
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA “tug-of-war” effect tunes Li-ion transport and enhances the rate capability of lithium metal batteries†
Han
Zhang
ab,
Ziqi
Zeng
*a,
Mengchuang
Liu
a,
Fenfen
Ma
c,
Mingsheng
Qin
ab,
Xinlan
Wang
ab,
Yuanke
Wu
ab,
Sheng
Lei
ab,
Shijie
Cheng
a and
Jia
Xie
 *a
*a
aState Key Laboratory of Advanced Electromagnetic Engineering and Technology, School of Electrical and Electronic Engineering, Huazhong University of Science and Technology, Wuhan 430074, Hubei, China. E-mail: xiejia@hust.edu.cn; ziqizeng@hust.edu.cn
bState Key Laboratory of Materials Processing and Die & Mould Technology, School of Materials Science and Engineering, Huazhong University of Science and Technology, Wuhan 430074, Hubei, China
cGuSu Laboratory of Materials, Suzhou 215123, Jiangsu, China
First published on 7th February 2023
Abstract
“Solvent-in-salt” electrolytes (high-concentration electrolytes (HCEs)) and diluted high-concentration electrolytes (DHCEs) show great promise for reviving secondary lithium metal batteries (LMBs). However, the inherently sluggish Li+ transport of such electrolytes limits the high-rate capability of LMBs for practical conditions. Here, we discovered a “tug-of-war” effect in a multilayer solvation sheath that promoted the rate capability of LMBs; the pulling force of solvent–nonsolvent interactions competed with the compressive force of Li+-nonsolvent interactions. By elaborately manipulating the pulling and compressive effects, the interaction between Li+ and the solvent was weakened, leading to a loosened solvation sheath. Thereby, the developed electrolytes enabled a high Li+ transference number (0.65) and a Li (50 μm)‖NCM712 (4 mA h cm−2) full cell exhibited long-term cycling stability (160 cycles; 80% capacity retention) at a high rate of 0.33C (1.32 mA cm−2). Notably, Li (50 μm)‖LiFePO4 (LFP; 17.4 mg cm−2) cells with a designed electrolyte reached a capacity retention of 80% after 1450 cycles at a rate of 0.66C. An 6 Ah Li‖LFP pouch cell (over 250 W h kg−1) showed excellent cycling stability (130 cycles, 96% capacity retention) under practical conditions. This design concept for an electrolyte provides a promising path to build high-energy-density and high-rate LMBs.
Introduction
Lithium metal batteries (LMBs) are considered as fruitful candidates for high-energy-density power source.1–3 However, typically LMBs express inferior electrochemical performance due to the low coulombic efficiency (CE) of the lithium metal anode (LMA) in nonaqueous electrolytes, which induces continuous consumption of both electrolyte and LMA.4–6 Among the various factors affecting the electrochemical performance of LMBs, the electrolyte plays a vital part in uncontrollable side reactions between the LMA and electrolytes.7,8 By fine-tuning electrolyte formulations, the solid electrolyte interphase (SEI) chemistry and Li morphology can be regulated to improve the CE and cyclability of LMBs.9,10 Electrolyte design has shown a great prospect for building better LMBs, including weakly solvating electrolytes (WSEs),11 high-concentration electrolytes (HCEs),12 and diluted high-concentration electrolytes (DHCEs).13 Although these promising electrolytes provide high CE (>99%) and derive robust SEI and cathode electrolyte interphase (CEI) on both the LMA and cathode, the limited rate performance of LMBs cannot meet the demand for commercialization.14–26 Specifically, ionic conductivity and the Li+ transference number (TLi+; ionic current is carried predominantly by Li+) are two critical parameters to measure the mobility of Li+ in electrolytes. The low mobility of Li+ will aggravate the concentration polarization in the electrolyte, thereby accelerating the formation of lithium dendrites. Therefore, high ionic conductivity and especially high TLi+ are indispensable requirements in developing electrolytes for high-rate LMBs.27–30For WSEs, rational tuning of molecule structures is an effective method to achieve high performance in LMBs.31–33 However, a weak solvating solvent with insufficient solvation ability leads to low solubility towards salts, ultimately leading to a poor capability of Li+ transport. As another highly competitive candidate, a DHCE expresses superiority in terms of viscosity compared with a HCE and shows great potential for practical application. Despite the sufficient solvation ability of the main solvent in a DHCE, the absence of free solvent and the tough constraining force between the Li+-solvent in a strong solvation structure makes it difficult to support fast Li+ transport ability.34–38 Inspired from WSEs and DHCEs, it is critical to achieve a balance between insufficient solvation and sufficient solvation in ideal electrolytes for fast Li+ transport.39–41 Thus, new strategies are required to innovate electrolyte design for maintaining the capability of solvating Li+ but simultaneously weakening fetters on Li+ of strong solvation structure. Considering the universality of non-coordinating solvents inevitably affect the interactions strength of Li+-solvent through intermolecular force, manipulating non-coordinating solvents with diverse interaction capabilities may be an effective strategy to relieve the strong bonding of Li+-solvents.42,66 Up to now, a loosened solvation structure by fine-tuning of non-coordinating solvents are desirable yet not well studied to realize both reversible Li plating/stripping and fast Li+ transfer.
In this work, we discovered a “tug-of-war” effect in multilayer solvation sheaths. That is, direct solvent–nonsolvent and indirect Li+–nonsolvent interactions were leveraged to tune the binding energy between Li+ and the main solvent. As a paradigm, a non-coordinating solvent (1,3,5-trifluorobenzene (F3B)) was introduced into a fluorobenzene (FB)-based DHCE (denoted “DHCE2”), which delivered a high TLi+ (0.65) without altering the primary solvation structure of Li+ to achieve a CE up to 99.2% under 3 mA cm−1. Notably, different abilities of intermolecular interaction between F3B and FB to the solvent weakened the interaction between Li+ and the main solvent, as evidenced by the spectroscopy (Raman, nuclear magnetic resonance (NMR), pulsed field gradient nuclear magnetic resonance (PFG-NMR)) and ab initio molecular dynamics (AIMD). Consequently, a loosened solvation sheath was achieved in DHCE2 for improving the kinetics of Li+ transfer. A superb rate capability using DHCE2 was achieved in a LMB cell employing a Li-NCM712 cathode with an areal capacity of 2 mA h cm−2. Moreover, a Li-NCM712 full cell was evaluated under practical conditions of high-NCM712 loading (4 mA h cm−2) and thin Li (50 μm), which sustained 160 cycles with 80% capacity retention at a high rate (0.33C; 1.32 mA cm−2) in DHCE2 while only 50 cycles were obtained in the control. Impressively, a Li (50 μm)–LiFePO4 (LFP; 17.4 mg cm−2) coin cell with a lean electrolyte (3 g A h−1) using such an electrolyte achieved over 1450 cycles at a rate of 0.66 C. Meanwhile, the 6 Ah Li‖LFP pouch cell delivered energy density of over 250 W h kg−1 and outstanding lifespan under practical conditions (lean electrolyte: 3 g A h−1; high areal capacity: 3.4 mA h cm−2; limited Li: 50 μm; high discharge current: 1.2 A). Overall, we demonstrated a tug-of-war effect that could be utilized to facilitate Li+ transport in a DHCE, shedding a new direction for building high-rate LMBs under practical conditions.
Results and discussion
The critical aim in this work was to improve rate capability of well-performing DHCE electrolytes. Despite high stability towards Li metal anodes, hydrofluoroether-based DHCE has the drawbacks of insufficient kinetics. Previously, we showed that F3B could assist a FB-based DHCE in obviously boosting thermodynamic and interfacial stabilities,43 but also significantly enhanced the high-rate capability compared with other DHCEs, yet the mechanism of improved kinetics was not investigated systematically. Therefore, F3B was introduced tentatively into a FB-based DHCE with an increased dosage gradient, denoted as DHCE1, DHCE2, and DHCE5 (DHCE: 1.87 g of LiFSI was dissolved in 1.5 ml of DME and 3 ml of FB; DHCE1: 1.87 g of LiFSI was dissolved in 1.5 ml of DME, 2.9 ml of FB, and 0.1 ml of F3B; DHCE2: 1.87 g of LiFSI was dissolved in 1.5 ml of DME, 2.8 ml of FB, and 0.2 ml of F3B; DHCE5: 1.87 g of LiFSI was dissolved in 1.5 ml of DME, 2.5 ml of FB, and 0.5 ml of F3B) to achieve in-depth understanding of the functions of F3B noncoordinating solvents for high-rate DHCE applications. The ionic conductivity (a key indicator for the diffusion of ions in bulk electrolytes) was first measured at room temperature (Fig. 1a and Table S1†). Different DHCEs delivered an almost identical ionic conductivity, demonstrating that F3B as the additive barely affected the ionic conductivity of DHCEs. Moreover, the TLi+ of various electrolytes is shown in Fig. 1b and Figure S1.† As the amount of F3B increased, the TLi+ first rose and then declined. Noticeably, DHCE2 delivered the highest TLi+ up to 0.65, indicating the fastest transport ability of Li+. Based on calculations (Fig. 1c), the Li+ conductivity in DHCE2 was higher than FSI− conductivity, suggesting a superb Li+ transfer capability. To better evaluate the effect of Li+ migration on electrochemical performance, the rate performance of Li-NCM712 was measured (Fig. 1d). DHCE2 showed significantly improved rate capability in the Li-NCM712 cell compared with that of other DHCEs, demonstrating superior Li+ diffusion kinetics, which is promising for the construction of high-energy-density and high-power-density LMBs. Beyond use of a potentiostatic polarization method to measure TLi+, PFG-NMR spectroscopy was also employed to examine Li+ transference number (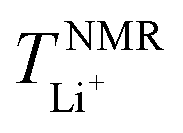 ). As shown in Fig. 1e, the lithium transference number (TLi+ = DLi+/(DLi+ + DFSI−)) was much higher in DHCE2 compared with that in other DHCEs. The transference number plays a critical part in determining the morphology and cycling stability of lithium.44,45 The Sand's capacity using the model proposed by Bai was also calculated using the following equation,46
). As shown in Fig. 1e, the lithium transference number (TLi+ = DLi+/(DLi+ + DFSI−)) was much higher in DHCE2 compared with that in other DHCEs. The transference number plays a critical part in determining the morphology and cycling stability of lithium.44,45 The Sand's capacity using the model proposed by Bai was also calculated using the following equation,46where J is the current density, Dapp is the diffusion coefficient, Zc is the charge number of the cation, C0 is the bulk concentration, F is the Faraday constant, and t+ is the cation transference number. Fig. 1f shows the Sand's capacity of DHCE and DHCE2 with respect to the current density. In this case, DHCE2 showed the highest Sand's capacity. This finding implied that cells with DHCE2 were less prone to ion depletion, which is one of the main driving forces for uncontrollable dendrite growth. This work points to a new direction to promote Li+ migration in a DHCE, which has been rarely explored. To understand the role of F3B in enhancing Li+ transfer in a DHCE, a series of DHCEs with an increased F3B gradient were used to analyse the solvation structure of FSI− by Raman vibrational spectroscopy (Fig. 2a). The heterogeneous ion-rich domain networks at 720–780 cm−1 were defined as a contact ion pair (CIP), small aggregate (AGG), and nanometric aggregate (n-AGG), suggesting that the addition of F3B to DHCE barely affected the Li+ primary solvation sheath (Li+-FSI−-DME). Interestingly, F3B with a lower dielectric constant (ε = 2)47 was likely to favour the formation of n-AGG. With an increase in F3B content in a DHCE, the peak area ratio assigned to n-AGG increased to 14.3% in DHCE2. According to previous research,48 ion migration in n-AGG is correlated to “hopping” conduction and may have a positive effect on improving Li+ transport. Besides, n-AGG may lead to the formation of an anion-derived SEI. Notably, with excess F3B in DHCE5, the signal of n-AGG disappeared. Despite DHCE5 sharing the same solvation structure with DHCE, the TLi+ was higher than that of DHCE (Fig. 1b). Therefore, we speculated that the function of F3B was more than driving the formation of n-AGG. Other intermolecular forces (e.g., solvent–solvent interaction) probably influenced the Li+ transfer.
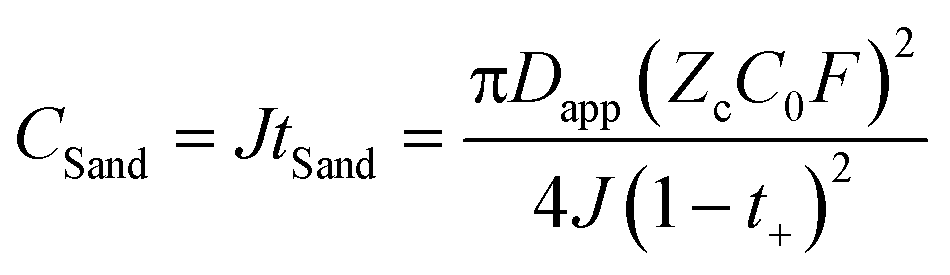
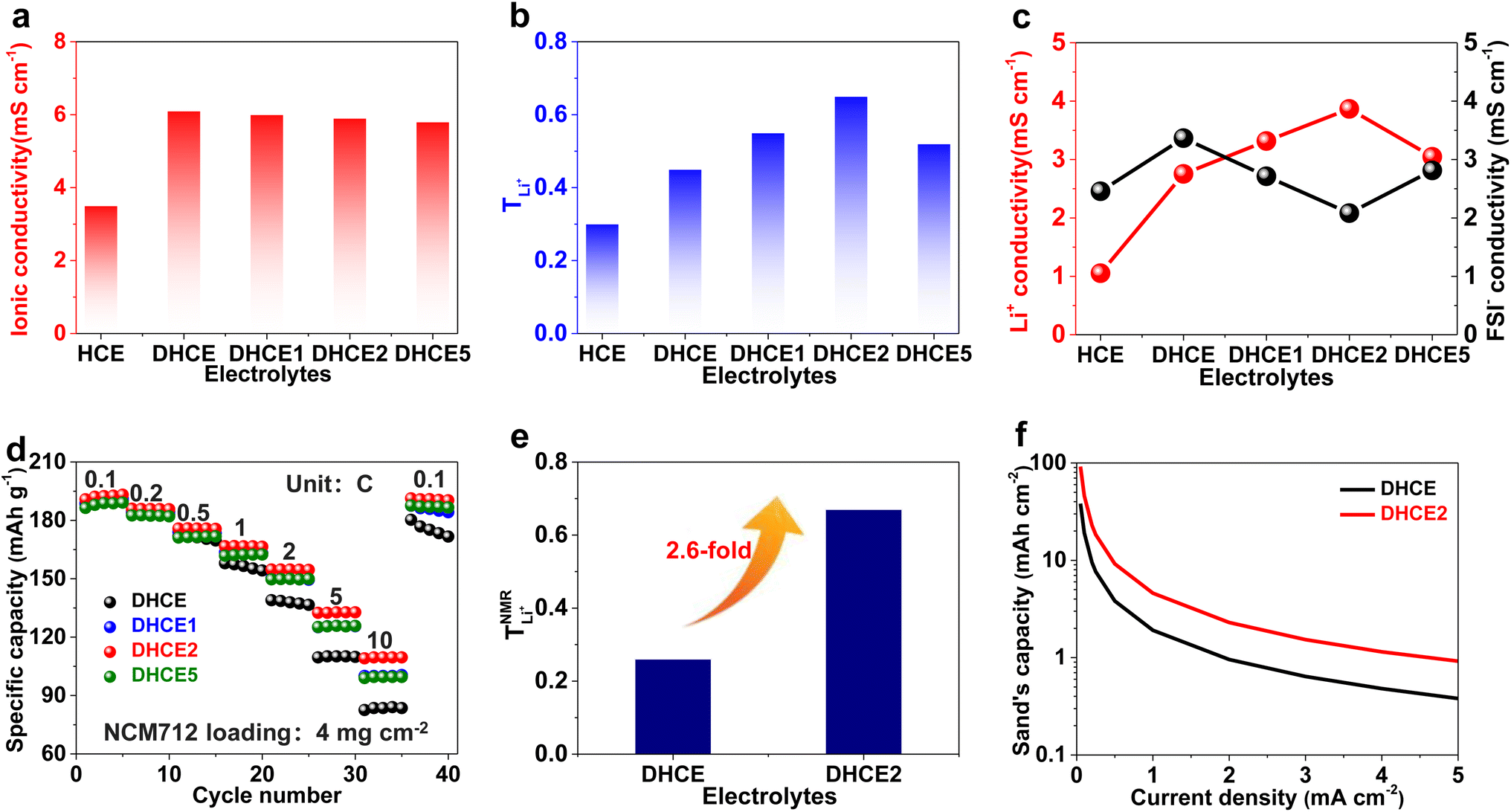 | ||
| Fig. 1 (a) Ionic conductivity of different electrolytes. (b) Li+ transference number of different electrolytes. (c) Li+ and FSI− conductivity of different electrolytes. (d) Rate performance of Li-NCM712 cells in different electrolytes. (e) Li+ transference number obtained through pulsed field gradient NMR spectroscopy for different electrolytes. (f) Sand's capacity of different electrolytes versus current density. | ||
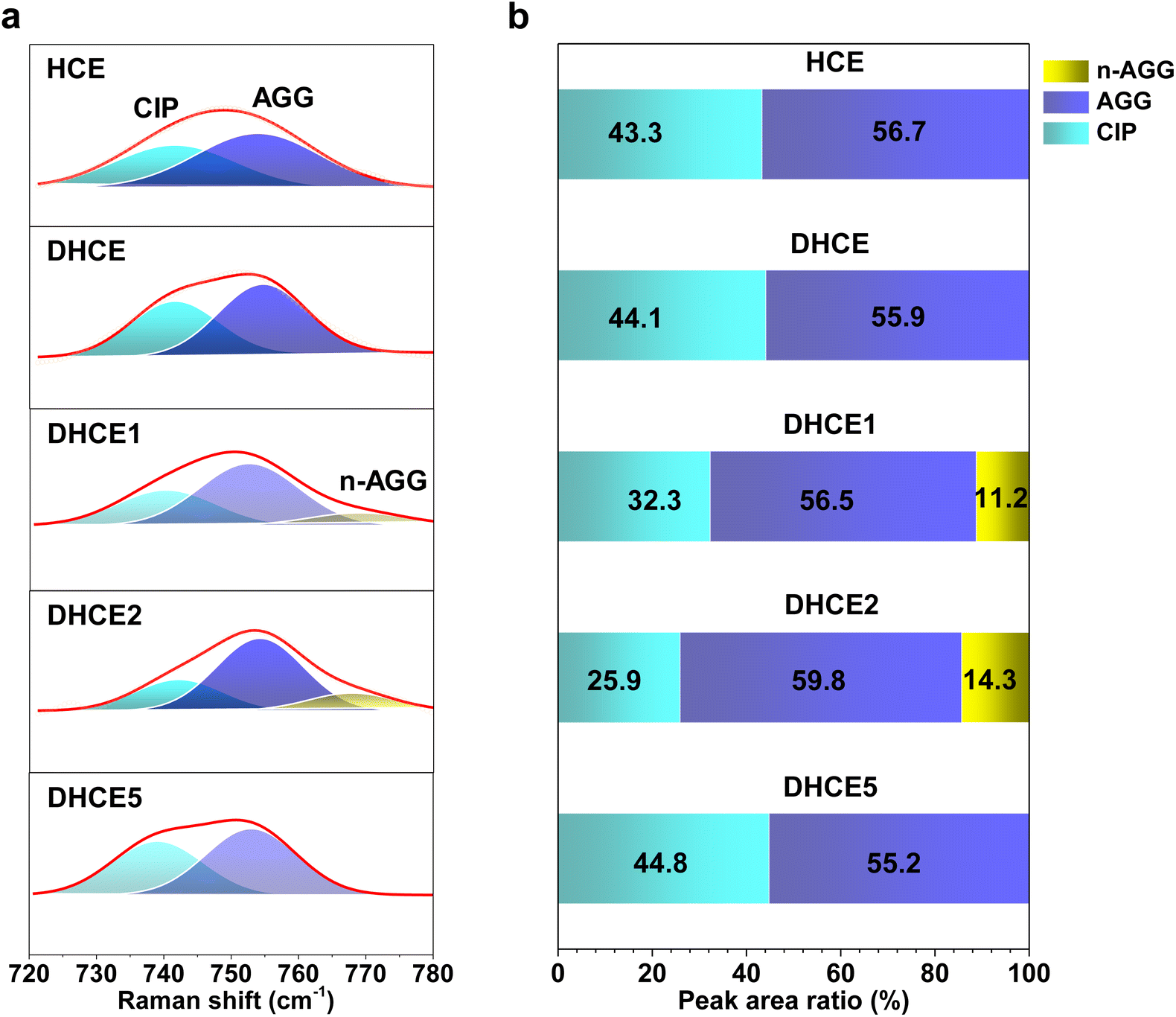 | ||
| Fig. 2 (a) Raman spectra of FSI− in various aggregate clusters. (b) Peak area ratio of CIP, AGG, and n-AGG in HCE, DHCE, DHCE1, DHCE2, and DHCE5 obtained from the fitting results of Raman spectra. | ||
Furthermore, NMR spectroscopy was applied to study the elaborate solvation environment around the Li+ and solvent–solvent interactions (Fig. 3a). From DHCE to DHCE1 and DHCE2, 7Li signal showed an obvious downfield shift, indicating a deshielding effect around Li+ and perhaps formation of a loosened solvation sheath, leading to improved Li+ transfer kinetics. Notably, the broadened signal of Li+ in DHCE2 suggested a disorder in the local environment of Li+, as evidenced by n-AGG formation. However, DHCE5 showed a shielding effect around Li+ compared with DHCE2, but expressed a deshielding effect compared with DHCE, possibly verifying a moderately loosened solvation sheath. As shown in Fig. 3b, the 1H NMR chemical shift revealed an opposite change towards 7Li NMR, demonstrating that the interaction between Li+-DME was first weakened and then strengthened with an increase in F3B. Correspondingly, the interactions between Li+ and FSI− were first enhanced and then weakened, as evidenced by 19F NMR spectroscopy (Fig. 3c). As shown in Fig. S2,† the 19F NMR spectra of FB and F3B in these DHCE systems were highly consistent with the above analysis. These results demonstrated that the non-coordinating F3B was essential for regulating the strong interaction of Li+-FSI−-DME. To further confirm this speculation, the 1H chemical shifts of FB, F3B and DME mixtures were acquired to demonstrate intermolecular interactions. Fig. S3a and b† suggest the existence of molecular interactions between FB and F3B, FB and DME, as well as F3B and DME. Even with mixing of all three solvents (FB, F3B and DME), the molecular interaction was not counteracted. After solvation of the Li salt, 1H NMR spectroscopy showed an identical variation tendency (Fig. S3c and d†), which suggested that the solvent–solvent interactions were preserved despite the presence of cations. To describe the relative strength of intermolecular forces qualitatively, identical amounts of FB and F3B were added to DME and then 1H NMR spectroscopy was done (Fig. 3d). The peaks of DME had a higher field shift after adding FB compared with F3B, which directly indicated a stronger molecular interaction between DME and FB than that between DME and F3B. The results from NMR spectroscopy corresponded closely to our conjecture that different interaction abilities between F3B and FB could loosen the Li+-FSI−-DME solvation structure.
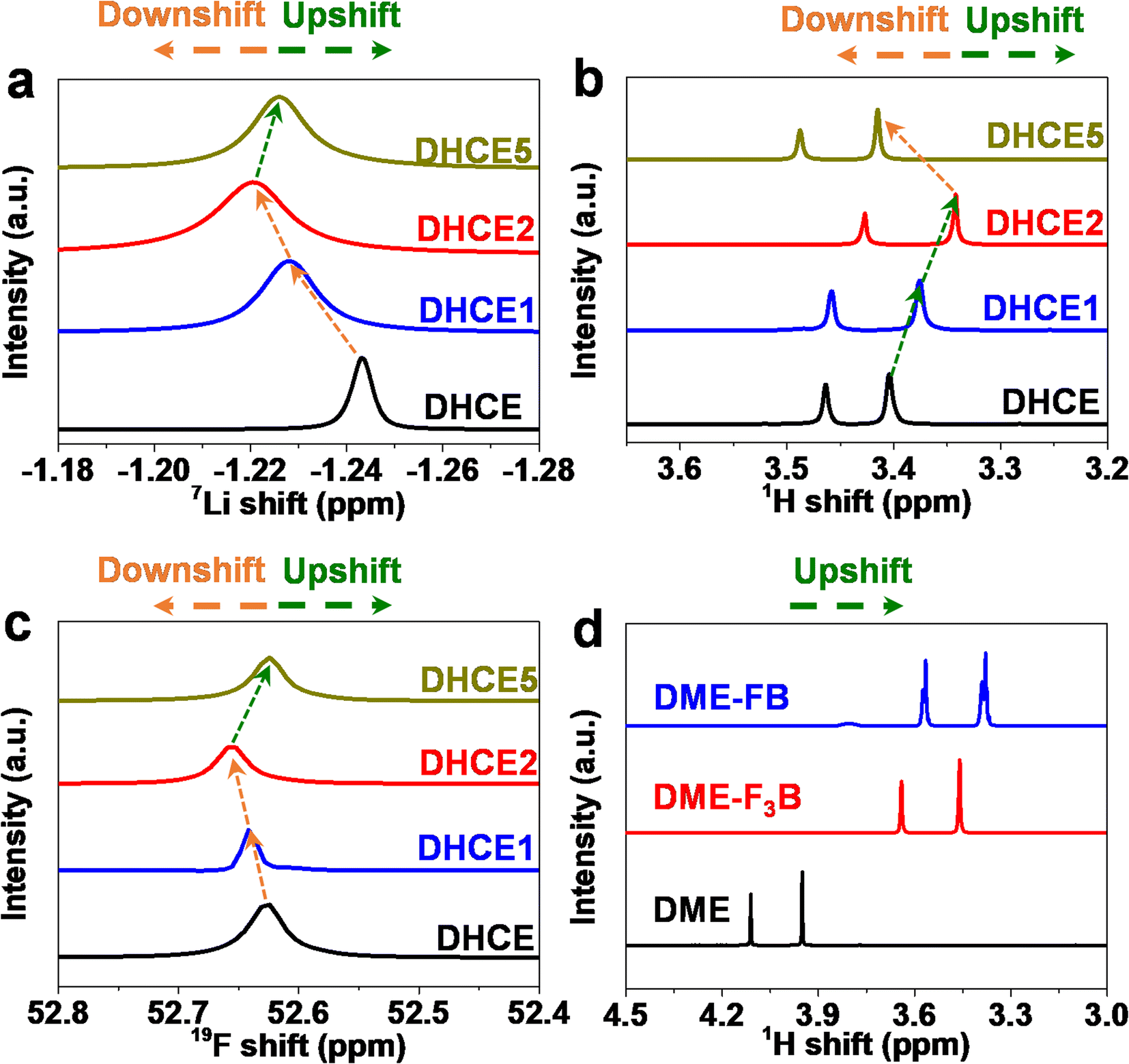 | ||
| Fig. 3 (a) 7Li NMR spectroscopy, (b) 1H NMR spectroscopy, and (c) FSI− in 19F NMR spectroscopy of different electrolytes. (d) 1H NMR spectroscopy of a mixture of different solvents. | ||
To further confirm the effect of F3B additives, DHCE and DHCE2 were described using AIMD simulations. Snapshots of simulated electrolytes are shown in Fig. 4a–f. The AIMD trajectory was used to obtain the radial distribution functions of Li–ODME and Li–OFSI− and F(FB/F3B)–H(DME) pairs. A sharp peak at 1.92 Å was associated with the Li–ODME pairs for the DHCE systems. In comparison, a weaker peak prolonged to 1.96 Å in the DHCE2 system (Fig. 4g), suggesting that Li+ was loosely coordinated with DME. Meanwhile, a mild peak at 2.15 Å was attributed to the Li–OFSI− pairs for the DHCE, shifting to 1.95 Å in DHCE2 (Fig. 4h).
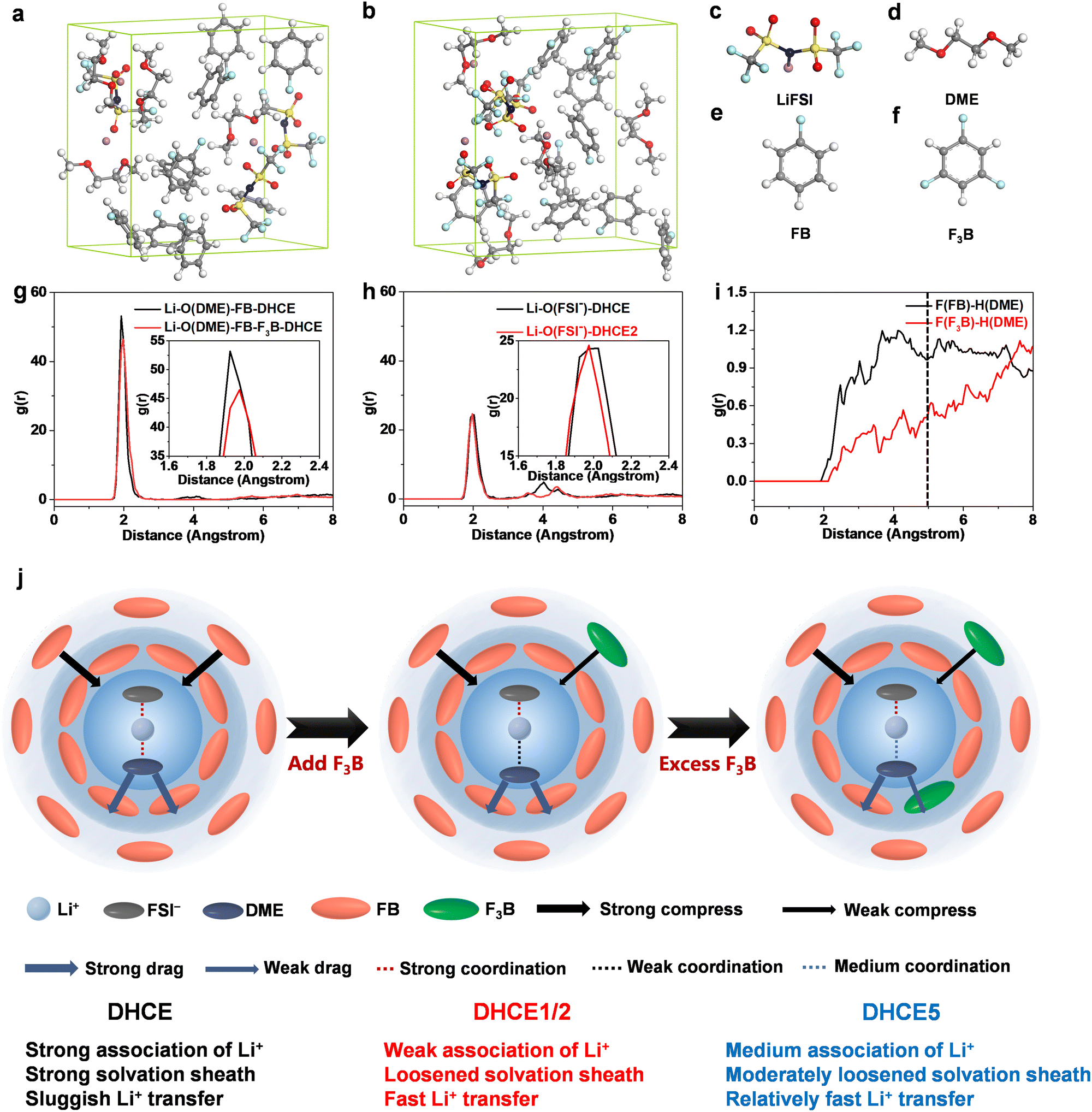 | ||
| Fig. 4 AIMD simulation snapshots of selected electrolytes at 300 K: (a) DHCE, (b) DHCE2, and (c–f) ball-and-stick model. (g–i) Radial distribution functions of Li–O(DME), Li–O(FSI−), and F(FB/F3B)–H(DME) pairs calculated from AIMD trajectories at 300 K of DHCE and DHCE2. (j) Solvation sheath model in DHCE, DHCE1/2, and DHCE5 (schematic). | ||
This result indicated that interactions between Li+ and FSI− were slightly strengthened in DHCE2 through weakening the interactions between Li+ and DME, which was consistent with the result of 19F NMR spectroscopy. Moreover, the RDF of F(FB/F3B)–H(DME) pairs in Fig. 4i revealed that, within a radius of 5 Å, FB was predominant, This result confirmed that FB may be close to the primary solvation structure, whereas F3B is away from the primary solvation structure with high probability, which further supports the idea of a stronger molecular interaction between DME and FB than that between DME and F3B. The different locations of FB and F3B outside the primary solvation sheath seem to be the main reason for promoting Li+ transport capability. Hence, we propose a possible multilayer solvation sheath model. Based on the probability of the diluent appearing at different locations outside the primary solvation structure, the diluent in DHCE is simplified as two layers: inner layer (close to the primary solvation structure) and outer layer (away from the primary solvation structure). This hypothesis is similar to theories in previous reports on the formation of multilayer solvation sheaths in a DHCE using non-coordinating solvents.49 The inner layer of FB is likely to interact with DME through intermolecular forces, which was demonstrated in our recent report,42 showing that FB “pulls” DME (denoted as “strong drag effect”). The outer layer of FB probably is attracted to Li+ according to classical Coulomb force theory due to the electron-rich π-conjugated system of FB showing a strong compressive effect on the primary solvation sheath, which is also consistent with our previous result.17 It has been suggested that FB has different effects on the primary solvation structure depending on its location. The pristine DHCE possesses a strong solvation sheath and sluggish Li+ transfer, suggesting that the compressive effect exceeds the drag effect towards Li+-FSI−-DME. In DHCE1 and DHCE2, owing to the reduced density of the electron cloud of the benzene ring and lower dielectric constant arising from the polyfluoride substitution, F3B likely appears at the outer layer to replace FB and expresses a weakened compressive effect compared with FB, while the inner layer of FB with a strong drag effect remains. The compressive effect was outperformed by the drag effect towards Li+-FSI−-DME in DHCE1 and DHCE2, leading to a loosened solvation sheath and fast Li+ transfer. Further increase in F3B (DHCE5) led to excess F3B beginning to reach the inner layer, weakening the drag effect, thus leading to a moderately loosened solvation sheath and relatively fast Li+ transfer. In short, non-coordinating solvents with diverse interactions were manipulated to alter the constraints on Li+ and accelerate its transport kinetics. A summary of current understanding on the intermolecular force of F3B on Li+-FSI−-DME (based on a solvation unit) is illustrated schematically in Fig. 4j.
Linear sweep voltammetry (LSV) was conducted to determine the reduction and oxidation potentials of HCE, DHCE and DHCE2 (Fig. S4a and b†). In addition to LiFSI, the reduction potential of F3B was higher than that of FB and DME. As shown in Fig. S4c,† the calculated LUMO level of F3B was −1.03 eV, which was significantly lower than that of FB (−0.83 eV) and DME (0.22 eV), further demonstrating the facile reduction of F3B rather than FB and DME on the surface of Li metal to form a LiF-rich protective layer. Benefiting from the lower HOMO level in F3B (−7.64 eV) compared with FB (−7.08 eV) and DME (−7.18 eV), DHCE2 showed better anti-oxidation. These results indicate that DHCE2 possibly enabled compatibility with the LMA and high-voltage cathode materials. Li plating/stripping reversibility is crucial for realistic LMBs, and can be reflected by Li/Cu half cells. A comparison of the CE during long-term cycling at 0.5 mA cm−2 and 0.5 mA h cm−2 is shown in Fig. S5a–d.† Remarkably, a superior average CE of 99.4% was achieved over 800 cycles in DHCE2. Under the test condition of 3 mA cm−2 and 1 mA h cm−2, the Li–Cu half-cell using DHCE showed an average CE of 98.8% and held steady for 200 cycles, followed by a sharp drop. In DHCE2, the CE could be further promoted to 99.2% and remained stable for over 250 cycles (Figs. 5a, S5e and f†). Hence, DHCE2 showed an excellent compatibility and high-rate capability with the Li metal anode. Furthermore, the cycling performance of a Li-NCM712 (4 mg cm−2) coin cell with different electrolytes at 1 C is shown in Fig. 5b. The cycle life could be further correlated with the average voltage gap between charge and discharge (Fig. S6†). After the formation cycles, Li-NCM712 with DHCE2 displayed stable cycling over 500 cycles (80% retention) and low overpotentials, while the control shows rapid capacity decay with sharply increased polarization. In addition, Li-NCM712 battery with medium loading (2 mA h cm−2) is examined (Figs. 5c and S7†). The cell with DHCE exhibited continuous capacity decay. In contrast, an improved cyclability with more than a tripled lifetime was obtained by using DHCE2 (300 cycles, 80% retention). Moreover, the rate performance of Li-NCM712 (2 mA h cm−2) in different electrolytes was evaluated (Figs. 5d and S8†). Using DHCE2, the cell delivered specific discharge capacities of 191, 183, 172, 158, 138, 123 and 111 mA h g−1 at 0.1, 0.2, 0.5, 1, 2, 3 and 4C, respectively. Impressively, under a high rate (4C), the specific discharge capacity of Li-NCM712 in DHCE2 was doubled compared with that of DHCE (58 mA h g−1). Therefore, the developed electrolytes exhibited a prominent high-rate capability even at high mass loading, which holds great promise for practical application. The activation energy (Ea) was studied via temperature-dependent EIS ranging from −10 °C to 20 °C (Fig. S9†). The Ea was evaluated from the slopes of Arrhenius plots with the Arrhenius equation, R−1 = A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−Ea/RT), where A denotes the frequency factor, R is the ideal gas constant and T denotes the absolute temperature. The Ea for Li/Li symmetric cells in DHCE2 was calculated to be 15.2 kJ mol−1, which was less than that of FB-DHCE (20.5 kJ mol−1), thereby demonstrating kinetics improvement after adding F3B. Also, an Li-NCM712 battery with ultra-high areal capacity (4 mA h cm−2) was used to measure the rate performance. The cell with DHCE2 displayed a great rate capability, thanks to improved Li+ transport kinetics, which far exceeded those of the reference electrolytes DHCE (Figs. S10 and S11†). Moreover, an Li-NCM712 battery with ultra-high areal capacity (4 mA h cm−2) and limited Li (50 μm) was assembled. Figs. 5e and S12† show that the Li-NCM cell with DHCE2 maintained 70% capacity retention after 160 cycles at a high rate (0.33C; 1.32 mA cm−2). The cell with FB-DHCE obtained 70% capacity retention after 60 cycles. To test the practicability of developed electrolytes in high-voltage LMBs, an Li-NCM712 full cell was built with an ultra-high areal capacity (4 mA h cm−2), low N/P ratio (2.3) and low E/S ratio (3 g A h−1). As shown in Figs. S13† and S14,† the battery using DHCE exhibited unstable cycling and rapid fading. In contrast, the cycling stability was improved significantly in DHCE2, where 70% capacity was retained after 110 cycles. Compared with other reports of advanced electrolytes in NCM-based LMBs under challenging conditions,11,15,33,50–56 DHCE2 showed an unprecedented cycling performance at a high rate (Fig. 5f). Li-NCM712 pouch cells with DHCE2 were also constructed to verify the results under challenging conditions (NCM712: 4 mA h cm−2, 200 μm Li, and 3 g A h−1 electrolyte). Such pouch cells delivered an outstanding capacity retention of 80% over 88 cycles (Figs. 5g, S15† and Table S2†). To pursue better electrochemical performance, we reduced the “free” DME molecules to the conventional DHCE concentration (LiFSI:DME:diluter = 1:1.2:3 by mol) for the study. First, we applied FB-DCHE (LiFSI:DME:FB = 1:1.2:3 by mol) and FB-F3B-DHCE (LiFSI:DME:FB:F3B = 1:1.2:2.8:0.2 by mol) to evaluate the effect of F3B. To assess the durable interphase stability and high-rate performance of DHCE, LMBs with LFP cathodes were measured. A superior rate performance of the Li-LFP cell could be achieved in FB-F3B-DHCE (Fig. 6a). Beyond low mass loading, the Li metal anode was also coupled with commercial LFP (17.4 mg cm−2) to evaluate the rate performance, and further verification of kinetics enhancement by adding F3B (Figs. S16 and S17†). As shown in Figs. 6b and S18,† excellent cycling performance (80% capacity after 1450 cycles) was realized in FB-F3B-DHCE with high cathode loading (17.4 mg cm−2), thin Li (50 μm), lean electrolyte (3 g A h−1) and high rate (0.33C charging/0.66C discharging). This is the first time that LMBs have achieved such prolonged cycling performances in stringent conditions. Furthermore, a high-capacity lithium metal pouch cell was constructed to verify its potential practicality (Figs. 6c and S19†). A 15-layer (8 × 8.5 cm) Li‖LFP pouch cell was assembled and measured under practical conditions: LFP areal capacity of 3.4 mA h cm−2, E/C ratio of 3 g A h−1, and a thin lithium foil of thickness 50 μm (N/P ratio of 2.9). The cell parameters are given in Fig. 6c. This Li‖LFP pouch cell offered a cell-level specific energy of 250 W h kg−1 with a capacity of 6 A h at 0.2C (1.2 A). Long-term cycling of 130 cycles with 96% capacity retention was achieved using FB-F3B-DHCE. The overall performance was one of the best among recently reported Ah-level practical lithium metal pouch cells (Fig. 6d).22,57–67
exp(−Ea/RT), where A denotes the frequency factor, R is the ideal gas constant and T denotes the absolute temperature. The Ea for Li/Li symmetric cells in DHCE2 was calculated to be 15.2 kJ mol−1, which was less than that of FB-DHCE (20.5 kJ mol−1), thereby demonstrating kinetics improvement after adding F3B. Also, an Li-NCM712 battery with ultra-high areal capacity (4 mA h cm−2) was used to measure the rate performance. The cell with DHCE2 displayed a great rate capability, thanks to improved Li+ transport kinetics, which far exceeded those of the reference electrolytes DHCE (Figs. S10 and S11†). Moreover, an Li-NCM712 battery with ultra-high areal capacity (4 mA h cm−2) and limited Li (50 μm) was assembled. Figs. 5e and S12† show that the Li-NCM cell with DHCE2 maintained 70% capacity retention after 160 cycles at a high rate (0.33C; 1.32 mA cm−2). The cell with FB-DHCE obtained 70% capacity retention after 60 cycles. To test the practicability of developed electrolytes in high-voltage LMBs, an Li-NCM712 full cell was built with an ultra-high areal capacity (4 mA h cm−2), low N/P ratio (2.3) and low E/S ratio (3 g A h−1). As shown in Figs. S13† and S14,† the battery using DHCE exhibited unstable cycling and rapid fading. In contrast, the cycling stability was improved significantly in DHCE2, where 70% capacity was retained after 110 cycles. Compared with other reports of advanced electrolytes in NCM-based LMBs under challenging conditions,11,15,33,50–56 DHCE2 showed an unprecedented cycling performance at a high rate (Fig. 5f). Li-NCM712 pouch cells with DHCE2 were also constructed to verify the results under challenging conditions (NCM712: 4 mA h cm−2, 200 μm Li, and 3 g A h−1 electrolyte). Such pouch cells delivered an outstanding capacity retention of 80% over 88 cycles (Figs. 5g, S15† and Table S2†). To pursue better electrochemical performance, we reduced the “free” DME molecules to the conventional DHCE concentration (LiFSI:DME:diluter = 1:1.2:3 by mol) for the study. First, we applied FB-DCHE (LiFSI:DME:FB = 1:1.2:3 by mol) and FB-F3B-DHCE (LiFSI:DME:FB:F3B = 1:1.2:2.8:0.2 by mol) to evaluate the effect of F3B. To assess the durable interphase stability and high-rate performance of DHCE, LMBs with LFP cathodes were measured. A superior rate performance of the Li-LFP cell could be achieved in FB-F3B-DHCE (Fig. 6a). Beyond low mass loading, the Li metal anode was also coupled with commercial LFP (17.4 mg cm−2) to evaluate the rate performance, and further verification of kinetics enhancement by adding F3B (Figs. S16 and S17†). As shown in Figs. 6b and S18,† excellent cycling performance (80% capacity after 1450 cycles) was realized in FB-F3B-DHCE with high cathode loading (17.4 mg cm−2), thin Li (50 μm), lean electrolyte (3 g A h−1) and high rate (0.33C charging/0.66C discharging). This is the first time that LMBs have achieved such prolonged cycling performances in stringent conditions. Furthermore, a high-capacity lithium metal pouch cell was constructed to verify its potential practicality (Figs. 6c and S19†). A 15-layer (8 × 8.5 cm) Li‖LFP pouch cell was assembled and measured under practical conditions: LFP areal capacity of 3.4 mA h cm−2, E/C ratio of 3 g A h−1, and a thin lithium foil of thickness 50 μm (N/P ratio of 2.9). The cell parameters are given in Fig. 6c. This Li‖LFP pouch cell offered a cell-level specific energy of 250 W h kg−1 with a capacity of 6 A h at 0.2C (1.2 A). Long-term cycling of 130 cycles with 96% capacity retention was achieved using FB-F3B-DHCE. The overall performance was one of the best among recently reported Ah-level practical lithium metal pouch cells (Fig. 6d).22,57–67
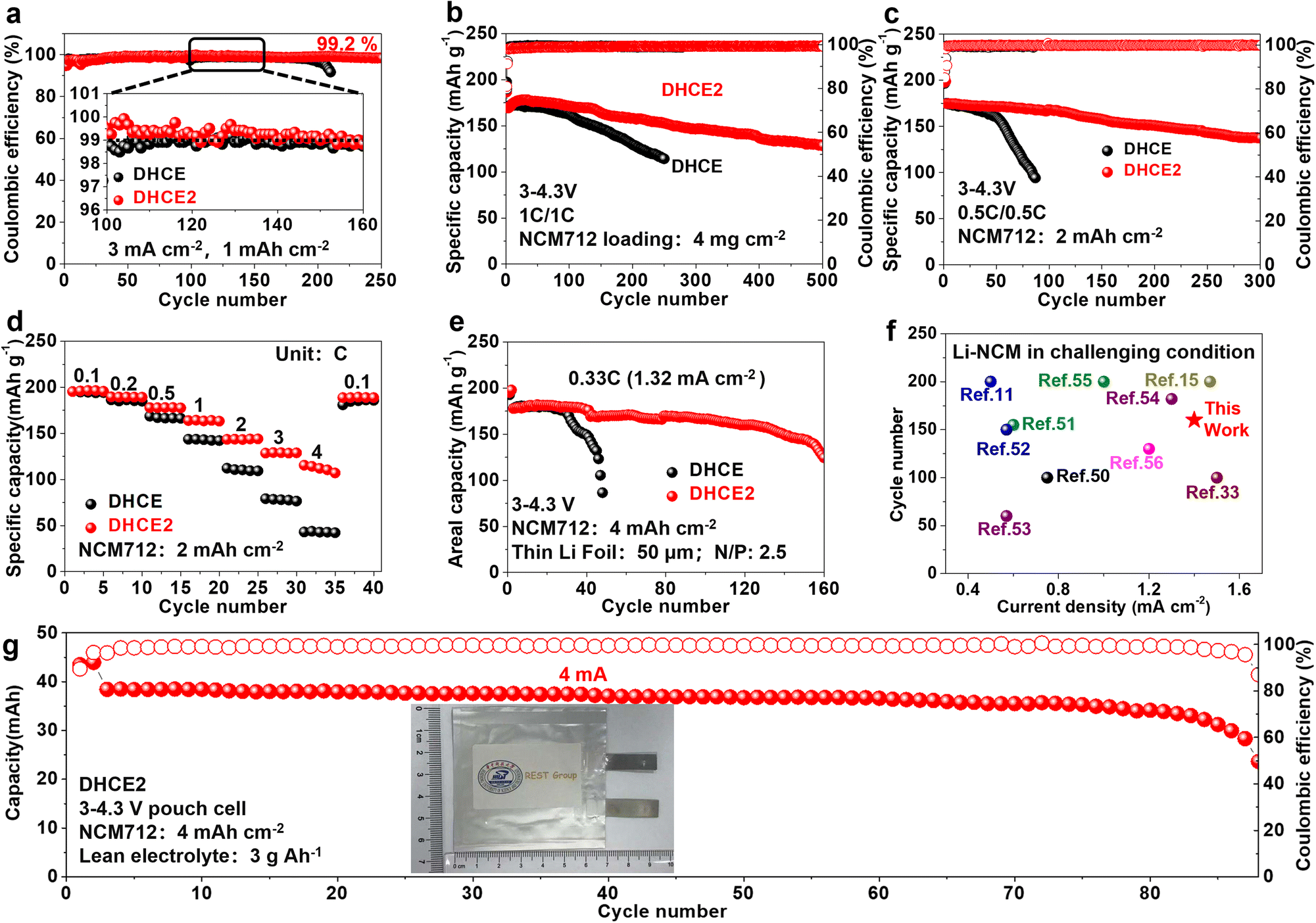 | ||
| Fig. 5 (a) Li–Cu tests toward different electrolytes. Long-term cycling performance of Li-NCM712 cells. (b) NCM712 loading: 4 mg cm−2. (c) Rate performance and (d) cycling performance of Li-NCM712 cells under different electrolytes (Li-NCM712: 3–4.3 V; areal capacity: 2 mA h cm−2). (e) Cycling performance of a Li-NCM712 coin cell under challenging conditions (NCM712: 4 mA h cm−2, 3–4.3 V, 50 μm Li). (f) Comparison of the electrochemical performance of Li-NCM cells in practical conditions. (g) Long-term cycling performance of a Li-NCM712 pouch cell (NCM712: 4 mA h cm−2, 3–4.3 V, 200 μm Li, 3 g A h−1 electrolyte). | ||
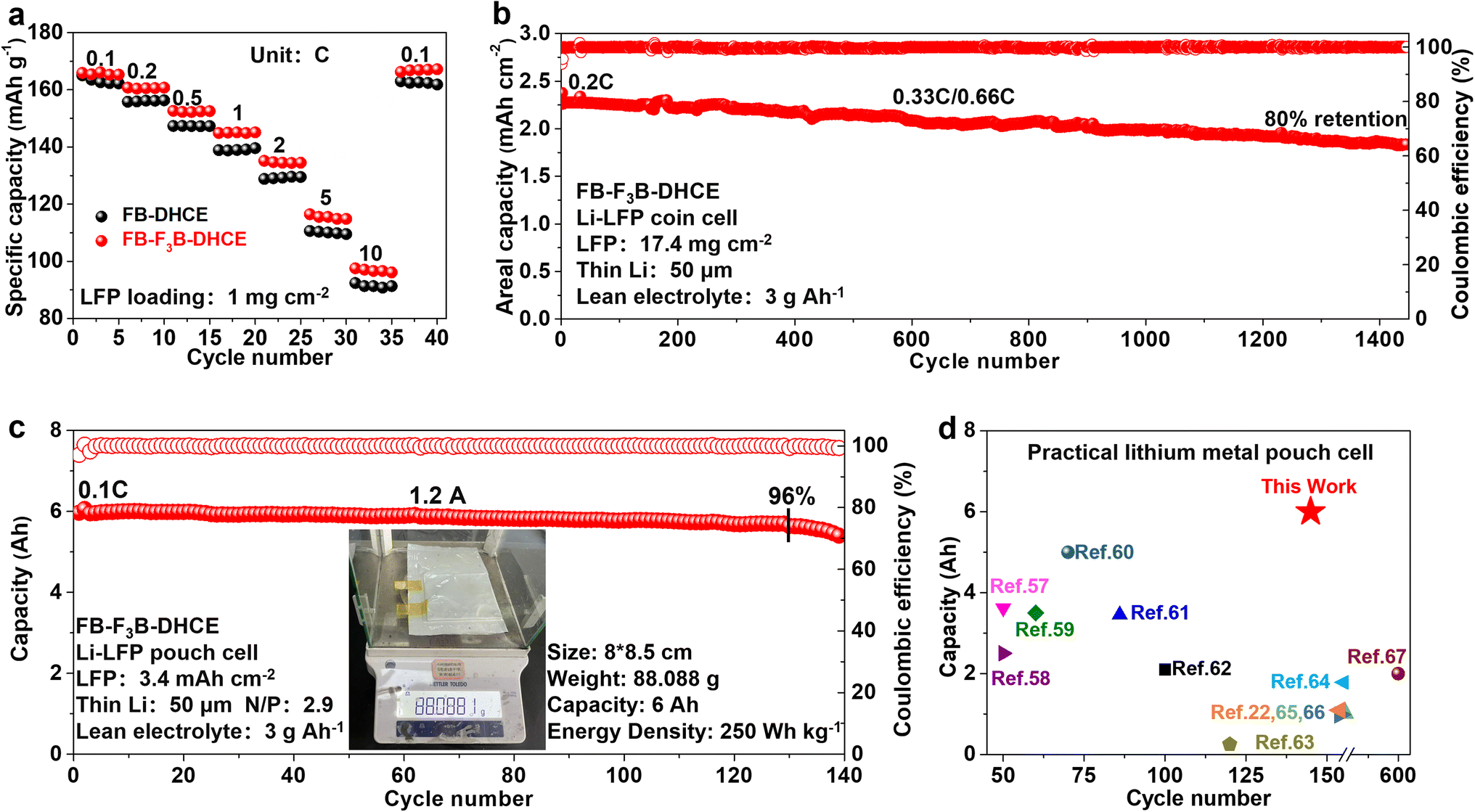 | ||
| Fig. 6 (a) Rate performance towards FB-DHCE and FB-F3B-DHCE. (b) Cycling performance of Li-LFP coin cells in FB-F3B-DHCE (LFP loading: 17.4 mg cm−2; thin Li: 50 μm; lean electrolyte: 3 g A h−1). (c) Cycling performance of a 6-Ah-grade lithium metal pouch cell under realistic conditions in FB-F3B-DHCE (insert: optical photo of the pouch cell with mass parameter). (d) Comparison of battery performance with other reported practical lithium metal pouch cells. | ||
Conclusions
A unique tug-of-war effect was disclosed, which allowed manipulation of the pulling and compressive force in a multilayer solvation sheath, achieving a loosened solvation sheath, and affording high-rate LMB. This effect was evidenced by spectroscopy (Raman, NMR, PFG-NMR) and AIMD, thereby revealing the underlying mechanisms of improved Li+ transfer kinetics. Consequently, Li-NMC712 full batteries under a challenging condition (NCM712: 4 mA h cm−2; Li: 50 μm; high current density: 1.32 mA cm−2) showed stable cycling over 160 times. An Li-LFP coin cell under harsh conditions delivered a prominent cycling performance (1450 cycles; 80% retention) at a high discharge rate of 0.66C. A practical 6-Ah-grade Li-LFP pouch cell with a specific energy of 250 W h kg−1 delivered 130 cycles with a capacity retention of 96%. This work opens a new direction for using intermolecular force to improve Li+ transport to develop novel DHCEs with high-rate capability for high-performance LMBs.Data availability
All the data supporting this study are included in the manuscript and the ESI.†Author contributions
J. Xie and Z. Zeng proposed the concept. H. Zhang prepared the materials, collected relevant data, and wrote the manuscript. All authors participated in data analyses and manuscript discussion.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported financially by the National Key Research and Development Program of China (2022YFB2404800) and the National Natural Science Foundation of China (22008082, 21975087). The authors gratefully acknowledge the Analytical and Testing Center of HUST for allowing us to use its facilities. The authors thank the Shiyanjia Research Team (https://www.shiyanjia.com/) for help with NMR spectroscopy.Notes and references
- J. Liu, Z. Bao, Y. Cui, E. J. Dufek, J. B. Goodenough, P. Khalifah, Q. Li, B. Y. Liaw, P. Liu, A. Manthiram, Y. S. Meng, V. R. Subramanian, M. F. Toney, V. V. Viswanathan, M. S. Whittingham, J. Xiao, W. Xu, J. Yang, X.-Q. Yang and J.-G. Zhang, Nat. Energy, 2019, 4, 180–186 CrossRef CAS.
- F. Duffner, N. Kronemeyer, J. Tübke, J. Leker, M. Winter and R. Schmuch, Nat. Energy, 2021, 6, 123–134 CrossRef CAS.
- Y. Cao, M. Li, J. Lu, J. Liu and K. Amine, Nat. Nanotechnol., 2019, 14, 200–207 CrossRef CAS.
- S. Cao, X. He, L. Nie, J. Hu, M. Chen, Y. Han, K. Wang, K. Jiang and M. Zhou, Adv. Sci., 2022, 9, 2201147 CrossRef CAS.
- M.-Y. Zhou, X.-Q. Ding, J.-F. Ding, L.-P. Hou, P. Shi, J. Xie, B.-Q. Li, J.-Q. Huang, X.-Q. Zhang and Q. Zhang, Joule, 2022, 6, 2122–2137 CrossRef CAS.
- C. Fang, J. Li, M. Zhang, Y. Zhang, F. Yang, J. Z. Lee, M.-H. Lee, J. Alvarado, M. A. Schroeder, Y. Yang, B. Lu, N. Williams, M. Ceja, L. Yang, M. Cai, J. Gu, K. Xu, X. Wang and Y. S. Meng, Nature, 2019, 572, 511–515 CrossRef CAS.
- H. Wang, Z. Yu, X. Kong, W. Huang, Z. Zhang, D. G. Mackanic, X. Huang, J. Qin, Z. Bao and Y. Cui, Adv. Mater., 2021, 33, 2008619 CrossRef CAS.
- Y.-X. Zhan, P. Shi, C.-B. Jin, Y. Xiao, M.-Y. Zhou, C.-X. Bi, B.-Q. Li, X.-Q. Zhang and J.-Q. Huang, Adv. Funct. Mater., 2022, 32, 2206834 CrossRef CAS.
- A. J. Louli, A. Eldesoky, R. Weber, M. Genovese, M. Coon, J. deGooyer, Z. Deng, R. T. White, J. Lee, T. Rodgers, R. Petibon, S. Hy, S. J. H. Cheng and J. R. Dahn, Nat. Energy, 2020, 5, 693–702 CrossRef CAS.
- C. V. Amanchukwu, Z. Yu, X. Kong, J. Qin, Y. Cui and Z. Bao, J. Am. Chem. Soc., 2020, 142, 7393–7403 CrossRef CAS.
- Z. Yu, H. Wang, X. Kong, W. Huang, Y. Tsao, D. G. Mackanic, K. Wang, X. Wang, W. Huang, S. Choudhury, Y. Zheng, C. V. Amanchukwu, S. T. Hung, Y. Ma, E. G. Lomeli, J. Qin, Y. Cui and Z. Bao, Nat. Energy, 2020, 5, 526–533 CrossRef CAS.
- J. Qian, W. A. Henderson, W. Xu, P. Bhattacharya, M. Engelhard, O. Borodin and J.-G. Zhang, Nat. Commun., 2015, 6, 6362 CrossRef CAS.
- X. Cao, X. Ren, L. Zou, M. H. Engelhard, W. Huang, H. Wang, B. E. Matthews, H. Lee, C. Niu, B. W. Arey, Y. Cui, C. Wang, J. Xiao, J. Liu, W. Xu and J.-G. Zhang, Nat. Energy, 2019, 4, 796–805 CrossRef CAS.
- Z. Zeng, V. Murugesan, K. S. Han, X. Jiang, Y. Cao, L. Xiao, X. Ai, H. Yang, J.-G. Zhang, M. L. Sushko and J. Liu, Nat. Energy, 2018, 3, 674–681 CrossRef CAS.
- W. Xue, M. Huang, Y. Li, Y. G. Zhu, R. Gao, X. Xiao, W. Zhang, S. Li, G. Xu, Y. Yu, P. Li, J. Lopez, D. Yu, Y. Dong, W. Fan, Z. Shi, R. Xiong, C.-J. Sun, I. Hwang, W.-K. Lee, Y. Shao-Horn, J. A. Johnson and J. Li, Nat. Energy, 2021, 6, 495–505 CrossRef CAS.
- X. Cao, L. Zou, B. E. Matthews, L. Zhang, X. He, X. Ren, M. H. Engelhard, S. D. Burton, P. Z. El-Khoury, H.-S. Lim, C. Niu, H. Lee, C. Wang, B. W. Arey, C. Wang, J. Xiao, J. Liu, W. Xu and J.-G. Zhang, Energy Storage Mater., 2021, 34, 76–84 CrossRef.
- Z. Jiang, Z. Zeng, X. Liang, L. Yang, W. Hu, C. Zhang, Z. Han, J. Feng and J. Xie, Fluorobenzene, Adv. Funct. Mater., 2021, 31, 2005991 CrossRef CAS.
- X. Ren, L. Zou, X. Cao, M. H. Engelhard, W. Liu, S. D. Burton, H. Lee, C. Niu, B. E. Matthews, Z. Zhu, C. Wang, B. W. Arey, J. Xiao, J. Liu, J.-G. Zhang and W. Xu, Joule, 2019, 3, 1662–1676 CrossRef CAS.
- Z. Jiang, Z. Zeng, W. Hu, Z. Han, S. Cheng and J. Xie, Energy Storage Mater., 2021, 36, 333–340 CrossRef.
- D.-J. Yoo, S. Yang, K. J. Kim and J. W. Choi, Angew. Chem., Int. Ed., 2020, 59, 14869 CrossRef CAS.
- S. Chen, J. Zheng, L. Yu, X. Ren, M. H. Engelhard, C. Niu, H. Lee, W. Xu, J. Xiao, J. Liu and J.-G. Zhang, Joule, 2018, 2, 1548–1558 CrossRef CAS.
- C. Niu, H. Lee, S. Chen, Q. Li, J. Du, W. Xu, J.-G. Zhang, M. S. Whittingham, J. Xiao and J. Liu, Nat. Energy, 2019, 4, 551–559 CrossRef CAS.
- H. Zhang, Z. Zeng, F. Ma, X. Wang, Y. Wu, M. Liu, R. He, S. Cheng and J. Xie, Adv. Funct. Mater., 2022, 2212000 Search PubMed.
- J. Zheng, G. Ji, X. Fan, J. Chen, Q. Li, H. Wang, Y. Yang, K. C. DeMella, S. R. Raghavan and C. Wang, Adv. Energy Mater., 2019, 9, 1803774 CrossRef.
- S. Chen, J. Zheng, D. Mei, K. S. Han, M. H. Engelhard, W. Zhao, W. Xu, J. Liu and J.-G. Zhang, Adv. Mater., 2018, 30, 1706102 CrossRef.
- X. Cao, P. Gao, X. Ren, L. Zou, M. H. Engelhard, B. E. Matthews, J. Hu, C. Niu, D. Liu, B. W. Arey, C. Wang, J. Xiao, J. Liu, W. Xu and J.-G. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2020357118 CrossRef CAS.
- J. Chen, X. Fan, Q. Li, H. Yang, M. R. Khoshi, Y. Xu, S. Hwang, L. Chen, X. Ji, C. Yang, H. He, C. Wang, E. Garfunkel, D. Su, O. Borodin and C. Wang, Nat. Energy, 2020, 5, 386–397 CrossRef CAS.
- J. Chen, Q. Li, T. P. Pollard, X. Fan, O. Borodin and C. Wang, Mater. Today, 2020, 39, 118–126 CrossRef.
- C. V. Amanchukwu, X. Kong, J. Qin, Y. Cui and Z. Bao, Adv. Energy Mater., 2019, 9, 1902116 CrossRef CAS.
- J.-G. Zhang, W. Xu, J. Xiao, X. Cao and J. Liu, Chem. Rev., 2020, 120, 13312–13348 CrossRef CAS.
- T. D. Pham and K.-K. Lee, Small, 2021, 17, 2100133 CrossRef CAS.
- T. D. Pham, A. Bin Faheem, J. Kim, H. M. Oh and K.-K. Lee, Small, 2022, 18, 2107492 CrossRef CAS.
- Z. Yu, P. E. Rudnicki, Z. Zhang, Z. Huang, H. Celik, S. T. Oyakhire, Y. Chen, X. Kong, S. C. Kim, X. Xiao, H. Wang, Y. Zheng, G. A. Kamat, M. S. Kim, S. F. Bent, J. Qin, Y. Cui and Z. Bao, Nat. Energy, 2022, 7, 94–106 CrossRef CAS.
- N. Yao, X. Chen, X. Shen, R. Zhang, Z.-H. Fu, X.-X. Ma, X.-Q. Zhang, B.-Q. Li and Q. Zhang, Angew. Chem., Int. Ed., 2021, 60, 21473 CrossRef CAS.
- X.-Q. Zhang, X.-B. Cheng, X. Chen, C. Yan and Q. Zhang, Adv. Funct. Mater., 2017, 27, 1605989 CrossRef.
- H. Shi, J. Qin, K. Huang, P. Lu, C. Zhang, Y. Dong, M. Ye, Z. Liu and Z.-S. Wu, Angew. Chem., Int. Ed., 2020, 59, 12147–12153 CrossRef CAS.
- J. Wu, Z. Rao, X. Liu, Y. Shen, C. Fang, L. Yuan, Z. Li, W. Zhang, X. Xie and Y. Huang, Adv. Mater., 2021, 33, 2007428 CrossRef CAS.
- X. Fan, X. Ji, L. Chen, J. Chen, T. Deng, F. Han, J. Yue, N. Piao, R. Wang, X. Zhou, X. Xiao, L. Chen and C. Wang, Nat. Energy, 2019, 4, 882–890 CrossRef CAS.
- X. Liu, X. Shen, H. Li, P. Li, L. Luo, H. Fan, X. Feng, W. Chen, X. Ai, H. Yang and Y. Cao, Adv. Energy Mater., 2021, 11, 2003905 CrossRef CAS.
- A. Nakanishi, K. Ueno, D. Watanabe, Y. Ugata, Y. Matsumae, J. Liu, M. L. Thomas, K. Dokko and M. Watanabe, J. Phys. Chem. C, 2019, 123, 14229–14238 CrossRef CAS.
- S. Perez Beltran, X. Cao, J.-G. Zhang, P. Z. El-Khoury and P. B. Balbuena, J. Mater. Chem. A, 2021, 9, 17459–17473 RSC.
- M. Qin, M. Liu, Z. Zeng, Q. Wu, Y. Wu, H. Zhang, S. Lei, S. Cheng and J. Xie, Adv. Energy Mater., 2022, 2201801 CrossRef CAS.
- H. Zhang, Z. Zeng, R. He, Y. Wu, W. Hu, S. Lei, M. Liu, S. Cheng and J. Xie, Energy Storage Mater., 2022, 393–402 CrossRef CAS.
- H. J. S. Sand, London, Edinburgh, Dublin Philos. Mag. J. Sci., 1901, 45–79 CrossRef CAS.
- Z. Wu, H. Liu, J. Holoubek, C. Anderson, L. Shi, H. Khemchandani, D. Lu, D. Liu, C. Niu, J. Xiao and P. Liu, ACS Energy Lett., 2022, 7, 2701–2710 CrossRef CAS.
- P. Bai, J. Li, F. R. Brushett and M. Z. Bazant, Energy Environ. Sci., 2016, 9, 3221–3229 RSC.
- L. Reynolds, J. A. Gardecki, J. V. Frankland, M. L. Horng and M. Maroncelli, J. Phys. Chem., 1996, 100, 10337–10354 CrossRef CAS.
- Z. Yu, N. P. Balsara, O. Borodin, A. A. Gewirth, N. T. Hahn, E. J. Maginn, K. A. Persson, V. Srinivasan, M. F. Toney, K. Xu, K. R. Zavadil, L. A. Curtiss and L. Cheng, ACS Energy Lett., 2022, 7, 461–470 CrossRef CAS.
- N. Sun, R. Li, Y. Zhao, H. Zhang, J. Chen, J. Xu, Z. Li, X. Fan, X. Yao and Z. Peng, Adv. Energy Mater., 2022, 2200621 CrossRef CAS.
- N. Piao, X. Ji, H. Xu, X. Fan, L. Chen, S. Liu, M. N. Garaga, S. G. Greenbaum, L. Wang, C. Wang and X. He, Adv. Energy Mater, 2020, 10, 1903568 CrossRef CAS.
- Q.-K. Zhang, X.-Q. Zhang, L.-P. Hou, S.-Y. Sun, Y.-X. Zhan, J.-L. Liang, F.-S. Zhang, X.-N. Feng, B.-Q. Li and J.-Q. Huang, Adv. Energy Mater, 2022, 12, 2200139 CrossRef CAS.
- D. Ruan, L. Tan, S. Chen, J. Fan, Q. Nian, L. Chen, Z. Wang and X. Ren, ChemRxiv, 2022,(preprint) DOI:10.26434/chemrxiv-2022-kh1qq.
- L. Tan, S. Chen, Y. Chen, J. Fan, D. Ruan, Q. Nian, L. Chen, S. Jiao and X. Ren, Angew. Chem., Int. Ed, 2022, 61, e202203693 CAS.
- Y. Chen, Z. Yu, P. Rudnicki, H. Gong, Z. Huang, S. C. Kim, J.-C. Lai, X. Kong, J. Qin, Y. Cui and Z. Bao, J. Am. Chem. Soc., 2021, 143, 18703–18713 CrossRef CAS.
- T. Li, X.-Q. Zhang, N. Yao, Y.-X. Yao, L.-P. Hou, X. Chen, M.-Y. Zhou, J.-Q. Huang and Q. Zhang, Angew. Chem., Int. Ed., 2021, 60, 22683–22687 CrossRef CAS PubMed.
- Y. Zhang, Y. Wu, H. Li, J. Chen, D. Lei and C. Wang, Nat. Commun., 2022, 13, 1297 CrossRef CAS PubMed.
- W. Deng, W. Dai, X. Zhou, Q. Han, W. Fang, N. Dong, B. He and Z. Liu, ACS Energy Lett., 2021, 6, 115–123 CrossRef CAS.
- K. Huang, S. Bi, B. Kurt, C. Xu, L. Wu, Z. Li, G. Feng, X. Zhang and J. Gu, Angew. Chem., Int. Ed., 2021, 60, 19232–19240 CrossRef CAS.
- X.-Q. Zhang, T. Li, B.-Q. Li, R. Zhang, P. Shi, C. Yan, J.-Q. Huang and Q. Zhang, Angew. Chem., Int. Ed., 2020, 59, 3252–3257 CrossRef CAS.
- P. Zhao, Y. Li, S. Chen, H. Fan, Y. Feng, L. Hu, Y. Zhang, Q. Nie, H. Pei, C. Yang, J. Deng, C. Bao and J. Song, Adv. Energy Mater, 2022, 12, 2200568 CrossRef CAS.
- Y.-H. Tan, G.-X. Lu, J.-H. Zheng, F. Zhou, M. Chen, T. Ma, L.-L. Lu, Y.-H. Song, Y. Guan, J. Wang, Z. Liang, W.-S. Xu, Y. Zhang, X. Tao and H.-B. Yao, Adv. Mater., 2021, 33, 2102134 CrossRef CAS.
- Z. Liu, D. Guo, W. Fan, F. Xu and X. Yao, ACS Mater. Lett., 2022, 4, 1516–1522 CrossRef CAS.
- X.-Q. Zhang, X. Chen, X.-B. Cheng, B.-Q. Li, X. Shen, C. Yan, J.-Q. Huang and Q. Zhang, Angew. Chem., Int. Ed., 2018, 57, 5301–5305 CrossRef CAS.
- C. Zhu, C. Sun, R. Li, S. Weng, L. Fan, X. Wang, L. Chen, M. Noked and X. Fan, ACS Energy Lett., 2022, 7, 1338–1347 CrossRef CAS.
- Y. Gao, M. Guo, K. Yuan, C. Shen, Z. Ren, K. Zhang, H. Zhao, F. Qiao, J. Gu, Y. Qi, K. Xie and B. Wei, Adv. Energy Mater, 2020, 10, 1903362 CrossRef CAS.
- G. S. Mattei, Z. Li, A. A. Corrao, C. Niu, Y. Zhang, B. Liaw, C. C. Dickerson, J. Xiao, E. J. Dufek and P. G. Khalifah, Chem. Mater., 2021, 33, 2378–2386 CrossRef CAS.
- C. Niu, D. Liu, J. A. Lochala, C. S. Anderson, X. Cao, M. E. Gross, W. Xu, J.-G. Zhang, M. S. Whittingham, J. Xiao and J. Liu, Nat. Energy, 2021, 6, 723–732 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc06620c |
| This journal is © The Royal Society of Chemistry 2023 |