
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructurally well-defined conjugated meso-aminoporphyrin oligomers analogous to polyanilines†
Ken-ichi
Yamashita
 *abc,
Shouichi
Takeuchi
a and
Ken-ichi
Sugiura
*a
*abc,
Shouichi
Takeuchi
a and
Ken-ichi
Sugiura
*a
aDepartment of Chemistry, Graduate School of Science and Engineering, Tokyo Metropolitan University, 1-1 Minami-Osawa, Hachioji, Tokyo 192-0397, Japan. E-mail: sugiura@porphyrin.jp
bDepartment of Chemistry, Graduate School of Science, Osaka University, 1-1 Machikaneyama, Toyonaka, Osaka 560-0043, Japan. E-mail: yamashita-k@chem.sci.osaka-u.ac.jp
cInnovative Catalysis Science Division, Institute for Open and Transdisciplinary Research Initiatives (ICS-OTRI), Osaka University, Suita, Osaka 565-0871, Japan
First published on 16th February 2023
Abstract
Polyaniline, which is formed by the oxidative polymerization of aniline, is a widely explored conducting polymer with several stable oxidation states, and can be applied in advanced materials, including sensing devices and electrochemical catalysts. The marriage of polyanilines with the diverse chemistry of porphyrins is expected to confer new properties, including a combination of electrical, optical, magnetic and chemical properties. Herein, we demonstrate that meso-aminodiarylporphyrin, a porphyrin analogue of aniline, undergoes oxidative oligomerization in an acidic solution under an oxygen atmosphere to yield stable oligomeric products that are analogous to fully oxidized polyanilines. The so-formed oligomers are composed of the same number of electron-rich porphyrinoid and electron-deficient quinoid moieties, and they exhibit a broad electronic absorption band in the near infrared (NIR) region, which is attributable to intramolecular charge transfer (ICT) transition from electron-rich porphyrinoid moieties to electron-deficient quinoid ones. The quinoid moieties in the oligomers could be reversibly reduced using sodium ascorbate to obtain all-porphyrinoid oligomers that resemble fully reduced polyanilines. The fully reduced oligomers do not exhibit the NIR ICT band. Furthermore, three types of partially reduced tetramers consisting of a single quinoid moiety were also obtained, among which two interconverted in solution. Their interconversion was significantly accelerated in the presence of a protic solvent. This result is consistent with the high electron conductivity of partially oxidized polyanilines following their protonation.
Introduction
π-Conjugated conductive polymers have been intensively studied over several decades.1–3 One of the fundamental strategies for preparing them is to polymerize a π-conjugated monomer with an aromatic moiety (e.g., benzene, thiophene, and pyrrole). Polyanilines (PANIs) are among the most popular conducting polymers, and their structure and redox behaviour have been studied extensively.4–12 PANIs are synthesized by the chemical or electrochemical oxidation of aniline, and they have three stable redox states (Scheme 1): a fully reduced state (leucoemeraldine), a fully oxidized state (pernigraniline), and a partially oxidized state (emeraldine). In addition, protonated states of PANIs in all these redox states can be obtained by the protonation of the bridged nitrogen atoms. The electrochemical and spectroscopic properties of PANIs in these different states differ significantly. For example, leucoemeraldines are colourless, while emeraldines and pernigranilines are strongly coloured owing to intramolecular charge transfer (ICT) from benzenoid to quinoid moieties in their structures. Moreover, protonated emeraldines are known to have the highest conductivity. As their characteristic states are readily interconvertible under external stimuli, PANIs can be applied in advanced materials, including sensing devices and electrochemical catalysts.13–16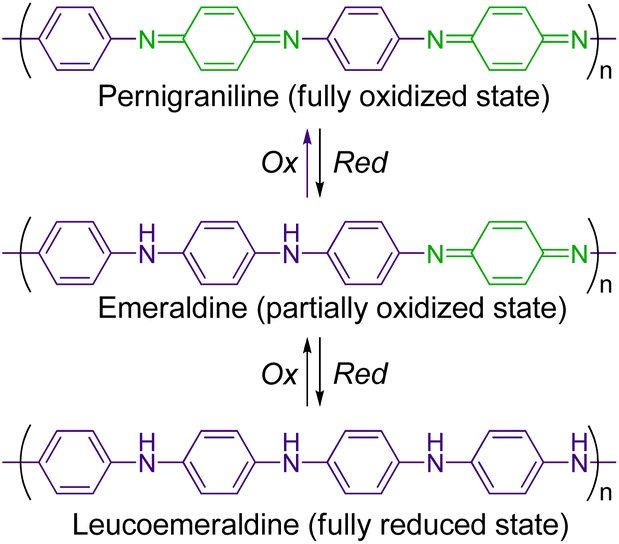 | ||
| Scheme 1 Structure and redox behaviour of polyanilines (PANIs). | ||
For understanding the structure–property relationship of PANIs and/or their precise chemical modification, structurally well-defined monodisperse oligoanilines with various end functional groups have also been developed.10,17–22 The replacement of the benzene ring with larger moieties affects their properties. Accordingly, analogous polymers of oligophenylenes, naphthalene, anthracene, and pyrene have been explored.23–29
Porphyrins are representative expanded aromatic molecules with an 18π aromatic character. Recently, π-conjugated linear porphyrin arrays (oligomers) have attracted considerable attention because of their unique properties.30,31 Owing to their highly π-conjugated structure, oligomeric porphyrins exhibit narrow HOMO–LUMO gaps and exhibit characteristic optical properties, such as intense visible (vis)-near infrared (NIR) electronic absorption and nonlinear optical properties. Butadiyne-linked porphyrin oligomers32,33 and triply-fused porphyrin oligomers34,35 are representative examples of such porphyrins.
The marriage of PANIs to the chemistry of porphyrins and their metal complexes, represented by their strong light absorption and emission, redox, catalytic and magnetic properties, promises the development of new hybrid properties of conductive polymers. In 1987, two research groups reported the electrochemical oxidation of meso-tetrakis(aminophenyl)porphyrins to obtain electroactive polymer films; however, their chemical structures were not well defined.36,37 Harvey et al. reported conjugated copolymers containing meso-diethynylporphyrin moieties38 and Gust et al. conducted the electrochemical polymerization of meso-(4-aminophenyl)diarylporphyrins to obtain PANI-like linear porphyrin polymers.39,40 However, the nearly orthogonal orientation between the porphyrin moieties and phenylene linkers disrupted effective π-conjugation; therefore, only fully reduced forms were obtained.
Replacing all benzene rings in PANI with porphyrin rings would be the simplest strategy for obtaining conjugated porphyrin polymers (oligomers) analogous to PANI. For this, meso-aminoporphyrins, porphyrin analogues of anilines, are promising monomer candidates. To the best of our knowledge, the oxidative polymerization/oligomerization of meso-aminoporphyrins has not been reported to date. On the other hand, Arnold et al. reported the copper-catalysed oxidative dimerization of Ni or Zn complexes of meso-aminotriarylporphyrins to obtain azo-linked dimers.41–43 In addition, nitrogen-bridged porphyrin dimers have also been synthesized by palladium-catalysed coupling reactions.44–47
Recently, we investigated the synthesis and properties of meso-heteroatom-substituted porphyrins48–50 and nonaromatic quinoidal porphyrins,49,51–53 and reported the facile synthesis of free-base meso-aminoporphyrins.50 Herein, we demonstrate that free-base meso-aminoporphyrin 1 undergoes aerobic oxidation under ambient acidic conditions to afford structurally well-defined π-conjugated meso-aminoporphyrin oligomers 2(n), which can be regarded as structural analogues of fully oxidized PANI (Scheme 2). Analogous structures to 2(n) have only been reported for the dimer as a Ni-complex41 and, very recently, the trimers as Ni-complexes.54 We also present the switching of their oxidation states, which is characteristically observed in PANIs.
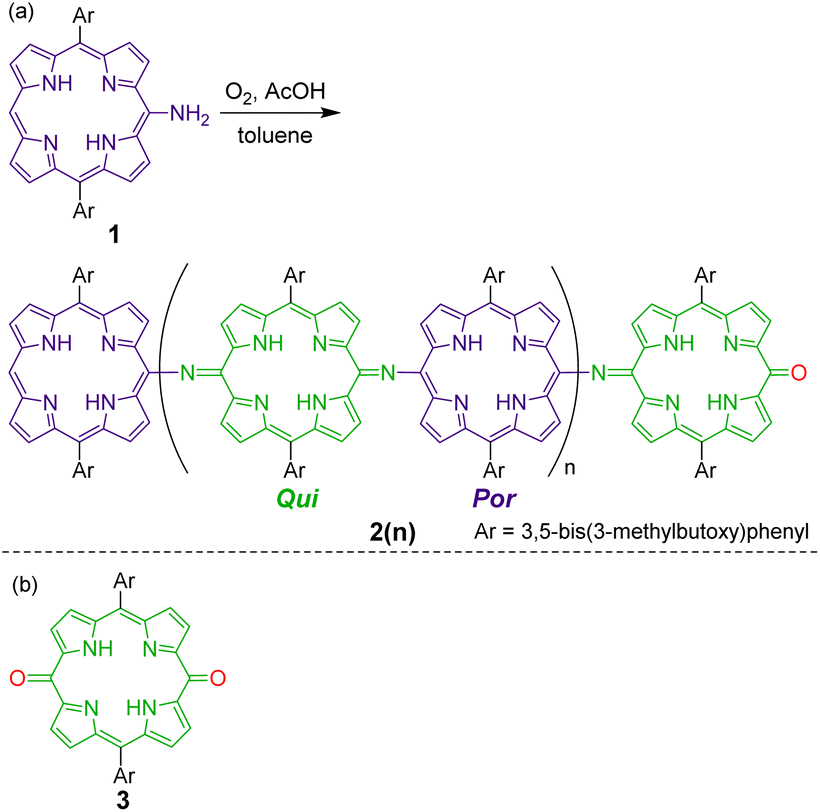 | ||
| Scheme 2 (a) Oxidative oligomerization of a meso-aminoporphyrin 1 that affords fully oxidized polyaniline-type oligomers 2(n). (b) Chemical structure of a monomeric quinone 3 as a reference compound. | ||
Results and discussion
Oligomerization of meso-aminoporphyrin
Meso-aminoporphyrin 1 remains stable in typical organic solvents, such as toluene and chloroform, even at high temperatures.50 However, we serendipitously discovered that 1 spontaneously oligomerizes in the presence of a weak acid, such as acetic acid (Scheme 2). When a toluene–AcOH solution of 1 was stirred at 100 °C under an oxygen atmosphere, 1 disappeared completely in a few hours because of the formation of the oligomeric products of 1. MALDI-TOF mass spectrometry of the crude product indicated the formation of a dimeric product of 1 as well as even-numbered oligomeric species (Fig. S3†). We isolated each oligomer using a combination of silica gel column chromatography and a recycling GPC-HPLC system. Based on NMR, mass, and elemental analyses and X-ray diffraction studies, the obtained oligomers were identified to be linear oligomers of 1, i.e., 2(n), with a structure analogous to fully oxidized PANIs; that is, half of the porphyrin moieties were oxidized to a quinoid structure, and the porphyrinoid moieties and quinoid moieties were linked alternately via imine bonds.
Table 1 summarizes the relationship between the isolated yields of 2(n) and reaction conditions. When the reaction was performed under dilute conditions (solvent, toluene![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :AcOH at 2:1 ratio) at 60 °C, dimer 2(0) was selectively formed (entry 1). Upon increasing the concentration of 1 (entries 3, 5, and 7) and/or increasing the reaction temperature (entries 2, 4, 6, and 8), the yields of higher oligomers (e.g., 2(1), 2(2), and 2(3)) increased, whereas that of 2(0) decreased. Under a specific condition (entry 8), traces of higher oligomers, up to docosamer 2(9), were detected.
:AcOH at 2:1 ratio) at 60 °C, dimer 2(0) was selectively formed (entry 1). Upon increasing the concentration of 1 (entries 3, 5, and 7) and/or increasing the reaction temperature (entries 2, 4, 6, and 8), the yields of higher oligomers (e.g., 2(1), 2(2), and 2(3)) increased, whereas that of 2(0) decreased. Under a specific condition (entry 8), traces of higher oligomers, up to docosamer 2(9), were detected.
| Entry | [1] (mM) | Solvent | Temp. (°C) | Time (h) | Yielda (%) | |||
|---|---|---|---|---|---|---|---|---|
| 2(0) | 2(1) | 2(2) | 2(3) | |||||
| a Isolated yields. ND: not detected. b Reaction was performed under a N2 atmosphere. c Full recovery of 1. d Undefined products were obtained. e Traces of higher oligomers, up to 2(9), were detected. | ||||||||
| 1 | 10 | Toluene–AcOH (2:1) |
60 | 26 | 82 | 5 | ND | ND |
| 2 | 10 | Toluene–AcOH (2:1) |
100 | 4 | 45 | 22 | 1 | ND |
| 3 | 50 | Toluene–AcOH (2:1) |
60 | 23 | 64 | 13 | 3 | ND |
| 4 | 50 | Toluene–AcOH (2:1) |
100 | 3 | 33 | 23 | 5 | ND |
| 5 | 100 | Toluene–AcOH (2:1) |
60 | 15 | 54 | 19 | 4 | 1 |
| 6 | 100 | Toluene–AcOH (2:1) |
100 | 3 | 20 | 20 | 9 | 3 |
| 7 | 200 | Toluene–AcOH (2:1) |
60 | 15 | 44 | 16 | 7 | 2 |
| 8 | 200 | Toluene–AcOH (2:1) |
100 | 3 | 18 | 22 | 10 | 3e |
| 9b | 10 | Toluene–AcOH (2:1) |
100 | 4 | NDc | ND | ND | ND |
| 10 | 10 | Toluene | 100 | 24 | NDc | ND | ND | ND |
| 11 | 10 | DMF | 100 | 24 | NDc | ND | ND | ND |
| 12 | 10 | Toluene–TFA (2:1) |
60 | 26 | NDc | ND | ND | ND |
| 13 | 10 | AcOH | 100 | 5 | NDd | ND | ND | ND |
When the reaction was carried out in a nitrogen atmosphere (oxygen-free condition), 2(n) was not formed (entry 9). Moreover, 1 was recovered completely. The results indicate that O2 is required for the observed oxidative oligomerization reactions. Further, no reaction was observed in the absence of acetic acid (entries 10 and 11), which indicates that an acidic condition is required for the oligomerization of 1. However, no reaction was observed by the use of trifluoroacetic acid (TFA), i.e., stronger acid (entry 12). Further, the reaction in neat acetic acid afforded undefined oligomeric products (entry 13).
Structural characterization of the oligomers of 1
Although the crystallization of 2(n) for single-crystal X-ray crystallography failed, we could obtain single crystals of the dinickel complex of the dimer, Ni-2(0), and analyse its crystal structure (Fig. 1). In the crystal of Ni-2(0), the CmesoPor–Nimino bond length is 1.357(9) Å, the CmesoQui![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Nimino bond length is 1.312(9) Å, and the bending angle of the imine moiety (CmesoPor–NiminoCmesoQui) is 125.4(6)°. The bending angle indicates partial stacking of the two monomer units in the structure. Both moieties were found to have a ruffled conformation, with the degree of ruffling of the porphyrinoid moiety being greater than that of the quinoid one (mean plane deviations defined by 24 core atoms are 0.31 and 0.49 Å for porphyrinoid and quinoid moieties, respectively). The intramolecular Ni⋯Ni distance was found to be 8.411(4) Å.
Nimino bond length is 1.312(9) Å, and the bending angle of the imine moiety (CmesoPor–NiminoCmesoQui) is 125.4(6)°. The bending angle indicates partial stacking of the two monomer units in the structure. Both moieties were found to have a ruffled conformation, with the degree of ruffling of the porphyrinoid moiety being greater than that of the quinoid one (mean plane deviations defined by 24 core atoms are 0.31 and 0.49 Å for porphyrinoid and quinoid moieties, respectively). The intramolecular Ni⋯Ni distance was found to be 8.411(4) Å.
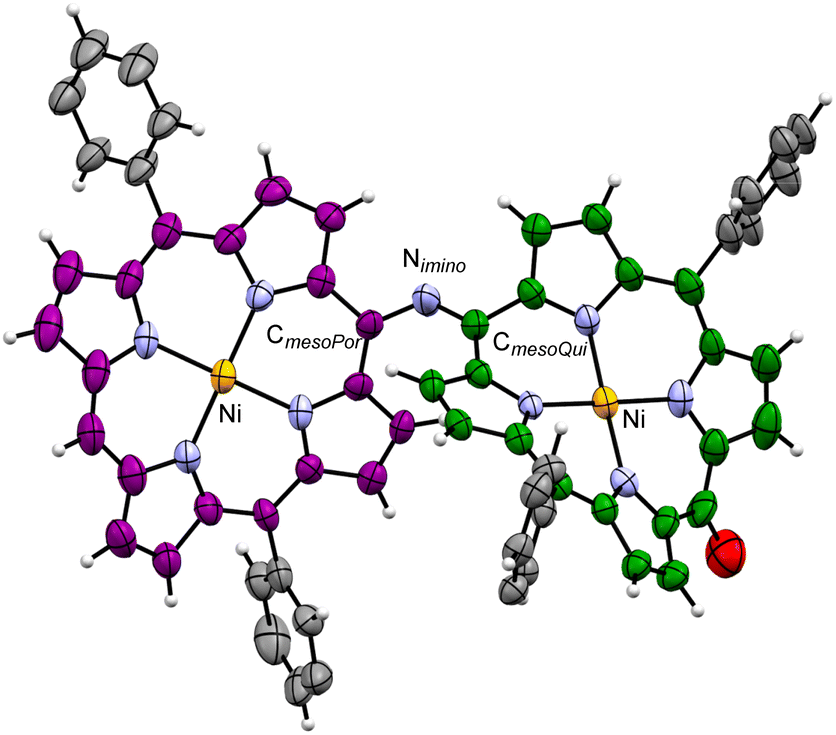 | ||
| Fig. 1 Thermal ellipsoid representation (40% probability level) of the crystal structure of Ni-2(0) (CPor = purple, CQui = green, CAr = grey, H = white, N = blue, O = red, and Ni = orange). Substituents on the aryl groups and solvent molecules are omitted for clarity. | ||
Fig. 2 shows the 1H NMR spectra of 2(n) (n = 0, 1, 2, and 3) in CDCl3 recorded at 323 K. The spectrum of 2(0) has characteristic signals of the aromatic porphyrinoid moiety: four doublet signals for β-protons at approximately 9.12–8.84 ppm, one singlet signal for the meso-proton (Hpor-meso) at 9.86 ppm, and one singlet signal for internal NH protons (NHpor) at −2.08 ppm. On the other hand, most of the signals for β-protons of the nonaromatic quinoid moiety (Hqui-meso) were considerably broadened. These broad signals sharpened at elevated temperatures and split at low temperatures (Fig. S8†). These changes indicate that the decrease in temperature slows the rotational motion along the NCQui double bond and/or the tautomerization of the NH proton in the quinoid unit49 enough to allow the NMR signal to differentiate. On the other hand, the characteristic signal of the internal NH protons of the quinoid moiety (NHqui) was clearly observed at 13.4 ppm. The integral ratio of each proton signal (especially Hpor-meso, Hpor-β, NHpor, and NHqui) was consistent with the porphyrinoid:quinoid ratio in the molecule.
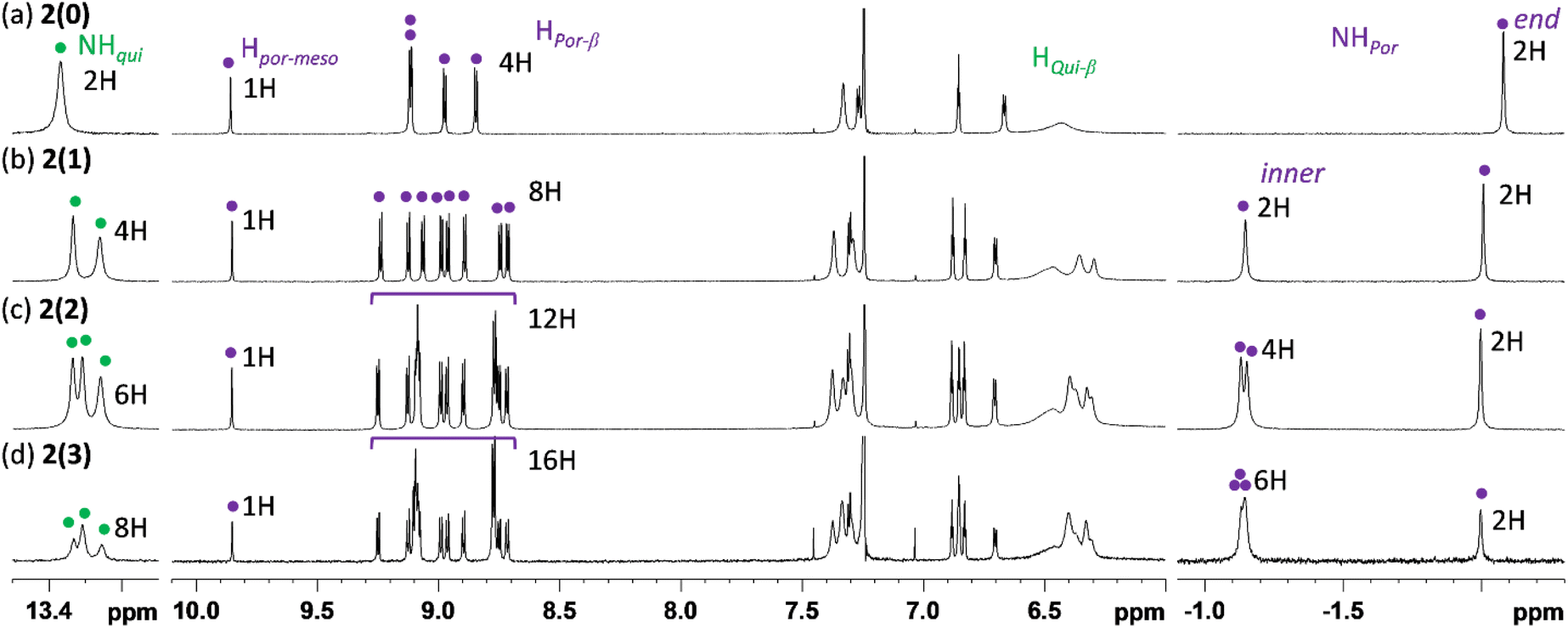 | ||
| Fig. 2 1H NMR (500 MHz, CDCl3, 323 K) spectra of (a) 2(0), (b) 2(1), (c) 2(2), and (d) 2(3). | ||
Similar to the case of 2(0), the ratio of the number of porphyrinoid moieties to quinoid moieties in 2(n) (n = 1, 2, and 3) was also determined to be 1:1 through 1H NMR analyses. It should be noted that internal NH signals for the end monoiminoporphyrin moiety and the inner diiminoporphyrin moieties could also be unambiguously distinguished (end: ca. −2.0 ppm, inner: ca. −1.1 ppm).
Fig. S9† presents the 1H NMR spectrum of Ni-2(0) in CDCl3 recorded at various temperatures. A notable feature of Ni-2(0) is that the eight quinoid β-protons were observed to be inequivalent in the NMR spectra recorded below room temperature. This result indicates that rotational motion along the NCQui double bond is mostly regulated below room temperature and it enabled us to assign most of the signals using 2D NMR techniques (1H COSY and 1H ROESY, Fig. S10 and S11†). The results revealed that two signals for quinoid β-protons appear more upfield (5.5 and 3.6 ppm) than those of others (7.7–6.4 ppm). Considering the molecular structure of Ni-2(0), these two protons are located above the porphyrinoid moieties owing to the bending of the imine bond and are therefore strongly affected by the shielding effect of the diatropic ring current of the aromatic porphyrinoid moieties.
Optical properties
The electronic absorption spectra of 2(n) (Fig. 3, top panel) consisted of a sharp and intense peak at ∼427 nm, assignable to the Soret band of the porphyrin unit, and a characteristic broad band between 750 and 1200 nm. As this broad NIR band is absent in the spectra of the monomer components, i.e., 1 and 3, it was assigned to ICT from electron-rich porphyrinoid moieties to electron-deficient quinoid ones. The peak maximum of the ICT band was almost the same for 2(1), 2(2), and 2(3) and was red-shifted relative to that of 2(0), indicating a narrower HOMO–LUMO gap of 2(1), 2(2), and 2(3) compared to that of 2(0). This can be attributed to the contribution of inner diiminoporphyrin moieties with higher HOMO energies than those of the end monoiminoporphyrin moieties. This inference is further supported by electrochemical measurements and theoretical calculations (vide infra).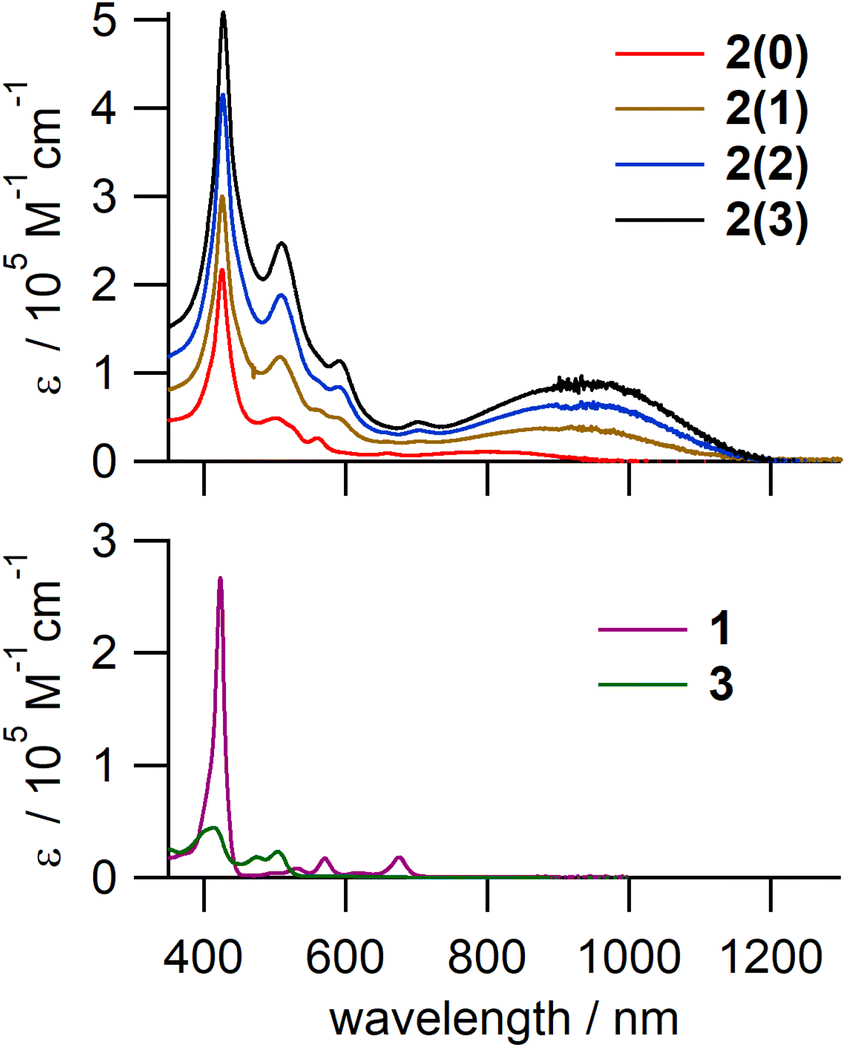 | ||
| Fig. 3 UV-vis-NIR absorption spectra of 2(n) and the corresponding monomers, 1 and 3, in toluene. | ||
Electrochemical properties
To evaluate the oxidation/reduction potentials and HOMO–LUMO gaps of the oligomers, the electrochemical properties of 2(0), Ni-2(0), and 2(1) were investigated by conducting cyclic voltammetry in CH2Cl2 containing 0.1 M nBu4NPF6 (Table 2 and Fig. S12†). 2(0) exhibited the first reversible oxidation wave at 0.42 V and the first reversible reduction waves at −0.98 V, respectively. The first reduction peak was slightly split in two when measured in a different solvent (−0.87 and −0.99 V in nPrCN). This suggests that the first reduction event is a two-electron process. Considering the redox potential of monomers 1 and 3, it can be inferred that the oxidation of 2(0) occurred at the porphyrinoid moiety, whereas reduction occurred at the quinoid moiety. Further, the first oxidation wave of 2(1) was observed at 0.12 V, which is lower by 0.30 V compared to that of 2(0). This suggests a higher HOMO level of 2(1) derived from the inner diiminoporphyrin moiety. The first reduction wave of 2(1) was observed at −0.91 V, which is higher by 0.07 V compared to that of 2(0). For Ni-2(0), two reversible reduction waves were observed at −0.82 and −1.00 V, and the first reduction potential is higher by 0.16 V compared to that of 2(0). This trend is also seen in the first reduction potentials of related monomer quinone reported previously.52 The first oxidation of Ni-2(0) was irreversibly observed.The HOMO–LUMO gaps estimated by the difference between EOx1 and ERed1 are 1.34 eV (1.08 × 104 cm−1, 925 nm) and 1.03 eV (8.30 × 103 cm−1, 1204 nm) for 2(0) and 2(1), respectively. These are narrower than those of monomers 1 and 3 and consistent with the peak maximum of the ICT band in the UV-vis-NIR spectra.
Theoretical calculations
To gain further insights into the electronic states of 2(n), density functional theory (DFT) calculations were performed on 2′(0) and 2′(1) (simplified derivatives in which alkoxy substituents on the phenyl rings were replaced by hydrogen atoms). Selected MOs and the molecular orbital (MO) energy diagrams of 2′(0) and 2′(1) are shown in Fig. 4 and Fig. S18,† respectively. As predicted from the electronic properties of each moiety, the HOMO and LUMO of 2′(0) were dominated by its porphyrinoid and quinoid moieties, respectively. The HOMO of 2′(1) was dominated by the inner porphyrinoid moiety of the molecular chain, whereas HOMO−1 was dominated by the end porphyrinoid moiety. The energy level of HOMO−1 of 2′(1) is 0.1 eV higher than that of the HOMO of 2′(1) while being almost the same as that of the HOMO of 2′(0). Both the LUMO and LUMO+1 of 2′(1) are localized on the quinoid moieties, but the difference between the two quinoid moieties is not as pronounced as in the case of the porphyrinoid moieties.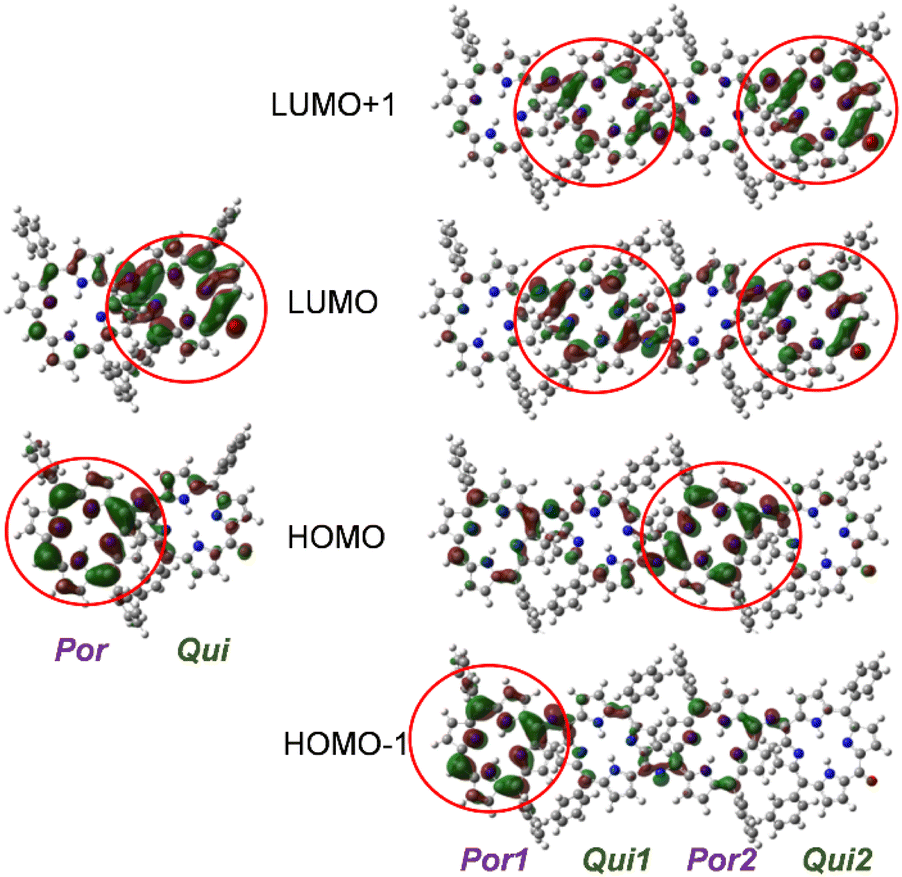 | ||
| Fig. 4 Frontier orbitals 2′(0) and 2′(1) (B3LYP-D3/6-31G(d,p)). | ||
Time-dependent (TD) DFT calculations were also performed to assign the UV-vis-NIR absorption bands (Tables S3 and S4, and Fig. S19 and S20†). The results suggest that the broad absorption band of 2(n) corresponds to the S0 → S1 transition due to the HOMO–LUMO transition, i.e., the ICT transition. The transition energies of 2′(0) and 2′(1) are 826 and 1070 nm, respectively. This difference is due to the higher energy level of the HOMO of 2′(1), and is consistent with the experimental absorption.
Plausible reaction mechanism for the oxidative oligomerization of 1
The reaction mechanism of the oligomerization of 1 might essentially be the same as that of the oxidative oligomerization of aniline to PANIs, and it might involve nucleophilic substitution reactions involving porphyrin π–cation radicals55,56 (Scheme 3). Because of its low oxidation potential, 1 would undergo single-electron oxidation by dioxygen to form the corresponding cation radical 1˙+. According to the DFT calculations of 1′˙+ (Fig. S21†), the carbon atoms at the meso-positions have a high spin density. Therefore, the subsequent nucleophilic attack of the amino group of 1 occurs at the free meso-position of 1˙+ to generate the all-porphyrinoid dimeric products. This intermediate dimer should readily undergo single-electron oxidation to form a cation radical, which either undergoes further nucleophilic attack by 1 to form a trimeric product or single-electron oxidation to form 2(0) after the replacement of the unstable terminal imine (CNH) with the carbonyl (CO) group.
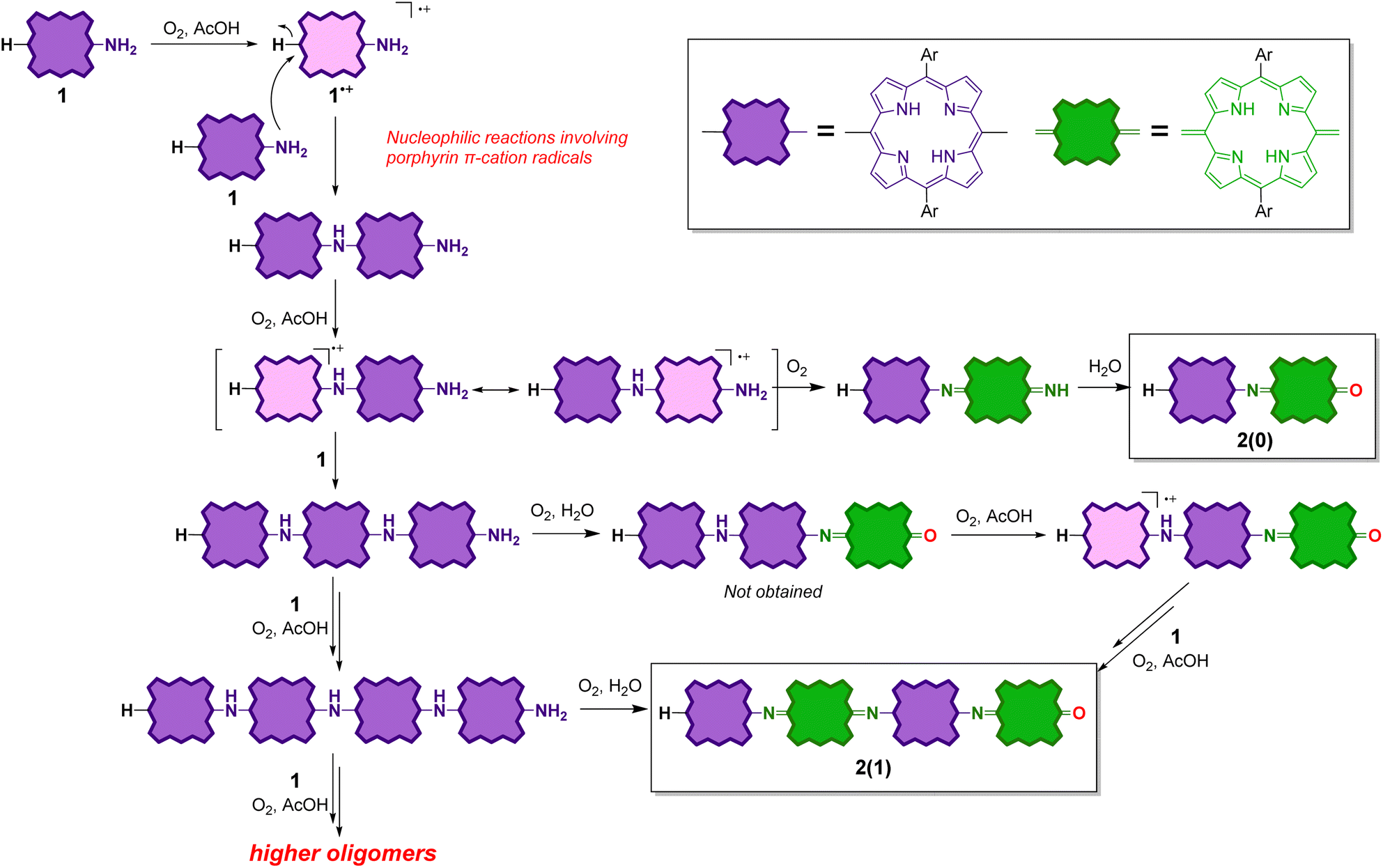 | ||
| Scheme 3 Plausible reaction mechanism for the oligomerization of 1. | ||
Considering the higher first oxidation potential of 2(n) relative to that of 1, the porphyrinoid moieties next to the quinoid moieties are considered to be less susceptible to oxidation under the reaction conditions. Therefore, no further elongation of 2(n) is possible. On the other hand, an odd-numbered oligomer contains at least one porphyrinoid moiety without quinoid moieties on both sides and is therefore prone to oxidize under the reaction conditions. Therefore, further elongation of an odd-numbered oligomer is possible. Scheme 3 shows an example of the elongation of the trimer to tetramer.
Experimental results (Table 1) show that the oligomerization reaction proceeds in the presence of weak acids such as acetic acid (and from preliminary experiments, formic acid), but the role of weak acids is not yet clear. In the case of using trifluoracetic acid (TFA), the reaction does not proceed at all and 1 is completely recovered. In the presence of TFA, the colour of 1 turns green, indicating the protonation of the inner nitrogen atoms of porphyrin macrocycles. N-Protonation of porphyrins results in a significant increase in their oxidation potential.57,58 This is consistent with the experimental results that 1 does not undergo oxidative oligomerization reactions in the presence of TFA. On the other hand, no significant change in the colour of 1 is observed in acetic acid, suggesting that the inner nitrogen atoms of 1 are not protonated. Therefore, the hydrogen bonding of an acetic acid molecule to the amino group on 1 may induce the oxidation of 1 by oxygen.
Reduction of the fully oxidized dimer
Because quinoid moieties can be readily reduced by mild reducing agents,47,52 the chemical reduction of 2(0) and 2(1) was examined to obtain reduced PANI-type derivatives (Schemes 4 and 5). When 2(0) was treated with sodium ascorbate in a polar solvent (e.g., CDCl3/DMSO-d6), the initial brown solution turned green. In the electronic absorption spectrum of the resulting solution, the broad ICT band was not observed; instead, two new peaks appeared at 650–700 nm. The 1H NMR spectrum of the reduced product (Fig. S13†) exhibited eight doublet peaks in the 9.0–7.6 ppm range and two singlet peaks at −0.6 and −1.6 ppm; the characteristic peaks of the quinoid moiety had disappeared (e.g., the broad singlet at 13.3 ppm). The NMR results indicate that the quinoid moiety in 2(0) was reduced to porphyrinoid, resulting in all-porphyrinoid dimer 4a. The characteristic singlet signal at 11.4 ppm was assigned to the bridge NH proton.44,45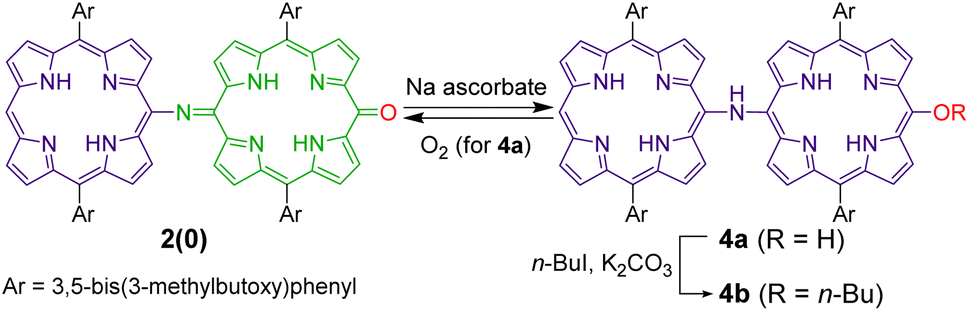 | ||
| Scheme 4 Redox switching between 2(0) and 4a, and etherification of the hydroxyl group of 4a. | ||
Further, the colour of the reduced sample turned brown upon standing in an aerobic atmosphere within several hours. The 1H NMR spectrum of the resultant solution indicated that the reduced dimer 4a had reverted to the original oxidized form, 2(0) by aerobic oxidation. The switching of the oxidation state of the dimer could be repeated at least twice (Fig. S13†).
The air-sensitive reduced dimer 4a was stabilized by the in situ alkylation of its terminal hydroxyl group.49 The in situ alkylation of 4a using 1-iodobutane resulted in butoxy-substituted dimer 4b in 79% yield (from 2(0)). 4b was sufficiently stable against aerobic oxidation to be purified by silica gel column chromatography.
The structure of 4b was determined using 1H NMR, high-resolution electrospray ionisation mass spectroscopy (ESI-HRMS), and elemental analyses. The 1H NMR spectrum of 4b (Fig. 5) in CDCl3 revealed the disappearance of the characteristic NH and β signals of the quinoid moiety and the presence of two porphyrinoid moieties. The signal of the bridge NH proton was observed at 10.7 ppm.
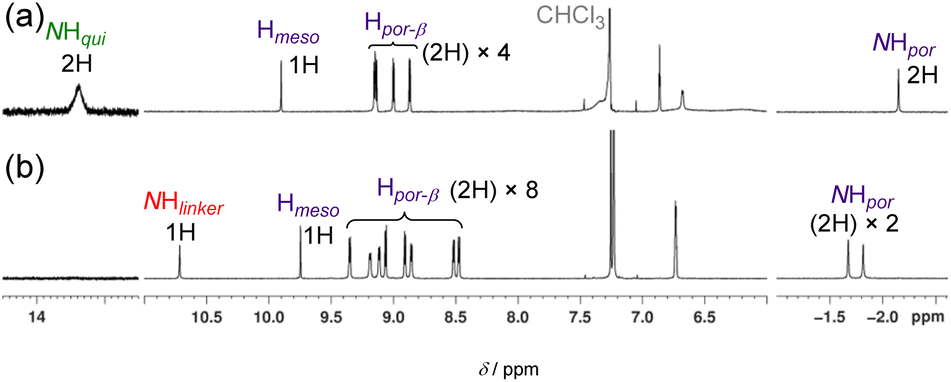 | ||
| Fig. 5 1H NMR (500 MHz, CDCl3) spectra of (a) 2(0) and (b) 4b. | ||
In the UV-vis-NIR absorption spectrum of 4b (Fig. 6), the characteristic ICT band of 2(0) could no longer be observed; the spectrum contained broad Soret and Q bands. In addition, 4b exhibited broad fluorescence in the 680–1000 nm range with a peak maximum of 738 nm.
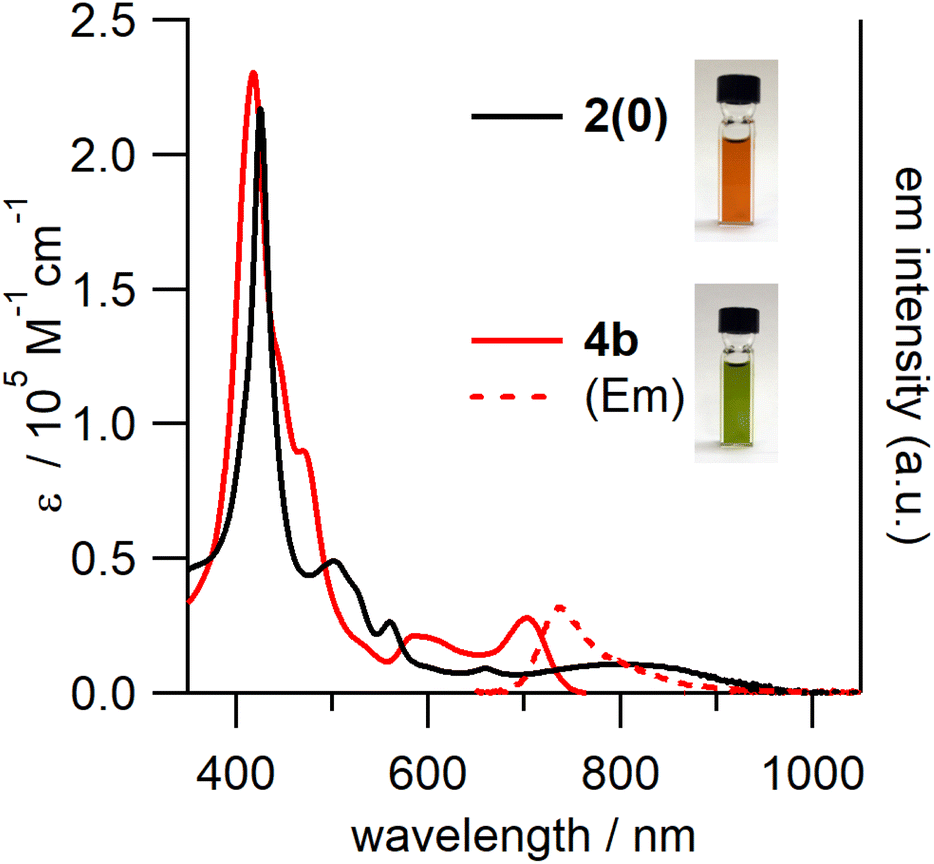 | ||
| Fig. 6 UV-vis-NIR absorption spectra of 2(0) and 4b and the fluorescence spectrum of 4b (excited at 430 nm) in toluene. | ||
Reduction of the fully oxidized tetramer
The reduction of tetramer 2(1) was also conducted in the same manner as 2(0) (Scheme 5). The reduction of 2(1) with sodium ascorbate resulted in fully reduced tetramer 5a as the sole product. The 1H NMR spectrum of this product is shown in Fig. S14.† Similar to reduced dimer 4a, 5a was unstable against aerobic oxidation; it readily oxidized during the workup process to form partially oxidized tetramer 6 with one quinoid moiety at the chain end. It is noteworthy that 6 was moderately stable in air; the oxidation of the inner porphyrinoid moiety in 6 to generate tetramer 2(1) in the solution required more than two days. This result indicates that the inner porphyrinoid moieties in 5a and 6 are less susceptible to air oxidation than the end porphyrinoid moiety. This inference is reasonable because meso-bis(N-arylamino)porphyrins are isolable,54,55,59,60 whereas meso-diaminoporphyrin readily undergoes aerobic oxidation to the corresponding quinone.50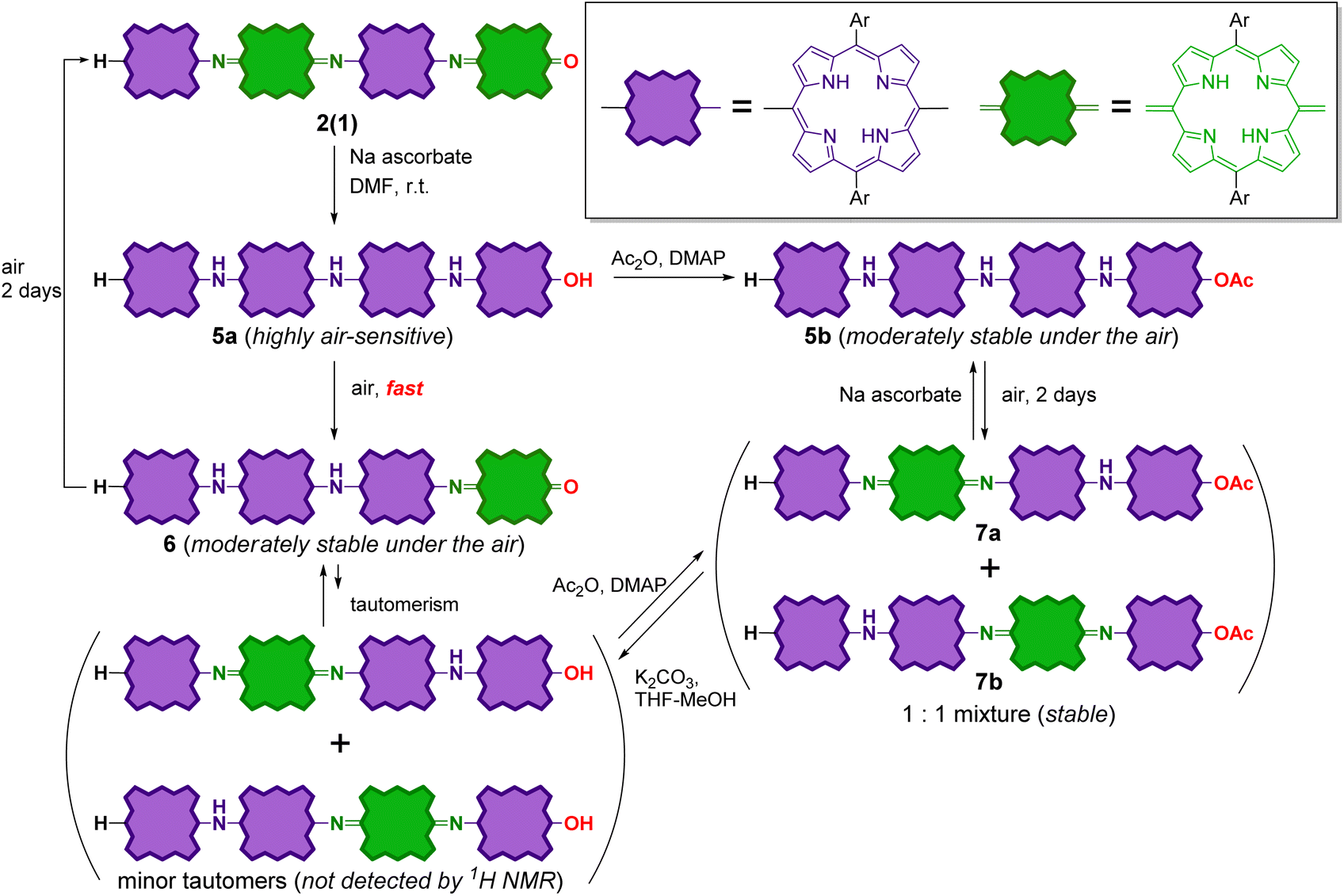 | ||
| Scheme 5 Redox switching of the tetramer. | ||
Further, the end hydroxyl group in fully reduced tetramer 5a was acetylated to stabilize it. The crude acetylated product of 5a contained the desired product, 5b along with two partially oxidized products, 7a and 7b (1:1 mixture) with one inner quinoid moiety. 5b and the mixture of 7a and 7b could be readily isolated by column chromatography. Similar to partially oxidized tetramer 6, 5b was moderately stable in air, and its aerobic oxidation in solution to afford a mixture of 7a and 7b required more than two days. 7 could be reduced with sodium ascorbate to obtain 5b again. That is, the interconversion between 5b and 7 was reversible.
It should also be noted that 6 and 7 could also be reversibly interconverted. When 6 was treated with acetic anhydride, a mixture of 7a and 7b was formed. Under the same reaction conditions, no reaction proceeded in the cases of fully oxidized forms 2(0) and 2(1). The removal of the terminal acetyl group in 7a and 7b (1:1 mixture) by basic hydrolysis resulted in the formation of 6 as the sole product. Therefore, the interconversion between 6 and 7 can be rationalized by considering the equilibrium between 6 and its tautomers with end hydroxyl groups, whose NMR signals were below the lower limit of detection (Scheme 5).
The structures of the reduced tetramers were confirmed by 1H NMR (Fig. S13†). The UV-vis-NIR absorption spectra of 2(1), 5b, and the mixture of 7a and 7b are shown in Fig. 7. Similar to reduced dimer 4, the fully reduced tetramer, 5b did not exhibit the characteristic ICT band in the NIR region; the spectrum contained strong Q bands at ∼780 nm. However, the partially reduced tetramers (7a and 7b) exhibited an ICT band, but the extinction coefficient of this band was smaller than that of 2(1) owing to the decrease in the number of quinoid moieties in the partially reduced molecules.
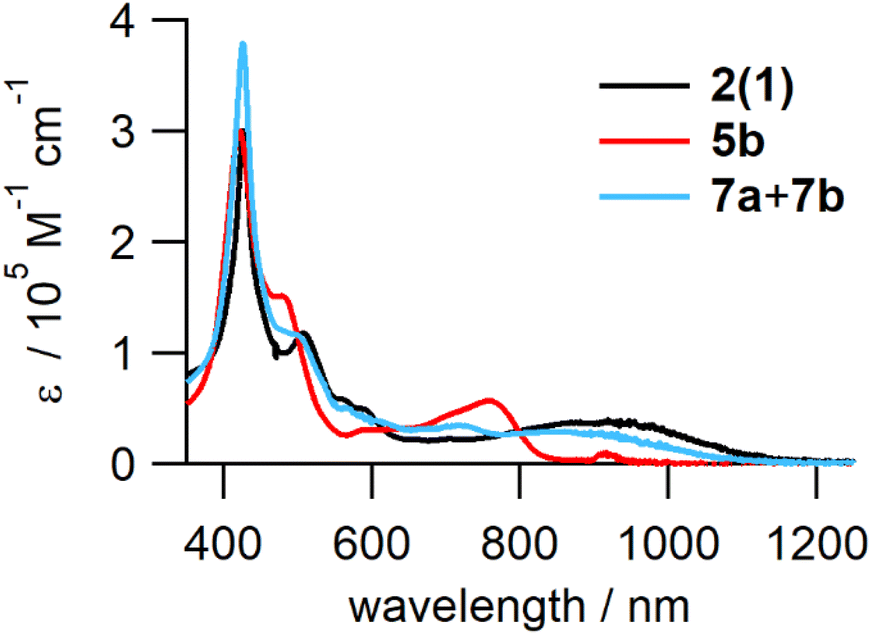 | ||
| Fig. 7 UV-vis-NIR absorption spectra of 2(1), 5b, and the mixture of 7a and 7b in toluene. | ||
Solvent-dependent dynamic behaviours of the partially reduced tetramers
Considering the reaction mechanism for the formation of 7 from 6, the position of the quinoid moieties in the partially reduced oligomers is considered to change dynamically in the solution. This quinoid transfer within the molecule involves intramolecular (proton-coupled) electron transfer. It should therefore be related to the electronic conductivity of the partially oxidized PANI analogue. Therefore, the interconversion of the partially reduced tetramers, 7a and 7b in solution was evaluated by 1H 1D-exchange spectroscopy (EXSY; Fig. 8). A pair of sharp singlet signals of the meso-proton of the end porphyrinoid moieties at ∼9.8 ppm was selected for EXSY measurements. In CDCl3 or CDCl3/CD3CN (2:1), no correlation was observed within 0.4–1.0 s of mixing time (tm) at 300 K (Fig. 8a). This result indicates that the interconversion rates were significantly low in these solvents. In contrast, a clear correlation was observed in CDCl3/CD3OD (2:1) (Fig. 8). The interconversion of 7a and 7b not only involved electron transfer but also proton transfer via the bridged nitrogen atoms. Considering that the conductivity of partially oxidized PANI was significantly enhanced in acidic media owing to N-protonation,4–10 the enhanced interconversion between 7a and 7b can be rationally explained by the hydrogen-bonding of CD3OD to the bridged nitrogen atoms.
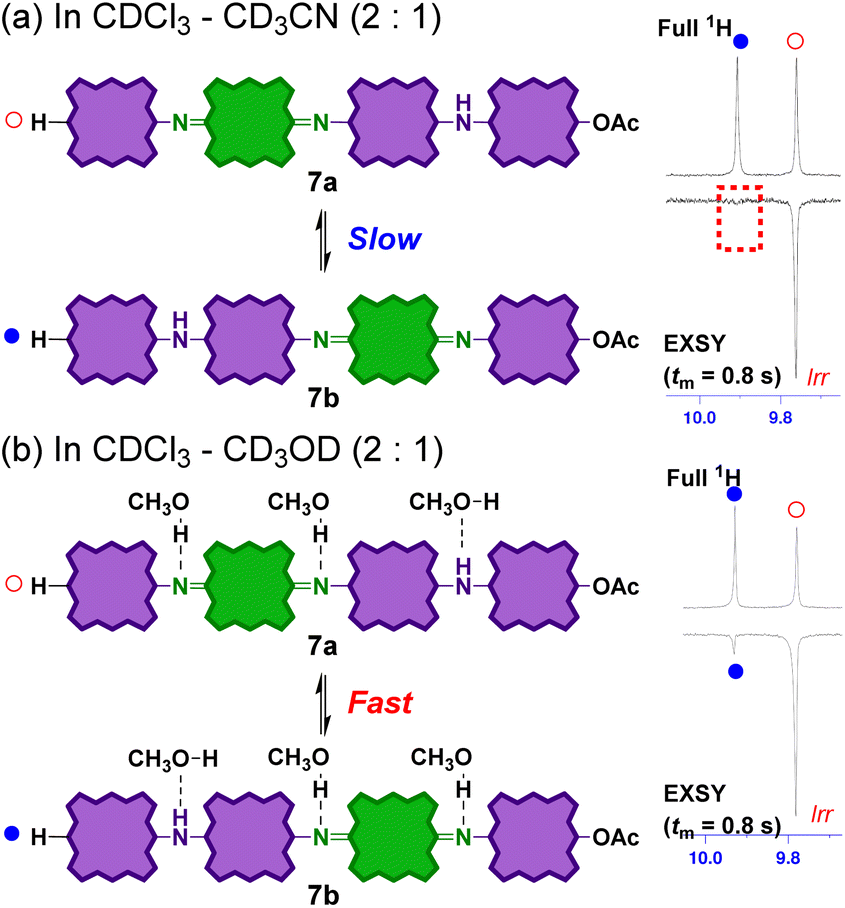 | ||
| Fig. 8
1H 1D EXSY NMR spectra (500 MHz, 300 K) of an equilibrium mixture of 7a and 7b in (a) CDCl3–CD3CN (2:1) and (b) CDCl3–CD3OD (2:1). Mixing time tm = 0.8 s. | ||
To estimate the activation barrier of this interconversion, the interconversion rate constants were determined by conducting 1H 1D EXSY measurements (see Fig. S15 and S16, and Table S2†) at different temperatures (278, 298, and 318 K). The interconversion rate constant increased at an elevated temperature. The Eyring plot of the rate is shown in Fig. S17,† from which the kinetic parameters were determined as ΔH‡ = 26.9 ± 5.5 kJ mol−1 (6.4 ± 1.3 kcal mol−1), ΔS‡ = −169 ± 19 J mol−1 K−1 (−40.4 ± 4.4 cal mol−1 K−1), and ΔG‡298 K = 77.3 ± 7.8 kJ mol−1 (18.5 ± 1.9 kcal mol−1).
Conclusions
We presented the synthesis and reversible redox switching of novel linear π-conjugated meso-aminoporphyrin oligomers, which are analogous to PANIs. The meso-aminoporphyrin undergoes oxidative oligomerization in the presence of dioxygen and an acid catalyst to afford oligomers with a structure similar to that of fully oxidized PANI (pernigraniline). The obtained fully oxidized oligomers have narrow HOMO–LUMO gaps and broad NIR electronic absorption assignable to ICT from porphyrinoid to quinoid moieties (HOMO–LUMO transition). The fully oxidized oligomers could be reversibly reduced with sodium ascorbate to obtain the corresponding reduced oligomers that do not exhibit the ICT absorption but the emission in the NIR region. Finally, we also demonstrated that the internal transfer of the quinoid moiety in the partially reduced tetramer was significantly enhanced in the presence of a protic solvent. We believe that these results are directly related to the high electronic conductivities of partially oxidized PANIs by N-protonation.The meso-aminoporphyrin oligomers discussed herein are a novel type of π-conjugated porphyrin oligomers/polymers, whose properties can be switched by redox stimulation. In the future, we plan to obtain meso-aminoporphyrin polymer films to explore their properties, such as their conductivity with a view to developing applications for sensors using their NIR absorption/luminescence properties, functional electrodes and electrochemical catalysts. A study on modulating the properties of meso-aminoporphyrin oligomers by metal insertion is also in progress.
Data availability
All necessary data have been included in the manuscript and in the ESI.†Author contributions
K. Y. and K. S. supervised and administrated this project. S. T. discovered the reactions and prepared the compounds. S. T. also conducted the basic NMR, MS, spectroscopic, and electrochemical analyses. K. Y. conducted X-ray diffraction analyses, EXSY NMR analyses, and DFT calculations. K. Y. and S. T. wrote the manuscript, and K. S. reviewed and edited it.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Prof. Dr Motoko S. Asano (Gunma University) for the helpful discussion. The computation was performed using Research Center for Computational Science, Okazaki, Japan (Projects: 20-IMS-C195, and 21-IMS-C218).Notes and references
- D. Kumar and R. C. Sharma, Eur. Polym. J., 1998, 34, 1053–1060 CrossRef CAS.
- A. B. Kaiser, Rep. Prog. Phys., 2001, 64, 1–49 CrossRef CAS.
- S. Tajik, H. Beitollahi, F. G. Nejad, I. S. Shoaie, M. A. Khalilzadeh, M. S. Asl, Q. Van Le, K. Zhang, H. W. Jang and M. Shokouhimehr, RSC Adv., 2020, 10, 37834–37856 RSC.
- A. G. MacDiarmid and A. J. Epstein, Faraday Discuss. Chem. Soc., 1989, 88, 317–332 RSC.
- A. J. Epstein and A. G. MacDiarmid, Synth. Met., 1995, 69, 179–182 CrossRef CAS.
- Fundamentals and Emerging Applications of Polyaniline, ed. M. Mozafari and N. P. S. Chauhan, Elsevier, 2019 Search PubMed.
- E. M. Geniès, A. Boyle, M. Lapkowski and C. Tsintavis, Synth. Met., 1990, 36, 139–182 CrossRef.
- J. Stejskal and R. G. Gilbert, Pure Appl. Chem., 2002, 74, 857–867 CrossRef CAS.
- N. Gospodinova and L. Terlemezyan, Prog. Polym. Sci., 1998, 23, 1443–1484 CrossRef CAS.
- Z. Wei and C. F. J. Faul, Macromol. Rapid Commun., 2008, 29, 280–292 CrossRef CAS.
- K. M. Molapo, P. M. Ndangili, R. F. Ajayi, G. Mbambisa, S. M. Mailu, N. Njomo, M. Masikini, P. Baker and E. I. Iwuoha, Int. J. Electrochem. Sci., 2012, 7, 11859–11875 CAS.
- Z. A. Boeva and V. G. Sergeyev, Polym. Sci., Ser. C, 2014, 56, 144–153 CrossRef CAS.
- S. K. Dhawan, D. Kumar, M. K. Ram, S. Chandra and D. C. Trivedi, Sens. Actuators, B, 1997, 40, 99–103 CrossRef CAS.
- G. Wu, K. L. More, C. M. Johnston and P. Zelenay, Science, 2011, 332, 443–447 CrossRef CAS.
- M. B. Regasa, T. R. Soreta, O. E. Femi, P. C. Ramamurthy and S. Kumar, J. Mol. Recognit., 2020, 33, 1–11 CrossRef.
- Z. Li and L. Gong, Materials, 2020, 13, 548 CrossRef CAS.
- R. Willstätter and C. W. Moore, Ber. Dtsch. Chem. Ges., 1907, 40, 2665–2689 CrossRef.
- R. A. Singer, J. P. Sadighi and S. L. Buchwald, J. Am. Chem. Soc., 1998, 120, 213–214 CrossRef CAS.
- Y. Yu, H. Mao, L. Chen, X. Lu, W. Zhang and Y. Wei, Macromol. Rapid Commun., 2004, 25, 664–668 CrossRef CAS.
- I. Różalska, P. Kułyk and I. Kulszewicz-Bajer, New J. Chem., 2004, 28, 1235–1243 RSC.
- R. Eelkema and H. L. Anderson, Macromolecules, 2008, 41, 9930–9933 CrossRef CAS.
- D. Qiu, Y. Cheng and L. Wang, Dalton Trans., 2009, 3247 RSC.
- A. H. Arévalo, H. Fernández, J. J. Silber and L. Sereno, Electrochim. Acta, 1990, 35, 741–748 CrossRef.
- D. K. Moon, K. Osakada, T. Maruyama, K. Kubota and T. Yamamoto, Macromolecules, 1993, 26, 6992–6997 CrossRef CAS.
- B. C. Roy, M. Dutta Gupta, L. Bhowmik and J. K. Ray, Bull. Mater. Sci., 2003, 26, 633–637 CrossRef CAS.
- M. Horie, I. Yamaguchi and T. Yamamoto, Macromolecules, 2006, 39, 7493–7501 CrossRef CAS.
- W. A. Badawy, K. M. Ismail and S. S. Medany, Electrochim. Acta, 2006, 51, 6353–6360 CrossRef CAS.
- N. Oyama, K. Hirabayashi and T. Ohsaka, Bull. Chem. Soc. Jpn., 1986, 59, 2071–2080 CrossRef CAS.
- T. Ohsaka, K. Hirabayashi and N. Oyama, Bull. Chem. Soc. Jpn., 1986, 59, 3423–3429 CrossRef CAS.
- H. L. Anderson, Chem. Commun., 1999, 2323–2330 RSC.
- T. Tanaka and A. Osuka, Chem. Soc. Rev., 2015, 44, 943–969 RSC.
- H. L. Anderson, S. J. Martin and D. D. C. Bradley, Angew. Chem., Int. Ed. Engl., 1994, 33, 655–657 CrossRef.
- P. N. Taylor, J. Huuskonen, R. T. Aplin, H. L. Anderson, J. Huuskonen, G. Rumbles and E. Williams, Chem. Commun., 1998, 909–910 RSC.
- A. Tsuda and A. Osuka, Science, 2001, 293, 79–82 CrossRef CAS.
- A. Tsuda, H. Furuta and A. Osuka, Angew. Chem., Int. Ed., 2000, 39, 2549–2552 CrossRef CAS.
- A. Bettelheim, B. A. White, S. A. Raybuck and R. W. Murray, Inorg. Chem., 1987, 26, 1009–1017 CrossRef CAS.
- K. A. Macor, Y. O. Su, L. A. Miller and T. G. Spiro, Inorg. Chem., 1987, 26, 2594–2598 CrossRef CAS.
- S. Lamare, S. M. Aly, D. Fortin and P. D. Harvey, Chem. Commun., 2011, 47, 10942 RSC.
- P. A. Liddell, M. Gervaldo, J. W. Bridgewater, A. E. Keirstead, S. Lin, T. A. Moore, A. L. Moore and D. Gust, Chem. Mater., 2008, 20, 135–142 CrossRef CAS.
- M. Gervaldo, P. A. Liddell, G. Kodis, B. J. Brennan, C. R. Johnson, J. W. Bridgewater, A. L. Moore, T. A. Moore and D. Gust, Photochem. Photobiol. Sci., 2010, 9, 890 CrossRef CAS.
- L. J. Esdaile, P. Jensen, J. C. McMurtrie and D. P. Arnold, Angew. Chem., Int. Ed., 2007, 46, 2090–2093 CrossRef CAS PubMed.
- B. Bašić, J. C. McMurtrie and D. P. Arnold, Eur. J. Org. Chem., 2010, 4381–4392 Search PubMed.
- B. Bašić, J. C. McMurtrie and D. P. Arnold, J. Porphyrins Phthalocyanines, 2010, 14, 481–493 CrossRef.
- L. J. Esdaile, M. O. Senge and D. P. Arnold, Chem. Commun., 2006, 2, 4192–4194 RSC.
- A. M. V. M. Pereira, M. G. P. M. S. Neves, J. A. S. Cavaleiro, C. Jeandon, J. Gisselbrecht, S. Choua and R. Ruppert, Org. Lett., 2011, 13, 4742–4745 CrossRef CAS.
- K. Merahi, A. M. V. M. Pereira, C. Jeandon, L. Ruhlmann, J. A. S. Cavaleiro, M. G. P. M. S. Neves, M. Orio, P. Turek, S. Choua and R. Ruppert, J. Porphyrins Phthalocyanines, 2016, 20, 1233–1243 CrossRef CAS.
- D. Shimizu, Y. Ide, T. Ikeue and A. Osuka, Angew. Chem., Int. Ed., 2019, 58, 5023–5027 CrossRef CAS.
- K. Yamashita, K. Kataoka, M. S. Asano and K. Sugiura, Org. Lett., 2012, 14, 190–193 CrossRef CAS.
- K. Yamashita, D. Hirano, M. S. Asano and K. Sugiura, Chem. Lett., 2014, 43, 1049–1051 CrossRef CAS.
- K. Yamashita, K. Kataoka, S. Takeuchi and K. Sugiura, J. Org. Chem., 2016, 81, 11176–11184 CrossRef CAS.
- K. Yamashita, S. Sakamoto, A. Suzuki and K. Sugiura, Chem.–Asian J., 2016, 11, 1004–1007 CrossRef CAS.
- K. Yamashita, D. Hirano and K. Sugiura, Eur. J. Inorg. Chem., 2020, 3507–3516 CrossRef CAS.
- K. Yamashita, D. Hirano, K. Fujimaki and K. Sugiura, Chem.–Asian J., 2020, 15, 3037–3043 CrossRef CAS.
- K. Wang, B. Xiao, L. Xu, M. Zhou, T. Tanaka, A. Osuka and J. Song, Chin. Chem. Lett., 2022, 33, 4545–4548 CrossRef CAS.
- D.-M. Shen, C. Liu, X.-G. Chen and Q.-Y. Chen, J. Org. Chem., 2009, 74, 206–211 CrossRef CAS.
- C. H. Devillers, A. K. D. Dime, H. Cattey and D. Lucas, Chem. Commun., 2011, 47, 1893–1895 RSC.
- J. H. Fuhrhop, K. M. Kadish and D. G. Davis, J. Am. Chem. Soc., 1973, 95, 5140–5147 CrossRef CAS.
- G. B. Maiya and V. Krishnan, Inorg. Chem., 1985, 24, 3253–3257 CrossRef CAS.
- Y. Chen and X. P. Zhang, J. Org. Chem., 2003, 68, 4432–4438 CrossRef CAS.
- K. Ladomenou, T. Lazarides, M. K. Panda, G. Charalambidis, D. Daphnomili and A. G. Coutsolelos, Inorg. Chem., 2012, 51, 10548–10556 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc06387e |
| This journal is © The Royal Society of Chemistry 2023 |