
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalyst-free synthesis of diverse fluorescent polyoxadiazoles for the facile formation and morphology visualization of microporous films and cell imaging†
Junyao
Xie
 a,
Niu
Niu
ab,
Xinyao
Fu
c,
Xiang
Su
a,
Dong
Wang
a,
Anjun
Qin
c,
Ting
Han
*a and
Ben Zhong
Tang
*d
a,
Niu
Niu
ab,
Xinyao
Fu
c,
Xiang
Su
a,
Dong
Wang
a,
Anjun
Qin
c,
Ting
Han
*a and
Ben Zhong
Tang
*d
aCenter for AIE Research, Shenzhen Key Laboratory of Polymer Science and Technology, Guangdong Research Center for Interfacial Engineering of Functional Materials, College of Materials Science and Engineering, Shenzhen University, Shenzhen, 518060, China. E-mail: hanting@szu.edu.cn
bCollege of Physics and Optoelectronic Engineering, Shenzhen University, Shenzhen, 518060, China
cState Key Laboratory of Luminescent Materials and Devices, Guangdong Provincial Key Laboratory of Luminescence from Molecular Aggregates, Center for Aggregation-Induced Emission, AIE Institute, South China University of Technology, Guangzhou, 510640, China
dSchool of Science and Engineering, Shenzhen Institute of Aggregate Science and Technology, The Chinese University of Hong Kong, Shenzhen, Guangdong 518172, China. E-mail: tangbenz@cuhk.edu.cn
First published on 21st December 2022
Abstract
The development of facile polymerizations toward functional heterocyclic polymers is of great significance for chemistry and materials science. As an important class of heterocyclic polymers, polyoxadiazoles (PODs) have found applications in various fields. However, the synthetic difficulties of PODs greatly restrict their structural diversity and property investigation. Herein, we report a series of catalyst-free multicomponent polymerizations (MCPs) that can facilely synthesize functional PODs with well-defined and diversified topological structures from commercially available or readily accessible aldehydes, carboxylic acids, secondary amines, and (N-isocyanimino)triphenylphosphorane at room temperature. Unlike conventional Ugi polycondensations, the present Ugi-type MCPs can in situ generate oxadiazole moieties in polymer backbones. The obtained PODs possess good solubility, high thermal and morphological stability, and excellent film-forming ability. The introduction of aggregation-induced emission (AIE) moieties together with the inherent structural features of PODs endow these polymers with multiple functionalities. The AIE-active linear PODs can form fluorescent microporous films with stable and ordered structures based on the simple breath figure patterning method, and the self-assembly morphologies can be directly visualized by fluorescence microscopy in a high-contrast and sensitive manner. Moreover, both the linear and hyperbranched AIE-active PODs possess excellent biocompatibility, good lysosome specificity, and excellent photobleaching resistance, which enable them to serve as promising lysosome-specific fluorescent probes in biological imaging.
Introduction
The development of facile polymerization methods for the construction of functional synthetic polymers with unique structures and attractive properties is of great academic and industrial significance. Among various functional polymers, heterocyclic polymers have gained tremendous attention for decades due to their wide applications in diverse fields.1–5 The presence of heterocycles or fused heterocycles in polymer backbones could endow such polymers with excellent thermal and mechanical performance, unique electronic and photophysical properties, high chemical stability, and stimuli-responsive characteristics. To date, a large variety of functional heterocyclic polymers have been developed, such as polyimides, polybenzothiadiazoles, polypyrroles, polyquinolines, polythiophenes, and polybenzimidazoles. As an important class of heterocyclic polymers, polyoxadiazoles (PODs) with conjugated heterocyclic rings have been widely studied in heat-resistant materials, gas separation, fuel cells, acid sensors, polymer electroluminescent devices, electron and proton conducting materials, etc., owing to their excellent film-forming ability and high chemical and thermal stability, as well as the specific properties determined by the oxadiazole ring.6–11 By contrast, the photoluminescence (PL)-related applications such as fluorescence-based visualization and biological imaging have been significantly less explored for oxadiazole-based polymers, which might be due to the poor fluorescence of conjugated PODs in the aggregated state arising from their rigid polymer backbones and strong intermolecular π–π interactions.12 Most application scenarios in real life require the PL materials to be used as solid films or in aqueous media. Therefore, the development of functional PODs with efficient aggregate-state fluorescence and the exploration of their PL-related advanced applications are highly desirable.The creation of polymer materials with new structures and desired properties often relies on expedient synthetic strategies. Traditional synthetic methods toward PODs typically include two types. One is the two-step method, in which a polyhydrazide was first prepared through the polymerization of a dicarboxylic acid chloride with a dihydrazide at low temperature in a polar solvent such as N-methylpyrrolidone (NMP), and then the polyhydrazide underwent the cyclodehydration reaction at high temperature using dehydrating agents to afford PODs (Fig. 1A, method I).10,13 This method can effectively control the structures of POD products, but the synthetic procedures are tedious and the starting materials are limited and relatively high-cost. The other method can directly afford POD derivatives through the polycyclocondensation reactions of dicarboxylic acids and hydrazine sulphate in the presence of poly(phosphoric acid) (PPA) or oleum (Fig. 1A, method II).6,14 However, this one-step method still needs to be conducted under heating conditions and hydrazine sulphate is toxic and difficult to handle. Moreover, these two methods are not very conducive to the flexible design and adjustment of POD structures, and the POD structure and the polyhydrazide block could simultaneously exist in the polymeric products. These synthetic difficulties greatly restrict the structural diversity and property investigation of PODs.
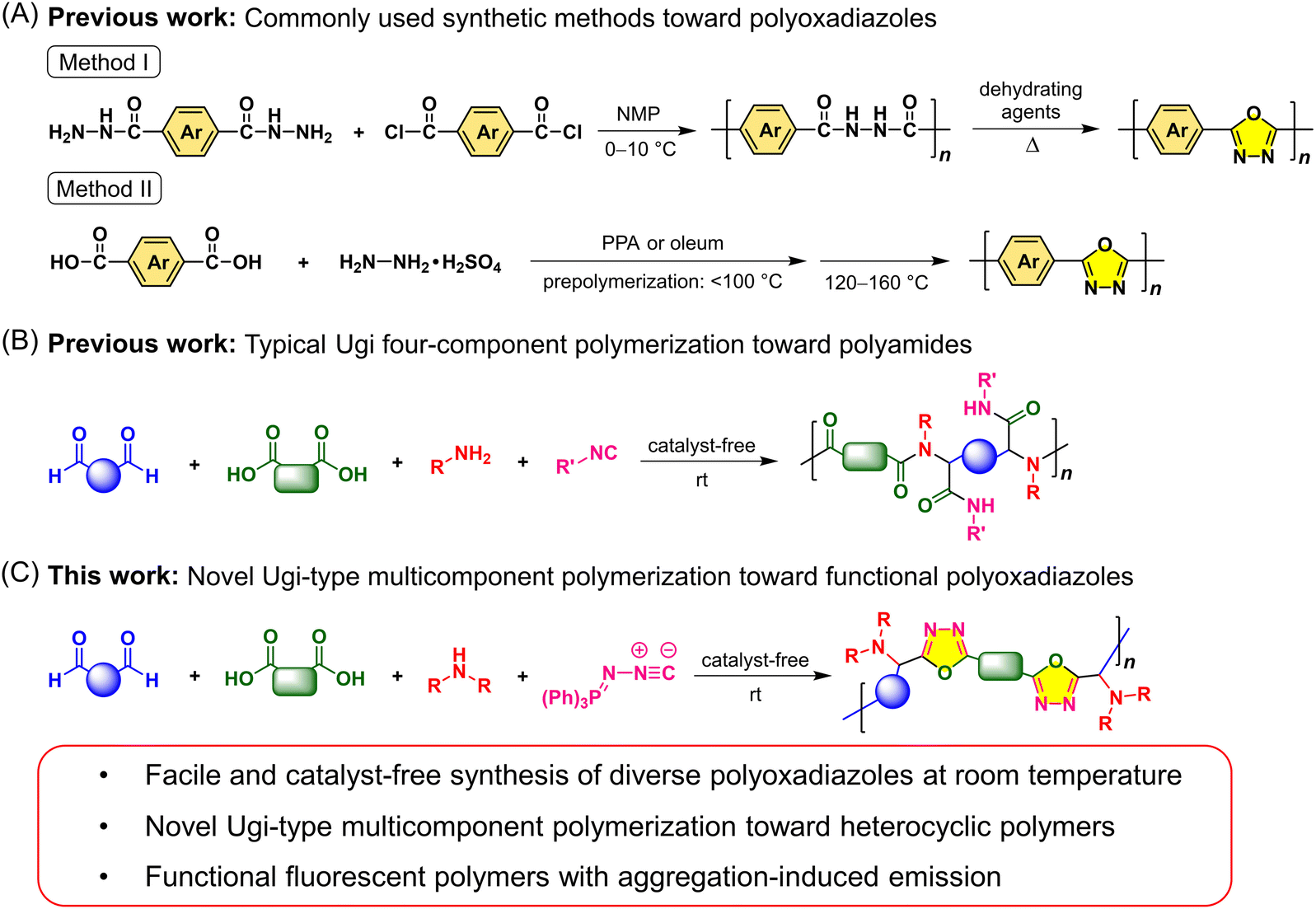 | ||
| Fig. 1 Comparison between (A and B) the previous work on the synthesis of polyoxadiazole and the typical Ugi polymerization and (C) the novel Ugi-type polymerization toward fluorescent polyoxadiazoles developed in this work. | ||
In recent years, multicomponent polymerizations (MCPs) have emerged as powerful tools for the synthesis of structurally well-defined and complex polymers with the merits of simple operation, mild reaction conditions, good functional-group tolerance, and high efficiency. The structural diversity and functionalities of MCP products can be readily tuned by changing the structures or combinations of multiple monomers.15 Benefiting from the advantages of MCPs, various multifunctional polymers that are difficult or troublesome to obtain by traditional methods were constructed.16–23 As one of the well-known MCPs, Ugi polymerization of acids, amines, isocyanides, and aldehydes or ketones has attracted tremendous attention since its first report in 2014 (Fig. 1B).24–26 By rationally designing the monomer structures or combinations, a series of Ugi-type MCPs have been developed for the preparation of polyamides and polypeptoids.27–33 However, the product structures of Ugi-type polycondensations are generally limited to diverse polyamide derivatives with linear structures. Examples of the synthesis of functional heterocyclic polymers with different topological structures by Ugi-type polycondensations are still very rare.
MCPs are generally developed from appropriate multicomponent reactions (MCRs). Recently, a facile and efficient Ugi-type MCR method for the synthesis of oxadiazole derivatives has caught our attention. As shown in Scheme S1,† the catalyst-free MCR of an aromatic aldehyde, carboxylic acid, secondary amine, and (N-isocyanimino)triphenylphosphorane can afford 2,5-disubstituted 1,3,4-oxadiazoles at room temperature by a sequence of the Ugi four-component reaction and an intramolecular aza-Wittig ring closure.34 The introduction of such a unique heterocyclic skeleton into polymer main chains is envisioned to endow the corresponding polymer materials with a variety of interesting properties and advanced functionalities. For instance, the non-conjugated backbone structures with distorted conformations and multiple large substituents might inherently block the formation of strong interchain π–π interactions, which would be conducive to the improvement of solubility and aggregate-state fluorescence of oxadiazole-based polymers. Moreover, the readily available starting materials, mild reaction conditions, and simple operation would make the corresponding polymerization method a feasible tool for the flexible design and functionalization of diverse POD structures, thereby providing new possibilities for the study on the structure–property relationship and applications of functional heterocyclic polymer materials.
With these concerns in mind, herein we successfully develop this MCR into a novel Ugi-type MCP tool to benefit the synthesis of diverse functional POD derivatives (Fig. 1C). Taking advantage of the wide monomer scope and various monomer combinations of the MCP method, a series of fully substituted PODs with well-defined and diversified topological structures are facilely synthesized from commercially available or readily accessible aldehydes, carboxylic acids, secondary amines, and (N-isocyanimino)triphenylphosphorane at room temperature without using any catalyst. Different from the typical Ugi polycondensations, the use of (N-isocyanimino)triphenylphosphorane as the isocyanide input and a secondary amine as the amine input is critical for the present Ugi-type MCP strategy, which allows the imino-anhydride intermediate to undergo an intramolecular aza-Wittig reaction rather than the Mumm rearrangement.35 The obtained PODs possess good solubility, high thermal and morphological stability, and excellent film-forming ability. By appropriately introducing fluorescent moieties with aggregation-induced emission (AIE) characteristics into the polymer structures, the resulting PODs showed efficient aggregate-state fluorescence and can be used in different PL-related advanced applications. The obtained AIE-active linear POD was demonstrated to possess interesting self-assembly properties and its thin films can readily form fluorescent patterns with stable and ordered porous structures on rainy days or based on the simple breath figure (BF) patterning method. These self-assembly morphologies can be directly visualized by fluorescence microscopy in a sensitive, on-site and real-time manner. To the best of our knowledge, this is the first report of the application of PODs for the fabrication of fluorescent microporous thin films using the BF method. Besides, the application potential of the fluorescent nanoparticles (NPs) of PODs in biological imaging was explored, demonstrating that both the linear and hyperbranched AIE-active PODs can serve as promising lysosome-specific fluorescent probes.
Results and discussion
Polymerization
To develop the Ugi-type polymerization route for the facile and efficient synthesis of PODs, we first optimized the polymerization conditions using the commercially available and inexpensive terephthalaldehyde (1a), terephthalic acid (2a), dibenzylamine (3a), and (N-isocyanimino)triphenylphosphorane (4) as the model monomer combination. The polymerizations were all carried out at room temperature in a one-pot manner without using any catalyst or additive. As summarized in Table 1, the effects of various polymerization parameters on the polymerization efficiency were systematically investigated, including solvent, reactant concentration, monomer feed ratio, reaction atmosphere, and time course.|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Solvent | [1a]b (M) | [1a]/2a]/[3a]/[4] | Atmosphere | Yield (%) | M n (MALLS) | M w (MALLS) | Đ |
| a Unless otherwise noted, the polymerizations were carried out at room temperature for 24 h. b Monomer concentration was calculated considering the volume of both the solvent and the amine liquid. c Absolute molecular weights were determined by GPC using a multi-angle laser light scattering (MALLS) detector in THF. Dispersity (Đ) = Mw/Mn. d Unidentified structures exist in the polymeric products. e The reaction proceeded for 36 h. f The reaction proceeded for 48 h. Abbreviation: DCM = dichloromethane; DMSO = dimethyl sulfoxide; DMAc = N,N-dimethylacetamide; THF = tetrahydrofuran. | ||||||||
| 1d | DCM | 0.10 | 1.0/1.0/2.4/2.4 | N2 | 62 | 29![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 300 |
45900 |
1.6 |
| 2 | DMSO | 0.10 | 1.0/1.0/2.4/2.4 | N2 | 74 | 24800 |
37100 |
1.5 |
| 3d | DMF | 0.10 | 1.0/1.0/2.4/2.4 | N2 | 55 | 29400 |
57100 |
1.9 |
| 4d | DMAc | 0.10 | 1.0/1.0/2.4/2.4 | N2 | 52 | 12500 |
20800 |
1.7 |
| 5d | THF | 0.10 | 1.0/1.0/2.4/2.4 | N2 | 57 | 77400 |
140000 |
1.8 |
| 6d | THF/MeOH | 0.10 | 1.0/1.0/2.4/2.4 | N2 | 32 | 27100 |
40700 |
1.5 |
| 7 | DMSO | 0.18 | 1.0/1.0/2.4/2.4 | N2 | 58 | 14900 |
24200 |
1.6 |
| 8 | DMSO | 0.22 | 1.0/1.0/2.4/2.4 | N2 | 59 | 12800 |
20000 |
1.6 |
| 9 | DMSO | 0.34 | 1.0/1.0/2.4/2.4 | N2 | 66 | 9100 | 13400 |
1.5 |
| 10 | DMSO | 0.47 | 1.0/1.0/2.4/2.4 | N2 | 68 | 8500 | 11700 |
1.4 |
| 11 | DMSO | 0.10 | 1.0/1.0/2.0/2.0 | N2 | 64 | 9900 | 14000 |
1.4 |
| 12 | DMSO | 0.10 | 1.0/1.0/3.0/3.0 | N2 | 50 | 7900 | 11800 |
1.5 |
| 13 | DMSO | 0.10 | 1.0/1.0/2.4/2.4 | Air | 55 | 31300 |
72200 |
2.3 |
| 14e | DMSO | 0.10 | 1.0/1.0/2.4/2.4 | N2 | 76 | 25000 |
50100 |
2.0 |
| 15f | DMSO | 0.10 | 1.0/1.0/2.4/2.4 | N2 | 69 | 19200 |
34800 |
1.8 |
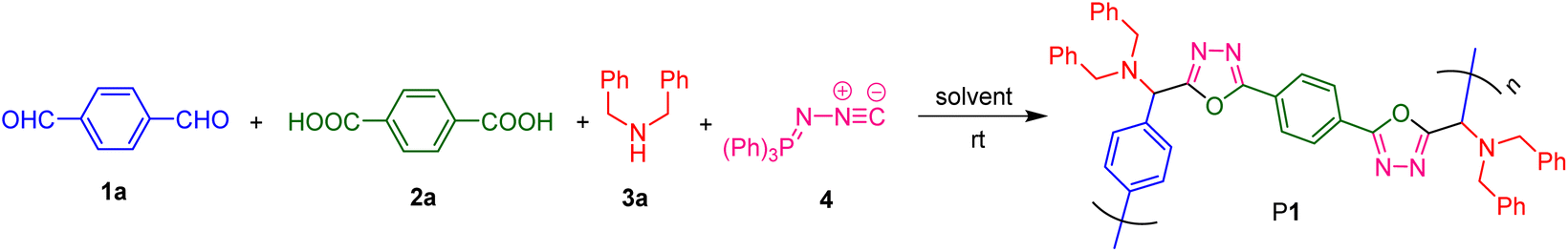
In the first attempt, the polymerization of 1a, 2a, 3a, and 4 was carried out in dichloromethane (DCM) under N2 at a monomer concentration of [1a] = 0.1 M and the monomer molar ratio of [1a]:[2a]:[3a]:[4] was 1.0:1.0:2.4:2.4. Under such conditions, a high molecular weight polymer with an absolute number-average molecular weight (Mn) of 293000 and an absolute weight-average molecular weight (Mw) of 45900 was produced in 62% yield after 24 h (Table 1, entry 1). However, there exist some unidentified structures in the product obtained from the polymerization in DCM. We then tried to carry out the polymerization reaction in DMSO, DMF, DMAC, THF, and the THF/MeOH mixture with a volume ratio of 1:1 (Table 1, entries 2–6). All these solvents can afford polymeric products, but only DMSO solvent can produce the purest polymeric product with the desired structure of the POD derivative (P1). Therefore, DMSO was chosen as the optimal solvent for the following investigation. Next, the effect of monomer concentration on polymerization was examined. As listed in entries 7–10 of Table 1, when the concentration of 1a was gradually increased from 0.10 M to 0.47 M without changing the monomer feed ratio, both the reaction yield and the molecular weights of the product were decreased. Thus, the influence of the monomer feed ratio was then studied at a monomer concentration of [1a] = 0.10 M. The results shown in entries 2, 11 and 12 of Table 1 suggested that neither decreasing nor increasing the monomer ratio can give better polymerization results. We thus chose [1a]:[2a]:[3a]:[4] = 1.0:1.0:2.4:2.4 as the optimal monomer feed ratio. Considering that no air-sensitive monomer or catalyst was involved in this room temperature MCP, the polymerization was next carried out in air to further simplify the experimental procedure. The polymerization result indicated that this MCP is tolerant to air (entry 13, Table 1). Compared with the product obtained under nitrogen (entry 2, Table 1), the polymerization yield was decreased although the molecular weights were obviously increased under air. Finally, the time course of the polymerization was investigated under N2 with the aim of further improving the reaction yield (Table 1, entries 2, 14 and 15). The results suggested that no significant improvement was achieved by prolonging the reaction time. Therefore, 24 h was adopted as the preferred reaction time for the MCP of 1a, 2a, 3a, and 4.
To explore the monomer scope of this catalyst-free MCP and meanwhile to enrich the structural diversity and functionalities of polymeric products, we then performed the polymerizations using various readily accessible dialdehydes, aromatic dicarboxylic acids, and secondary aliphatic diamines as monomers (Fig. 2A). The polymerization results shown in Fig. 2B suggested that this MCP can be applied to a wide variety of monomers. Under the optimized conditions, the MCPs of 1a–d, 2a and b, 3a–c, and 4 generally proceeded smoothly at room temperature under N2, producing a series of linear PODs P1–P6 with high absolute Mws of up to 83800 in moderate yields of up to 74%. Both aromatic and aliphatic dialdehydes were suitable for this polymerization method, and the MCP of tetraphenylethylene (TPE)-containing aromatic dialdehyde (1d), aromatic dicarboxylic acid with alkoxy chain (2b), dibenzylamine (3a), and 4 can afford the TPE-functionalized linear POD with a Mw of 10200 and a narrow dispersity of 1.5. However, when 4,4′-([2,2′-bipyridine]-5,5′-diyl)dibenzoic acid (2c) was adopted to react with monomers 1d, 3a and 4, a polymeric product with poor solubility and an impure structure was obtained, possibly due to the poor solubility and reactivity of 2c for this MCP. The other obtained polymers (P1–P6) all possess good solubility. They can be well dissolved in commonly used organic solvents such as THF, DMSO, DCM, CHCl3, and DMF.
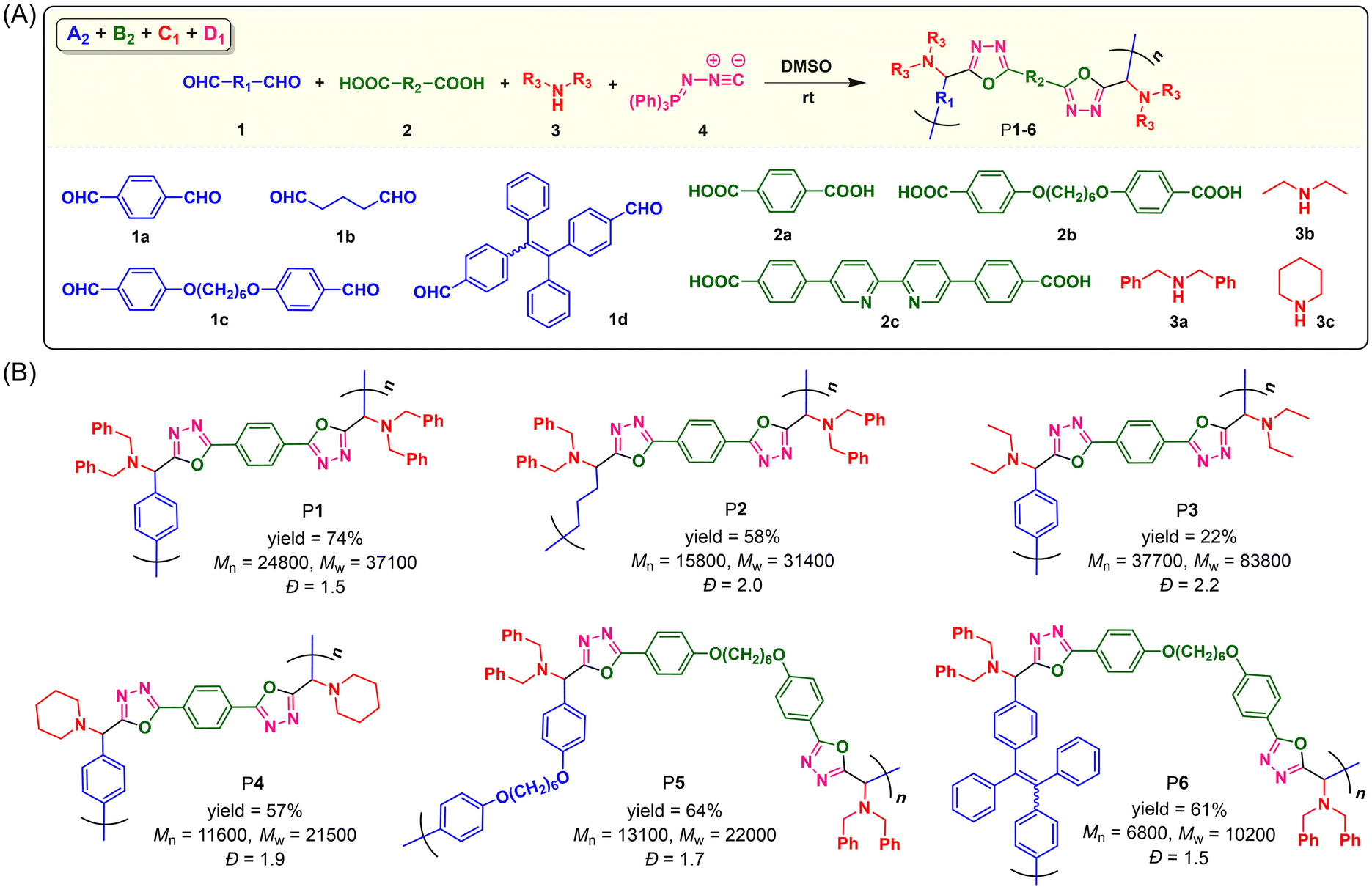 | ||
| Fig. 2 (A) Catalyst-free multicomponent polymerizations of dialdehydes, dicarboxylic acids, secondary amines, and (N-isocyanimino)triphenylphosphorane. (B) Chemical structures, yields, molecular weights, and the dispersity of polymers P1–6. The polymerizations were carried out in DMSO at room temperature under N2 for 24 h, [1] = 0.10 M, [1]/[2]/[3]/[4] = 1/1/2.4/2.4. Absolute molecular weights were determined by GPC using a multi-angle laser light scattering (MALLS) detector in THF. Dispersity (Đ) = Mw/Mn. | ||
In addition to the synthesis of linear PODs based on the monomer combination of A2 + B2 + C1 + D1, we further explored the applicability of this MCP strategy using trifunctional and tetrafunctional monomers for the construction of hyperbranched polymers. As shown in Fig. 3, triphenylamine (TPA)-containing tricarboxylic acid (2d) and TPE-containing tetracarboxylic acid (2e) were synthesized to react with dialdehyde 1a or 1d, dibenzylamine 3a, and 4. Based on the above-mentioned optimized conditions, we first tried the monomer combination strategy of A2 + B3 + C1 + D1 by polymerizing 1a (A2), 2d (B3), 3a (C1), and 4 (D1) in DMSO at room temperature under N2 at a monomer concentration of [1a] = 0.10 M for 24 h. According to the Flory statistics,36,37 the mole ratio of monomers could determine the gel point of such an “A2 + B3”-type polymerization system, and theoretically the problem of gelation could be overcome by using an equal molar ratio of A2 and B3.38 Therefore, the polymerization was carried out with the monomer ratio of [1a]:[2d]:[3a]:[4] = 1.0:1.0:2.4:2.4. However, a partially soluble polymer was obtained in a yield of 50% (Table S1,† entry 1). A similar phenomenon was also observed when the TPE-containing dialdehyde 1d was used to replace 1a (Table S1,† entry 2). By changing the reaction condition to [1a]:[2d]:[3a]:[4] = 1.0:1.0:2.0:2.0 in a higher monomer concentration under air, high molecular weight hb-P1 (Mn = 66400 and Mw = 124100) with good solubility was obtained in 66% yield after reacting for 24 h (Fig. 3A). Decreasing the reaction time from 24 h to 3 h led to a significant drop in molecular weights (Table S1,† entries 3 and 4). Under similar conditions, the polymerization of 1d, 2d, 3a and 4 produced soluble hb-P2 with a Mw of 8100 in 73% yield after 3 h (Table S1,† entry 5). Encouraged by these results, we then investigated the feasibility of the A2 + B4 + C1 + D1 monomer strategy to synthesize another TPE-containing hyperbranched POD (hb-P3) (Fig. 3B). As depicted in entries 6 and 7 of Table S1,† partially soluble polymeric products were obtained from the polymerization of 1a, tetracarboxylic acid 2e, 3a, and 4 under the optimal conditions for linear polymers, while a soluble polymer with a high Mw of 64500 was generated under air after reacting for 3 h. The successful synthesis of hb-P1–hb-P3 demonstrated the general applicability of this catalyst-free MCP for the facile synthesis of hyperbranched polymers with diversified chemical and topological structures.
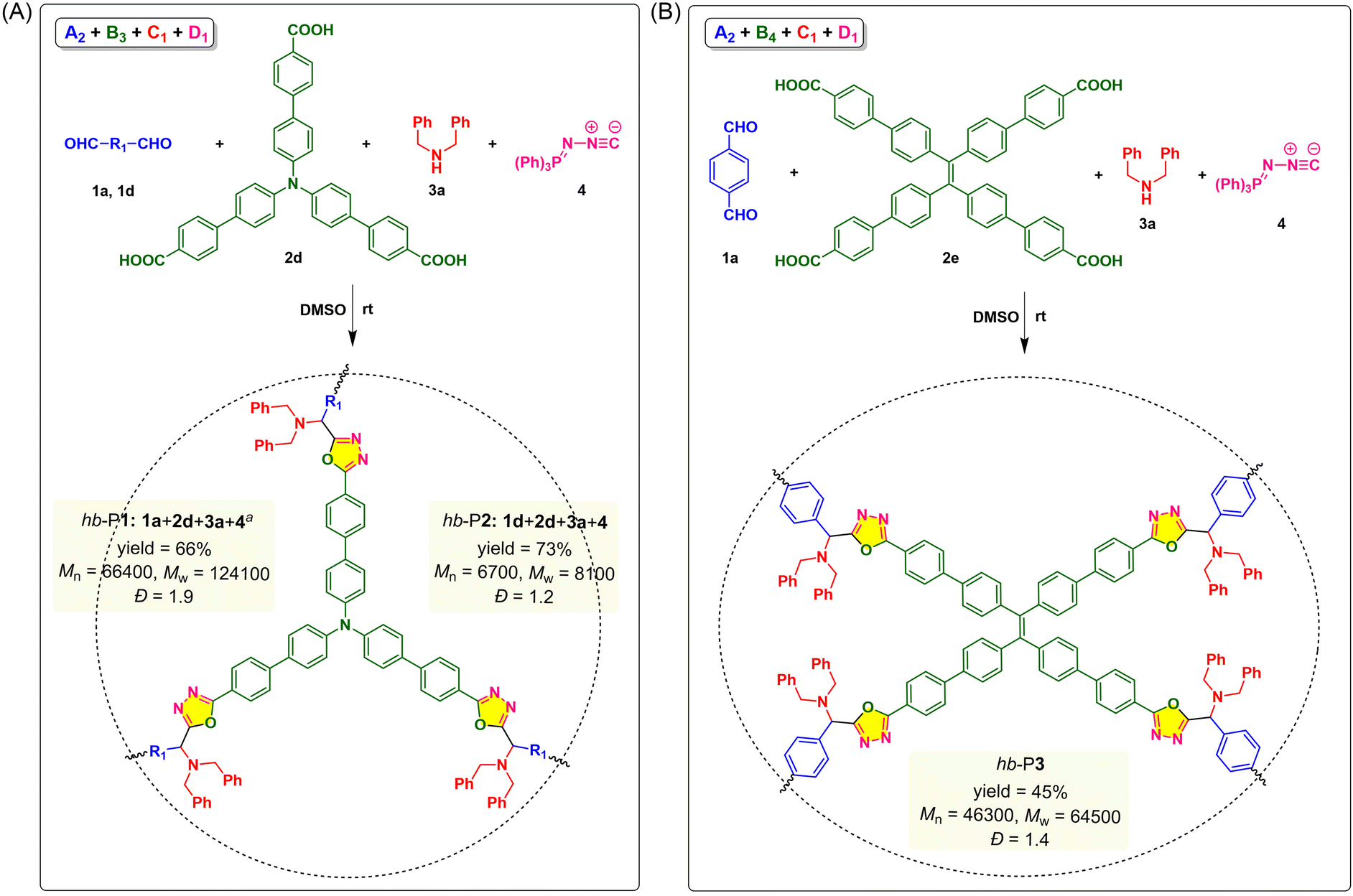 | ||
| Fig. 3 Catalyst-free multicomponent polymerizations of aldehydes, carboxylic acids, secondary amines, and (N-isocyanimino)triphenylphosphorane through (A) A2 + B3 + C1 + D1 and (B) A2 + B4 + C1 + D1 manners. The polymerizations were carried out in DMSO at room temperature in air for 3 h, [1] = 0.18 M, [1]/[2]/[3]/[4] = 1.0/1.0/2.0/2.0. Absolute molecular weights were determined by GPC using a multi-angle laser light scattering (MALLS) detector in THF. Dispersity (Đ) = Mw/Mn. aReaction time = 24 h. | ||
Structural characterization and thermal properties
To assist the structural characterization of the obtained polymers, model compound 5 was synthesized according to the synthetic route shown in Scheme S2.† The structures of model compound 5 and polymers were fully characterized and confirmed by IR, 1H NMR, and 13C NMR analysis. By comparing the characterization results of the polymers with those of 5 and the monomers, the polymeric products were demonstrated to possess the expected oxadiazole-containing structures. Detailed characterization data can be found in the ESI.† The IR, 1H NMR and 13C NMR spectra of P1, model compound 5, and the corresponding monomers (1a, 2a, 3a, 4) were compared and discussed here as an example. As shown in Fig. S1,† the peaks associated with the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations of 1a and 2a at 1685 and 1670 cm−1 and the broad absorption band corresponding to the O–H stretching vibration of COOH in 2a all disappeared in the IR spectra of P1. Meanwhile, the characteristic peaks of the N–H and C
O stretching vibrations of 1a and 2a at 1685 and 1670 cm−1 and the broad absorption band corresponding to the O–H stretching vibration of COOH in 2a all disappeared in the IR spectra of P1. Meanwhile, the characteristic peaks of the N–H and C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N stretching vibrations of 3a and 4 at 3320 and 2070 cm−1, respectively, were also not detected after the polymerization. The IR spectrum of P1 resembled that of model compound 5 to a large extent. These results clearly demonstrated the occurrence of this multicomponent polymerization.
N stretching vibrations of 3a and 4 at 3320 and 2070 cm−1, respectively, were also not detected after the polymerization. The IR spectrum of P1 resembled that of model compound 5 to a large extent. These results clearly demonstrated the occurrence of this multicomponent polymerization.
More structural details could be revealed by the NMR spectra. As depicted in Fig. 4A–F, the 1H NMR spectrum of P1 showed no resonance of the –CHO proton of 1a at δ 10.15. The resonance peaks associated with the –COOH proton of 2a at δ 13.28 and the –NH– proton of 3a at δ 2.59 also completely disappeared after the polymerization. Instead, a new peak emerged at δ 5.33 and δ 5.36 in the spectra of 5 and P1, respectively, which suggested the presence of newly formed methine groups (protons e and e′) in their structures. Regarding the 13C NMR results (Fig. 4G–L), no resonance signal related to the CO groups of 1a and 2a at δ 193.07 or 166.73 was observed in the 13C NMR spectrum of P1. Moreover, the carbon resonance of the isocyanide group in 4 at δ 121.16 disappeared and two adjacent peaks emerged at δ 164.77 and 163.89 for P1 after the polymerization, which was in good accordance with the similar resonance peaks for 5 at δ 164.52 and 164.30 (carbons i and j). These results indicated the complete conversion from CO and isocyanide to CN bonds in the newly formed oxadiazole rings. Besides, a new peak associated with the methane carbon atom (positions f and f′) was observed at 59.18 and 59.10, respectively, in the 13C NMR spectra of 5 and P1. Similarly, the IR, 1H NMR and 13C NMR results (Fig. S2–S7†) of the other polymers, including both the linear polymers (P2–P6) and the hyperbranched polymers (hb-P1–hb-P3) also showed the above-mentioned features. All these results confirmed the successful synthesis of PODs with the expected structures as depicted in Fig. 2 and 3.
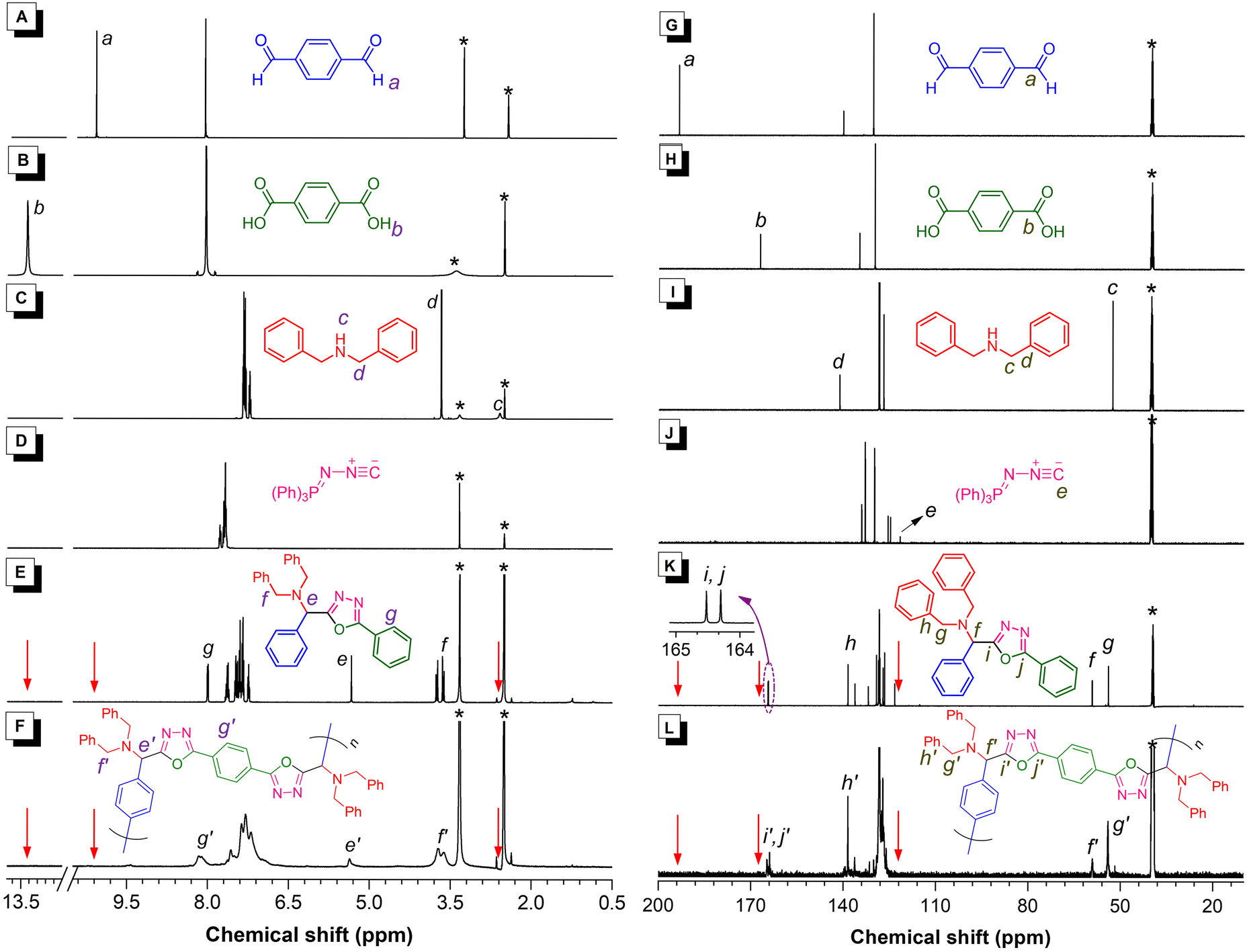 | ||
| Fig. 4 (A–F) 1H NMR spectra of (A) 1a, (B) 2a, (C) 3a, (D) 4, (E) model compound 5, and (F) P1 in DMSO-d6. (G–L) 13C NMR spectra of (G) 1a, (H) 2a, (I) 3a, (J) 4, (K) model compound 5, and (L) P1 in DMSO-d6. The solvent peaks are marked with asterisks. | ||
The thermal stability of the resulting polymers was characterized by thermogravimetric analysis (TGA) under a nitrogen atmosphere at a heating rate of 10 °C min−1. The TGA results depicted in Fig. S8† suggested that all these polymers possess good thermal stability. The decomposition temperature (Td) at 5% weight loss of P1–P6 and hb-P1–hb-P3 located in the range of 237−300 °C and 257−272 °C, respectively, and their char yields reached up to 60 wt% at 600 °C. The glass transition temperatures (Tg) of these polymers were then measured using the differential scanning calorimetry (DSC) analysis. As summarized in Fig. S9,† the linear polymers P1–P6 showed a similar Tg ranging from 96 to 98 °C, whereas the hyperbranched polymers hb-P1–P3 showed much higher Tg values varying from 163 to 199 °C, possibly due to the presence of more complex intra- and inter-chain interactions among the hyperbranched polymer chains.
Photophysical properties
The absorption spectra of the obtained PODs in Fig. S10† suggested that P1–P6 absorbed little light in the visible spectral region due to the weak electronic conjugation in their polymer backbones, and their maximum absorption wavelengths (λabs,max) were ranging from 281 to 330 nm. The λabs,max of hyperbranched polymers (hb-P1–hb-P3) varied from 317 nm to 372 nm, among which hb-P1 and hb-P2 showed much longer absorption wavelengths due to the existence of a typical electron-donating moiety (TPA) in their structures.In the past two decades, luminogens with AIE characteristics have received tremendous attention as functional fluorescent materials due to their advantages of efficient aggregate-state fluorescence and excellent photostability.39,40 As an archetypical AIE luminogen, TPE is non-emissive in dilute solution but becomes highly fluorescent upon aggregation.41 TPA is also a well-known fluorophore that is widely used for the design of functional fluorescent materials.42 The incorporation of TPE and/or a TPA moiety in PODs could endow these polymers with interesting PL behaviors. We thus systematically investigated the PL properties of the TPE- and/or TPA-containing PODs in THF solutions and THF/water mixtures with different water fractions (fw). As depicted in Fig. 5A–C, due to the presence of the TPE moiety in their polymer skeletons, P6 and hb-P3 showed typical AIE features. With the gradual addition of water into their THF solutions, the PL intensity of P6 and hb-P3 was steadily enhanced and reached the maximum at a fw of 80% and 70%, respectively. The maximum PL intensity of P6 aggregates was 9.4-fold higher than that of its pure THF solution, while a 3.8-fold increase in PL intensity was observed for hb-P3 after aggregation. The fluorescence quantum yields (Φf) of P6 and hb-P3 in different states further confirmed their AIE characteristics (Table S2†). The weakly emissive THF solution of P6 with a Φf of 0.6% increased to 4.5% when fw reached 80%, whereas Φf of green-emissive hb-P3 increased from 3.9% (pure THF solution) to 14.1% (fw = 70%). The remarkably higher PL efficiencies of the hyperbranched polymer hb-P3 in THF solution and aggregated states than those of the linear polymer P6 might be ascribed to the inherently stronger restriction of the intramolecular motions of TPE units in the hyperbranched polymer chains as well as the presence of much richer intra-/inter-chain interactions upon aggregation.43,44
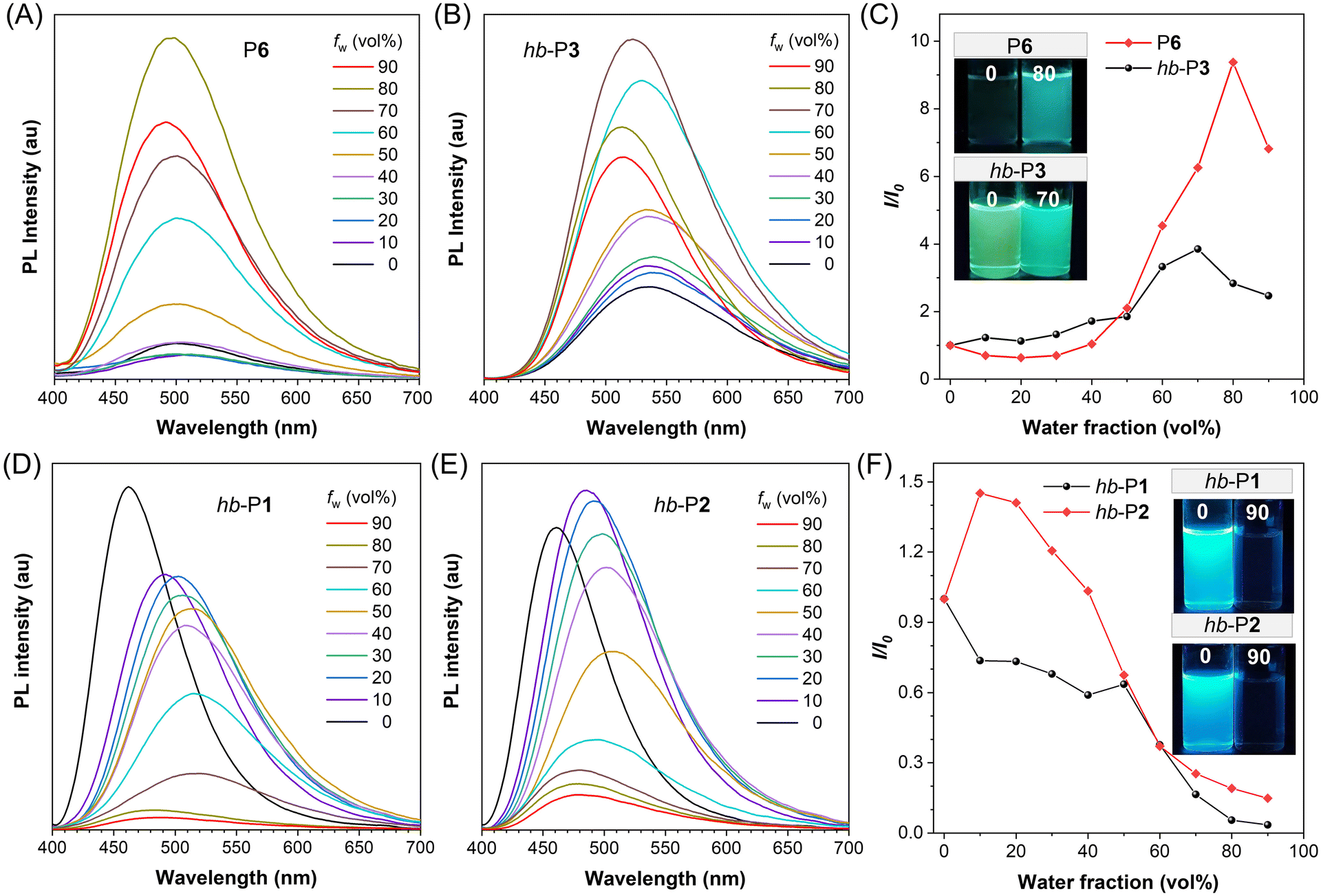 | ||
| Fig. 5 (A, B, D and E) Photoluminescence (PL) spectra of (A) P6, (B) hb-P3, (C) hb-P1, and (D) hb-P2 in THF/water mixtures with different water fractions (fw). Solution concentration: 10 μM. Excitation wavelength = 325 nm for P6; 370 nm for hb-P1; 360 nm for hb-P2; 300 nm for hb-P3. (C and F) Plot of the relative PL intensity (I/I0) versus the composition of the aqueous mixture of P6 and hb-P1–hb-P3. I0 = intensity at fw = 0%. Inset: fluorescence photographs of the THF solutions of polymers P6 and hb-P1–hb-P3 and their aggregates in THF/water mixtures with different fws. All fluorescence photographs were taken under UV irradiation at 365 nm. | ||
In contrast, the TPA-containing polymer hb-P1 showed an aggregation-caused quenching effect with strong blue fluorescence in the solution state (Fig. 5D and F). A similar phenomenon was also observed for hb-P2 (Fig. 5E and F), which indicates that the introduction of TPE is not effective to endow hb-P1 with AIE properties. The UV-vis absorption spectra of hb-P1 and hb-P2 in different states were then compared with the PL spectrum of TPE. As depicted in Fig. S11,† the PL spectrum of TPE overlapped with the absorption spectrum of hb-P2 aggregates to a larger degree than those of hb-P2 solution and hb-P1. We thus speculated that the unexpected weak fluorescence of hb-P2 in the aggregated state might result from the energy transfer from the excited state of the TPE luminogen to the ground state of the TPA-containing conjugated moiety.45 Because the TPE moiety of hb-P2 is linked with the TPA-phenyl-oxadiazole moiety in a non-conjugated manner and meanwhile TPE is non-emissive in the solution state, it is reasonable that the PL properties of hb-P2 solution are similar to those of hb-P1 solution.
Formation and visualization of microporous film morphologies
Attracted by the excellent film-forming capability and potential self-assembly properties of PODs as reported in the literature,46 we then investigated the film-forming and assembly behaviors of the obtained polymers with P3 as an example. As depicted in Fig. S12,† the dilute chloroform solution of P3 at a concentration of 0.01 mg mL−1 could form very thin films after evaporation at room temperature. Interestingly, when the solution concentration was increased to 0.1 mg mL−1, an ordered microporous structure was observed in the thin film under a scanning electron microscope (SEM, Fig. 6A–C). Inspired by this serendipity, we further characterized the film morphology of the AIE-active linear polymer (P6) to achieve fluorescent microporous films. Different from P3, the evaporation of the chloroform solution of P6 with a concentration of 0.1 mg mL−1 generated a fluorescent thin film with a non-porous structure (Fig. S13†). However, as evidenced by the SEM and fluorescence images in Fig. 6D–I, increasing the solution concentration of P6 to 1.0 mg mL−1 can produce fluorescent microporous films, and these porous structures can be readily observed by fluorescence microscopy. Considering that these microporous films can only be obtained on rainy days, we speculated that the formation of such a pattern might also result from a BF process at high humidity (≥80%) in air. | ||
| Fig. 6 (A–C) SEM images of the porous thin films prepared by the solvent evaporation of the chloroform solution of P3 (0.1 mg mL−1) on silicon wafers. (D–F) SEM images and (G–I) fluorescence images of the porous thin films formed by the solvent evaporation of the chloroform solution of P6 (1.0 mg mL−1) on silicon wafers. Excitation wavelength = 400–440 nm. All these films were prepared on rainy days with an air humidity of above 80%. | ||
Polymers that can form stable and ordered microporous films have attracted wide interest in the past few decades due to their potential applications in various fields, such as separation, optoelectronic devices, electrochemical energy systems, cell or bacteria culture, tissue engineering, etc.47–52 The combination of a porous structure and PL performance could further impart materials with potential fluorescence-sensing capabilities. As a commonly used method for the preparation of ordered microporous or honeycomb-structured films, the BF patterning method with water droplets as the sacrificial templates has been demonstrated to be effective for diverse polymeric materials, including amphiphilic copolymers, star polymers, hyperbranched polymers, polymeric polyion materials, and organic–inorganic hybrids.53–55 Compared with other templating and lithography methods, the BF approach shows the advantages of simplicity, low cost, spontaneous occurrence, and large area applicability. However, the BF method has rarely been applied for the fabrication of luminescent porous films using homogeneous polymer systems.56 Motivated by the microporous self-assembly morphology of P6 obtained at high humidity, we next tried to employ the standard static BF process to obtain fluorescent thin films with stable and ordered microporous arrays.
Fig. 7A and S14† show the experimental set-up for the preparation of ordered porous thin films of P6 by the static BF process. First, a silicon wafer was placed and sealed in a vessel saturated with water vapor. Then the solution of P6 was cast onto the silicon wafer through the plastic wrap using a syringe, and the pinhole on the plastic wrap was immediately sealed with transparent tape. Finally, the sealed vessel was placed at room temperature for 30 min to allow the spontaneous interfacial self-assembly of P6 solution in a humid environment. As depicted in Fig. 7B–D, depending on the way in which the humid environment was established, fluorescent honeycomb-patterned films with an adjustable pore size and uniformity could be obtained. In an initial attempt, the experimental device was sealed at room temperature for 2 h to make the water vapor saturated before casting the chloroform solution (1.0 mg mL−1) of P6 on the substrate (method I). This method can produce an efficiently fluorescent thin film with a microporous structure, but the shape and size uniformity of these pores needed to be further improved (Fig. 7B). Then we tried to first seal the whole device at room temperature for 30 min and then heat the bottom of the vessel at around 70 °C for 2 min to accelerate the saturation of water vapor (method II). Under this condition, a fluorescent microporous film with more ordered morphology and a uniform pore size (diameter = 10.2 ± 1.5 μm; sample size = 200) was produced (Fig. 7C). On the other hand, when the experimental device was only heated at around 70 °C for 2 min before adding the polymer solution (method III), the obtained porous film possessed obviously poorer porosity and a lower film-pore fluorescence contrast (Fig. 7D). Therefore, method II was chosen to be the optimal approach to create a humid environment for the BF patterning of P6 films.
 | ||
| Fig. 7 (A) Schematic illustration of the experimental set-up and the formation mechanism for the preparation of porous films of P6 by the static breath figure process. (B–D) Fluorescence images of the film morphology formed by the chloroform solution of P6 (1.0 mg mL−1) using different methods of establishing a humid environment. (E–G) Fluorescence images showing the influence of organic solvents on the film morphology of P6. Solution concentration: 1.0 mg mL−1. (H–J) Fluorescence images showing the influence of the concentration of the chloroform solution of P6 on the film morphology. Excitation wavelength = 400–440 nm. | ||
As illustrated in Fig. 7A, the formation of porous P6 films through the BF process was supposed to include several steps: (1) evaporation of organic solvent to reduce the air/solution interface temperature below the dew point; (2) condensation of water vapor on the cold solution surface to form water droplets together with the self-organization of water droplets into an ordered droplet array as a patterning template; (3) complete evaporation of the solvent and water droplets to produce the porous polymer film on the substrate. The brighter peripheries around the non-emissive pores suggested that the polymer solute tends to accumulate or precipitate in the interface between polymer solution and water droplets during the solvent evaporation. Taking advantage of the efficient solid-state fluorescence of P6, the effects of other preparation conditions on the formation of BF morphology for this novel POD system were further investigated by directly visualizing the morphology using a fluorescence microscope. The results shown in Fig. 7B–G suggested that uniform microporous structures could only be obtained from the chloroform solution of P6. Other commonly used solvents in the BF method, including toluene, THF, and DCM, all showed negative effects for P6 to form porous films possibly due to the relatively poor solubility of P6 in toluene, the good water miscibility of THF, and the inappropriate solvent volatility of DCM for this polymer system. The screening results on the concentration of the chloroform solution of P6 suggested that the best BF morphology was obtained at 1.0 mg mL−1 (Fig. 7B). Decreasing the solution concentration to 0.5 mg mL−1 led to an irregular and disordered porous structure (Fig. 7H), while increasing the concentration to 2.0 and 5.0 mg mL−1 significantly reduced the porosity and the pore size (Fig. 7I and J). Such results are consistent with the proposed mechanism for the formation of porous polymer films. The polymer concentration could affect the viscosity and density of the casting solution, thereby influencing the condensation process of water droplets on the solution surface.53,54
The BF structures of P6 films were also characterized by SEM (Fig. S15†), which further confirmed the conclusion drawn from the fluorescence images. Compared with traditional characterization methods such as SEM and TEM for the study of BF formation, the fluorescence imaging technique is much simpler, more sensitive and informative, and less expensive. The obtained BF morphology can be directly observed under a fluorescence microscope without tedious sample treatment procedures. In addition to linear polymers, we also tried to fabricate BF patterns using the hyperbranched AIE polymer (hb-P3). However, hb-P3 cannot generate regular porous films under the above-mentioned fabrication conditions, indicating that the topological polymer structures might greatly influence the formation capability of microporous BF structures.
Cell imaging
Considering the catalyst-free characteristic of this MCP and the advantages of AIE polymers as fluorescence imaging reagents,57 we then explored the application of the obtained AIE-active PODs in cell imaging. The cell staining capability of P6 was first evaluated using a confocal laser scanning microscope (CLSM). As shown in Fig. S16,† bright blue fluorescence was observed in 4T1 cells after incubation with P6, indicating its good cell staining ability. Closer inspection suggested that P6 might specifically stain the lysosomes of 4T1 cells, which was further verified by the co-staining result of P6 with a commercial lysosome imaging probe (LysoTracker Red, LTR; Fig. S17†). However, the cytotoxicity of P6 needed to be further improved as suggested by the 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyltetrazolium bromide (MTT) cell-viability assay result (Fig. S18†). To solve this issue, P6 was prepared into NPs through a nanoprecipitation method with a biocompatible amphiphilic copolymer DSPE-PEG2000 as the encapsulation matrix. The obtained P6 NPs indeed showed much better biocompatibility toward 4T1 cells (Fig. 8A). Similarly, the biocompatibility of the AIE-active hyperbranched polymer hb-P3 NPs was also found to be much better than that of its DMSO stock solution (Fig. S19† and 8A). No significant change was observed in the cell viability after incubation with P6 NPs or hb-P3 NPs at a high concentration of up to 50 μg mL−1. The cell viability can remain about 94% and 85%, respectively, for P6 NPs and hb-P3 NPs at a concentration of 100 μg mL−1. The cell imaging experiments were subsequently conducted by costaining 4T1 cells with LTR and polymer NPs. As depicted in Fig. 8C–J, the staining regions of P6 NPs and hb-P3 NPs overlapped with those of LTR to a large extent. The Pearson correlation coefficient of P6 NPs and hb-P3 NPs for 4T1 cells was calculated to be 0.79 and 0.83, respectively. These results indicated that these polymer NPs showed good specificity for lysosomes. Furthermore, both P6 NPs and hb-P3 NPs showed excellent photostability. There was no remarkable signal loss after continuous irradiation at 405 nm excitation for 60 scans (Fig. 8B and S20†). The excellent biocompatibility, good lysosome specificity, and excellent photobleaching resistance enable the obtained AIE-active PODs to function as promising lysosome-specific fluorescent probes in biological imaging.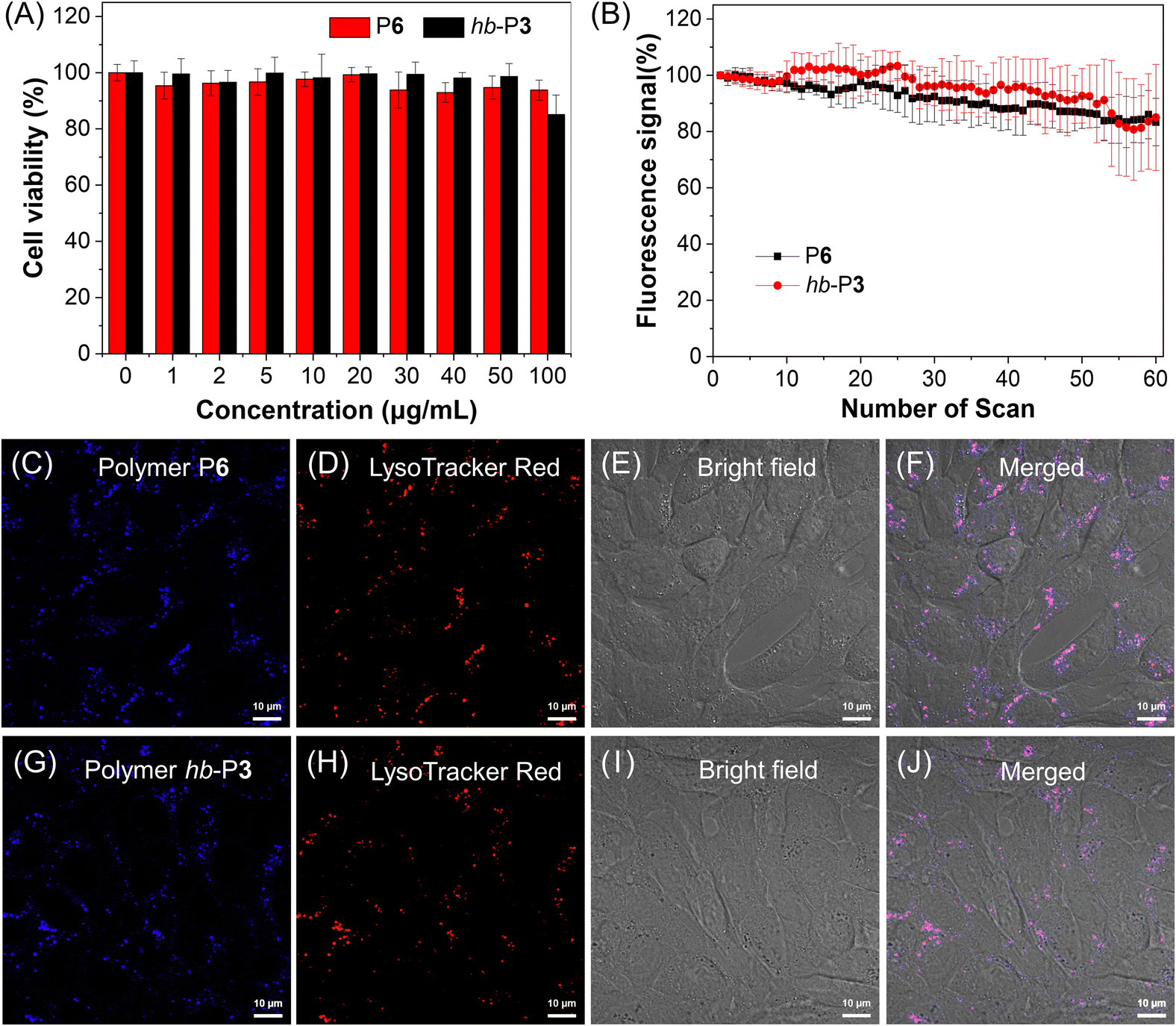 | ||
| Fig. 8 (A) Cell viability of 4T1 cells in the presence of P6 and hb-P3 nanoparticles (NPs) at different concentrations. (B) Change in the fluorescence signal from 4T1 cells stained with P6 and hb-P3 NPs (20 μg mL−1, 4 h) with the number of scans. (C–F) Confocal images of 4T1 cells stained with (C) P6 NPs (20 μg mL−1, 4 h) and (D) LysoTracker Red (LTR, 1 μM, 30 min) and (E) the bright-field image as well as (F) their merged image. (G–J) Confocal images of 4T1 cells stained with (G) hb-P3 NPs (20 μg mL−1, 4 h) and (H) LysoTracker Red (LTR, 1 μM, 30 min) and (I) the bright-field image as well as (J) their merged image. Scale bar = 10 μm. Excitation wavelength: 405 nm for polymers and 561 nm for LTR; emission filter: 420–650 nm for polymers and 650–700 nm for LTR. | ||
Conclusions
In summary, we have developed a novel Ugi-type MCP tool for the facile synthesis of functional PODs with diverse topological structures and attractive properties. Unlike conventional Ugi polycondensations, the present MCPs can in situ generate oxadiazole moieties in polymer backbones because of the remarkably different reaction mechanism arising from the use of (N-isocyanimino)triphenylphosphorane and secondary amines as the monomers. Compared with traditional synthetic methods toward PODs, this catalyst-free MCP strategy enjoys the advantages of readily available monomers, simple operation, mild reaction conditions, and well-defined structures. The structures of the resulting PODs can be flexibly adjusted by adopting different monomer combinations, including A2 + B2 + C1 + D1, A2 + B3 + C1 + D1, and A2 + B4 + C1 + D1. Through these MCPs, a series of linear and hyperbranched PODs with high molecular weights and good solubility in common organic solvents were readily prepared at room temperature. The produced POD structures are hard to achieve by other polymerization methods. All the obtained polymers possess good thermal and morphological stability and excellent film-forming ability. The inherent structural features of these non-conjugated PODs and the appropriate introduction of AIE moieties into polymer backbones endow the resulting polymers with efficient aggregate-state fluorescence. Taking advantage of the good film-forming ability, self-assembly properties, and bright solid-state fluorescence of the AIE-active linear PODs, fluorescent thin films with stable and ordered microporous structures were prepared based on the simple BF patterning method. The influence factors for PODs to form ordered microporous films were systematically investigated and the BF morphologies can be directly visualized by fluorescence microscopy in a simple, sensitive, on-site and real-time manner, which could facilitate further study on the applications of such fluorescent porous thin films in different fields. In addition to the direct visualization of self-assembly morphologies, the efficient aggregate-state fluorescence of the obtained AIE polymers also enables them to find applications in biological imaging. The NPs of both the linear AIE-active PODs and the hyperbranched ones can function as promising lysosome-specific fluorescent probes with excellent biocompatibility, good lysosome specificity, and excellent photobleaching resistance. The new and facile synthetic strategy, diverse polymer structures, systematic study of the structure–property relationship, and fascinating functionalities of the obtained polymers in this work will provide informative insights for researchers from different areas to further design, synthesize, and explore the advanced applications of diversified functional heterocyclic polymer materials.Data availability
All of the experimental data associated with this study are available in the ESI.†Author contributions
J. X. performed the synthesis, structural characterization, thermal and photophysical measurements, and fabrication of the microporous films. N. N performed the cell imaging studies. X. F. performed the measurement of absolute molecular weights. X. S., D. W., and A. Q. participated in the experimental design, data analysis and discussion. T. H. and B. Z. T. conceived and designed the experiments and supervised the overall project. J. X., T. H., and B. Z. T. wrote and revised the paper with the contribution of all other authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant no. 21905176 and 22271197), the Natural Science Foundation for Distinguished Young Scholars of Guangdong Province (grant no. 2020B1515020011), and the Science and Technology Plan of Shenzhen (grant no. JCYJ20190808142403590, JCYJ20220531102601003, and JCYJ20190808153415062). The authors also acknowledge the Instrumental Analysis Center of Shenzhen University.Notes and references
- J. Li, J. Wang, Z. Wu, S. Tao and D. Jiang, Angew. Chem., Int. Ed., 2021, 60, 12918–12923 CrossRef CAS PubMed.
- F. Vidal and F. Jäkle, Angew. Chem., Int. Ed., 2019, 58, 5846–5870 CrossRef CAS PubMed.
- M. Zhang, S. Dai, S. Chandrabose, K. Chen, K. Liu, M. Qin, X. Lu, J. M. Hodgkiss, H. Zhou and X. Zhan, J. Am. Chem. Soc., 2018, 140, 14938–14944 CrossRef CAS PubMed.
- T. Wang, N. Zhang, W. Bai and Y. Bao, Polym. Chem., 2020, 11, 3095–3114 RSC.
- L. Lu, W. Chen, T. Xu and L. Yu, Nat. Commun., 2015, 6, 7327 CrossRef CAS PubMed.
- H. Maab and S. Pereira Nunes, J. Membr. Sci., 2013, 445, 127–134 CrossRef CAS.
- J. Xu, N. M. Srivatsa Bettahalli, S. Chisca, M. K. Khalid, N. Ghaffour, R. Vilagines and S. P. Nunes, Desalination, 2018, 432, 32–39 CrossRef CAS.
- Z. Yuan, X. Li, Y. Duan, Y. Zhao and H. Zhang, J. Membr. Sci., 2015, 488, 194–202 CrossRef CAS.
- Y. Yu, H. Gao, J. Zhu, D. Li, F. Wang, C. Jiang, T. Zhong, S. Liang and M. Jiang, Chin. Chem. Lett., 2021, 32, 203–209 CrossRef CAS.
- G.-S. Liou, S.-H. Hsiao, W.-C. Chen and H.-J. Yen, Macromolecules, 2006, 39, 6036–6045 CrossRef CAS.
- M. Li, V. Karanikola, X. Zhang, L. Wang and M. Elimelech, Environ. Sci. Technol. Lett., 2018, 5, 266–271 CrossRef CAS.
- B. S. Gaylord, S. Wang, A. J. Heeger and G. C. Bazan, J. Am. Chem. Soc., 2001, 123, 6417–6418 CrossRef CAS PubMed.
- D. Gomes, S. P. Nunes, J. Carlos Pinto and C. Borges, Polymer, 2003, 44, 3633–3639 CrossRef CAS.
- D. Gomes, J. Roeder, M. L. Ponce and S. P. Nunes, J. Power Sources, 2008, 175, 49–59 CrossRef CAS.
- H. Xue, Y. Zhao, H. Wu, Z. Wang, B. Yang, Y. Wei, Z. Wang and L. Tao, J. Am. Chem. Soc., 2016, 138, 8690–8693 CrossRef CAS PubMed.
- Z. Zhang, Y.-Z. You, D.-C. Wu and C.-Y. Hong, Macromolecules, 2015, 48, 3414–3421 CrossRef CAS.
- L. V. Kayser, M. Vollmer, M. Welnhofer, H. Krikcziokat, K. Meerholz and B. A. Arndtsen, J. Am. Chem. Soc., 2016, 138, 10516–10521 CrossRef CAS PubMed.
- X. Wu, H. Lin, F. Dai, R. Hu and B. Z. Tang, CCS Chem., 2020, 2, 191–202 CrossRef CAS.
- Y. Hu, N. Yan, X. Liu, L. Pei, C. Ye, W.-X. Wang, J. W. Y. Lam and B. Z. Tang, CCS Chem., 2022, 4, 2308–2320 CrossRef CAS.
- T. Han, H. Deng, Z. Qiu, Z. Zhao, H. Zhang, H. Zou, N. L. C. Leung, G. Shan, M. R. J. Elsegood, J. W. Y. Lam and B. Z. Tang, J. Am. Chem. Soc., 2018, 140, 5588–5598 CrossRef CAS PubMed.
- H. Kim, K. T. Bang, I. Choi, J. K. Lee and T. L. Choi, J. Am. Chem. Soc., 2016, 138, 8612–8622 CrossRef CAS PubMed.
- J. Peng, N. Zheng, P. Shen, Z. Zhao, R. Hu and B. Z. Tang, Chem, 2022, 8, 2301–2316 CAS.
- P. Liu, W. Fu, P. Verwilst, M. Won, J. Shin, Z. Cai, B. Tong, J. Shi, Y. Dong and J. S. Kim, Angew. Chem., Int. Ed., 2020, 59, 8435–8439 CrossRef CAS PubMed.
- A. Sehlinger, P.-K. Dannecker, O. Kreye and M. A. R. Meier, Macromolecules, 2014, 47, 2774–2783 CrossRef CAS.
- P. Stiernet and A. Debuigne, Polym. Sci., 2022, 128, 101528 CAS.
- B. Yang, Y. Zhao, Y. Wei, C. Fu and L. Tao, Polym. Chem., 2015, 6, 8233–8239 RSC.
- N. Gangloff, D. Nahm, L. Döring, D. Kuckling and R. J. Luxenhofer, J. Polym. Sci., A: Polym. Chem., 2015, 53, 1680–1686 CrossRef CAS.
- X. Zhang, S. Wang, J. Liu, Z. Xie, S. Luan, C. Xiao, Y. Tao and X. Wang, ACS Macro Lett., 2016, 5, 1049–1054 CrossRef CAS PubMed.
- Y. Tao, J. Chen, S. Wang and Y. Tao, Acta Polym. Sin., 2020, 51, 738–743 CAS.
- A. Al Samad, J. De Winter, P. Gerbaux, C. Jérôme and A. Debuigne, Chem. Commun., 2017, 53, 12240–12243 RSC.
- M. Hartweg and C. R. Becer, Green Chem., 2016, 18, 3272–3277 RSC.
- A. Sehlinger, R. Schneider and M. A. Meier, Macromol. Rapid Commun., 2014, 35, 1866–1871 CrossRef CAS PubMed.
- J. Zheng, C. Fu, Z. Chen, P. Zhang, R. Zhao, L. Ding, H. Liu and K. Deng, Polym. Adv. Technol., 2022, 33, 1978–1988 CrossRef CAS.
- A. Ramazani and A. Rezaei, Org. Lett., 2010, 12, 2852–2855 CrossRef CAS PubMed.
- H. G. O. Alvim, E. N. da Silva Júnior and B. A. D. Neto, RSC Adv., 2014, 4, 54282–54299 RSC.
- P. J. Flory, Principles of Polymer Chemistry, Cornell University Press, Ithaca, NY, 1953 Search PubMed.
- S. Unal and T. E. Long, Macromolecules, 2006, 39, 2788–2793 CrossRef CAS.
- B. Yao, T. Hu, H. Zhang, J. Li, J. Z. Sun, A. Qin and B. Z. Tang, Macromolecules, 2015, 48, 7782–7791 CrossRef CAS.
- T. Han, D. Yan, Q. Wu, N. Song, H. Zhang and D. Wang, Chin. J. Chem., 2021, 39, 677–689 CrossRef CAS.
- J. Yang, M. Fang and Z. Li, Aggregate, 2020, 1, 6–18 CrossRef.
- Z. Zhao, J. W. Y. Lam and B. Z. Tang, J. Mater. Chem. A, 2012, 22, 23726–23740 RSC.
- H.-J. Yen and G.-S. Liou, Prog. Polym. Sci., 2019, 89, 250–287 CrossRef CAS.
- J. Guan, C. Shen, J. Peng and J. Zheng, J. Phys. Chem. Lett., 2021, 12, 4218–4226 CrossRef CAS PubMed.
- Q. Li and Z. Li, Acc. Chem. Res., 2020, 53, 962–973 CrossRef CAS PubMed.
- H. Deng, Z. He, J. W. Y. Lam and B. Z. Tang, Polym. Chem., 2015, 6, 8297–8305 RSC.
- J. Zhu, Z. Dong, S. Lei, L. Cao, B. Yang, W. Li, Y. Zhang, J. Liu and J. Shen, Angew. Chem., Int. Ed., 2015, 54, 3097–3101 CrossRef CAS PubMed.
- J. Liang, Z. Song, S. Wang, X. Zhao, Y. Tong, H. Ren, S. Guo and Q. T. Y. Liu, ACS Appl. Mater. Interfaces, 2020, 12, 52992–53002 CrossRef CAS PubMed.
- J. Li and Z. He, Environ. Sci. Water Res. Technol., 2015, 1, 355–362 RSC.
- Y. Tao, H. Liu, H.-Y. Kong, T.-X. Wang, H. Sun, Y. J. Li, X. Ding, L. Sun and B.-H. Han, Angew. Chem., Int. Ed., 2022, 61, e202205796 CAS.
- J. R. Varcoe, P. Atanassov, D. R. Dekel, A. M. Herring, M. A. Hickner, P. A. Kohl, A. R. Kucernak, W. E. Mustain, K. Nijmeijer, K. Scott, T. Xu and L. Zhuang, Energy Environ. Sci., 2014, 7, 3135–3191 RSC.
- L. Chen, C. Yan and Z. Zheng, Mater. Today, 2018, 21, 38–59 CrossRef CAS.
- T. Kuang, S. Chen, Z. Gu, Z. Shen, A. Hejna, M. Saeb, F. Chen, M. Zhong and T. Liu, Hybrid Mater., 2022, 5, 1376–1384 CAS.
- Y. Dou, M. Jin, G. Zhou and L. Shui, Membranes, 2015, 5, 399–424 CrossRef CAS PubMed.
- H. Bai, C. Du, A. Zhang and L. Li, Angew. Chem., Int. Ed., 2013, 52, 12240–12255 CrossRef CAS PubMed.
- R. Hu, J. W. Y. Lam, M. Li, H. Deng, J. Li and B. Z. Tang, J. Polym. Sci., A: Polym. Chem., 2013, 51, 4752–4764 CrossRef CAS.
- J. Li, Y. Li, C. Y. K. Chan, R. T. K. Kwok, H. Li, P. Zrazhevskiy, X. Gao, J. Z. Sun, A. Qin and B. Z. Tang, Angew. Chem., Int. Ed., 2014, 53, 13518–13522 CrossRef CAS PubMed.
- P. Xiao, K. Ma, M. Kang, L. Huang, Q. Wu, N. Song, J. Ge, D. Li, J. Dong, L. Wang, D. Wang and B. Z. Tang, Chem. Sci., 2021, 12, 13949–13957 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc05960f |
| This journal is © The Royal Society of Chemistry 2023 |