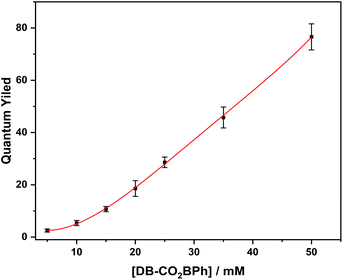 |
| | Fig. 8 Quantum yield values of DB-CO2BPh at 5, 10, 15, 20, 25, 35, 50 mM in MeCN solution. | |
Quantum chain reaction in nanocrystalline suspensions
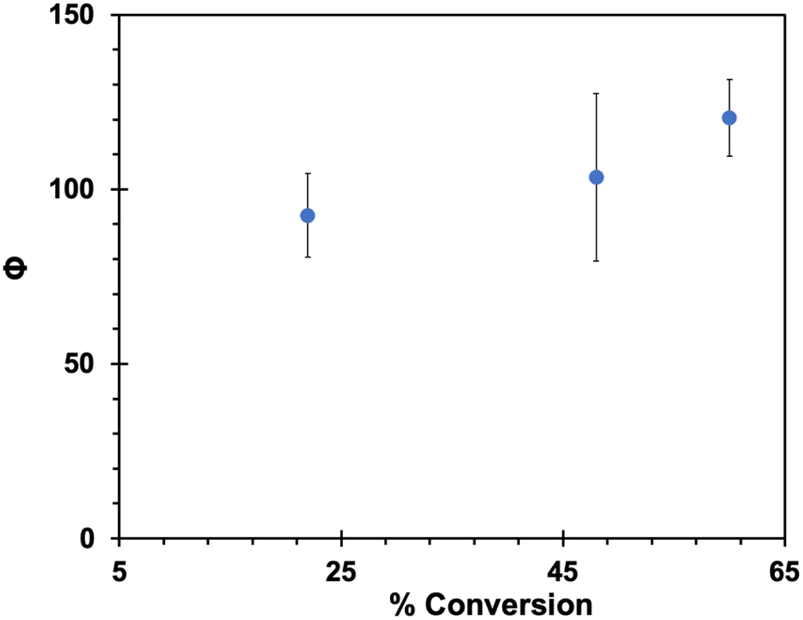
Aqueous nanocrystalline suspensions of DB-CO2BPh and DCK were generated by the re-precipitation method.50 Sample loadings were adjusted to generate optically dense suspensions of DB-CO2BPh and DCK that were optically matched, as indicated by a UV-Vis immersion probe. Suspensions were irradiated in 3 mL Pyrex tubes at 312 nm. Initial experiments revealed complete conversion of DB-CO2BPh at the shortest irradiation time of 10 seconds. A modified setup required suspensions of DB-CO2BPh and DCK to be irradiated with the former placed behind a 5 × 5 cm neutral density filter with a 1% transmission at 312 nm, so that the photon dose reaching the Dewar benzene was 100 times weaker (images of the setup can be found in page S25 of the ESI†). The results shown in Fig. 9 reveal that quantum yields of product formation in the suspension sample have values that range from ca. ΦHB-CO2BPh = 90–120, as the extent of reaction increases from 22% to 60% conversion. An increase in the observed quantum yield as a function of accumulated product suggests that the HB-CO2BPh product may be a better absorber and potentially a more effective sensitizer. Alternatively, it is also possible that the efficiency of the solid-state adiabatic reaction is improved once the crystals of the reactant are perturbed by the presence of the photoproduct.
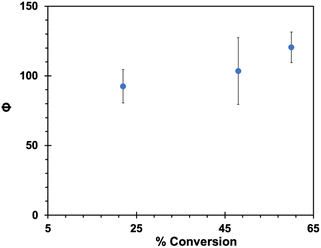 |
| | Fig. 9 Quantum yields of formation of HB-CO2BPh in the solid state using suspensions prepared by the re-precipitation method. | |
While the large reactivity and high quantum yields observed in Fig. 9 support the expectation of an efficient quantum chain reaction, quantum yields on the order of 90–120 are far from the optimum if we assume a radiative triplet-state lifetime on the order of a few milliseconds (Fig. 4, inset) with a fast adiabatic reaction, and exciton hopping in the picosecond time scale. If an adiabatic reaction in the nanosecond time scale were the limiting factor, one should expect chain lengths as high as 106, given by the number of reactions that can occur within a few milliseconds. Notably, all our attempts towards the optical detection of the proposed chain carrier 3HB*-CO2BPh in the solid state led to no signal detection under conditions where nanocrystals of 4-AcOBPh, used as a model compound, gave a strong signal with a relatively short 1.2 μs lifetime (ESI, page S18†). The lack of an observable transient suggests that all 90 to 100 reactions in the chain take place within the ca. 8 ns laser pulse, indicating that a quenching event must be the chain-limiting factor. In fact, we have previously shown that crystalline benzophenones are susceptible of efficient self-quenching by a reductive charge transfer mechanism.51 It was shown that nanocrystalline 4,4′-diamino-benzophenone, with strong electron donating aromatic rings (σ+ = −1.3), has a triplet lifetime of only ca. 6.2 ps at 298 K. On the other end, electron poor 4,4′-benzophenone dicarboxylic acid (σ+ = 0.42) has a triplet lifetime of ca. 97 μs, which is seven orders of magnitude greater. Self-quenching by the neighboring benzophenones in HB-CO2BPh is also evident in solution by comparing the lifetimes of freeze-pump-thawed samples of the former with those of 4-AcOBPh. A lifetime of 14.6 μs (ESI, page S19†) obtained for the isolated ketone at 298 K in acetonitrile is reduced to 1.9 μs for the bichromophoric compound (ESI, page S20†). This suggests that self-quenching also accounts for the 1.2 μs lifetime of nanocrystalline 4-AcOBPh, which has a σ+ = −0.19 and fits well in the previously published Hammet plot with a reaction constant ρ = −2.85.51
An alternative explanation based on the quality of the sample was explored by X-ray powder diffraction analysis of centrifuged and dried suspension samples of DB-CO2BPh. The results revealed a largely amorphous powder, suggesting that the main sample population in suspension is not crystalline, even though one is able to find crystalline specimens by scanning electron microscopy (Fig. 2). To determine whether the use of a higher quality, more crystalline sample can have a greater effect on the quantum chain, we carried out experiments to compare the extent of reaction between polycrystalline powders suspended in surfactant-containing water, and the nanocrystalline suspensions generated by the re-precipitation method. While aggregate sizes are different, and microcrystals are prone to multiple excitations and triple-triplet annihilation, we reasoned that a greater conversion in suspended polycrystalline powders would qualitatively confirm a greater efficiency for an exciton-mediated quantum chain amplification. Thus, suspended polycrystalline powder samples were prepared by dispensing about 10 mg of DB-CO2BPh into 3 mL of a vortexing submiceller CTAB solution; nanocrystalline suspensions generated by the re-precipitation method could be obtained with loading values up to ca. 2 mg in 3 mL of submiceller CTAB. While stirring, the two sample types were irradiated using an immersion 302 nm pen lamp. Analyzing the total amount of product generated under such similar conditions revealed that ground crystals suspended in water have quantum yields of ΦQC ≈ 300, indicating reactivity that is ca. 3–4 times greater than that of the mainly amorphous sample obtained by the re-precipitation method. This result qualitatively confirms that exciton-mediated energy transfer in the crystalline phase has the potential of increasing the quantum amplification in the triplet-sensitized reaction of Dewar benzenes.
Conclusions
Taking advantage of a crystalline tetramethyl Dewar benzene diester with 4-hydroxybenzophenone groups acting as intramolecular triplet sensitizers, we were able to show that triplet quantum chain reactions in the crystalline state display quantum yields of product formation in the range of ΦQC = 100–300, which are greater than those measured as a function of concentration in acetonitrile solution (ΦQC = 5–76), but significantly smaller than those expected based on assumed fast adiabatic reactions, efficient energy transfer, and long triplet lifetimes. The adiabatic and triplet state nature of the valence-bond isomerization reaction was confirmed in solution by taking advantage of laser flash photolysis, where the same transient was observed from both the Dewar and the Hückel benzene isomers. The lack of an observable transient in the solid state was interpreted in terms of a quantum chain that occurs within the ca. 8 ns laser pulse, suggesting a quenching mechanism as a limiting factor for the solid state quantum chain. Another contributing factor for the lower than expected quantum chain was unveiled when we showed that samples obtained by re-precipitation are formed as semicrystalline aggregates with coexisting amorphous and crystalline phases. With work in progress, we are addressing the roles of excited-state quenching and crystallinity to find conditions where exciton-mediated reactions can produce quantum chain reactions with values that extend into the thousands or millions of reactions per photon.
Data availability
Data supporting this manuscript is available within the ESI.†
Author contributions
E. Rivera and I. Paul carried out synthesis, photochemical experiments, and nanocrystal characterization. E. Rivera and J. Fajardo, Jr. carried out spectroscopic and kinetic measurements. M. Garcia-Garibay directed this project and all authors contributed to the preparation and revision of the manuscript and have given approval for publication.
Conflicts of interest
There are no conflict of interest to declare.
Acknowledgements
This work was supported by NSF grants CHE-1855342 and CHE-2154210. ER acknowledges the UCLA Graduate Division for a Cota Robles Award and JFJ a UCLA Chancellor's Postdoctoral Fellowship.
Notes and references
-
N. J. Turro, V. Ramamurthy and J. C. Scaiano, Modern Molecular Photochemistry of Organic Molecules, U Science, Sausalito, CA, 2010 Search PubMed.
- J. C. Johnson, A. J. Nozik and J. Michl, Triplet Yield from Singlet Fission in a Thin Film of 1,3-Diphenylisobenzofuran, J. Am. Chem. Soc., 2010, 132, 16302–16303 CrossRef CAS PubMed.
- S. Jin, H.-J. Son, O. K. Farha, G. P. Wiederrecht and J. T. Hupp, Energy Transfer from Quantum Dots to Metal-Organic Frameworks for Enhances Light Harvesting, J. Am. Chem. Soc., 2013, 135, 955–958 CrossRef CAS PubMed.
- M. K. Manna, S. Shokri, G. P. Wiederrecht, D. J. Gosztola and A. J. Ayitou, New perspectives for triplet-triplet annihilation-based photon upconversion using all-organic energy donor & acceptor chromophores, Chem. Commun., 2018, 54, 5809–5818 RSC.
- D. N. Congreve, J. Lee, N. J. Thompson, E. Hontz, S. R. Yost, P. D. Reusswig, M. E. Bahlke, S. Reineke, V. Voorhis and M. A. Baldo, External quantum efficiency above 100% in a singlet-exciton-fission-based organic photovoltaic cell, Science, 2013, 340, 334–337 CrossRef CAS PubMed.
- M. F. Joubert, Photon avalanche upconversion in rare earth laser materials, Opt. Mater., 1999, 11, 181–203 CrossRef CAS.
- J. C. Scaiano, L. J. Johnston, W. G. McGimpsey and D. Weir, Photochemistry of organic reaction intermediates: novel reaction paths induced by two-photon laser excitation, Acc. Chem. Res., 1988, 21, 22–29 CrossRef CAS.
- T. N. Singh-Rachford and F. N. Castellano, Photon upconversion based on sensitized triplet–triplet annihilation, Coord. Chem. Rev., 2010, 254, 2560–2573 CrossRef CAS.
- K. Miyata, F. S. Conrad-Burton, F. L. Geyer and X.-Y. Zhu, Triplet Pair States in Singlet Fission, Chem. Rev., 2019, 119, 4261–4292 CrossRef CAS PubMed.
- M. B. Smith and J. Michl, Singlet Fission, Chem. Rev., 2010, 110, 6891–6936 CrossRef CAS PubMed.
- H. L. Hyndman, B. M. Monroe and G. S. Hammond, Mechanisms of photochemical reactions in solution. LX. Photochemical isomerization of 2,4-hexadiene via a quantum-chain mechanism”, J. Am. Chem. Soc., 1969, 91, 2852–2859 CrossRef CAS.
- N. J. Turro, J. Mcvey, V. Ramamurthy and P. Lechtken, Adiabatic Photoreactions of Organic Molecules, Angew. Chem., Int. Ed. Engl., 1979, 18, 572–586 CrossRef.
- T. Forster, Diabatic and Adiabatic Processes in Photochemistry, Pure Appl. Chem., 1970, 24, 443–449 CrossRef.
- T. Forster, Excimers, Angew. Chem., Int. Ed. Engl., 1969, 8, 333 CrossRef.
- N. J. Turro, P. Lechtken, A. Lyons, R. R. Hautala, E. Carnahan and T. J. Katz, Molecular photochemistry. LXIV. Photochemical generation of electronically excited organic products in adiabatic pericyclic photoreactions. Unexpected propensity toward spin inversion in a retrocycloaddition and in a valence isomerization, J. Am. Chem. Soc., 1973, 95, 2035–2037 CrossRef CAS.
- J. Saltiel, D. E. Townsend and A. Sykes, Quantum chain process in the sensitized cis-trans photoisomerization of 1,3 dienes, J. Am. Chem. Soc., 1973, 95, 5968–5973 CrossRef CAS.
- J. Saltiel, J. D'Agostino, E. D. Megarity, L. Metts, K. R. Neuberger, M. Wrighton and O. C. Zafiriou, The cis-trans photoisomerization of olefins, Org. Photochem., 1973, 3, 1–113 CAS.
- W. A. Yee, S. J. Hug and D. S. Kliger, Direct and sensitized photoisomerization of 1,4-diphenylbutadienes, J. Am. Chem. Soc., 1988, 110, 2164–2169 CrossRef CAS.
- S. Ganapathy and R. S. H. Liu, Photoisomerization of polyenes. 30. Quantum chain processes in photoisomerization of the all-trans, 7-cis, and 11-cis isomers of retinal, J. Am. Chem. Soc., 1992, 114, 3459–3464 CrossRef CAS.
- M. Sundahl and O. Wennerstrom, Catalysis of a photochemical reaction; a cis-trans isomerization proceeding
by a quantum chain process, J. Photochem. Photobiol., A, 1996, 98, 117–120 CrossRef CAS.
- M. Brink, H. Jonson and M. Sundahl, Catalysis of triplet state cis-trans isomerizations making a quantum chain process more efficient, J. Photochem. Photobiol., A, 1998, 112, 149–153 CrossRef CAS.
- D. G. Whitten and J. A. Mercer-Smith, Photosensitization o Stilbene Isomerization by Palladium and Platinum Porphyrins, an Intermolecular Quantum Chain Process, J. Am. Chem. Soc., 1978, 110, 2620–2625 Search PubMed.
- V. R. Gopal, M. A. Reddy and V. J. Rao, Wavelength Dependent Trans to Cis and Quantum Chain Isomerizations of Anthrylethylene Derivatives, J. Org. Chem., 1995, 60, 7966–7973 CrossRef CAS.
- J. Saltiel, S. Wang, D. Ko and D. A. Gormin, Cis-Trans Photoisomerization of the 1,6-Diphenyl-1,3,5-hexatrienes in the Triplet State. The Quantum Chain Mechanism and the Structure of the Triplet State, J. Phys. Chem. A, 1998, 102, 5383–5392 CrossRef CAS.
- K. Tokumaru and T. Arai, Factors Determining the Adiabatic or the Diabatic Pathway of the Photoisomerization of Unsaturated Bonds, Bull. Chem. Soc. Jpn., 1995, 68, 1065–1087 CrossRef CAS.
- V. P. Kazakov, A. I. Voloshin and N. M. Shavaleev, Chemiluminescence in visible and infrared spectral regions and quantum chain reactions upon thermal and photochemical decomposition of adamantylideneadamantane-1,2-dioxetane in the presence of chelates Pr(dpm)3 and Pr(fod)3, J. Photochem. Photobiol., 1998, 11, 177–186 CrossRef.
- P. Lechtken, A. Yekta and N. J. Turro, Tetramethyl-1,2-dioxetane. Mechanism for an autocatalytic decomposition. Evidence for a quantum chain reaction, J. Am. Chem. Soc., 1973, 95, 3027–3028 CrossRef CAS.
- N. J. Turro, N. E. Schore and A. Yekta, Quantum chain processes. Novel procedure for measurement of quenching parameters. Evidence that exothermic triplet-triplet energy transfer is not diffusion limited and an estimation of the efficiency of exothermic quenching in a solvent cage, J. Am. Chem. Soc., 1974, 96, 1936–1938 CrossRef CAS.
- N. J. Turro and W. H. Waddell, Quantum chain processes. Direct observation of high quantum yields in the direct and photosensitized excitation of tetramethyl-1,2-dioxetane, Tetrahedron Lett., 1975, 25, 2069–2072 CrossRef.
- G. B. Schuster, N. J. Turro, H. Steinmetzer, A. P. Schaap, G. Faler, W. Adam and J. C. Liu, Adamantylideneadamantane-1,2-dioxetane. Chemiluminescence and decomposition kinetics of an unusually stable 1,2-dioxetane, J. Am. Chem. Soc., 1975, 97, 7110–7117 CrossRef CAS.
- T. Wilson and A. P. Schaap, Chemiluminescence from cis-diethoxy-1,2-dioxetane. Unexpected effect of oxygen, J. Am. Chem. Soc., 1971, 93, 4126–4136 CrossRef CAS.
- N. J. Turro, P. Lechtken, N. E. Schore, G. Schuster, H. Steinmetzer and A. Yekta, Tetramethyl-1,2-dioxetane. Experiments in chemiexcitation, chemiluminescence, photochemistry, chemical dynamics, and spectroscopy, Acc. Chem. Res., 1974, 7, 97–105 CrossRef CAS.
- R. V. Carr, B. Kim, J. K. McVey, N. C. Yang, W. Gerhartz and J. Michl, Photochemistry and photophysics of 1,4-dewarnaphthalene, Chem. Phys. Lett., 1976, 39, 57–60 CrossRef CAS.
- N. C. Yang, R. V. Carr, E. Li, J. K. McVey and S. A. Rice, 2,3-Naphtho-2,5-bicyclo[2.2.0]hexadiene, J. Am. Chem. Soc., 1976, 96, 2297–2298 CrossRef.
- G. Kuzmanich, A. Natarajan, K. K. Chin, M. Veerman, C. J. Mortko and M. A. Garcia-Garibay, Solid-state photodecarbonylation of diphenylcyclopropenone: a quantum chain process made possible by ultrafast energy transfer, J. Am. Chem. Soc., 2008, 130, 1140–1141 CrossRef CAS PubMed.
- G. Kuzmanich, M. N. Gard and M. A. Garcia-Garibay, Photonic amplification by a singlet state quantum chain reaction in the photodecarbonylation of crystalline diarylcyclopropenones, J. Am. Chem. Soc., 2009, 131, 11606–11614 CrossRef CAS PubMed.
- R. C. Powel and A. G. Soos, Singlet exciton energy transfer in organic solids, J. Lumin., 1975, 11, 1–45 CrossRef.
-
C. E. Swenberg and N. E. Gaecintov, in Organic Molecular Photophysics, ed. J. B. Birks, John Wiley and Sons, London, 1973, vol. 1 Search PubMed.
- Z. S. Yoon, M.-C. Yoon and D. Kim, Excitonic Coupling in covalently linked multiporphyrin systems by matrix diagonalization, J. Photochem. Photobiol., C, 2005, 6, 249–263 CrossRef CAS.
-
C. Swenberg and M. Pope, Electronic Processes in Organic Crystals and Polymers, Oxford University Press, 1999 Search PubMed.
- Z. Soos and R. Powell, Generalized Random-Walk Model for Singlet-Exciton Energy Transfer, Phys. Rev. B: Solid State, 1972, 6, 4035–4046 CrossRef CAS.
- S. C. Doan, G. Kuzmanich, M. N. Gard, M. A. Garcia-Garibay and B. J. Schwartz, Ultrafast spectroscopic observation of a quantum chain reaction: The photodecarbonylation of nanocrystalline diphenylcyclopropenone, J. Phys. Chem. Lett., 2012, 3, 81–86 CrossRef CAS.
- A. Ryasnyanskiy and I. Biaggio, Triplet exciton dynamics in rubrene single crystals, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 193203 CrossRef.
- N. J. Turro, V. Ramamurthy and T. J. Katz, Energy Storage and Release. Direct and sensitized photoreactions of Dewar benzene and prismane, Nouv. J. Chim., 1978, 1, 363–365 Search PubMed.
- P. B. Merkel, Y. Roh, J. P. Dinnocenzo, D. R. Robello and S. Farid, Highly Efficient Triplet Chain Isomerization of Dewar Benzenes: Adiabatic Rate Constants from Cage Kinetics, J. Phys. Chem. A, 2007, 111, 1188–1199 CrossRef CAS PubMed.
- L. Ferrar, M. Mis, J. P. Dinnocenzo, S. Farid, P. B. Merkel and D. R. Robello, Quantum Amplified Isomerization in Polymers Based on Triplet Chain Reactions, J. Org. Chem., 2008, 73, 5683–5692 CrossRef CAS PubMed.
- M. Tomasulo, S. L. Kaanumal, S. Sortino and F. M. Raymo, Synthesis and Properties of Benzophenone–Spiropyran and Naphthalene–Spiropyran Conjugates, J. Org. Chem., 2007, 72, 595–605 CrossRef CAS PubMed.
-
The bimolecular diffusion rate constant in acetonitrile is 6.83 × 109 M−1 s−1, CRC Handbook of Chemistry and Physics, ed. R. C. Weast, CRC Press, Inc., Boca Raton, FL, 68th edn, 1987–1988 Search PubMed.
- M. Veerman, M. J. E. Resendiz and M. A. Garcia-Garibay, Large Scale Photochemical Reactions of Nanocrystalline suspensions: A Promising Green Chemistry Method, Org. Lett., 2006, 8, 2615–2617 CrossRef CAS PubMed.
- H. Kasai, H. S. Nalwa, H. Oikawa, S. Okada, H. Matsuda, N. Minami, A. Kakuta, K. Ono, A. Mukoh and H. Nakanishi, A Novel Preparation Method of Organic Microcrystals, Jpn. J. Appl. Phys., 1992, 31, L1132–L1134 CrossRef CAS.
- G. Kuzmanich, S. Simoncelli, M. N. Gard, F. Spanig, B. L. Henderson, D. M. Guldi and M. A. Garcia-Garibay, Excited State Kinetics in Crystalline Solids: Self-Quenching in Nanocrystals of 4,4’-Disubstituted Benzophenone Triplets Occurs by a Reductive Quenching Mechanism, J. Am. Chem. Soc., 2011, 43, 17296–17306 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures and analytical data, nanocrystal characterization and photochemical protocols. CCDC 2084271. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc01074k |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence and
Miguel A.
Garcia-Garibay
and
Miguel A.
Garcia-Garibay