
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVisible-light-induced selective defluoroalkylations of polyfluoroarenes with alcohols†
Wengang
Xu
 *a,
Qi
Shao
b,
Congjian
Xia
b,
Qiao
Zhang
a,
Yadi
Xu
a,
Yingguo
Liu
*c and
Mingbo
Wu
*ab
*a,
Qi
Shao
b,
Congjian
Xia
b,
Qiao
Zhang
a,
Yadi
Xu
a,
Yingguo
Liu
*c and
Mingbo
Wu
*ab
aCollege of New Energy, China University of Petroleum (East China), Qingdao, Shandong Province 266580, P. R. China. E-mail: chmxw@upc.edu.cn; wumb@upc.edu.cn
bCollege of Chemical Engineering, State Key Laboratory of Heavy Oil Processing, China University of Petroleum (East China), Qingdao, Shandong Province 266580, P. R. China
cDivision of Molecular Catalysis and Synthesis, Henan Institute of Advanced Technology, Zhengzhou University, Zhengzhou, Henan Province 450001, P. R. China. E-mail: liuyg@zzu.edu.cn
First published on 21st December 2022
Abstract
To provide α-polyfluoroarylalcohols, a novel protocol for the selective defluoroalkylation of polyfluoroarenes with easily accessible alcohols was reported via the cooperation of photoredox and hydrogen atom transfer (HAT) strategies with the assistance of Lewis acids under visible light irradiation. The protocol featured broad scope, excellent regioselectivity for both C–H and C–F bond cleavages, and mild conditions. Mechanistic studies suggested that the reaction occurred through Lewis acid-promoted HAT to provide an alkyl radical and sequential addition to polyfluoroarenes. Impressively, the regioselectivity for C–F cleavage was verified with the Fukui function. The feasibility and application of this protocol on fluoroarene synthesis were well illustrated by gram-scale synthesis under both batch and flow conditions, late-stage decoration of bioactive compounds, and further transformations of the fluoroarylalcohols.
Introduction
Fluoroarenes are important motifs exhibiting wide applications in pharmaceuticals,1–6 agrochemicals,7,8 and materials.9 Among the newly released top 200 pharmaceuticals in 2021,10 over 10% of them contain aryl C–F bonds including eight with polyfluoroarenes. However, the synthetic methods for polyfluoroarenes were still limited to traditional methods, such as direct fluorination with highly reactive fluorine sources, leading to poor selectivity.11–15 Selective C–F bond activation of readily available poly- or perfluoroarenes to construct novel C–C bonds provided an alternative method for fluoroarene synthesis.16–21 However, due to the inertness of C–F bonds, the majority of the reports on the transformation of C–F bonds of polyfluoroarenes into C–C bonds occurred with strong nucleophiles such as Grignard, zinc, and copper reagents via directed transition metal catalysis to ensure the regioselectivity for C–F bond activations. The preinstalled proximal directing or activating groups and highly reactive reaction partners largely undermined the feasibility of these strategies.With the significant development of visible-light-induced organic transformations in recent years,22–32 photocatalytic chemo- and regioselective conversion of C–F bonds in polyfluoroarenes to C–C bonds via defluoroalkylation has drawn significant attention.33,34 Weaver reported a series of C–F functionalizations of polyfluoroarenes via photoreduction of polyfluoroarenes and sequential radical addition to unsaturated C–C bonds present in alkenes,35 alkynes,36 and arenes.37 On the other hand, the Hashmi group developed a visible-light-induced defluoroalkylation reaction to achieve the α-fluoroarylation of N-dialkylanilines through a radical–radical coupling pathway.38 Recently, the Ritter group reported a defluoroalkylation of polyfluoroarenes using aliphatic carboxylic acids as the alkylation reagent via photocatalytic decarboxylation for alkyl radical formations.39 However, the facile and efficient defluoroalkylations of polyfluoroarenes via direct coupling of C(sp3)–H and C–F bonds are still underdeveloped.
α-Polyfluoroarylalcohols are important skeletons in drugs and bioactive compounds (Fig. 1a).4 However, the synthetic methods were still limited to the nucleophilic addition of polyfluoroaryl precursors such as Grignard40,41 and silane reagents42 to aldehydes or ketones with or without transition metal catalysis (Fig. 1b). Recently, Luo, Radius and Marder reported a transition metal-free, base-promoted additions of polyfluoroarylboronates to aldehydes and ketones,43 based on their achievements in nickel-catalyzed defluoroborylation of polyfluoroarenes44,45 and palladium-catalyzed dechloroborylation of chlorine-substituted polyfluoroarenes.46 The usage of highly reactive or not easily accessible fluoroaryl reagents dramatically restricted the application of these methodologies and the variety of the products. Meanwhile, alcohols were one of the most abundant and accessible starting materials for organic transformations.47 Using alcohols as alkylation reagents via photocatalytic hydrogen atom transfer (HAT) of their α-C–H bonds has witnessed significant progress in recent years. Elegant studies from Macmillan's group using alcohol as the alkyl radical precursor provided an efficient methodology to construct C–C bonds via radical addition to Michael acceptors48 or cross-coupling with aryl halides with the cooperation of nickel catalysis.49 Nevertheless, the defluoroalkylations of polyfluoroarenes using alcohols as the alkyl source via HAT were rarely reported and challenging, maybe because of the nucleophilicity of alcohols. Nucleophilic aromatic substitution (SNAr) readily occurred with polyfluoroarenes to provide defluoroetherification products under basic conditions.50 In 1991, Pahor et al. reported a seminal study on the defluoroalkylation of polyfluoroarenes with alcohols using benzophenone as the stoichiometric HAT reagent under neutral conditions and UV irradiation,51 albeit with poor selectivity, low efficiency, and limited scope for both substrates (Fig. 1c). Since then, no significant progress has been made on this transformation. Considering the development of photocatalytic selective C–F activation of polyfluoroarenes and HAT reactions of alcohols and our continuous interest in fluoroarene synthesis,52 herein we hypothesized that with the assistance of Lewis acids, the activations of α-C–H of alcohols could be facilitated via the cooperation of photoredox and HAT catalysis to produce an alkyl radical, which would react with polyfluoroarenes to deliver valuable α-polyfluoroaryl alcohols (Fig. 1d).
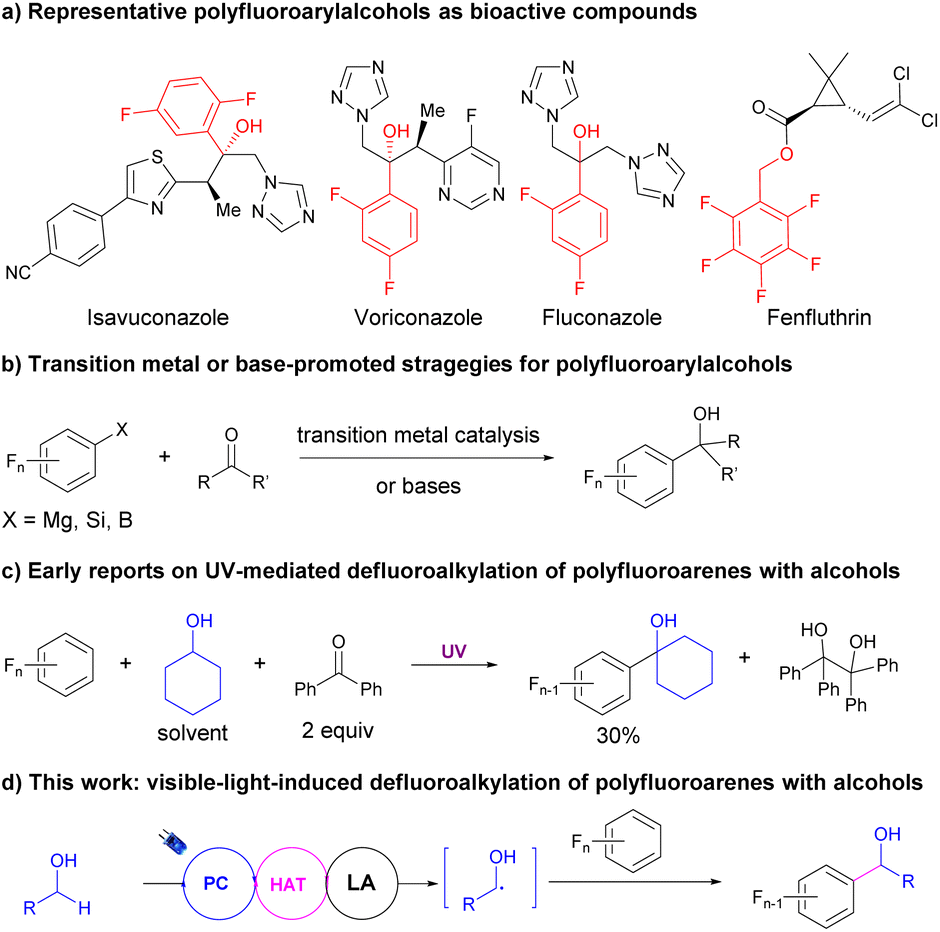 | ||
| Fig. 1 Applications and synthetic strategies of polyfluoroarylalcohols. | ||
Results and discussion
The optimization of the reaction conditions was initiated using methyl 2,3,4,5,6-pentafluorobenzoate (1a) and hexan-1-ol (2a) as the reaction partners, and a catalytic system containing photoredox catalysts (PCs), HAT catalysts, Lewis acids, and bases (Table 1). After substantial exploration, we achieved the optimal conditions for defluoroalkylation with a combination of 4-CzIPN (3 mol%), quinuclidine (30 mol%), ZnCl2 (1.5 equiv.) and K3PO4 (1.0 equiv.) in DMSO under blue LED irradiation to afford polyfluoroarylalcohol products (3aa) in 87% isolated yields (Table 1, entry 1). Using precious transition metal complexes such as Ir(ppy)3, Ir(ppy)2(dtbbpy)PF6, and Ir[dF(CF3)ppy]2(dtbbpy)PF6 instead of 4-CzIPN as photoredox catalysts, the yields for 3aa were dramatically decreased (entry 2–4). Other solvents including MeCN, EtOAc, DMF, or DMA led to sluggish reaction mixtures, providing 3aa only in less than 15% yields (entry 5). Control experiments exhibited the necessity for the presence of light, 4-CzIPN, and quinuclidine (entry 6). Excluding ZnCl2 sharply lowered the yields to 28% (entry 7). The absence of bases led to comparable yields of 3aa (entry 8). Notably, without bases, the usage of ZnCl2 could be decreased to less than the stoichiometric amount (entry 9).|
|
||
|---|---|---|
| Entry | Deviation | Yieldb (%) |
| a Reaction conditions: methyl 2,3,4,5,6-pentafluorobenzoate (1a, 0.20 mmol), hexan-1-ol (2a, 0.30 mmol), 4-CzIPN (3 mol%), quinuclidine (30 mol%), ZnCl2 (0.30 mmol), DMSO (4 mL), 18 W blue LED irradiation, room temperature, 24 h. b Yields of 3aa were determined by analysis of the crude 1H NMR spectra using 1,3,5-trimethoxybenzene as an internal standard. c Isolated yields. | ||
| 1a | No | 87c |
| 2 | Ir(ppy)3 (2 mol%) instead of 4-CzIPN | 0 |
| 3 | PC1 (1 mol%) instead of 4-CzIPN | 6 |
| 4 | PC2 (1 mol%) instead of 4-CzIPN | 70 |
| 5 | MeCN, EtOAc, DMF, or DMA as solvent | <15 |
| 6 | No light, 4-CzIPN, or quinuclidine | 0 |
| 7 | Without ZnCl2 | 28 |
| 8 | Without K3PO4 | 86 |
| 9 | ZnCl2 (50 mol%) and without K3PO4 | 70 |

After the establishment of the optimized conditions, we continued to evaluate the scope of polyfluoroarenes (1) for defluoroalkylations with hexan-1-ol (2a) (Scheme 1). Perfluoroarenes including hexafluorobenzene and pentafluoropyridine worked well to produce the monoalkylation products (3ba–3ca) in good to excellent yields with specific regioselectivities. Mono and di-alkylation of perfluoro-1,1′-biphenyl (3da–3ea) could be selectively achieved by adjusting the equivalents of alcohols. Defluoroalkylation of pentafluorobenzene occurred smoothly to give the product (3fa) in 57% yields. Pentafluoroarenes bearing electron-withdrawing groups i.e., CF3 were exceptional candidates for this transformation (3ga). Quite a few functional groups such as acetyl, ester, alkynyl, chloro, bromo, amide, pyrrolidinone, and piperidinone were well tolerated and produced the corresponding alkyltetrafluorobenzene derivatives (3ha–3qa) in good to excellent yields. 1,2,3,4-Tetrafluorobenzene was defluoroalkylated exclusively at the 2-position (3ra), albeit in a lower yield. Nevertheless, adding a trifluoromethyl group or an ester group led to much higher yields for the defluoroalkylated products (3sa–3ta) of tetrafluoroarenes, albeit with lower regioselectivity for 3sa. Trifluoroarenes bearing ester groups with different substitution patterns both underwent defluoroalkylation selectively to produce the desired products (3ua–3va) in 95% and 44% yields, respectively. Interestingly, the selectivity for C–F cleavage in 1f and 1r was different but complementary with transition-metal catalysis.53,54 In addition, no directing effect of the proximal groups such as ester and amide groups, was exhibited for the C–F bond activations. This protocol was also amendable to natural products such as D-menthol and (+)-fenchol derived pentafluoroarenes and gave the tetrafluoroarene products (3wa–3xa) in appreciable yields.
 | ||
| Scheme 1 Photocatalytic selective defluoroalkylation of polyfluoroarenes. aAll cited yields are isolated yields. The product was produced as a single isomer unless otherwise noted. b2a (5.0 equiv.) was used. c2a (1.5 equiv.) was used. | ||
The applications of this strategy on C–H fluoroarylation of various alcohols (2) with methyl 2,3,4,5,6-pentafluorobenzoate (1a) were also explored (Scheme 2). Simple linear alkyl alcohols such as methanol, ethanol, and 1-decanol were fluoroarylated at the α-position of alcohols (3ab–3ad) in 49–92% yields. Sterically hindered β-alkyl substituted alcohols were also applicable for fluoroarylation (3ae–3af). Functional groups such as fluoro, chloro, CF3, and OTBS were well incorporated to provide the tetrafluoroarylalcohols (3ag–3aj) in 44–75% yields. Gratefully, aryl substituted alcohols performed the alkylation exclusively at the α-position of alcohols instead of benzylic positions to give the fluoroarylated products (3ak–3am) in moderate yields. Notably, the alcohols with sterically hindered α-substituents were workable for fluoroarylation to afford the products (3an–3ao) in 41–87% yields. α-Cycloalkyl methanols including cyclohexyl, cyclopentyl, and cyclobutyl were successful candidates for fluoroarylation to afford products (3ap–3as) in good to excellent yields. To our delight, the fluoroarylation of acyclic and cyclic secondary alcohols such as isopropanol, cyclohexanol, and even 2-adamantanol occurred smoothly to deliver the desired products (3at–3av) in 55–78% yields. Diols including 3-methylbutane-1,3-diol, ethane-1,2-diol and propane-1,3-diol worked well for selective mono-fluoroarylation to give fluoroarylated diols (3aw–3ay) in good yields. The bioactive lauryl alcohol was also a suitable candidate to provide the fluoroarylation products (3az) quantitively.
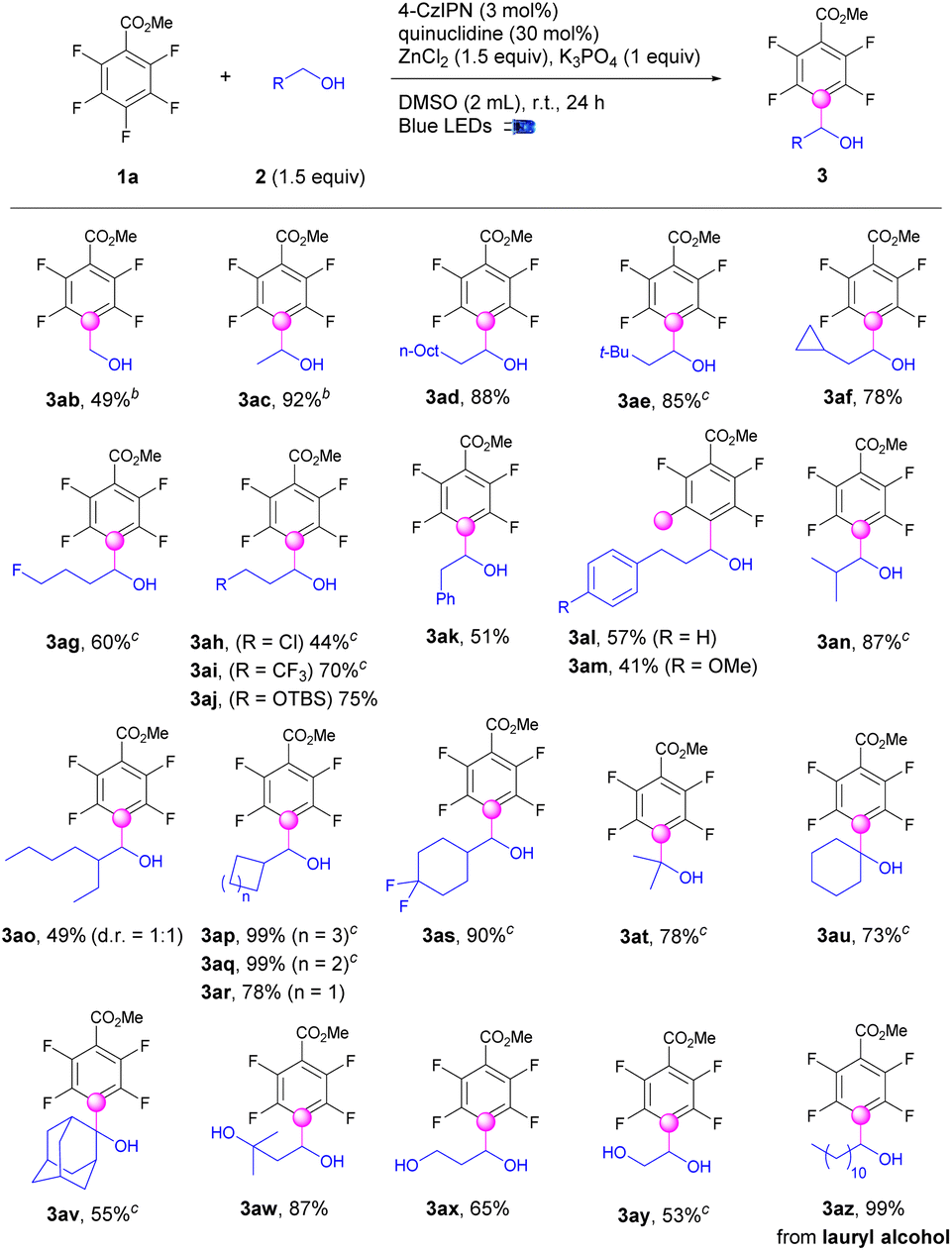 | ||
| Scheme 2 Photocatalytic selective fluoroarylations of alcohols. aAll cited yields are isolated yields. The product was produced as a single isomer unless otherwise noted. b2 (5.0 equiv.) was used. c2 (3.0 equiv.) was used. | ||
Impressively, the protocol was further applicable for direct fluoroarylations of complex natural products and pharmaceutical derivatives with alcohol functionalities (Scheme 3). Epiandrosterone, an anabolic steroid, which is the source of testosterone and other sex hormones, could be directly polyfluoroarylated to afford the products (4) in excellent yields. The fluoroarylation of β-cholestanol was also successful to give the derivative (5) in moderate yields. Lithocholic acid known as one of the bile acids was efficiently decorated with a fluoroaryl group after esterification (6). Alcohols linked with pharmaceutical moieties, such as ibuprofen, probenecid, and oxaprozin, could be fluoroarylated successfully to deliver the complex α-polyfluoroarylalcohols (7–9) in good yields with excess amounts of inexpensive polyfluoroarenes.
 | ||
| Scheme 3 Photo-mediated selective fluoroarylation of bioactive alcohols. aAll cited yields are isolated yields. The product was produced as a single isomer unless otherwise noted. b1a (3.0 equiv.) was used. | ||
To test the feasibility of this transformation, the gram-scale synthesis of α-fluoroaryl alcohols was performed which afforded the products (3aa) in 76% yields under standard conditions for 3 days (Scheme 4). This product 3aa was further reduced by LiAlH4 to deliver fluoroaryl-diols (10) in 99% yields, which were important precursors for the synthesis of insecticides.55 The hydroxyl groups in the products could be transformed into bromine (11) using PBr3 in 77% yields.56 The oxidation of the alcohols to ketone products (12) occurred smoothly with PDC in 88% yields.57 The combination of Tf2O and NEt3 could dehydroxylate the fluoroarylalcohols to afford the E-fluoroarylalkenes exclusively in 62% yields (13),58 which could not be achieved by previous reported photocatalytic defluoroalkenylation strategies.36 To our delight, the continuous-flow conditions were applicable for a modified protocol utilizing 4CzIPN (1 mol%), quinuclidine (20 mol%) and ZnCl2 (50 mol%) under base-free conditions, to produce 3aa with higher efficiency compared with that for batch conditions.
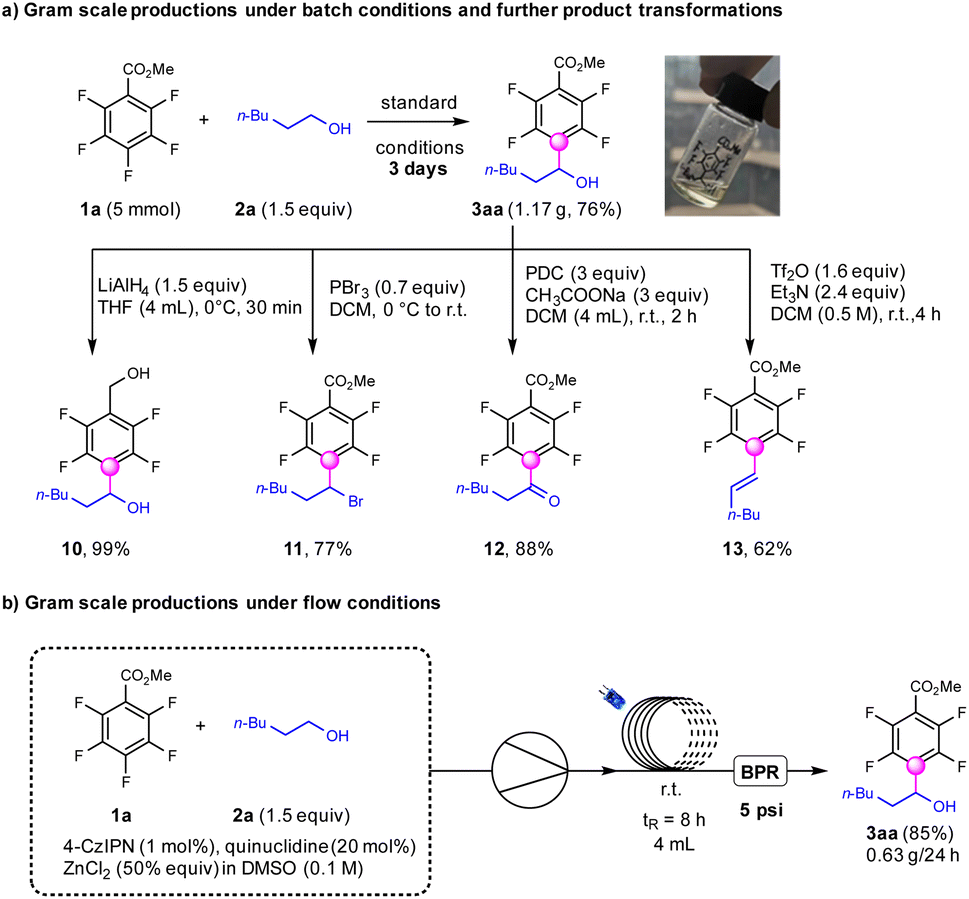 | ||
| Scheme 4 Gram scale productions and further product transformations. | ||
To elucidate the mechanism for defluoroalkylation, we performed a series of experiments (Scheme 5). No desired product was obtained with the addition of TEMPO suggesting a radical mechanism (Scheme 5a). In the presence of benzyl acrylate, the yield of 3aa was decreased to 9% accompanied by the formation of 5-pentyldihydrofuran-2(3H)-one, which indicated the presence of an alcohol α-carbon radical (Scheme 5b).48 Meanwhile, when adding radical trappers for polyfluoroaryl radicals, such as alkynes36 and trimethoxybenzene,37 no radical addition products were observed with no significant influence on the yields of 3aa. Stern–Volmer quenching studies indicate that the quenching effect of quinuclidine was much stronger than that of polyfluoroarenes and alcohols (see the ESI†). In addition, the stronger C–F bonds of pentafluorochlorobenzene (1y) instead of the C–Cl bond were selectively activated to produce the defluoroalkylation products (3ya) under standard conditions with moderate regioselectivity (Scheme 5c), indicating that the reaction might not involve the single electron reduction of the polyfluoroarenes.59 CV testing on the substrates showed that the reductive potential of the polyfluoroarenes (Ered = −2.67 V vs. Ag/AgCl in DMSO) was much lower than EPCn/PCn−1 (−1.21 V vs. SCE in CH3CN), while the oxidative potential of quinuclidine (Eox = 1.14 V vs. Ag/AgCl in DMSO) was slightly lower than EPCn*/PCn−1 (1.35 V vs. SCE in CH3CN).60 Above all, the photoredox cycle could be initiated via reductive quenching of the excited photocatalyst with quinuclidine. D4-methanol (2b-D) led to tetrafluoroarylated methanol (3ab-D) in moderate yields with no H–D exchanges at the α-position, while the deuterium atom on the hydroxyl group was exclusively exchanged to hydrogen (Scheme 5d), indicating the formation of an alkoxide intermediate. The intermolecular competition reaction of 2b and 2b-D with 1a was conducted to figure out a KIE value of 5.8, suggesting that the C–H activation via HAT could be the rate-controlling step.61 Finally, quantum yields (0.31) were determined to exclude the radical chain mechanism.62
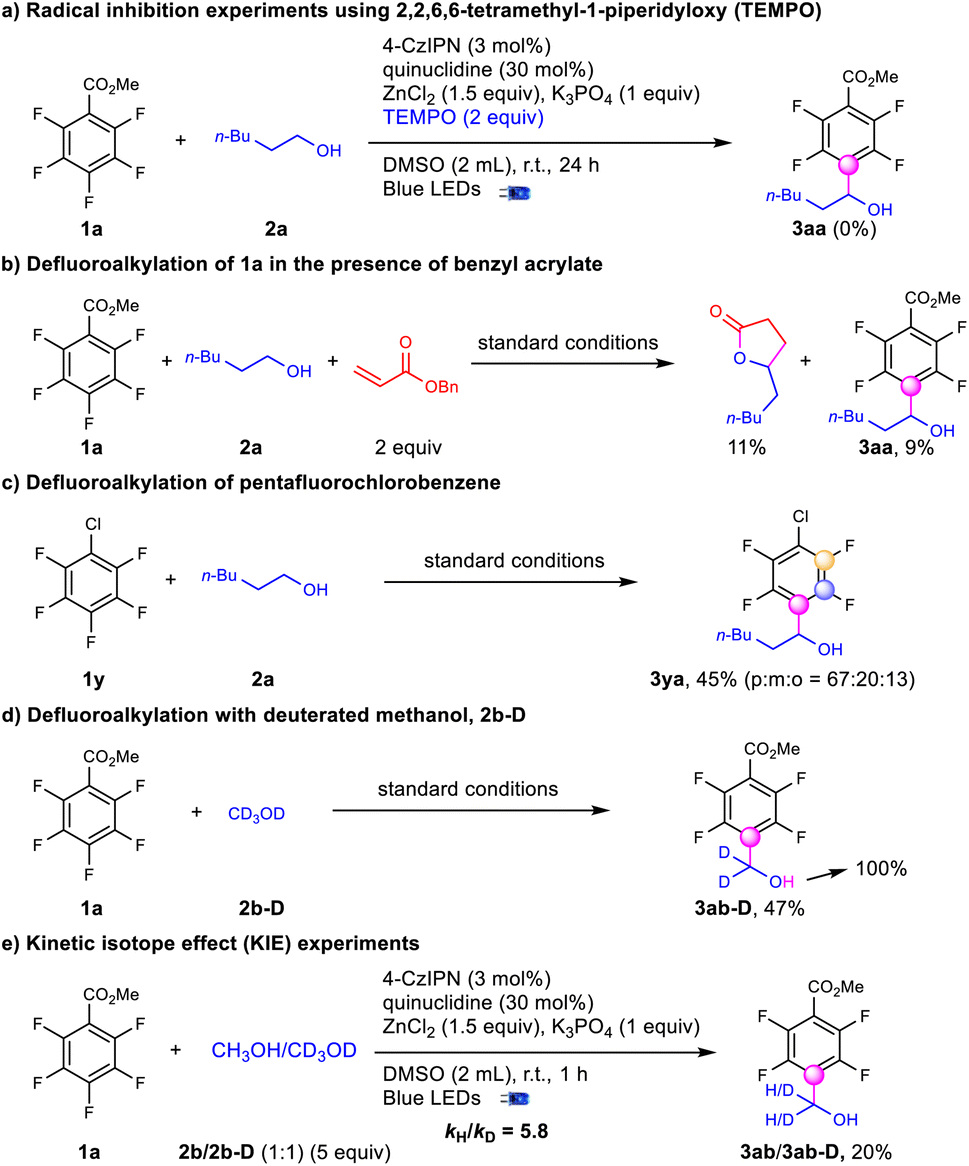 | ||
| Scheme 5 Experiments to elucidate the reaction mechanism. | ||
Plausible catalytic cycles were proposed based on all the above observations (Scheme 6). As illustrated in Scheme 6, the PC (4-CzIPN) was first photo-excited to PC* under blue LED irradiation. Reductive quenching occurred for PC* with quinuclidine to deliver a quinuclidine radical cation and PCn−1. This radical cation abstracted the hydrogen atom from the alkoxide zinc intermediate I, which is the product of the reaction between alcohol and ZnCl2,49 to give the alkyl radical II. Radical addition of II then occurred with polyfluoroarenes to deliver the aryl-lradical III. Followed by the reduction of III with PCn−1, the polyfluoroaryl anion IV was produced along with the recovery of the PC. The resulting intermediate IV would extrude the fluoride to afford the final product 3,63 followed by the sequential zinc-proton exchange. To illustrate the regioselectivity for C–F bond cleavage in polyfluoroarenes, the orbital-weighted Fukui index (OW_f+) was computed for representative polyfluoroarene substrates,64–67 including 1a, 1f–1g, 1r, and 1t–1v (Fig. 2). The orbital weighted Fukui function could analyze the contribution of inner orbitals to the reactivity and predict accurate regioselectivity for C–F cleavage of polyfluoroarenes.68 The isosurface images of OW_f+ are shown in Fig. 2. Interestingly, the defluoroalkylation occurred specifically on the carbon atoms (C1) exhibiting the highest OW_f+ values in these substrates, which was in good accord with the nucleophilic carbon radical addition mechanism and provided a straightforward strategy to predict the regioselectivity for these types of radical addition reactions to polyfluoroarenes.
 | ||
| Scheme 6 Proposed mechanism for defluoroalkylation of polyfluoroarenes. | ||
 | ||
| Fig. 2 The orbital-weighted Fukui index (OW_f+) of the representative substrates. | ||
Conclusions
In summary, we have established a feasible protocol for the selective defluoroalkylation of polyfluoroarenes with alcohols utilizing a cooperative catalytic system combining photocatalysts, HAT catalysts, and Lewis acids. Varieties of polyfluoroaryl alcohols were successfully constructed via selective C–F activation of polyfluoroarenes and C–H activation of alcohols. Notably, the strategy was applicable to direct late-stage decoration of natural products and bioactive alcohols and could also be conducted under continuous flow conditions efficiently to facilitate promising large-scale productions. With the preservation of the hydroxy group, the further transformation led to broad functionalities and will see broad applications in the synthesis of value-added fluoroaryl compounds.Data availability
All experimental and characterization data, as well as NMR spectra are available in the ESI.†Author contributions
M. W., W. X. and Q. S. designed this project. W. X., Q. S., H. Z., C. X., Q. Z. and Y. X. conducted the experiments, analyzed the data, and prepared the ESI.† Y. L. performed the DFT calculations. M. W. and W. X. analyzed the data and prepared the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We appreciate the help of Dr Zhang Hongwei from the China University of Petroleum (East China) in NMR analysis and Prof. Wu Junliang from Zhengzhou University in helpful discussion. We are grateful for the financial support from the National Natural Science Foundation of China (22101301), Fundamental Research Funds for China University of Petroleum (East China) (Grant No. 27RA2014007), China Postdoctoral Science Foundation (Grant No. 31CZ2019010 and 05FW2014001), Major Scientific and Technological Innovation Project of Shandong Province (Grant No. 2020CXGC010402) and Shan-dong Province Natural Science Foundation (Grant No. ZX20210037). Dr Yingguo Liu is thankful for the financial support from the International Postdoctoral Exchange Fellowship Program (Talent-Introduction Program, No. YJ20210304), Youth Talent Program of Science & Technology Think Tanks by the China Association for Science and Technology (No. 20220615ZZ07110054), and China Postdoctoral Science Foundation (No. 2022M722865).Notes and references
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- W. K. Hagmann, J. Med. Chem., 2008, 51, 4359–4369 CrossRef CAS.
- D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308–319 RSC.
- J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS.
- E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315–8359 CrossRef CAS PubMed.
- Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Aceña, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422–518 CrossRef CAS PubMed.
- P. Jeschke, ChemBioChem, 2004, 5, 570–589 CrossRef CAS PubMed.
- P. Jeschke, Pest Manage. Sci., 2017, 73, 1053–1066 CrossRef CAS.
- M. L. Tang and Z. Bao, Chem. Mater., 2011, 23, 446–455 CrossRef CAS.
- https://njardarson.lab.arizona.edu/sites/njardarson.lab.arizona.edu/files/Top%20200%20Pharmaceuticals%202021V2.pdf, 2022.
- C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2015, 54, 3216–3221 CrossRef CAS PubMed.
- C. N. Neumann and T. Ritter, Acc. Chem. Res., 2017, 50, 2822–2833 CrossRef CAS.
- Q. Cheng and T. Ritter, Trends Chem., 2019, 1, 461–470 CrossRef CAS.
- R. Szpera, D. F. J. Moseley, L. B. Smith, A. J. Sterling and V. Gouverneur, Angew. Chem., Int. Ed., 2019, 58, 14824–14848 CrossRef CAS.
- E. Nobile, T. Castanheiro and T. Besset, Angew. Chem., Int. Ed., 2021, 60, 12170–12191 CrossRef CAS PubMed.
- H. Amii and K. Uneyama, Chem. Rev., 2009, 109, 2119–2183 CrossRef CAS PubMed.
- T. Fujita, K. Fuchibe and J. Ichikawa, Angew. Chem., Int. Ed., 2019, 58, 390–402 CrossRef CAS.
- N. A. LaBerge and J. A. Love, in Organometallic Fluorine Chemistry, ed. T. Braun and R. P. Hughes, Springer International Publishing, Cham, 2015, pp. 55–111, DOI:10.1007/3418_2015_90.
- T. Ahrens, J. Kohlmann, M. Ahrens and T. Braun, Chem. Rev., 2015, 115, 931–972 CrossRef CAS PubMed.
- J. L. Kiplinger, T. G. Richmond and C. E. Osterberg, Chem. Rev., 1994, 94, 373–431 CrossRef CAS.
- J. Weaver and S. Senaweera, Tetrahedron, 2014, 70, 7413–7428 CrossRef CAS.
- D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77–80 CrossRef CAS.
- M. A. Ischay, M. E. Anzovino, J. Du and T. P. Yoon, J. Am. Chem. Soc., 2008, 130, 12886–12887 CrossRef CAS.
- J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC.
- J. Xuan and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828–6838 CrossRef CAS.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS.
- D. Ravelli, S. Protti and M. Fagnoni, Chem. Rev., 2016, 116, 9850–9913 CrossRef CAS.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- L. Marzo, S. K. Pagire, O. Reiser and B. König, Angew. Chem., Int. Ed., 2018, 57, 10034–10072 CrossRef CAS.
- R. C. McAtee, E. J. McClain and C. R. J. Stephenson, Trends Chem., 2019, 1, 111–125 CrossRef CAS.
- R. Cannalire, S. Pelliccia, L. Sancineto, E. Novellino, G. C. Tron and M. Giustiniano, Chem. Soc. Rev., 2021, 50, 766–897 RSC.
- J. Wang, H. Gao, C. Shi, G. Chen, X. Tan, X. Chen, L. Xu, X. Cai, B. Huang and H. Li, Chem. Commun., 2021, 57, 12203–12217 RSC.
- B. Niu, K. Sachidanandan, B. G. Blackburn, M. V. Cooke and S. Laulhé, Org. Lett., 2022, 24, 916–920 CrossRef CAS.
- A. Singh, J. J. Kubik and J. D. Weaver, Chem. Sci., 2015, 6, 7206–7212 RSC.
- A. Singh, C. J. Fennell and J. D. Weaver, Chem. Sci., 2016, 7, 6796–6802 RSC.
- S. Senaweera and J. D. Weaver, J. Am. Chem. Soc., 2016, 138, 2520–2523 CrossRef CAS.
- J. Xie, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2017, 56, 7266–7270 CrossRef CAS PubMed.
- X. Sun and T. Ritter, Angew. Chem., Int. Ed., 2021, 60, 10557–10562 CrossRef CAS PubMed.
- M. Abarbri, F. Dehmel and P. Knochel, Tetrahedron Lett., 1999, 40, 7449–7453 CrossRef CAS.
- X. Jia, J. Wang, X. Ding, J. Yang, N. Li, N. Zhao and Z. Huang, J. Org. Chem., 2015, 80, 10874–10882 CrossRef CAS.
- G.-F. Du, F. Xing, C.-Z. Gu, B. Dai and L. He, RSC Adv., 2015, 5, 35513–35517 RSC.
- Z. Liu, G. K. Kole, Y. P. Budiman, Y.-M. Tian, A. Friedrich, X. Luo, S. A. Westcott, U. Radius and T. B. Marder, Angew. Chem., Int. Ed., 2021, 60, 16529–16538 CrossRef CAS.
- J. Zhou, M. W. Kuntze-Fechner, R. Bertermann, U. S. D. Paul, J. H. J. Berthel, A. Friedrich, Z. Du, T. B. Marder and U. Radius, J. Am. Chem. Soc., 2016, 138, 5250–5253 CrossRef CAS.
- Y.-M. Tian, X.-N. Guo, M. W. Kuntze-Fechner, I. Krummenacher, H. Braunschweig, U. Radius, A. Steffen and T. B. Marder, J. Am. Chem. Soc., 2018, 140, 17612–17623 CrossRef CAS.
- Y. P. Budiman, S. Lorenzen, Z. Liu, U. Radius and T. B. Marder, Chem.–Eur. J., 2021, 27, 3869–3874 CrossRef CAS.
- B. K. Chi, J. K. Widness, M. M. Gilbert, D. C. Salgueiro, K. J. Garcia and D. J. Weix, ACS Catal., 2022, 12, 580–586 CrossRef CAS PubMed.
- J. L. Jeffrey, J. A. Terrett and D. W. C. MacMillan, Science, 2015, 349, 1532–1536 CrossRef CAS PubMed.
- J. Twilton, M. Christensen, D. A. DiRocco, R. T. Ruck, I. W. Davies and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2018, 57, 5369–5373 CrossRef CAS PubMed.
- D. Prescher, V. E. Platonov, K. V. Dvornikova, J. Schulze, O. I. Osina and G. G. Yakobson, J. Fluorine Chem., 1985, 29, 204 CrossRef.
- B. Šket, M. Zupan, N. Zupančič and B. Pahor, Tetrahedron, 1991, 47, 5029–5042 CrossRef.
- W. Xu, Q. Zhang, Q. Shao, C. Xia and M. Wu, Asian J. Org. Chem., 2021, 10, 2454–2472 CrossRef CAS.
- Y. M. Tian, X. N. Guo, M. W. Kuntze-Fechner, I. Krummenacher, H. Braunschweig, U. Radius, A. Steffen and T. B. Marder, J. Am. Chem. Soc., 2018, 140, 17612–17623 CrossRef CAS PubMed.
- B. Cui, S. Jia, E. Tokunaga and N. Shibata, Nat. Commun., 2018, 9, 4393 CrossRef PubMed.
- Y. Shono, K. Ujihara, T. Iwasaki, M. Sugano, T. Mori, T. Matsunaga and N. Matsuo, Pestic. Chem., 2007, 149–158, DOI:10.1002/9783527611249.ch16.
- X.-L. Su, L. Ye, J.-J. Chen, X.-D. Liu, S.-P. Jiang, F.-L. Wang, L. Liu, C.-J. Yang, X.-Y. Chang, Z.-L. Li, Q.-S. Gu and X.-Y. Liu, Angew. Chem., Int. Ed., 2021, 60, 380–384 CrossRef CAS PubMed.
- H. Hagiwara, K. Hamano, M. Nozawa, T. Hoshi, T. Suzuki and F. Kido, J. Org. Chem., 2005, 70, 2250–2255 CrossRef CAS PubMed.
- S. Sato, K. Sakata, Y. Hashimoto, H. Takikawa and K. Suzuki, Angew. Chem., Int. Ed., 2017, 56, 12608–12613 CrossRef CAS PubMed.
- S. M. Senaweera, A. Singh and J. D. Weaver, J. Am. Chem. Soc., 2014, 136, 3002–3005 CrossRef CAS PubMed.
- J. Luo and J. Zhang, ACS Catal., 2016, 6, 873–877 CrossRef CAS.
- E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066–3072 CrossRef CAS PubMed.
- M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426–5434 RSC.
- W. Xu, H. Jiang, J. Leng, H.-W. Ong and J. Wu, Angew. Chem., Int. Ed., 2020, 59, 4009–4016 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS.
- Y. Ma, J. Liang, D. Zhao, Y.-L. Chen, J. Shen and B. Xiong, RSC Adv., 2014, 4, 17262–17264 RSC.
- J. Klein, P. Fleurat-Lessard and J. Pilmé, J. Comput. Chem., 2021, 42, 840–854 CrossRef CAS PubMed.
- H. Huang, L. Liu, J. Wang, Y. Zhou, H. Hu, X. Ye, G. Liu, Z. Xu, H. Xu, W. Yang, Y. Wang, Y. Peng, P. Yang, J. Sun, P. Yan, X. Cao and B. Z. Tang, Chem. Sci., 2022, 13, 3129–3139 RSC.
- R. Pino-Rios, O. Yañez, D. Inostroza, L. Ruiz, C. Cardenas, P. Fuentealba and W. Tiznado, J. Comput. Chem., 2017, 38, 481–488 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc06290a |
| This journal is © The Royal Society of Chemistry 2023 |